

Abstract
Key points
Hyperexcitability and hypersynchrony of neuronal networks are thought to be linked to the generation of epileptic activity in both humans and animal models.
Here we show that human epileptic postoperative neocortical tissue is able to generate two different types of synchronies in vitro.
Epileptiform bursts occurred only in slices derived from epileptic patients and were hypersynchronous events characterized by high levels of excitability.
Spontaneous population activity emerged in both epileptic and non‐epileptic tissue, with a significantly lower degree of excitability and synchrony, and could not be linked to epilepsy.
These results help us to understand better the role of excitatory and inhibitory neuronal circuits in the generation of population events, and to define the subtle border between physiological and pathological synchronies.
Abstract
Interictal activity is a hallmark of epilepsy diagnostics and is linked to neuronal hypersynchrony. Little is known about perturbations in human epileptic neocortical microcircuits, and their role in generating pathological synchronies. To explore hyperexcitability of the human epileptic network, and its contribution to convulsive activity, we investigated an in vitro model of synchronous burst activity spontaneously occurring in postoperative tissue slices derived from patients with or without preoperative clinical and electrographic manifestations of epileptic activity. Human neocortical slices generated two types of synchronies. Interictal‐like discharges (classified as epileptiform events) emerged only in epileptic samples, and were hypersynchronous bursts characterized by considerably elevated levels of excitation. Synchronous population activity was initiated in both epileptic and non‐epileptic tissue, with a significantly lower degree of excitability and synchrony, and could not be linked to epilepsy. However, in pharmacoresistant epileptic tissue, a higher percentage of slices exhibited population activity, with higher local field potential gradient amplitudes. More intracellularly recorded neurons received depolarizing synaptic potentials, discharging more reliably during the events. Light and electron microscopic examinations showed slightly lower neuron densities and higher densities of excitatory synapses in the human epileptic neocortex. Our data suggest that human neocortical microcircuits retain their functionality and plasticity in vitro, and can generate two significantly different synchronies. We propose that population bursts might not be pathological events while interictal‐like discharges may reflect the epileptogenicity of the human cortex. Our results show that hyperexcitability characterizes the human epileptic neocortical network, and that it is closely related to the emergence of synchronies.
Keywords: Epilepsy, oscillation, neuronal circuits, neocortex, interictal discharges
Key points
Hyperexcitability and hypersynchrony of neuronal networks are thought to be linked to the generation of epileptic activity in both humans and animal models.
Here we show that human epileptic postoperative neocortical tissue is able to generate two different types of synchronies in vitro.
Epileptiform bursts occurred only in slices derived from epileptic patients and were hypersynchronous events characterized by high levels of excitability.
Spontaneous population activity emerged in both epileptic and non‐epileptic tissue, with a significantly lower degree of excitability and synchrony, and could not be linked to epilepsy.
These results help us to understand better the role of excitatory and inhibitory neuronal circuits in the generation of population events, and to define the subtle border between physiological and pathological synchronies.
Introduction
Epilepsies are characterized by the presence of interictal spikes detected on scalp electroencephalographic (EEG) recordings, which are considered to reflect hypersynchronous and widespread activation of neuronal circuits. Interictal discharges in both human and experimental focal epilepsies were described as high amplitude, fast EEG spikes followed by a slow wave (de Curtis & Avanzini, 2001). In vivo human experiments showed that neocortical interictal spikes were either generated locally or propagated from distant sites, both with an initial current sink and a considerable increase in cellular firing (Ulbert et al. 2004a).
Substantial effort has been made to reveal whether the resected human cortical tissue retains its ability to generate convulsive activity if maintained in vitro (for review see Avoli et al. 2005). Spontaneous synchronous discharges are known to be generated by slices prepared from the human epileptic neocortex (McCormick, 1989; Köhling et al. 1998; Gorji et al. 2002; Roopun et al. 2010; Pallud et al. 2014) or the hippocampal formation (Cohen et al. 2002; Wozny et al. 2005; Huberfeld et al. 2007, 2011; Wittner et al. 2009) in a physiological perfusion solution. When filtered as the EEG (1–100 Hz), the waveform of in vitro events (Cohen et al. 2002) resembled in vivo interictal discharges (Ulbert et al. 2004a; Fabó et al. 2008). Other similarities include the increase of cellular activity and fast oscillations seen in wide band (1–3000 Hz) filtering (Wittner et al. 2009; Simon et al. 2014). Intracellularly, synchronous population events were reflected as large, complex postsynaptic potentials with or without synchronous cell firing (Schwartzkroin & Knowles, 1984; McCormick, 1989; Wittner et al. 2009; Pallud et al. 2014), involving both excitatory and inhibitory signalling (Schwartzkroin & Haglund, 1986; Köhling et al. 1998; Cohen et al. 2002). These postsynaptic potentials were found more frequently in neurons deriving from the epileptogenic zone than in those from adjacent neocortical tissue (Schwartzkroin & Knowles, 1984). Since synchronous bursts could not be detected in non‐primate neocortical slices (Köhling et al. 1998), and healthy human control is lacking for ethical reasons, most groups believed that they might be epilepsy‐related phenomena (Cohen et al. 2002; Huberfeld et al. 2007, 2011; Roopun et al. 2010; Pallud et al. 2014). However, similar synchronous events were generated by healthy monkey hippocampal tissue, as detected in intracellular records (Schwartzkroin & Haglund, 1986), and could also be evoked in human non‐epileptic neocortical tissue by activating single pyramidal cells (Molnár et al. 2008; Szegedi et al. 2016), suggesting that synchronous population activity is not necessarily related to epileptic processes.
Impaired balance of excitation and inhibition is the most conventional theory underlying interictal spike generation. Most of our knowledge about the cellular and network basis of cortical hyperexcitability comes from animal models (for review see McCormick & Contreras, 2001). Human focal cortical epilepsy is extensively studied in the limbic structures, and a wide range of data are available concerning the cellular properties of neocortical neurons (for review see Avoli et al. 2005). Prolonged depolarizations, all‐or‐none and graded bursts could be evoked by electrical stimulation in a subset of human neocortical neurons located in the epileptogenic area verified either by electrocorticography (Prince & Wong, 1981; Avoli & Olivier, 1989), or by imaging techniques (Strowbridge et al. 1992; Williamson et al. 2003). Signs of network hyperexcitability were found in the 4‐aminopyridine model of ictal activity generated by human slices from patients with focal cortical dysplasia (for review see Avoli et al. 2005). Anatomical findings also support the presence of excess excitation in the epileptic neocortex. Higher numbers of excitatory axon terminals and a loss of inhibitory synapses were found in the neocortex of patients with temporal lobe epilepsy (Marco & DeFelipe, 1997), as well as perturbed densities of excitatory–inhibitory synapses in areas of focal cortical dysplasia (Alonso‐Nanclares et al. 2005). Although the cellular properties underlying hyperexcitability have been widely explored in the human neocortex, little is known about how an impaired balance of excitation and inhibition of the neuronal network contributes to epileptic hypersynchrony in humans.
The question of control tissue is always problematic in the case of human studies. Anatomical studies usually include human autopsy tissue as control (for example Tóth et al. 2010), while electrophysiological studies typically operate with healthy monkey (Schwartzkroin & Haglund, 1986) or rodent tissue (Köhling et al. 1998; Heinemann et al. 2000), sometimes together with its corresponding epilepsy model (for example Lehmann et al. 2000). Furthermore, sclerotic hippocampus is usually compared to non‐sclerotic hippocampus derived from epileptic patients (Isokawa & Fried, 1996; Kivi et al. 2000; Gabriel et al. 2004; Williamson et al. 2005), and epileptogenic neocortex surrounding epileptogenic lesions is compared to adjacent, non‐affected areas (Strowbridge et al. 1992; Williamson et al. 2003; Alonso‐Nanclares et al. 2005; Thom et al. 2005). The best control available for human epileptic neocortex, i.e. tissue from non‐epileptic patients, has been investigated in only one intracellular study so far (Prince & Wong, 1981).
In the present work, we used spontaneous population bursts emerging in human neocortical slices as a network model for synchrony to examine excitatory processes contributing to epileptic activity. We demonstrate that excess excitation is present at the network level in the human epileptic neocortex compared to tissue derived from tumour patients without any preoperative clinical or electrographic appearances of seizures. We show that human neocortical slices can generate two distinct types of synchronous activities. Based on their occurrence and network characteristics we conclude that synchronous population activity (SPA) does not seem to be related to epileptic processes, whereas interictal‐like discharges (IID) seem to be associated to epilepsy. The anatomical and physiological differences of neocortical networks between humans and animals are also discussed.
Methods
Patients
All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional and national research committees and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. All patients gave written consent, and our protocol was approved by the Hungarian Ministry of Health. The study was approved by the Regional and Institutional Committee of Science and Research Ethics of Scientific Council of Health (ETT TUKEB 20680–4/2012/EKU).
Epileptic patients
Epileptic tissue samples were obtained from 49 patients (Table 1). We obtained epileptic neocortical tissue from frontal (n = 15 patients), temporal (n = 25 patients), parietal (n = 5 patients) and occipital (n = 4 patients) lobes. Most of the patients (n = 26) suffered from focal cortical epilepsy for 19.6 ± 14.6 years on average. The scalp EEG showed the presence of interictal spikes in all these patients. The remaining 23 patients had brain tumours, and had recurrent epileptic seizures (n = 12 patients) for 6.0 ± 6.0 years on average, or had only one seizure (or status epilepticus) within 10 ± 22 months prior to their surgery (n = 11 patients) and were therefore considered to be epileptic. Seventeen patients suffering from epilepsy and tumour had tumours of glial origin. Three of these 17 patients were operated for their glial tumour earlier, and now underwent their second operation to resect necrotic brain tissue caused by radiotherapy. Four patients had carcinoma metastasis. Two patients had other types of tumour (see Table 1). Thirteen epileptic patients were diagnosed with cortical dysgenesis; they were epileptic for 20.2 ± 12.7 years on average. Focal cortical dysplasia was found in nine patients; two of them also suffered from hippocampal sclerosis. Four patients showed signs of other cortical dysgenesis. Hippocampal sclerosis was detected in eight patients, who were epileptic for 25.8 ± 16.7 years on average. The remaining five patients had cavernoma (n = 4) or viral encephalitis (n = 1), and were classified as ‘other’. The duration of epilepsy of these five patients was 8.0 ± 10.3 years. The epileptic patients were 24 females, 25 males, age range: 18–72 years, mean ± SD 40.1 ± 16.1 years.
Table 1.
Patient data
| Patient no. | Gender | Age | Diagnosis | Resected cortical region | Duration of epilepsy | Stage of epilepsy | Distance from tumour | Anatomy of obtained tissue |
|---|---|---|---|---|---|---|---|---|
| Epileptic patients | ||||||||
| E1 | F | 22 | Encephalitis | Parietal | 1 month | A | Normal/cell loss | |
| E2 | F | 21 | Focal cortical dysplasia with glioneural heterotopia | Frontal | 2 years | A | Dysgenetic | |
| E3 | F | 18 | Bilateral frontal polymicrogyria | Frontal | 5 years | A | Dysgenetic | |
| E5 | F | 18 | Focal cortical dysplasia II B | Temporal | 5 years | A | Dysgenetic | |
| E6 | M | 51 | Hippocampal sclerosis | Temporal | 50 years | A | Normal | |
| E8 | F | 33 | Focal cortical dysplasia II B | Occipital | 31 years | A | Normal + dysgenetic | |
| E10 | M | 21 | Hippocampal sclerosis | Temporal | 21 years | A | Normal | |
| E11 | M | 29 | Cavernous malformation | Temporal | 3 years | A | N/A | |
| E12 | M | 40 | Hippocampal sclerosis | Temporal | 35 years | A | Normal | |
| E13 | F | 53 | Hippocampal sclerosis | Temporal | 40 years | A | Normal | |
| E15 | M | 35 | Focal cortical dysplasia + hippocampal sclerosis | Temporal | 34 years | A | Normal | |
| E16 | M | 26 | Subependymal gliosis (dysgenesis) + hippocampal sclerosis | Temporal | 24 years | A | Normal | |
| E17 | F | 38 | Focal cortical dysplasia + hippocampal sclerosis | Frontal | 31 years | A | Normal | |
| E18 | F | 35 | Focal cortical dysplasia II B (with balloon cells) | Frontal | 30 years | A | Normal | |
| E21 | M | 56 | Haemangioma cavernosum | Temporal | 26 years | A | N/A | |
| E23 | F | 33 | Haemangioma cavernosum | Temporal | 5 years | A | Normal | |
| E25 | M | 53 | Hippocampal sclerosis | Temporal | 24 years | A | Normal | |
| E27 | M | 27 | Hippocampal sclerosis | Temporal | 2 years | A | Normal | |
| E29 | M | 18 | Focal cortical dysplasia | Parietal | 5 years | A | Dysgenetic | |
| E30 | M | 30 | Microdysgenesis + hippocampal sclerosis | Temporal | 17 years | A | Dysgenetic | |
| E32 | F | 31 | Focal cortical dysplasia II B (with balloon cells) | Parietal | 18 years | A | Dysgenetic | |
| E34 | F | 18 | Hippocampal sclerosis | Temporal | 4 years | A | N/A | |
| E35 | F | 37 | Cavernous malformation | Temporal | 6 years | A | N/A | |
| E39 | F | 36 | Hippocampal sclerosis | Temporal | 30 years | A | N/A | |
| E42 | M | 44 | Subpialis gliosis (dysgenesis) | Frontal | 22 years | A | N/A | |
| E45 | M | 48 | Focal cortical dysplasia II B | Frontal | 39 years | A | N/A | |
| Epileptic patients with tumour | ||||||||
| E4 | M | 24 | Ganglioglioma grade I | Temporal | 4 years | A | Distant | Normal |
| E14 | M | 53 | Ganglioglioma grade I | Parietal | 18 years | A | Close | Normal |
| E19 | F | 72 | Radionecrosis (1 year earlier: astrocytoma grade II) | Frontal | 2 years | C | Close | Normal |
| E20 | F | 33 | Glioblastoma grade IV | Frontal | 3 months | C | Distant | N/A |
| E24 | M | 71 | Glioblastoma grade IV | Frontal | 1 week (1 seizure) | B | Close | Normal |
| E26 | M | 44 | Anaplastic ganglioglioma grade III | Temporal | 3 months (1 seizure) | C | Distant | N/A |
| E28 | F | 60 | Anaplastic oligodendroglioma grade III, recidiva | Frontal | 10 years | B | Close | N/A |
| E31 | M | 31 | Complex dysembrioplastic neuroepithelial tumour | Temporal | 10 years | A | Distant | N/A |
| E33 | M | 63 | Lung adenocarcinoma metastaticum | Occipital | 2 weeks (1 seizure) | C | Close | N/A |
| E40 | M | 32 | Anaplastic oligoastrocytoma grade III | Frontal | 4 years | A | Distant | N/A |
| E41 | F | 26 | Radionecrosis (2 years earlier: pylocytic astrocytoma) | Temporal | 13 years | A | Close | N/A |
| E44 | F | 32 | Anaplastic astrocytoma grade III | Frontal | N/A (1 seizure) | C | Distant | N/A |
| E46 | F | 38 | Ganglioglioma grade I | Temporal | 10 years | A | Close | N/A |
| O42 | M | 72 | Glioblastoma multiforme | Frontal | 9 months (1 seizure) | A | Distant | Normal |
| T1 | F | 54 | Oligodendroglioma grade II | Frontal | 2 weeks (1 seizure) | B | Distant | N/A |
| T3 | F | 18 | Anaplastic ependymoma grade III | Frontal | 1 month | B | Distant | Normal |
| T5 | F | 35 | Oligodendroglioma grade III | Occipital | 1 month (1 seizure) | B | Close | Infiltrated |
| T9 | F | 48 | Lung small cell carcinoma metastaticum | Occipital | 2 weeks (1 status epilepticus) | B | Close | Infiltrated |
| T10 | M | 44 | Glioblastoma multiforme, astrocytoma grade IV | Temporal | 3 weeks | B | Distant | Normal |
| T13 | M | 53 | Radionecrosis (6 years earlier: anaplastic oligoastrocytoma grade III) | Frontal | 6 years (1 seizure) | B | Distant | N/A |
| T22 | M | 65 | Epidermoid carcinoma metastaticum | Occipital | 13 months (1 seizure) | C | Distant | Normal |
| T31 | M | 64 | Lung carcinoma metastaticum | Temporal | 3 months (1 seizure) | B | Distant | Normal |
| T46 | F | 68 | Glioblastoma grade IV | Temporal | 2 weeks | B | Close | N/A |
| Tumour patients | ||||||||
| T2 | M | 59 | Glioblastoma multiforme sarcomatosum | Temporal | D | Close | N/A | |
| T4 | F | 69 | Glioblastoma multiforme | Temporal | D | Distant | Normal | |
| T6 | M | 31 | Cavernoma, haematoma intracerebralis acuta | Frontal | D | Distant | Normal | |
| T7 | F | 58 | Glioblastoma multiforme, meningitis | Temporal | D | Close | Infiltrated | |
| T8 | F | 78 | Glioblastoma multiforme, astrocytoma grade IV | Temporal | D | Distant | Normal | |
| T11 | F | 57 | Glioblastoma multiforme grade IV | Occipital | D | Distant | Normal | |
| T12 | M | 59 | Glioblastoma, with oligodendroglioma fragments grade IV | Frontal | D | Close | Normal | |
| T14 | M | 67 | Lung anaplastic carcinoma metastaticum | Temporal | D | Close | N/A | |
| T15 | F | 67 | Meningioma grade I | Frontal | D | Distant | N/A | |
| T16 | F | 69 | Gastrointestinal adenocarcinoma metastaticum | Occipital | D | Close | N/A | |
| T17 | F | 74 | Glioblastoma multiforme grade IV | Parietal | D | Distant | Normal | |
| T18 | M | 68 | Melanoma malignum metastaticum | Parietal | D | Distant | Normal | |
| T19 | M | 69 | Lung adenocarcinoma metastaticum | Parietal | D | Close | N/A | |
| T20 | F | 59 | Breast carcinoma metastaticum | Frontal | D | Close | Infiltrated | |
| T21 | F | 69 | Kidney carcinoma metastaticum | Parietal | D | Distant | Normal | |
| T23 | F | 81 | Meningioma grade I | Frontal | D | Distant | Normal | |
| T24 | M | 55 | Lung adenocarcinoma metastaticum | Temporal | D | Distant | N/A | |
| T25 | F | 55 | Glioblastoma multiforme | Parietal | D | Distant | N/A | |
| T26 | F | 63 | Glioblastoma multiforme grade IV | Parietal | D | Close | N/A | |
| T27 | F | 61 | Lung carcinoma metastaticum | Frontal | D | Distant | N/A | |
| T28 | M | 49 | Kidney carcinoma metastaticum | Occipital | D | Close | N/A | |
| T29 | F | 62 | Lung adenocarcinoma metastaticum | Parietal | D | Close | N/A | |
| T32 | M | 79 | Glioblastoma multiforme grade III | Temporal | D | Close | N/A | |
| T33 | M | 45 | Anaplastic astrocytoma | Frontal | D | Close | N/A | |
| T34 | F | 64 | Haematoma | Frontal | D | Distant | N/A | |
| T35 | M | 60 | Glioblastoma grade IV | Temporal | D | Distant | N/A | |
| T38 | M | 82 | Stomach anaplastic carcinoma metastaticum | Temporal | D | Close | N/A | |
| T39 | M | 37 | Centralis neurocytoma grade II | Frontal | D | Distant | N/A | |
| T40 | M | 45 | Anaplastic astrocytoma grade III | Temporal | D | Close | N/A | |
| T42 | F | 59 | Breast carcinoma metastaticum | Frontal | D | Close | N/A | |
| T43 | M | 73 | Melanoma malignum metastaticum | Temporal | D | Distant | N/A | |
| T44 | F | 58 | Breast carcinoma metastaticum | Occipital | D | Distant | N/A | |
| T45 | F | 56 | Glioblastoma grade IV | Occipital | D | Distant | N/A | |
F, female; M, male; N/A, not available. Stage of epilepsy: A, pharmacoresistant epilepsy; B, treatable epilepsy; C, no need for medication; D, no epilepsy.
Non‐epileptic patients
Thirty‐three patients diagnosed with brain tumour but without epilepsy were included in this study (Table 1). These patients – as stated in their anamnesis – did not survive clinical manifestation of epileptic seizure before the date of their brain surgery. Preoperative clinical EEG recordings confirmed in eight of these patients that no electrographic signs of epileptic activity were present on their scalp EEG. We obtained non‐epileptic neocortical specimens from frontal (n = 10 patients), temporal (n = 11 patients), parietal (n = 7 patients) and occipital (n = 5 patients) lobes. Fourteen patients were diagnosed with tumours of glial origin: glioblastoma (n = 12) or anaplastic astrocytoma (n = 2). Fourteen patients had carcinoma metastasis. The remaining five patients were operated for other reasons (for details see Table 1). The distance of the obtained neocortical tissue from the tumour has been provided by the neurosurgeon, based on magnetic resonance (MR) images, intraoperative pictures and occasionally defined by navigational system. Non‐epileptic patients were 18 females, 15 males, age range: 31–82 years, mean ± SD 61.7 ± 11.7 years.
Tissue preparation
Tissue was transported from the operating room to the laboratory (located in the same building) in a cold, oxygenated solution containing (in mm): 248 d‐sucrose, 26 NaHCO3, 1 KCl, 1 CaCl2, 10 MgCl2, 10 d‐glucose and 1 phenol red, equilibrated with 5% CO2–95% O2. Neocortical slices of 500 μm thickness were cut with a vibrating blade microtome. They were transferred and maintained at 35–37°C in an interface chamber perfused with a standard solution containing (in mm): 124 NaCl, 26 NaHCO3, 3.5 KCl, 1 MgCl2, 1 CaCl2 and 10 d‐glucose, equilibrated with 5% CO2–95% O2.
Recordings
Intracellular recordings were made with microelectrodes that contained 2 m potassium acetate with a resistance of 50–100 MΩ. The data were obtained within 10–20 min of penetration. Signals were amplified with a BA‐1S amplifier (NPI Electronic GmbH, Tamm, Germany) operated in current‐clamp mode. In acceptable records, the membrane potential was more negative than −50 mV, input resistance was higher than 20 MΩ and action potentials were overshooting.
The extracellular local field potential gradient (LFPg) was recorded with a laminar multiple channel (24 channels, distance between contacts: 150 μm) microelectrode (Ulbert et al. 2001, 2004a, b; Fabó et al. 2008; Wittner et al. 2009), using a custom made 48‐channel voltage gradient amplifier of bandpass 0.01 Hz to 10 kHz. Signals were digitized with a 32‐channel, 16‐bit resolution analog‐to‐digital converter (National Instruments, Austin, TX, USA) at 20 kHz sampling rate, recorded with a home‐written routine in LabView7 (National Instruments). The linear 24‐channel microelectrode was placed perpendicular to the pial surface, and slices were mapped from one end to the other at every 300–400 μm.
Drugs
A‐type γ‐aminobutyric acid (GABAA) receptor‐mediated signalling was suppressed by bicuculline methiodide (10 μm). α‐Amino‐3‐hydroxyl‐5‐methyl‐4‐isoxazole‐propionate (AMPA)‐ and kainate (KA)‐type glutamate receptors were blocked using 2,3‐dihydroxy‐6‐nitro‐7‐sulfamoyl‐benzo(f)quinoxaline (NBQX; 5 μm), and N‐methyl‐d‐aspartate (NMDA)‐type receptors were blocked with dl‐2‐amino‐5‐phosphonovaleric acid (dl‐APV; 50 μm). Drugs were obtained from Tocris Bioscience (Izinta Kft, Budapest, Hungary).
Data analysis
Data were analysed with the Neuroscan Edit4.5 program (Compumedics Neuroscan, Charlotte, NC, USA), and home‐written routines for Matlab (The MathWorks, Natick, MA, USA). Current source density (CSD), an estimate of population transmembrane currents, and multiple unit activity (MUA) were calculated from the LFPg using standard techniques (Ulbert et al. 2001, 2004b; Wittner et al. 2009). Baseline correction (−150 to −50 ms) was applied to averaged LFPg, CSD and MUA. In the colour maps, CSD sinks are presented in red, sources in blue. Warm colours (red) depict MUA increases and cold colours (blue) depict MUA decreases compared to baseline.
Synchronous activity (SPA and IID) detection was performed on LFPg records after a double Hamming window spatial smoothing and a band‐pass filtering between 3 and 30 Hz (zero phase shift, 12 dB/octave). The peak of the LFPg transient was detected with a routine in Matlab, and was considered as time zero for further event‐triggered averaging. Events larger than 3× standard deviation were detected and included in the analyses. The location of SPAs/IIDs was determined in each case. The 24‐channel microelectrode covered all layers of the neocortex in almost all cases. Usually channels 1–8 were in the supragranular, channels 9–13 in the granular and channels 14–23 were in the infragranular layers. Channel positions were determined according to the thickness of the neocortex of the given patient, and corrected if necessary. Waveform analysis was performed on averaged synchronous activities with a home‐written C++ routine.
Ripple and fast ripple components of SPAs and IIDs were determined with the aid of routines written in Matlab, as follows. Original 20 kHz sampling rate records were down‐sampled to 2000 Hz. Wavelet analysis was applied on epochs containing 4096 sampling points (from −1000 to 1047 ms) with the LFPg peak of the SPA/IID at time zero (detected as above). Time–frequency analysis was performed between 0 and 800 Hz on the electrode channels where SPA was present, and baseline corrected to −300 to −100 ms. We systematically saw a peak around 200 Hz, and therefore we modified the conventional limit of 200 Hz for ripple frequency (Bragin et al. 1999) to 250 Hz. For each channel, the maximal power change (relative to the baseline) was determined within the range of 130–250 Hz (ripple frequency) and 300–800 Hz (fast ripple frequency) at time zero (i.e. at the time point of the LFPg peak of the SPAs/IIDs). The frequencies where the power showed the maximum were also determined. Both the ripple and fast ripple power and frequencies were averaged across channels, to receive one ripple and one fast ripple power and frequency parameter for each recording. This last step was needed for the comparison of recordings with population activities spreading to different numbers of channels.
Anatomy
Intracellularly recorded cells were labelled, processed and reconstructed in three dimensions as described earlier (Wittner et al. 2009).
Immunohistochemical procedures were used to verify the laminar structure and the possible tumour infiltration or signs of dysgenesis in the neocortex of 28 epileptic and 13 tumour patients. Either tissue blocks or neocortical slices following electrophysiological recording were fixed and processed as described earlier (Wittner et al. 2009). Neuronal cell bodies were stained with NeuN antibody (1:2000, EMD Millipore, Billerica, MA, USA, RRID: AB_2298772), astroglial cells were stained with glial fibrillary acidic protein antibody (1:2000, EMD Millipore, RRID: AB_94844), and perisomatic inhibitory cells were marked with parvalbumin antibody (1:7000, Swant, Bellinzona, Switzerland, RRID: AB_10000343). All antibodies were mouse monoclonal antibodies. Their specificity was tested by the manufacturer. Visualization of immunopositive elements was performed as described earlier (Wittner et al. 2009).
The total cell density and the synaptic connectivity were examined in regions which generated SPA and in neighbouring regions that did not show SPA. The following criteria were taken into consideration when choosing the samples for quantitative analysis: the site that generated SPA and the site that did not generate SPA should be part of the same slice. The NeuN‐ or the PV‐stained sections chosen for cell counting should contain the whole length of the slice, and the whole width of the cortex, including all layers. Two slices were chosen from both tumour (patients T17 and T23) and epileptic (patients E13 and E16) subjects for NeuN+ neuron counting. Four slices were chosen from three epileptic patients (E12, E13 and E15), and three slices from three non‐epileptic patients (T17, T20 and T23) for counting PV+ cell bodies. Two regions were marked including all layers: one where SPA was detected and another where recordings did not show SPA. The areas of all neocortical layers were measured, and NeuN‐ or PV‐positive somata were counted in both regions with the aid of the NeuroLucida system (MicroBrightField Inc., Williston, VT, USA), at a magnification of ×40.
After light microscopic examination, areas of interest were re‐embedded and sectioned for electron microscopy. Ultrathin serial sections were collected on Formvar‐coated single slot grids, stained with lead citrate, and examined with a Hitachi 7100 electron microscope. Electron microscopic analysis was performed only on specimens with high quality ultrastructural preservation. For the analysis of synaptic connectivity three slices from epileptic (E12, E13, E15) and three from non‐epileptic (T17, T20, T23) patients were selected. SPA occurred in the supragranular layers in all selected slices. Two blocks were re‐embedded from the supragranular layers (upper layer III) of each slice, one from the spot where SPA was recorded and the other from an area where SPA was not generated. Photos were systematically taken at ×20,000 magnification, from one side to the other side of the block without overlapping areas, to avoid multiple sampling of the same synapse. The number of the examined spots varied between 22 and 39 among the samples, with an area of 30.57 μm2 per spot. Neuronal and glial somata were excluded from the examined area, so all values are given relative to the neuropil. The values of the examined area from each block varied between 2200 and 3400 μm2. The number of asymmetrical (presumably excitatory) and symmetrical (presumably inhibitory) synapses were determined per 100 μm2 in each region.
Statistical analysis
Our data sets did not follow Gaussian distributions, and therefore non‐parametric tests were used. The Mann–Whitney U test was used when two groups were compared and Kruskal–Wallis ANOVA was used when more than two groups were compared. In the latter case, differences between groups were determined via post hoc tests provided by Matlab. In case of multiple testing, P‐values were corrected using the Bonferroni–Holm method.
For testing the effect of dl‐APV, we used the paired Wilcoxon's signed rank test. For contingency tables Fisher's exact test or the chi‐square test was used, if the expected values were low or high, respectively. P‐values below 0.05 were considered to be statistically significant.
Results
Occurrence of synchronous population activity
Synchronous population activity (SPA) was spontaneously generated in a standard bathing solution in slices from both epileptic (133/287 slices, 46.3% in 43/49 patients) and non‐epileptic patients (60/194 slices, 30.9% in 24/33 patients, significantly different, P < 0.05, Fig. 1; for patient data see Methods and Table 1). In several cases (9 activities, 3.1%, in 7/287 slices, in 5/49 epileptic patients), considerably larger and more complex bursts also emerged in the neocortex derived only from epileptic patients (Fig. 1 G). They resembled epileptiform population bursts induced by 4‐amino‐pyridine (Avoli et al. 1994), and were therefore named interictal‐like discharges (IID, see Fig. 1 G), and analysed separately. When separating IIDs from SPAs we considered every examined network feature of the synchronous activities: layer of emergence, recurrence frequency, local field potential gradient (LFPg), current source density (CSD) and multiple unit activity (MUA) amplitude (see later in detail). LFPg and CSD amplitude are related to each other, since CSD is the first derivative of the LFPg signal (see Methods). LFPg gives the difference in local field potential between two neighbouring electrode contacts, and might be misrepresentative in cases of synchronies spreading to higher numbers of contacts. In contrast, CSD amplitude gives an estimate of transmembrane current amplitude, independent from the spatial dimensions of the synchrony. Moreover, MUA amplitude can be misleading if a large amplitude single unit with related firing to the synchrony is visible on the trace. All these technical considerations were taken into account when establishing our categories. Although each property showed slightly overlapping values, combining all parameters gave a clear distinction between SPAs and IIDs. We combined CSD and MUA, to be able to visualize the examined features in a three‐dimensional plot (Fig. 1 J).
Figure 1. Network characteristics of SPAs in human neocortical slices.
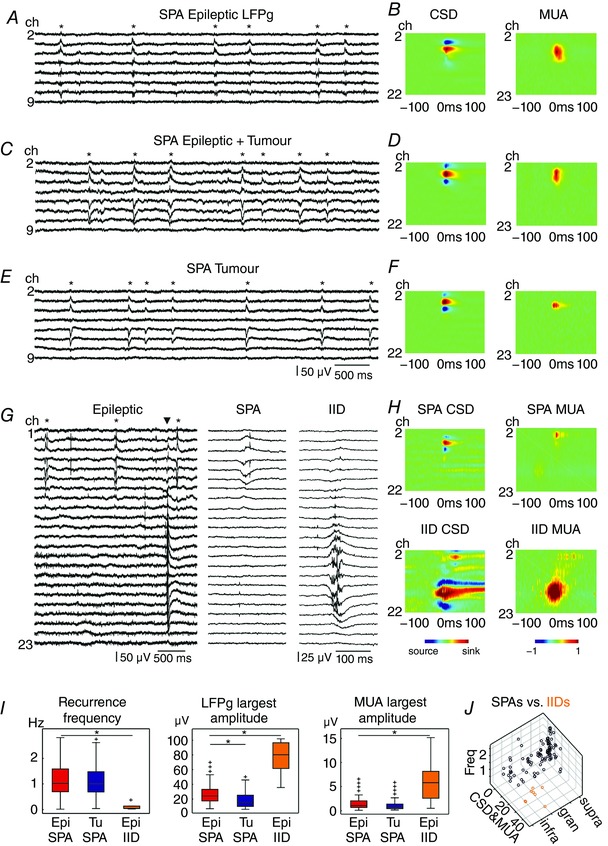
SPAs were observed in tissue from epileptic patients without tumour (A and B), epileptic patients with tumour (C and D) and tumour patients without epilepsy (E and F). Furthermore, IID was detected in the epileptic neocortex (G and H). Left panels show LFPg traces from eight (A, C and E) channels positioned in the supragranular layers, while G shows 23 channels covering the entire width of the neocortex, with simultaneously occurring SPA and IID. Asterisks label SPA events (A, C, E and G); the triangle (G) indicates the IID event. One SPA and IID event each is magnified on the right side (G). Colour maps (B, D, F and H) show the CSD and the change in MUA. In most SPA cases CSD consisted of a pair or triple of simultaneous sinks and sources and was similar in all three patient groups. Warm colours depict sinks, while cold colours indicate sources. MUA increase was detected during SPAs and IIDs in almost all cases. Warm colours show MUA increase, cold colours label MUA decrease. Note the higher amplitude of CSD and MUA in case of IIDs (colour scales are the same for all heat maps). I, the recurrence frequency and the MUA were similar for SPAs of epileptic and tumour patients. The LFPg amplitudes were significantly larger for SPAs in epileptic patients than in tumour patients (LFPg: P < 0.01). All of these network characteristics of IIDs were significantly different from SPAs detected in epileptic tissue (P < 0.01). *Significant difference. J, the values of recurrence frequency (Freq), the average of CSD and MUA (CSD&MUA), and the intracortical location of all SPAs (black circles) and IIDs (orange circles) from epileptic and tumour tissue are shown on a three‐dimensional plot. Epi, epileptic; Tu, tumour; supra, supragranular; gran, granular; infra, infragranular. [Color figure can be viewed at wileyonlinelibrary.com]
SPA occurrence was similar in slices derived from different lobes (Table 2; P > 0.1 and P > 0.8 in epileptic and tumour tissue, respectively), but was significantly higher in tissue resected from patients with epilepsy than in slices derived from non‐epileptic patients (P < 0.01). SPA was generated at a significantly lower rate in slices from epileptic patients with tumour (n = 23, 36.4 ± 29.0%, mean ± standard deviation) than from epileptic patients with dysgenesis (n = 13), hippocampal sclerosis (n = 8), or other associated symptoms (n = 5, varying from 50.3 ± 34.0% to 58.3 ± 28.6%, P < 0.05, Table 3). The specimens obtained from patients with dysgenesis were classified as affected (n = 46 slices from seven patients) or non‐affected (n = 26 slices from five patients) by the dysgenesis, based on our subsequent anatomical analysis. We found similar values of SPA emergence in the dysgenetic (n = 26/46 slices, 56.5%) and in the non‐dysgenetic (n = 14/26 slices, 53.9%) neocortex (chi‐square test; P > 0.8).
Table 2.
SPA occurrence in neocortical slices deriving from different lobes of the brain
| All epileptic patients | Epileptic patients without tumour | Epileptic patients with tumour | Tumour patients without epilepsy | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Region | n | Ratio of slices generating SPA (%, sum of slices with SPA/sum of all slices) | Ratio of slices generating SPA (%, by patient, mean ± SD) | n | Ratio of slices generating SPA (%, sum of slices with SPA/sum of all slices) | Ratio of slices generating SPA (%, by patient, mean ± SD) | n | Ratio of slices generating SPA (%, sum of slices with SPA/sum of all slices) | Ratio of slices generating SPA (%, by patient, mean ± SD) | n | Ratio of slices generating SPA (%, sum of slices with SPA/sum of all slices) | Ratio of slices generating SPA (%, by patient, mean ± SD) |
| Frontal lobe | 15 | 35.2 | 35.0 ± 29.4 | 6 | 37.1 | 42.7 ± 34.4 | 9 | 34.0 | 29.8 ± 26.5 | 10 | 34.0 | 34.2 ± 29.2 |
| Occipital lobe | 4 | 41.2 | 35.0 ± 27.4 | 1 | 66.7 | 66.7 ± 0.0 | 3 | 27.3 | 24.4 ± 21.4 | 5 | 36.4 | 31.7 ± 31.1 |
| Parietal lobe | 5 | 47.2 | 48.7 ± 25.4 | 3 | 63.6 | 66.1 ± 12.1 | 2 | 21.4 | 22.5 ± 3.5 | 7 | 33.3 | 30.3 ± 19.3 |
| Temporal lobe | 25 | 54.3 | 55.5 ± 27.7 | 16 | 59.3 | 58.5 ± 24.5 | 9 | 46.3 | 50.0 ± 33.7 | 11 | 25.4 | 24.3 ± 27.8 |
| Total | 49 | 46.6 | 46.8 ± 28.9 | 26 | 55.0 | 56.1 ± 25.9 | 23 | 37.1 | 36.4 ± 29.0 | 33 | 31.1 | 29.7 ± 26.3 |
Table 3.
Relationship of SPA occurrence and aetiology
| Patients | Aetiology | Number of patients | Ratio of slices generating SPA (%, sum of slices with SPA/sum of all slices) | Ratio of slices generating SPA (% by patient, mean ± SD) |
|---|---|---|---|---|
| Epileptic | Dysgenesis | 13 | 54.9 | 58.3 ± 28.6 |
| Hippocampal sclerosis | 8 | 57.1 | 56.1 ± 17.5 | |
| Other | 5 | 52.0 | 50.3 ± 34.0 | |
| Tumour | 23 | 37.1 | 36.4 ± 29.0 | |
| Total | 49 | 46.6 | 46.8 ± 28.9 | |
| Tumour | Glial tumour | 31 | 38.1 | 38.7 ± 28.6 |
| Carcinoma metastasis | 18 | 23.7 | 20.6 ± 21.3 | |
| Other tumour | 7 | 35.9 | 35.0 ± 29.0 | |
| Total | 56 | 33.5 | 32.4 ± 27.4 |
SPA generation was variable in patients with brain tumour, relative to their type of tumour (Table 3). The SPA occurrence rate in all tumour patients with or without preoperative epileptic seizures was 32.4 ± 27.4%. Higher, although not significant (P > 0.1), ratios were found in the cases with glial tumour (38.7 ± 28.6%, n = 31 patients), and other associated symptoms (35.0 ± 29.0%, n = 7 patients), than in brains with carcinoma metastasis (20.6 ± 21.3%, n = 12 patients). We verified how the distance of the specimen from the tumour affected SPA generation in our samples. In the cases where the resected neocortical tissue was close (< 30 mm) to the tumour, 25.0 ± 22.8% of the slices generated SPA (in n = 23 patients; for determination of the distance between the tumour and the resected neocortical tissue see Methods). When the examined tissue was at higher distances from the tumour (>30 mm, but in most cases >50 mm), SPA emerged in a higher percentage of the slices (36.1 ± 27.9%, in n = 31 patients), although the difference was not statistically significant (P > 0.1).
Next, we wished to examine whether the different stages of epilepsy affect the ability of the neocortex to generate SPAs. Therefore, we grouped our patients (Table 1) as follows: (A) patients with pharmacoresistant epilepsy; (B) patients with focal or grand mal seizures who are seizure free with medication (treatable epilepsy); (C) patients with one grand mal seizure or with occasional (provoked) seizures, and with no need for medication (these patients were operated for their tumour); (D) patients without preoperative seizures (no epilepsy). SPA occurrence was the highest in the group with pharmacoresistant epilepsy (group A, n = 33 patients): 57.0 ± 28.0%; it was lower in the group with treatable epilepsy (group B, n = 11 patients): 32.3 ± 13.9%. We found the lowest ratios in the group with no need for medication (group C, n = 5 patients): 11.7 ± 16.2%, and in the group without epilepsy (group D, n = 33 patients): 29.1 ± 26.4%. SPA occurrence was related to the stage of epilepsy, i.e. the ratio of slices generating SPA in group A was significantly higher than in group D (P < 0.001) or in group C (P < 0.01).
Our above analyses show that both the presence of tumour and the stage of the patient's epilepsy influence the generation of synchronies. In this study, we wished to identify epilepsy‐related phenomena, which are not affected by tumour formation or by differences in the stages of epilepsy. Thus, for the analysis of network and cellular properties of the synchronies, we made three groups as follows. (1) Patients with pharmacoresistant epilepsy and without tumour (which will be referred as ‘epileptic’). Note that this category largely overlaps with group A, but excludes patients with tumour and pharmacoresistant epilepsy. (2) Tumour patients without epilepsy (which will be referred as ‘tumour’); this category includes patients falling into group D. (3) The remaining patients suffering from both epilepsy and tumour were pooled into the heterogeneous ‘epileptic + tumour’ group (see Table 1). We analysed data deriving from these groups separately, and made comparisons only between ‘epileptic’ and ‘tumour’ patients to draw conclusions about changes related to epilepsy.
Characteristics of SPAs and IIDs
Several types of SPAs were separated by their location and extension within the neocortex (Fig. 2 A and Table 4). SPA occurred most frequently in the supragranular layers: 69.9% in epileptic, 65.2% in epileptic + tumour and 67.3% in tumour tissue (Fig. 2 D, see also (Köhling et al. 1998). Less often SPA could be detected in the granular and infragranular layers, and in a few cases SPA invaded the entire width of the neocortex. Interictal‐like discharges (IID) emerged mainly in the deeper layers of the neocortex: granular (n = 1) or in the granular + infragranular (n = 5) and infragranular (n = 2) layers (Fig. 2 A and D, and Table 4) in slices from epileptic patients, and in the supragranular + granular layers (n = 1) in one slice from a patient having epilepsy + tumour.
Figure 2. Different types of single and multiple SPAs.
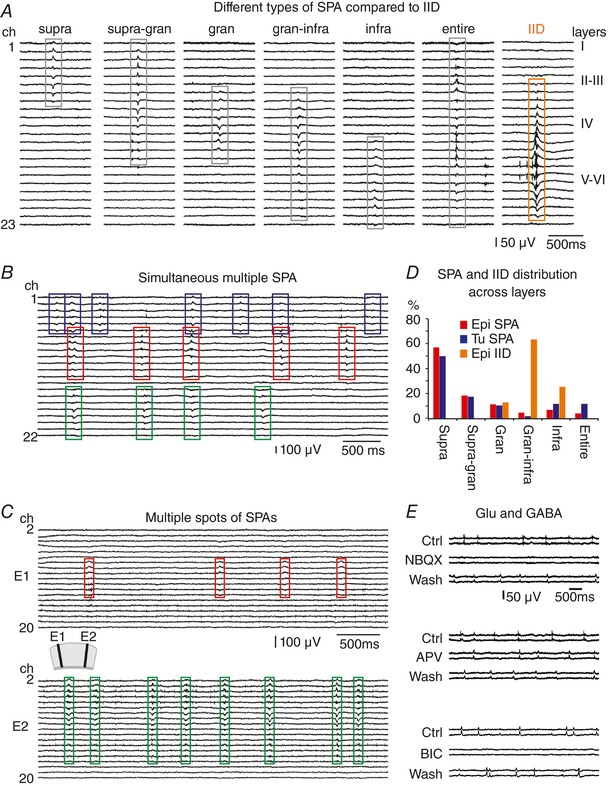
A, different types of SPAs were separated based on their location and extension across the neocortical layers. SPAs were found to be spread over supragranular (supra), supragranular + granular (supra‐gran), granular (gran), granular + infragranular (gran‐infra) or infragranular (infra) layers or over the entire width of the neocortex (entire). For comparison, an IID event is also shown. Multiple SPAs occurred more frequently in samples from epileptic and epileptic + tumour patients. We differentiated between simultaneous multiple SPAs at one recording site (B) and multiple spots of SPAs in the same slice (C). D, the prevalence of the different SPA types was similar in the epileptic and tumour patient groups. Most of the SPAs were generated in the supragranular layers, whereas IIDs emerged mainly in the deeper layers. (E) The AMPA/kainate‐type glutamate receptor antagonist NBQX blocks the emergence of SPAs (upper panel). The NMDA‐type glutamate receptor antagonist dl‐APV reduced the frequency of SPAs (middle panel). The GABAA receptor antagonist bicuculline also blocked the generation of SPAs (lower panel). All these effects were reversible. [Color figure can be viewed at wileyonlinelibrary.com]
Table 4.
SPA distribution across neocortical layers
| Epileptic patients without tumour | Epileptic patients with tumour | Tumour patients without epilepsy | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SPAs in neocortical layers | Single SPA | Multiple SPA | Total SPA (%) | IID | Single SPA | Multiple SPA | Total SPA (%) | IID | Single SPA | Multiple SPA | Total SPA (%) |
| Supra | 35 | 41 | 76 (56.3%) | 0 | 18 | 17 | 35 (49.3%) | 0 | 33 | 17 | 50 (53.8%) |
| Supra‐gran | 12 | 12 | 24 (17.8%) | 0 | 6 | 6 | 12 (16.9%) | 1 | 6 | 7 | 13 (14.0%) |
| Gran | 5 | 10 | 15 (11.1%) | 1 | 3 | 4 | 7 (9.9%) | 0 | 3 | 11 | 14 (15.1%) |
| Gran‐infra | 0 | 6 | 6 (4.4%) | 5 | 0 | 1 | 1 (1.4%) | 0 | 1 | 3 | 4 (4.3%) |
| Infra | 2 | 7 | 9 (6.7%) | 2 | 1 | 7 | 8 (11.3%) | 0 | 2 | 8 | 10 (10.8%) |
| Entire | 2 | 3 | 5 (3.7%) | 0 | 4 | 4 | 8 (11.3%) | 0 | 0 | 2 | 2 (2.2%) |
| Total number of SPAs or IIDs | 56 | 79 | 135 | 8 | 32 | 39 | 71 | 1 | 45 | 48 | 93 |
| Number of slices with SPAs or IIDs (% of slices with SPA/IID) | 56 (66.7%) | 28 (33.3%) | 84 (54.9%) | 6 (7.1%) | 32 (65.3%) | 16 (32.7%) | 49 (36.6%) | 1 (2.0%) | 45 (75%) | 15 (25%) | 60 (30.9%) |
| Total number of examined slices | 153 | 134 | 194 | ||||||||
Number of SPAs (and IIDs) are provided appearing as single or as part of multiple SPAs, in the different layers of the human neocortex. Supra, supragranular (layers I–III) layers; Supra‐gran, supragranular + granular (layer IV) layers; Gran, granular layer; Gran‐infra, granular + infragranular (layers V–VI) layers; Infra, infragranular layers; Entire, entire width of the neocortex, involving supragranular + granular + infragranular layers; see also Fig. 2.
As in a previous study (Köhling et al. 1998), we observed multiple independent SPAs in the same slice. We differentiated between simultaneous multiple SPAs (Fig. 2 B) or simultaneous SPA + IID (n = 2 cases, Fig. 1 G) at one recording site, and multiple spots of SPAs (Fig. 2 C). The ratio of slices exhibiting multiple SPAs was somewhat higher in epileptic (33.3%) and epileptic+tumour (32.7%) than in tumour patients (25.0%, Table 4).
Next, the network characteristics of SPAs and IIDs emerging in the human neocortex were analysed (Fig. 1 I and Table 5). We compared SPAs emerged in epileptic slices to SPAs generated in tumour slices, as well as to IIDs detected in epileptic tissue. The recurrence frequency of SPAs was 1.20 ± 0.71 Hz in epileptic (n = 46 SPAs) and 1.18 ± 0.63 Hz in tumour (n = 48 SPAs) tissue, respectively, while that of IIDs was significantly lower (0.10 ± 0.13 Hz, n = 8 IIDs, P < 0.0001). The largest amplitude on the local field potential gradient (LFPg) was significantly higher for IIDs (74.70 ± 21.99 μV) than for SPAs (23.22 ± 17.23 μV, P < 0.0001) in the neocortex of epileptic patients, and it was significantly lower for SPAs in tumour patients (18.40 ± 10.48 μV, P < 0.01, Table 5) than for SPAs in epileptic patients. Multiple unit activity (MUA), as an estimate of cellular firing, did not differ during SPAs in epileptic and tumour tissue (1.49 ± 1.46 vs. 1.29 ± 1.17 μV, respectively), but was significantly higher during IIDs (6.46 ± 4.57 μV, P < 0.01, Fig. 1 H and I). Similar to a previous study (Köhling et al. 1999), we found that the current source density (CSD) associated with the SPAs was very variable. However, in most of the cases a source–sink–source pattern was detected in the layer where the SPA occurred (Fig. 1 B, D and F). The CSD pattern was usually more complex, comprising several peaks, and significantly higher in amplitude during IIDs (Fig. 1 H, P < 0.0001).
Table 5.
Network properties of human neocortical SPAs and IIDs
| Synchronous activity type | Number of SPAs/IIDs analysed | Recurrence frequency (Hz) | Largest LFPg amplitude (μV) | Width at half of the maximal amplitude (ms) | Asymmetry at half of the maximal amplitude (right/left) | Largest CSD amplitude (μV) | Largest MUA amplitude (μV) | |
|---|---|---|---|---|---|---|---|---|
| Epileptic SPA | Total SPA | 46 | 1.03 [0.91 1.22] | 23.22 [18.59 27.01] | 26.35 [23.75 31.85] | 1.55 [1.45 1.77] | 14.30 [12.92 16.97] | 1.06 [0.77 1.12] |
| (1.20 ± 0.71) | (27.31 ± 17.23) | (34.09 ± 20.43) | (1.72 ± 0.64) | (16.76 ± 8.79) | (1.49 ± 1.46) | |||
| Supragran SPA | 38 | 1.04 [0.88 1.31] | 23.38 [18.82 28.72] | 26.35 [23.60 31.65] | 1.63 [1.46 1.91] | 14.30 [12.91 17.77] | 1.10 [0.93 1.42] | |
| (1.22 ± 0.67) | (28.25 ± 18.14) | (33.89 ± 21.53) | (1.79 ± 0.62) | (17.14 ± 9.03) | (1.65 ± 1.55) | |||
| Infragran SPA | 7 | 0.99 [0.13 1.74] | 17.67 [14.77 27.81] | 24.70 [22.75 39.30] | 1.09 [0.92 1.57] | 13.56 [8.66 18.35] | 0.75 [0.28 1.06] | |
| (1.06 ± 0.96) | (22.65 ± 12.87) | (31.53 ± 12.40) | (1.23 ± 0.54) | (14.86 ± 8.50) | (0.68 ± 0.38) | |||
| Tumour SPA | Total SPA | 48 | 1.06 [0.90 1.38] | 15.41 [12.82 20.47] | 23.05 [21.00 25.85] | 1.46 [1.20 1.59] | 10.08 [8.14 12.98] | 0.90 [0.68 1.17] |
| (1.18 ± 0.60) | (18.40 ± 10.48) | (24.98 ± 11.23) | (1.62 ± 0.78) | (12.05 ± 6.34) | (1.29 ± 1.17) | |||
| Supragran SPA | 36 | 1.08 [0.89 1.39] | 16.09 [12.74 20.61] | 23.70 [21.10 27.40] | 1.41 [1.15 1.57] | 11.06 [8.50 13.19] | 0.73 [0.57 1.09] | |
| (1.16 ± 0.58) | (18.64 ± 10.51) | (26.10 ± 11.98) | (1.58 ± 0.80) | (12.36 ± 6.46) | (1.11 ± 1.12) | |||
| Infragran SPA | 11 | 0.98 [0.57 1.81] | 13.08 [7.39 25.67] | 21.00 [17.70 27.55] | 1.65 [1.03 2.21] | 7.84 [6.45 15.43] | 1.76 [0.87 2.93] | |
| (1.17 ± 0.69) | (16.97 ± 11.07) | (21.58 ± 8.50) | (1.75 ± 0.78) | (10.70 ± 6.24) | (1.90 ± 1.22) | |||
| Epileptic IID | 8 | 0.05 [0.02 0.14] | 74.36 [60.16 100.34] | 37.00 [19.30 52.55] | 1.77 [1.30 2.62] | 42.97 [40.86 52.21] | 5.81 [1.66 8.94] | |
| (0.10 ± 0.13) | (74.70 ± 21.99) | (40.69 ± 20.19) | (1.92 ± 1.04) | (46.09 ± 10.05) | (6.46 ± 4.57) | |||
| Significant differences | Epi SPA > Epi IID P < 0.0001 |
Epi SPA > Tumour SPA P < 0.01 Epi supra > Tumour supra P < 0.01 Epi IID > Epi SPA P < 0.0001 |
Epi SPA > Tumour SPA P < 0.05 | n.s. |
Epi SPA > Tumour SPA P < 0.01 Epi supra > Tumour supra P < 0.05 Epi IID > Epi SPA P < 0.0001 |
Epi IID > Epi SPA P < 0.01 | ||
Data are medians [95% confidence interval] (mean ± SD), since none of the examined parameters showed Gaussian distribution. Epi, epileptic; n.s., non‐significant; Supragran, supra, supra‐gran and gran SPAs; Infragran, gran‐infra and infra SPAs.
A relationship between the recurrence frequency and the field potential amplitude has been previously observed in cases of population events in vitro (Papatheodoropoulos & Kostopoulos, 2002; Hofer et al. 2015), i.e. as the frequency of events increases as a consequence of higher levels of [K+]o, the field potential amplitude decreases. On the other hand, this relationship could not be demonstrated in cases of population bursts without changing the composition of the physiological bath solution (Hájos et al. 2013). To verify this possible correlation during human neocortical synchronies, we plotted the average LFPg amplitude of SPAs/IIDs against the mean recurrence frequency or the mean inter‐event interval. We could not find a considerable correlation between LFPg amplitude and frequency (R 2 = 0.0042 for SPA and R 2 = 0.006 for IIDs), or inter‐event interval (R 2 = 0.0011 for SPAs and R 2 = 0.0957 for IIDs). However, when plotting MUA amplitude against LFPg amplitude, we found a weak correlation for SPAs (R 2 = 0.3679), indicating that larger LFPg (reflecting synaptic input; Gulyás et al. 2010) is linked to larger MUA (reflecting output). No correlation could be demonstrated in cases of IIDs (R 2 = 0.0135). We also examined the relationship between the LFPg amplitude of every SPA/IID event and the length of its preceding inter‐event interval within each recording. In the majority of the cases (n = 65/102) no significant correlation was demonstrated. In about one‐third of the cases (n = 30/102), a significant positive correlation was shown (with very weak to moderate Spearman correlation coefficients), but we also found negative correlations (n = 7/102, with very weak to weak Spearman correlation coefficients). None of the IID activities were significant, possibly due to the low IID event numbers. Altogether, correlations could be either positive or negative in both epileptic and tumour tissue, but only positive correlations showed moderate strength (data not shown).
High frequency oscillations (HFOs) were examined in the range of ripples (130–250 Hz) and fast ripples (300–800 Hz) during SPAs in slices derived from both epileptic (n = 37 slices from 12 patients) and tumour patients (n = 33 slices from 9 patients; Fig. 3 and Table 6), as well as during IIDs in eight recordings from epileptic patients without tumour. Simultaneous multiple SPA was present in 8/37 and 11/33 recordings, and therefore 46 and 48 SPAs were examined in epileptic and tumour tissue, respectively. HFOs (mainly at ripple frequency) associated to SPAs were detected slightly (but not significantly) more frequently in slices from epileptic than from tumour patients (Table 6). Ripple frequency was significantly lower during SPAs in epileptic than in tumour tissue (P < 0.01). Fast ripple frequency was similar in epileptic and tumour tissue, and showed slightly lower values during IIDs. Ripple and fast ripple powers during SPAs did not differ in epileptic vs. tumour tissue, but were significantly higher during IIDs (P < 0.001 for both ripple and fast ripple powers, Table 6, Fig. 3).
Figure 3. High frequency oscillations during SPAs.
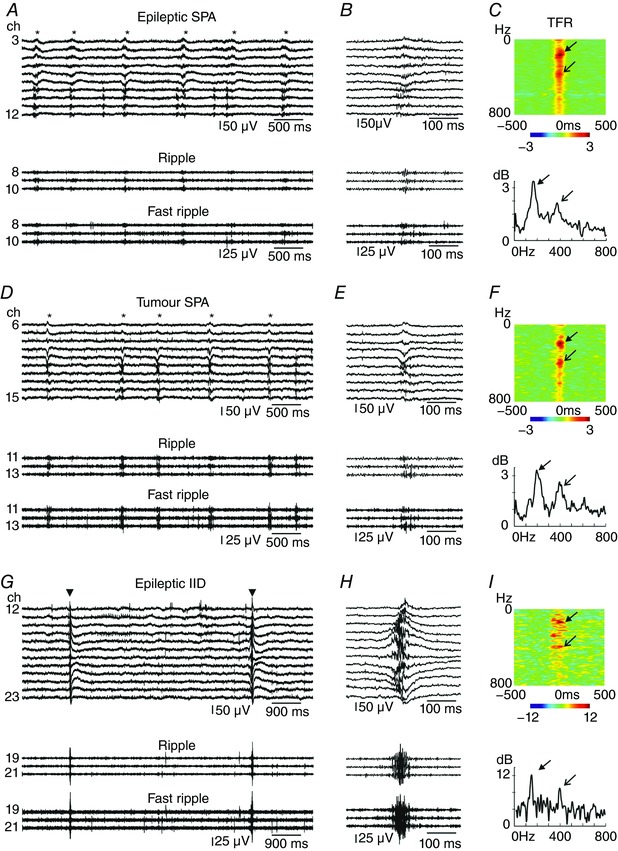
SPAs were more often accompanied by high frequency oscillations in epileptic (A–C) than in tumour (D–F) patients. High frequency oscillations were superimposed on IIDs (G–I) at similar ratios as on epileptic SPAs. Traces in both ripple (A and B, D and E, G and H middle trace) and fast ripple frequency band (A and B, D and E, G and H bottom trace) showed an increased activity during SPAs (asterisks on A and D, upper trace) and IIDs (triangles on G, upper trace). B and E show one magnified SPA event; H shows one IID event. Note the difference in the amplitude of high frequency oscillations between SPAs and IID. Arrows indicate peaks at ripple frequency, open end arrows mark the peaks at fast ripple frequency on the heat maps (C, F and I, upper panels) and on the line plots made at the LFPg peak (at time 0, lower panels) generated using wavelet analysis (C, F and I, lower panels). Time frequency analysis (TFR, C, F and I) shows the results obtained on channel 10 (A and B), channel 13 (D and E) and channel 19 (G and H), respectively. Warm colours depict an increase in frequency power, cold colours show a decrease. Note the scale differences between TFR plots. A and D: *labels the SPA events; G: inverted triangle shows IID events. [Color figure can be viewed at wileyonlinelibrary.com]
Table 6.
High frequency oscillations during SPA and IID
| Number of SPAs/IIDs (n) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total (n) | With HFO | Without HFO | With ripple activity | With fast ripple activity | Ripple frequency (Hz) | Ripple power | Fast ripple frequency (Hz) | Fast ripple power | ||
| Epileptic SPA | Total | 46 | 38 | 8 | 35 | 29 | 178.00 [161.00 193.00] | 1.77 [1.45 2.31] | 440.00 [400.00 512.00] | 1.44 [1.15 1.69] |
| (82.6%) | (17.4%) | (76.1%) | (63.0%) | (180.89 ± 37.39) | (2.07 ± 1.03) | (475.55 ± 130.24) | (1.75 ± 0.91) | |||
| Supragran SPA | 38 | 32 | 6 | 29 | 24 | 180.00 [164.00 193.00] | 1.70 [1.45 2.24] | 437.50 [400.00 534.00] | 1.43 [1.15 1.73] | |
| (84.2%) | (15.8%) | (76.3%) | (63.2%) | (184.14 ± 36.99) | (2.09 ± 1.06) | (486.75 ± 136.83) | (1.67 ± 0.91) | |||
| Infragran SPA | 7 | 6 | 1 | 6 | 5 | 161.50 [127.50 206.50] | 2.19 [0.82 2.84] | 471.00 [329.00 500.00] | 2.26 [0.91 3.07] | |
| (85.7%) | (14.3%) | (85.7%) | (71.4%) | (165.17 ± 38.57) | (1.95 ± 0.95) | (421.80 ± 81.94) | (2.15 ± 0.87) | |||
| Tumour SPA | Total | 48 | 33 | 15 | 30 | 26 | 198.00 [187.00 214.50] | 2.02 [1.61 2.87] | 462.50 [438.00 573.00] | 1.47 [1.31 2.20] |
| (68.8%) | (31.3%) | (62.5%) | (54.2%) | (208.83 ± 43.35) | (2.34 ± 1.08) | (508.81 ± 110.31) | (1.83 ± 0.80) | |||
| Supragran SPA | 36 | 24 | 12 | 21 | 19 | 194.00 [179.00 219.00] | 1.80 [1.42 2.71] | 522.00 [445.00 624.00] | 1.36 [1.20 1.69] | |
| (66.7%) | (33.3%) | (58.3%) | (52.8%) | (209.86 ± 48.24) | (2.17 ± 1.16) | (535.69 ± 113.84) | (1.60 ± 0.70) | |||
| Infragran SPA | 11 | 8 | 3 | 8 | 6 | 208.50 [197.00 248.00] | 3.15 [2.56 3.37] | 433.50 [381.00 488.00] | 2.77 [2.00 3.10] | |
| (72.7%) | (27.3%) | (72.7%) | (54.5%) | (208.75 ± 32.71) | (2.89 ± 0.68) | (434.17 ± 62.96) | (2.62 ± 0.67) | |||
| Epileptic IID | 8 | 7 | 1 | 6 | 7 | 147.95 [135.25 226.30] | 4.90 [3.69 6.04] | 397.50 [317.40 689.00] | 4.01 [3.37 4.23] | |
| (87.5%) | (12.5%) | (75.0%) | (87.5%) | (169.83 ± 52.33) | (4.88 ± 1.09) | (459.46 ± 168.76) | (4.11 ± 1.25) | |||
| Significant differences | n.s. | n.s. | n.s. | Epi SPA < Tumour SPA P < 0.01 | Epi SPA < Epi IID P < 0.001 | n.s. | Epi SPA < Epi IID P < 0.001 | |||
Ripple (130–250 Hz) and fast ripple (300–800 Hz) frequencies were examined in neocortical slices with SPA and IID. Data are medians [95% confidence interval] (mean ± SD). Epi, epileptic; n.s., non‐significant; Supragran, supra, supra‐gran and gran SPAs; Infragran, gran‐infra and infra SPAs.
Role of glutamate and GABA receptors in the generation of SPAs
To reveal the role of glutamate and GABA signalling in the generation of SPAs, we applied the AMPA/kainate glutamate receptor agonist NBQX (5 μm), or the GABAA receptor agonist bicuculline (10 μm) on human neocortical slices. As described previously (Köhling et al. 1998), SPAs were reversibly suppressed by blocking either AMPA/kainate receptors (n = 4 and n = 5 SPAs in slices from 3 tumour and 4 epileptic patients, respectively, Fig. 2 E) or GABAA receptors (n = 8 and n = 9 SPAs in slices from 6 tumour and 8 epileptic patients, respectively). The role of NMDA receptors was investigated with the application of its antagonist dl‐APV (50 μm), which significantly reduced the recurrence frequency of SPAs (n = 4 from 3 tumour patients, n = 5 from 4 epileptic patients) to 80.16 (58.63–99.17) % (P < 0.05), and diminished the LFPg and MUA amplitudes to 90.68 (81.81–105.05) and 86.78 (69.53–110.81) %, respectively (both 0.05 < P < 0.1, Fig. 2 E).
Intracellular correlates of SPAs
Putative pyramidal cells were intracellularly recorded in neocortical slices from epileptic (n = 17) and from tumour (n = 16) patients (Fig. 4, Table 7), simultaneously with the extracellular linear recordings.
Figure 4. Cellular responses of intracellularly recorded cells.
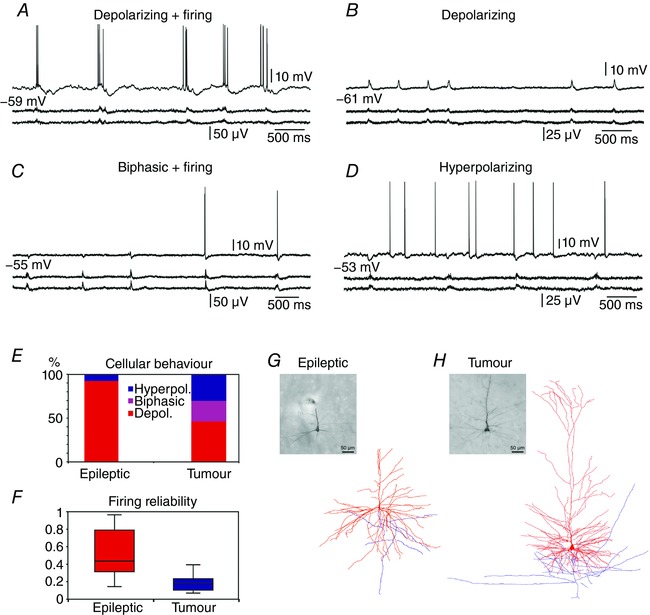
Human neocortical pyramidal cells showed various behaviours during SPAs (two lower traces) in intracellular records (upper trace): depolarizing (A and B), biphasic (C) or hyperpolarizing (D). In epileptic tissue, seven out of 11 depolarizing cells were also discharging (A) during SPA. In tumour tissue, four out of six depolarizing and all three biphasic cells (C) were also firing during SPA, although with significantly lower reliability (F) than neurons from epileptic tissue (P < 0.05). E, the ratio of depolarizing cells was significantly higher in epileptic tissue (P < 0.05), and biphasic cells were only found in tumour tissue. Light micrographs and three‐dimensional reconstructions show a layer V pyramidal cell from epileptic tissue (G) and a layer II pyramidal cell from tumour tissue (H). Note the very complex dendritic arborization (red) of human cells, and the truncated apical dendrite of the cell in (G). Axons are shown in blue. The same scale bar applies to the micrograph and the reconstruction. [Color figure can be viewed at wileyonlinelibrary.com]
Table 7.
Characteristics of intracellularly recorded cells
| Cell | Cortex | Cell location (anatomically identified, layer) | RMP (mV) | Spontaneous firing | SPA location (layer) | Response to SPA | Firing during SPA (% of events) |
|---|---|---|---|---|---|---|---|
| Cells from epileptic tissue | |||||||
| E3 Cell 1 | Frontal | Supra | −58.7 | Silent | No SPA | — | |
| E3 Cell 2 | Frontal | Infra (reconstructed, L5) | −55.5 | Firing | No SPA | — | |
| E5 Cell 1 | Frontal | Infra | −54.0 | Silent | No SPA | — | |
| E5 Cell 2 | Frontal | Infra | −54.9 | Silent | No SPA | — | |
| E8 Cell 1 | Occipital | Supra | −54.9 | Firing | Supra‐gran | Depolarizing + firing | 43.5% |
| E10 Cell 1 | Temporal | Infra (reconstructed, L5) | −52.9 | Firing | Supra | No response | |
| E13 Cell 2 | Temporal | Infra (identified, L5) | −58.0 | Silent | Infra | Depolarizing + firing | 14.3% |
| E13 Cell 3 | Temporal | Supra (identified, L3) | −68.0 | Silent | Supra | Depolarizing | |
| E15 Cell 1 | Temporal | Supra | −59.5 | Firing | Supra‐gran | Depolarizing + firing | 96.5% |
| E15 Cell 2 | Temporal | Supra | −59.8 | Firing | Supra | Depolarizing + firing | 55.6% |
| E15 Cell 3 | Temporal | Supra (identified, L2) | −67.4 | Silent | Supra‐gran | Depolarizing | |
| E16 Cell 1 | Temporal | Supra | −55.5 | Silent | Supra | Depolarizing | |
| Gran | No response | ||||||
| Gran‐infra | Depolarizing | ||||||
| E16 Cell 2 | Temporal | Supra | −52.4 | Silent | Supra | Depolarizing | |
| Gran | Depolarizing | ||||||
| Gran‐infra | Depolarizing | ||||||
| E16 Cell 3 | Temporal | Supra | −55.0 | Firing | Supra | Depolarizing + firing | 29.8% |
| Gran | No response | ||||||
| Gran‐infra | Depolarizing | ||||||
| E16 Cell 4 | Temporal | Infra (reconstructed, L5) | −75.0 | Silent | Supra | No response | |
| Gran | No response | ||||||
| Gran‐infra | Depolarizing + firing | 36.3% | |||||
| E17 Cell 1 | Temporal | Supra | −53.1 | Firing | Supra | Hyperpolarizing | |
| E17 Cell 2 | Temporal | Supra | −52.2 | Firing | Supra | Depolarizing + firing | 86.7% |
| Total (n = 17) | −58.3 ± 6.5 | 9 silent/8 firing |
11 depolarizing 1 hyperpolarizing |
51.8 ± 30.1% (7/12 cells firing) |
|||
| Cells from tumour tissue | |||||||
| T2 Cell 1 | Frontal | Supra | −53.1 | Firing | Supra | Hyperpolarizing | |
| T2 Cell 2 | Frontal | Supra | −54.6 | Firing | Supra | Biphasic + firing | 7.0% |
| T2 Cell 3 | Frontal | Supra (reconstructed, L2) | −71.0 | Firing | Gran | Depolarizing + firing | 11.1% |
| T8 Cell 1 | Temporal | Supra (identified, L3) | −64.2 | Silent | Supra | Hyperpolarizing | |
| T8 Cell 2 | Temporal | Supra | −64.1 | Firing | Supra | Biphasic + firing | 24.0% |
| T11 Cell 1 | Occipital | Supra | −50.1 | Firing | Supra | Depolarizing + firing | 21.7% |
| T14 Cell 1 | Frontal | Supra | −54.7 | Firing | Supra | Hyperpolarizing | |
| T14 Cell 2 | Frontal | Infra | −55.3 | Firing | Supra‐gran | Biphasic + firing | 17.6% |
| T15 Cell 2 | Frontal | Infra | −51.1 | Firing | No SPA | — | |
| T17 Cell 1 | Parietal | Supra | −52.0 | Silent | No SPA | — | |
| T17 Cell 2 | Parietal | Supra | −75.0 | Silent | Supra | Depolarizing | |
| T18 Cell 1 | Parietal | Supra | −68.0 | Firing | Supra | No response | |
| Gran | No response | ||||||
| Supra2 | Depolarizing + firing | 10.0% | |||||
| T23 Cell 1 | Parietal | Supra | −56.7 | Silent | Supra | Depolarizing | |
| T23 Cell 2 | Parietal | Supra | −61.9 | Firing | Supra | Depolarizing + firing | 39.3% |
| T26 Cell 1 | Parietal | Supra | −54.8 | Firing | Supra | Hyperpolarizing | |
| T38 Cell 1 | Parietal | Supra (identified, L3) | −50.2 | Firing | No SPA | — | |
| Total (n = 16) | −58.5 ± 7.8 | 5 silent/11 firing |
6 depolarizing 3 biphasic 4 hyperpolarizing |
18.7 ± 11.0% 7/13 cells firing |
|||
Supra: supragranular; supra‐gran: supragranular+granular; gran: granular; gran‐infra: granular+infragranular; infra: infragranular; L2, L3, L5: layer 2, 3, 5'.
Subsequent anatomical studies in cases of nine cells (six from epileptic and three from tumour tissue; see Table 7) confirmed that intracellularly recorded neurons were indeed pyramidal cells. All intracellularly filled cells showed the characteristics of pyramidal cells: a triangular cell body, a long and thick apical dendrite and numerous thin basal dendrites, covered by mainly thin and mushroom spines. Four well filled cells (one from tumour tissue, three from epileptic tissue, one located in layer II, three in layer V) were reconstructed in three dimensions (Fig. 4 G and H), and had an average total dendritic tree length of 34.04 ± 9.28 mm. We should note that the real dendritic length of our human neocortical pyramidal cells was even higher, since the apical dendrite of three cells was truncated because of the slice preparation procedure.
Both the resting membrane potential and the ratio of spontaneously firing/silent cells at resting membrane potential were similar in epileptic and tumour tissue (Table 7; Mann–Whitney U test, P = 0.69, and Fisher's exact test, P = 0.31, respectively).
As in previous studies (Köhling et al. 1998; Roopun et al. 2010; Pallud et al. 2014), different types of cellular behaviours were detected during SPA (Fig. 4 A–E). Cells in the epileptic slice preparations showed either depolarizing (n = 11/12 cells, 91.7%) or hyperpolarizing (n = 1/12 cells, 8.3%) responses to SPA. At resting potential, seven depolarizing cells (58.3% of responding cells) also fired during SPA, at 43.5% (29.8–86.7%) of the events (median (95% confidence interval); Fig. 4 F).
In the tumour tissue, we found neurons showing depolarizing (n = 6/13 cells, 46.2%, significantly different from epileptic samples, P < 0.05), hyperpolarizing (n = 4/13 cells, 30.7%) or biphasic responses (n = 3/13 cells, 23.1%), which consisted of a hyperpolarization followed by a depolarization. At resting potential, seven cells (53.8% of responding cells) also discharged during SPA. Four of these cells showed depolarizing, three showed biphasic responses to SPA, firing at 17.7% (10.0–24.0%) of the events (median (95% confidence interval); Fig. 4 F, significantly lower than in epileptic tissue, P < 0.05).
Anatomical examinations
When counting NeuN‐immunoreactive neurons, we found slightly lower cell densities in all layers in the epileptic (n = 14202 neurons from two patients) than in the non‐epileptic (n = 8633 neurons from two patients) neocortex (Fig. 5 A and B; Table 8). This is in accordance with previous findings showing that neuron numbers are lower in the epileptic neocortex affected by focal cortical dysplasia compared to the adjacent, non‐affected neocortex (Thom et al. 2005). Furthermore, neuron density was slightly higher in all layers in the regions where SPA was generated than in the area where SPA could not be detected, in specimens from both epileptic (SPA: 1820 ± 1338, no SPA: 1694 ± 1244 cells mm−2) and tumour patients (SPA: 2202 ± 1635, no SPA: 1834 ± 1374 cells mm−2, Table 8).
Figure 5. Anatomical data related to SPA generation.
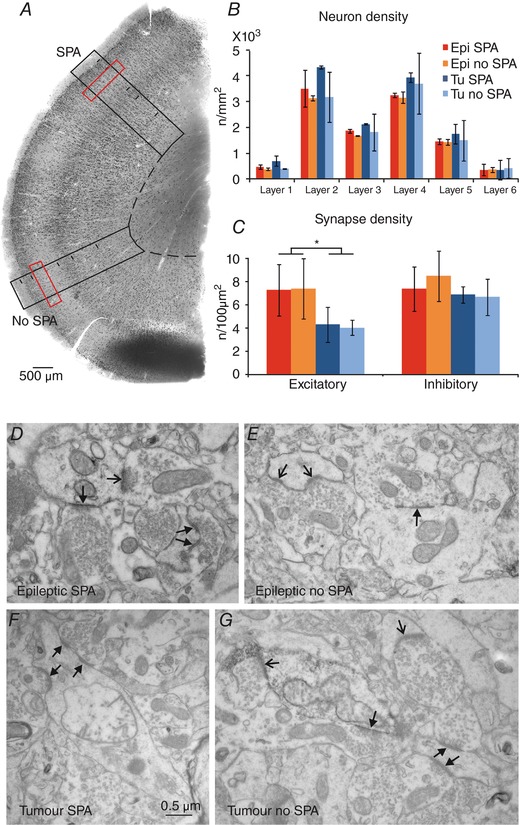
Neuron and synapse counting was performed on NeuN‐stained sections (A). Neuron density (B, black boxes on A) and synapse density (C, red boxes on A) were determined in areas where SPA was recorded and in areas of the same slice where SPA could not be detected. B, neuronal densities (n/mm2) were variable in the different layers of the neocortex of epileptic and tumour patients. Neuron density was lower in epileptic than in tumour tissue, and it was slightly higher in regions with SPA than in areas lacking SPA. C, density of asymmetrical (excitatory) and symmetrical (inhibitory) synapses (n/100 μm2) in epileptic and tumour tissue. No difference was found between areas with or without SPA. The density of excitatory synapses was higher in epileptic than in tumour tissue (P < 0.001). D–G, electron micrographs show asymmetrical (thin‐headed arrows) and symmetrical (triangular‐headed arrows) synapses from epileptic (D and E) and tumour (F and G) tissue, from areas with SPA (D and F) or without SPA (E and G). [Color figure can be viewed at wileyonlinelibrary.com]
Table 8.
Neuron density in the neocortex generating or not generating SPA
| Epileptic | Tumour | |||||
|---|---|---|---|---|---|---|
| Layer | Number of cells counted | Neuron density (n/mm2) SPA | Neuron density (n/mm2) No SPA | Number of cells counted | Neuron density (n/mm2) SPA | Neuron density (n/mm2) No SPA |
| I | 367 | 476 ± 81 | 389 ± 46 | 116 | 705 ± 199 | 398 ± 10 |
| II | 3055 | 3506 ± 707 | 3129 ± 98 | 939 | 4327 ± 57 | 3170 ± 965 |
| III | 3415 | 1866 ± 81 | 1683 ± 8 | 2727 | 2132 ± 27 | 1821 ± 717 |
| IV | 2890 | 3251 ± 77 | 3162 ± 227 | 1840 | 3938 ± 179 | 3700 ± 1172 |
| V | 3913 | 1462 ± 107 | 1428 ± 112 | 2292 | 1753 ± 391 | 1496 ± 778 |
| VI | 562 | 361 ± 223 | 372 ± 97 | 719 | 360 ± 356 | 421 ± 334 |
| Total | 14202 | 1820 ± 1338 | 1694 ± 1244 | 8633 | 2202 ± 1635 | 1834 ± 1374 |
To examine the changes of perisomatic inhibition (Del Rio & DeFelipe, 1994), we counted the parvalbumin (PV)‐positive interneurons in the human epileptic (n = 4) and non‐epileptic (n = 3) neocortical slices. We determined the density of PV‐stained interneurons in areas with and without SPA of the same slice. Density was very variable when comparing areas generating and areas lacking SPA. We found no correlation between the presence of SPA and the number of PV‐stained neurons. The density of PV‐positive cells was lower in areas with SPA than in regions without SPA in three slices from epileptic and in two slices from tumour patients, but it was higher in one and one slice derived from epileptic and tumour patients. On average, PV‐positive interneuron density was slightly lower in regions generating SPA than areas lacking SPA in epileptic patients (45.0 ± 11.4 cells mm−2 in regions with SPA vs. 54.1 ± 19.1 cells mm−2 in regions without SPA), while it was slightly higher in tumour patients (87.7 ± 26.9 cells mm−2 in regions with SPA vs. 78.1 ± 14.5 cells mm−2 in regions without SPA). As previously described (DeFelipe et al. 1993), the overall density of PV‐positive cells was significantly lower (P < 0.05) in epileptic (49.5 ± 15.4 cells mm−2) than in non‐epileptic (82.9 ± 20.0 cells mm−2) neocortex.
Synaptic reorganization has been found in epileptic tissue for both excitatory and inhibitory networks (Marco & DeFelipe, 1997). We examined the account of this phenomenon on the emergence of SPA by investigating 757 synapses in 5136 μm2 in epileptic (n = 3) and 679 synapses in 6144 μm2 in tumour (n = 3) samples. Asymmetrical (presumably excitatory) and symmetrical (presumably inhibitory) synapse numbers per unit area were determined at electron microscopic level. Synapse densities were similar in areas generating and not generating SPA in both epileptic and tumour specimens (Fig. 5 C–G and Table 9). Interestingly, the density of inhibitory synapses was not lower, but slightly higher in epileptic (7.9 ± 1.9 symmetrical synapses per 100 μm2) compared to non‐epileptic tissue (6.7 ± 1.1 synapses per 100 μm2), as could have been expected based on the lower numbers of PV‐positive cells. Moreover, the density of excitatory synapses and, thus, the total synaptic density were significantly higher in epileptic (7.3 ± 2.1 asymmetrical synapses per 100 μm2 and 15.2 ± 3.7 synapses per 100 μm2) than in non‐epileptic tissue (4.2 ± 1.0 asymmetrical synapses per 100 μm2 and 10.9 ± 1.8 synapses per 100 μm2, P < 0.0001).
Table 9.
Number of excitatory (asymmetrical) and inhibitory (symmetrical) synapses in areas generating or not generating SPA
| Excitatory synapses | Inhibitory synapses | All synapses | ||||||
|---|---|---|---|---|---|---|---|---|
| Sample | Examined area (μm2) | Number (% of total) | Number per 100 μm2 | Number (% of total) | Number per 100 μm2 | Number | Number per 100 μm2 | Ratio of excitatory/inhibitory synapses |
| Epileptic SPA | 2904 | 199 (49.4 ± 5.3%) | 7.3 ± 2.2 | 204 (50.6 ± 5.3%) | 7.3 ± 1.9 | 403 | 14.6 ± 3.9 | 0.99 ± 0.21 |
| Epileptic No SPA | 2232 | 165 (46.2 ± 9.2%) | 7.4 ± 2.6 | 189 (53.8 ± 9.2%) | 8.4 ± 2.2 | 354 | 15.8 ± 4.1 | 0.89 ± 0.31 |
| Tumour SPA | 3393 | 149 (37.8 ± 7.7%) | 4.3 ± 1.5 | 233 (62.2 ± 7.7%) | 6.8 ± 0.7 | 382 | 11.1 ± 2.0 | 0.62 ± 0.19 |
| Tumour No SPA | 2751 | 112 (38.0 ± 5.2%) | 4.0 ± 0.7 | 185 (62.0 ± 5.2%) | 6.6 ± 1.6 | 297 | 10.7 ± 2.0 | 0.62 ± 0.14 |
Discussion
Hyperexcitability in the human epileptic neocortex
The main goal of the present study was to explore how the excess excitation of the epileptic neuronal network contributes to the generation of synchronous population bursts. The spontaneously occurring SPAs served as an excellent model for the synchronous activity of neocortical neural assemblies. We found that the hyperexcitability of the human epileptic neocortex is manifested not only at the cellular (for review see Avoli et al. 2005), but also at the network level. In the epileptic compared to non‐epileptic neocortex, SPAs occurred in a higher proportion of slices, more multiple SPAs were detected, and the LFPg amplitude of SPAs was also higher. The higher percentage of depolarizing cells, discharging more frequently during SPA also demonstrate the hyperexcitability of the epileptic neuronal network (see also McCormick & Contreras, 2001). The increased numbers of excitatory synapses together with a slightly decreased neuronal density confirm the phenomenon of epileptic synaptic reorganization (Marco & DeFelipe, 1997), and provide further evidence for an impaired balance between excitatory and inhibitory signalling in the human neocortex. The decreased number of parvalbumin‐positive interneurons (staining perisomatic inhibitory basket and axo‐axonic cells) could indicate an impaired inhibition in the epileptic neocortex (see also DeFelipe et al. 1993). However, when we investigated the density of inhibitory synapses independent of their parvalbumin, contrary to a previous study (Marco & DeFelipe, 1997), we did not observe any loss, but a slightly increased number of inhibitory connections in the epileptic neocortex. This suggests that inhibitory circuits might also participate in the epileptic synaptic reorganization, as they do in the human hippocampus (Wittner et al. 2001).
High frequency oscillations were detected in the human neocortex during both normal and epileptic brain states (Blanco et al. 2010), and increased ripples and fast ripples were proposed to identify the epileptogenic zone (Worrell et al. 2008; Jacobs et al. 2012). Since in vitro slice preparations represent considerably altered conditions compared to in vivo human neocortical circuitry, solid conclusions cannot be made on the presence of high frequency oscillations. However, the same tendency could be observed in our samples as in vivo, i.e. higher numbers of SPAs and IIDs were associated with prominent HFOs in epileptic vs. tumour tissue. In addition, the HFO power of IIDs was about twice as large compared to that of SPAs, supporting the hypersynchronous nature of epileptic processes.
Our observations indicate that the emergence of population activity is related to the level of excitation and synchrony in the human neocortex, although both glutamatergic and GABAergic signalling participate in it (see the pharmacological results). The generation of SPAs in epileptic samples is linked to a higher degree of excitation compared to non‐epileptic tissue. The sprouting of excitatory connections, as well as the higher numbers of depolarizing and more reliably firing cells, contributes to the modification of the neocortical neuronal network, and seems to facilitate the emergence of SPAs (more slices exhibit SPA, more multiple SPAs). The emergence of IIDs is associated with an even more elevated level of excitation and synchrony, reflected in the significantly higher values of LFPg, MUA and HFO power values. Future studies are needed to define (if possible) the subtle border between physiological and pathological processes.
Complexity of the human neocortex
As in a recent study (Mohan et al. 2015), we found that the total dendritic length of human neocortical pyramidal cells (∼34 mm) is over three times as large as that of rodents (∼9–10 mm; Ascoli et al. 2007; Krieger et al. 2007; Chen et al. 2014). The exceptionally long dendritic tree of human neurons offers the possibility of receiving input from a very large number of synapses, and thus, may serve as the anatomical basis of the highly interconnected and reliable neocortical circuitry (Molnár et al. 2008). Human neocortical neurons show heterogeneous firing patterns during interictal spikes (Keller et al. 2010), supporting the complexity of distinct neuronal groups interacting to generate hypersynchronous discharges. The occurrence of multiple SPAs and simultaneous SPAs and IIDs in the same slice also indicate the presence of an exceptionally complex neuronal network able to induce different types of synchronies. Our results provide further evidence that the anatomical and physiological complexity of the human neocortex seems to highly exceed that of rodents. The ability of the neocortical neuronal network to spontaneously generate complex synchronies may contribute to cognitive functions characteristic of humans. The complexity of the neuronal circuit provides the potential of improved encoding capabilities (Fourcaud‐Trocme et al. 2003; Ilin et al. 2013; Eyal et al. 2014), which might have resulted in an evolutionary benefit for humans.
Two distinct types of synchronies generated by human neocortical slices
In the present study, we show that two types of spontaneous synchronous activities can emerge in human neocortical slice preparations. The initiation layers, occurrence rate and network characteristics provided a clear distinction between IIDs and SPAs.
In our records, spontaneous IIDs were generated only in slices from epileptic patients, mainly in the granular or infragranular layers, like in vivo interictal spikes (Ulbert et al. 2004a). The recurrence frequency of IIDs was ∼0.05 Hz, a value comparable to the 4‐amino‐pyridine induced epileptiform activity recorded in human neocortical slices (0.3 to 0.1 Hz; Avoli et al. 1994) and to spiking activity in the subiculum (∼0.15 Hz) of epileptic patients (Fabó et al. 2008). The large LFPg transient, the initial current sink and the considerably enhanced cell firing recalled the characteristics of interictal spikes detected in vivo (Ulbert et al. 2004a; Fabó et al. 2008), and those of the Mg2+‐free model of epileptic activity recorded in the human neocortex in vitro (Avoli et al. 1995). The remarkably high ripple and fast ripple powers during IIDs further indicate the hypersynchronous nature of these bursts. The striking similarity of IIDs to in vivo interictal spikes and to pharmacologically induced epileptiform events recorded in human slices suggest that this activity might be linked to epileptic processes. The fact that they could be recorded only in specimens originating from epileptic patients further supports this idea.
SPAs described in our samples were significantly different from IIDs, and showed remarkable similarities to population activity recorded in epileptic human hippocampal (Cohen et al. 2002; Wozny et al. 2005; Huberfeld et al. 2007; Wittner et al. 2009) and neocortical (Köhling et al. 1998; Roopun et al. 2010; Pallud et al. 2014) slices. The cellular responses of our intracellularly recorded neurons were also comparable to intracellular reflections of spontaneous network activity detected in human neocortical neurons (Schwartzkroin & Knowles, 1984; Schwartzkroin & Haglund, 1986). SPAs were recorded in slices derived from epileptic patients and from tumour patients with no preoperative clinical or electrographic manifestations of epileptic activity. Contrary to IIDs, SPAs were initiated mainly in the supragranular layers (see also Köhling et al. 1998; Pallud et al. 2014), and their recurrence frequency was one order of magnitude higher than that of IIDs. They were also characterized by a field potential transient with superimposed high frequency oscillations and increased cellular activity. However, all examined network properties (LFPg, CSD, MUA, high frequency oscillation power) gave significantly lower values compared to IIDs, suggesting that considerably lower numbers of neurons participate in these synchronous events.
Epileptic activity in human tissue in vitro?
The question whether human cortical tissue retains its epileptic activity has been investigated since the first in vitro human studies (in the 1970s, for review see Avoli et al. 2005). Spontaneous interictal‐like activity was very rarely observed, whereas ictal‐like activity could not be recorded in human preparations, even in tissue derived from brain regions chronically involved in seizure activity. However, both interictal and ictal‐like activities could be induced by appropriate pharmacological agents. The most obvious conclusions from this work were that deafferentation might be linked to the disappearance of epileptic activity, or that foci of epileptic activity are small, and thus difficult to detect (Köhling et al. 1998). The size of the tissue sample – and thus, the number of cells constituting the neuronal network – has been related to the emergence of synchronous activity in the rodent hippocampus (Wu et al. 2005), suggesting that a minimal number of neurons and connections (Wittner & Miles, 2007) are required to generate synchronous activity. A systematic study would be needed to reveal what is the smallest subset of a human cortical network which can retain its spontaneous epileptic activity. Knowledge of differences in neuron/glia numbers, synaptic connectivity, and receptor/transmitter systems in the epileptic cortex compared to non‐epileptic samples would also help to make an estimation of the smallest tissue sample able to generate epileptic activity in vitro.
The epileptogenicity of the in vitro spontaneous network events has been a debated question since the beginning of human in vitro research. The picture is notably complex with regard to different species and different cortical regions. Since healthy human samples are not available, researchers often use animal tissue as control. In vitro spontaneous neocortical network events could not be detected either in healthy rodents (Köhling et al. 1998; Avoli et al. 2005) or in primates (Schwartzkroin & Haglund, 1986). Complex population bursts could, however, be evoked in neocortical slices derived from non‐epileptic patients by activating single pyramidal cells (Molnár et al. 2008). Population activity arising in healthy monkey hippocampal slices was also considered to be proof that spontaneous synchronous bursts are not related to epilepsy (Schwartzkroin & Haglund, 1986). Accordingly, in the last two decades, work exploring the spontaneous network activity in the hippocampus of healthy rodents consistently correlated this type of event to sharp‐wave ripple complexes – the physiologically occurring phenomenon thought to participate in learning and memory consolidation processes (for review see Buzsáki, 2015). On the other hand, the behaviour of human neocortical neurons (Prince & Wong, 1981; Schwartzkroin & Knowles, 1984; Strowbridge et al. 1992; Avoli et al. 2003; Williamson et al. 2003), and the similarities in the wave form of in vitro events and in vivo interictal spikes (Cohen et al. 2002; Pallud et al. 2014) strengthened the idea that in vitro population bursts are linked to epilepsy. Neocortical population events were either compared to sharp waves – the clinical term for the short variant of interictal discharges (Köhling et al. 1998; Köhling et al. 1999) – or were called interictal‐like activity (Pallud et al. 2014). In the latter study network events were recorded in neocortical slices from tumour patients both with and without preoperative seizures. The patients suffered from gliomas known to be highly epileptogenic, and the authors related the emergence of these events to the epileptogenicity of the human peritumoural neocortex. In summary, some studies indicate that in vitro population activity is not related to epileptic processes (Schwartzkroin & Haglund, 1986; Molnár et al. 2008; Szegedi et al. 2016), whereas most authors believe in its epileptiform nature (Prince & Wong, 1981; Schwartzkroin & Knowles, 1984; Strowbridge et al. 1992; Köhling et al. 1998; Cohen et al. 2002; Avoli et al. 2003; Williamson et al. 2003; Pallud et al. 2014).
Our results strongly support the hypothesis that SPAs are not a direct reflection of neocortical epileptogenicity. SPAs detected in slices derived from tumour patients without having preoperative clinical and electrographic manifestations of epileptic seizures were very similar in all aspects to those recorded from tissue resected from epileptic patients. Furthermore, the higher neuronal density in the spots generating SPA also indicates its non‐pathological feature, since focal cortical epilepsies are linked to neuron loss (Thom et al. 2005). However, signs of epileptic hyperexcitability can be observed in human epileptic neocortical slices, since they can generate IIDs as well, a presumably epileptiform synchronous activity.
Additional information
Competing interests
None of the authors has any conflict of interest.
Author contributions
K.T. performed data acquisition and analysis (anatomy and electrophysiology), K.T.H. performed data acquisition, analysis and contributed to the design of the study (electrophysiology), Á.K. performed data acquisition and analysis (electrophysiology), L.E. provided neurological data and human tissue, A.B. provided human tissue, L.E. provided neurological data and human tissue, Z.J. provided neurological data, G.N. provided human tissue, A.S. provided neurological data, D.F. provided neurological data and designed the study, I.U. conceived and designed the study, wrote the article and L.W. designed the study, collected and analysed data (anatomy, electrophysiology), and wrote the article.
Funding
This study was supported by a Postdoctoral Fellowship of the Hungarian Academy of Sciences (to K.T.); by the Hungarian Brain Research Program – Grants No. KTIA_13_NAP‐A‐IV/1‐4,6 and KTIA 13 NAP‐A‐I/1 (to I.U.) and KTIA_NAP_13‐1‐2013‐0001 (to D.F.); by the EU FP7 Grant No. 600925 NeuroSeeker; and by Hungarian National Research Fund grants OTKA K119443 (to L.W.) and PD121123 (to K.T.).
Acknowledgements
The authors would like to thank to Ms K. Lengyel, Ms E. Szépné Simon and Mr Gy. Goda for technical assistance, and Dr Richard Miles for his helpful comments.
Edited by: Jaideep Bains & Katalin Toth
K. Tóth and K. T. Hofer contributed equally.
References
- Alonso‐Nanclares L, Garbelli R, Sola RG, Pastor J, Tassi L, Spreafico R & DeFelipe J (2005). Microanatomy of the dysplastic neocortex from epileptic patients. Brain 128, 158–173. [DOI] [PubMed] [Google Scholar]
- Ascoli GA, Donohue DE & Halavi M (2007). NeuroMorpho.Org: a central resource for neuronal morphologies. J Neurosci 27, 9247–9251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avoli M, Louvel J, Drapeau C, Pumain R & Kurcewicz I (1995). GABAA‐mediated inhibition and in vitro epileptogenesis in the human neocortex. J Neurophysiol 73, 468–484. [DOI] [PubMed] [Google Scholar]
- Avoli M, Louvel J, Mattia D, Olivier A, Esposito V, Pumain R & D'Antuono M (2003). Epileptiform synchronization in the human dysplastic cortex. Epileptic Disord 5(Suppl 2), S45–S50. [PubMed] [Google Scholar]
- Avoli M, Louvel J, Pumain R & Köhling R (2005). Cellular and molecular mechanisms of epilepsy in the human brain. Prog Neurobiol 77, 166–200. [DOI] [PubMed] [Google Scholar]
- Avoli M, Mattia D, Siniscalchi A, Perreault P & Tomaiuolo F (1994). Pharmacology and electrophysiology of a synchronous GABA‐mediated potential in the human neocortex. Neuroscience 62, 655–666. [DOI] [PubMed] [Google Scholar]
- Avoli M & Olivier A (1989). Electrophysiological properties and synaptic responses in the deep layers of the human epileptogenic neocortex in vitro. J Neurophysiol 61, 589–606. [DOI] [PubMed] [Google Scholar]
- Blanco JA, Stead M, Krieger A, Viventi J, Marsh WR, Lee KH, Worrell GA & Litt B (2010). Unsupervised classification of high‐frequency oscillations in human neocortical epilepsy and control patients. J Neurophysiol 104, 2900–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragin A, Engel J Jr, Wilson CL, Fried I & Buzsáki G (1999). High‐frequency oscillations in human brain. Hippocampus 9, 137–142. [DOI] [PubMed] [Google Scholar]
- Buzsáki G (2015). Hippocampal sharp wave‐ripple: A cognitive biomarker for episodic memory and planning. Hippocampus 25, 1073–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JR, Wang BN, Tseng GF, Wang YJ, Huang YS & Wang TJ (2014). Morphological changes of cortical pyramidal neurons in hepatic encephalopathy. BMC Neurosci 15, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clémenceau S, Baulac M & Miles R (2002). On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science 298, 1418–1421. [DOI] [PubMed] [Google Scholar]
- de Curtis M & Avanzini G (2001). Interictal spikes in focal epileptogenesis. Prog Neurobiol 63, 541–567. [DOI] [PubMed] [Google Scholar]
- DeFelipe J, Garcia Sola R, Marco P, del Rio MR, Pulido P & Ramon y Cajal S (1993). Selective changes in the microorganization of the human epileptogenic neocortex revealed by parvalbumin immunoreactivity. Cereb Cortex 3, 39–48. [DOI] [PubMed] [Google Scholar]
- Del Rio MR & DeFelipe J (1994). A study of SMI 32‐stained pyramidal cells, parvalbumin‐immunoreactive chandelier cells, and presumptive thalamocortical axons in the human temporal neocortex. J Comp Neurol 342, 389–408. [DOI] [PubMed] [Google Scholar]
- Eyal G, Mansvelder HD, de Kock CP & Segev I (2014). Dendrites impact the encoding capabilities of the axon. J Neurosci 34, 8063–8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabó D, Maglóczky Z, Wittner L, Pék A, Erőss L, Czirják S, Vajda J, Sólyom A, Rásonyi G, Szűcs A, Kelemen A, Juhos V, Grand L, Dombovári B, Halász P, Freund TF, Halgren E, Karmos G & Ulbert I (2008). Properties of in vivo interictal spike generation in the human subiculum. Brain 131, 485–499. [DOI] [PubMed] [Google Scholar]
- Fourcaud‐Trocme N, Hansel D, van Vreeswijk C & Brunel N (2003). How spike generation mechanisms determine the neuronal response to fluctuating inputs. J Neurosci 23, 11628–11640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel S, Njunting M, Pomper JK, Merschhemke M, Sanabria ER, Eilers A, Kivi A, Zeller M, Meencke HJ, Cavalheiro EA, Heinemann U & Lehmann TN (2004). Stimulus and potassium‐induced epileptiform activity in the human dentate gyrus from patients with and without hippocampal sclerosis. J Neurosci 24, 10416–10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorji A, Hohling JM, Madeja M, Straub H, Kohling R, Tuxhorn I, Ebner A, Wolf P, Panneck HW, Behne F, Lahl R & Speckmann EJ (2002). Effect of levetiracetam on epileptiform discharges in human neocortical slices. Epilepsia 43, 1480–1487. [DOI] [PubMed] [Google Scholar]
- Gulyás AI, Szabó GG, Ulbert I, Holderith N, Monyer H, Erdélyi F, Szabó G, Freund TF & Hájos N (2010). Parvalbumin‐containing fast‐spiking basket cells generate the field potential oscillations induced by cholinergic receptor activation in the hippocampus. J Neurosci 30, 15134–15145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hájos N, Karlócai MR, Németh B, Ulbert I, Monyer H, Szabó G, Erdélyi F, Freund TF & Gulyás AI (2013). Input‐output features of anatomically identified CA3 neurons during hippocampal sharp wave/ripple oscillation in vitro. J Neurosci 33, 11677–11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann U, Gabriel S, Jauch R, Schulze K, Kivi A, Eilers A, Kovacs R & Lehmann TN (2000). Alterations of glial cell function in temporal lobe epilepsy. Epilepsia 41, S185–S189. [DOI] [PubMed] [Google Scholar]
- Hofer KT, Kandrács A, Ulbert I, Pál I, Szabó C, Héja L & Wittner L (2015). The hippocampal CA3 region can generate two distinct types of sharp wave‐ripple complexes, in vitro. Hippocampus 25, 169–186. [DOI] [PubMed] [Google Scholar]
- Huberfeld G, Menendez de la Prida L, Pallud J, Cohen I, Le Van Quyen M, Adam C, Clémenceau S, Baulac M & Miles R (2011). Glutamatergic pre‐ictal discharges emerge at the transition to seizure in human epilepsy. Nat Neurosci 14, 627–634. [DOI] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Clémenceau S, Baulac M, Kaila K, Miles R & Rivera C (2007). Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci 27, 9866–9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilin V, Malyshev A, Wolf F & Volgushev M (2013). Fast computations in cortical ensembles require rapid initiation of action potentials. J Neurosci 33, 2281–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isokawa M & Fried I (1996). Extracellular slow negative transient in the dentate gyrus of human epileptic hippocampus in vitro. Neuroscience 72, 31–37. [DOI] [PubMed] [Google Scholar]
- Jacobs J, Staba R, Asano E, Otsubo H, Wu JY, Zijlmans M, Mohamed I, Kahane P, Dubeau F, Navarro V & Gotman J (2012). High‐frequency oscillations (HFOs) in clinical epilepsy. Prog Neurobiol 98, 302–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller CJ, Truccolo W, Gale JT, Eskandar E, Thesen T, Carlson C, Devinsky O, Kuzniecky R, Doyle WK, Madsen JR, Schomer DL, Mehta AD, Brown EN, Hochberg LR, Ulbert I, Halgren E & Cash SS (2010). Heterogeneous neuronal firing patterns during interictal epileptiform discharges in the human cortex. Brain 133, 1668–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivi A, Lehmann TN, Kovacs R, Eilers A, Jauch R, Meencke HJ, von Deimling A, Heinemann U & Gabriel S (2000). Effects of barium on stimulus‐induced rises of [K+]o in human epileptic non‐sclerotic and sclerotic hippocampal area CA1. Eur J Neurosci 12, 2039–2048. [DOI] [PubMed] [Google Scholar]
- Köhling R, Lucke A, Straub H, Speckmann EJ, Tuxhorn I, Wolf P, Pannek H & Oppel F (1998). Spontaneous sharp waves in human neocortical slices excised from epileptic patients. Brain 121, 1073–1087. [DOI] [PubMed] [Google Scholar]
- Köhling R, Qu M, Zilles K & Speckmann EJ (1999). Current‐source‐density profiles associated with sharp waves in human epileptic neocortical tissue. Neuroscience 94, 1039–1050. [DOI] [PubMed] [Google Scholar]
- Krieger P, Kuner T & Sakmann B (2007). Synaptic connections between layer 5B pyramidal neurons in mouse somatosensory cortex are independent of apical dendrite bundling. J Neurosci 27, 11473–11482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann TN, Gabriel S, Kovacs R, Eilers A, Kivi A, Schulze K, Lanksch WR, Meencke HJ & Heinemann U (2000). Alterations of neuronal connectivity in area CA1 of hippocampal slices from temporal lobe epilepsy patients and from pilocarpine‐treated epileptic rats. Epilepsia 41, S190–S194. [DOI] [PubMed] [Google Scholar]
- Marco P & DeFelipe J (1997). Altered synaptic circuitry in the human temporal neocortex removed from epileptic patients. Exp Brain Res 114, 1–10. [DOI] [PubMed] [Google Scholar]
- McCormick DA (1989). GABA as an inhibitory neurotransmitter in human cerebral cortex. J Neurophysiol 62, 1018–1027. [DOI] [PubMed] [Google Scholar]
- McCormick DA & Contreras D (2001). On the cellular and network bases of epileptic seizures. Annu Rev Physiol 63, 815–846. [DOI] [PubMed] [Google Scholar]
- Mohan H, Verhoog MB, Doreswamy KK, Eyal G, Aardse R, Lodder BN, Goriounova NA, Asamoah B, B Brakspear AB, Groot C, van der Sluis S, Testa‐Silva G, Obermayer J, Boudewijns ZS, Narayanan RT, Baayen JC, Segev I, Mansvelder HD & de Kock CP (2015). Dendritic and axonal architecture of individual pyramidal neurons across layers of adult human neocortex. Cereb Cortex 25, 4839–4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár G, Oláh S, Komlósi G, Füle M, Szabadics J, Varga C, Barzó P & Tamás G (2008). Complex events initiated by individual spikes in the human cerebral cortex. PLoS Biol 6, e222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallud J, Le Van Quyen M, Bielle F, Pellegrino C, Varlet P, Labussiere M, Cresto N, Dieme MJ, Baulac M, Duyckaerts C, Kourdougli N, Chazal G, Devaux B, Rivera C, Miles R, Capelle L & Huberfeld G (2014). Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci Transl Med 6, 244ra289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papatheodoropoulos C & Kostopoulos G (2002). Spontaneous, low frequency (approximately 2‐3 Hz) field activity generated in rat ventral hippocampal slices perfused with normal medium. Brain Res Bull 57, 187–193. [DOI] [PubMed] [Google Scholar]
- Prince DA & Wong RK (1981). Human epileptic neurons studied in vitro. Brain Res 210, 323–333. [DOI] [PubMed] [Google Scholar]
- Roopun AK, Simonotto JD, Pierce ML, Jenkins A, Nicholson C, Schofield IS, Whittaker RG, Kaiser M, Whittington MA, Traub RD & Cunningham MO (2010). A nonsynaptic mechanism underlying interictal discharges in human epileptic neocortex. Proc Natl Acad Sci USA 107, 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzkroin PA & Haglund MM (1986). Spontaneous rhythmic synchronous activity in epileptic human and normal monkey temporal lobe. Epilepsia 27, 523–533. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA & Knowles WD (1984). Intracellular study of human epileptic cortex: in vitro maintenance of epileptiform activity? Science 223, 709–712. [DOI] [PubMed] [Google Scholar]
- Simon A, Traub RD, Vladimirov N, Jenkins A, Nicholson C, Whittaker RG, Schofield I, Clowry GJ, Cunningham MO & Whittington MA (2014). Gap junction networks can generate both ripple‐like and fast ripple‐like oscillations. Eur J Neurosci 39, 46–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strowbridge BW, Masukawa LM, Spencer DD & Shepherd GM (1992). Hyperexcitability associated with localizable lesions in epileptic patients. Brain Res 587, 158–163. [DOI] [PubMed] [Google Scholar]
- Szegedi V, Paizs M, Csákvári E, Molnár G, Barzó P, Tamás G & Lamsa K (2016). Plasticity in single axon glutamatergic connection to GABAergic interneurons regulates complex events in the human neocortex. PLoS Biol 14, e2000237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom M, Martinian L, Sen A, Cross JH, Harding BN & Sisodiya SM (2005). Cortical neuronal densities and lamination in focal cortical dysplasia. Acta neuropathologica 110, 383–392. [DOI] [PubMed] [Google Scholar]
- Tóth K, Erőss L, Vajda J, Halász P, Freund TF & Maglóczky Z (2010). Loss and reorganization of calretinin‐containing interneurons in the epileptic human hippocampus. Brain 133, 2763–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbert I, Halgren E, Heit G & Karmos G (2001). Multiple microelectrode‐recording system for human intracortical applications. J Neurosci Methods 106, 69–79. [DOI] [PubMed] [Google Scholar]
- Ulbert I, Heit G, Madsen J, Karmos G & Halgren E (2004a). Laminar analysis of human neocortical interictal spike generation and propagation: current source density and multiunit analysis in vivo. Epilepsia 45 ( Suppl 4 ), 48–56. [DOI] [PubMed] [Google Scholar]
- Ulbert I, Maglóczky Z, Erőss L, Czirják S, Vajda J, Bognár L, Tóth S, Szabó Z, Halász P, Fabó D, Halgren E, Freund TF & Karmos G (2004b). In vivo laminar electrophysiology co‐registered with histology in the hippocampus of patients with temporal lobe epilepsy. Exp Neurol 187, 310–318. [DOI] [PubMed] [Google Scholar]
- Williamson A, Patrylo PR, Lee S & Spencer DD (2003). Physiology of human cortical neurons adjacent to cavernous malformations and tumors. Epilepsia 44, 1413–1419. [DOI] [PubMed] [Google Scholar]
- Williamson A, Patrylo PR, Pan J, Spencer DD & Hetherington H (2005). Correlations between granule cell physiology and bioenergetics in human temporal lobe epilepsy. Brain 128, 1199–1208. [DOI] [PubMed] [Google Scholar]
- Wittner L, Huberfeld G, Clémenceau S, Erőss L, Dezamis E, Entz L, Ulbert I, Baulac M, Freund TF, Maglóczky Z & Miles R (2009). The epileptic human hippocampal cornu ammonis 2 region generates spontaneous interictal‐like activity in vitro. Brain 132, 3032–3046. [DOI] [PubMed] [Google Scholar]
- Wittner L, Maglóczky Z, Borhegyi Z, Halász P, Tóth S, Erőss L, Szabó Z & Freund TF (2001). Preservation of perisomatic inhibitory input of granule cells in the epileptic human dentate gyrus. Neuroscience 108, 587–600. [DOI] [PubMed] [Google Scholar]
- Wittner L & Miles R (2007). Factors defining a pacemaker region for synchrony in the hippocampus. J Physiol 584, 867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worrell GA, Gardner AB, Stead SM, Hu S, Goerss S, Cascino GJ, Meyer FB, Marsh R & Litt B (2008). High‐frequency oscillations in human temporal lobe: simultaneous microwire and clinical macroelectrode recordings. Brain 131, 928–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozny C, Knopp A, Lehmann TN, Heinemann U & Behr J (2005). The subiculum: a potential site of ictogenesis in human temporal lobe epilepsy. Epilepsia 46 ( Suppl 5 ), 17–21. [DOI] [PubMed] [Google Scholar]
- Wu C, Luk WP, Gillis J, Skinner F & Zhang L (2005). Size does matter: generation of intrinsic network rhythms in thick mouse hippocampal slices. J Neurophysiol 93, 2302–2317. [DOI] [PubMed] [Google Scholar]