

Abstract
Key points
Optogenetics has emerged as a potential alternative to electrotherapy for treating heart rhythm disorders, but its applicability for terminating atrial arrhythmias remains largely unexplored.
We used computational models reconstructed from clinical MRI scans of fibrotic patient atria to explore the feasibility of optogenetic termination of atrial tachycardia (AT), comparing two different illumination strategies: distributed vs. targeted.
We show that targeted optogenetic stimulation based on automated, non‐invasive flow‐network analysis of patient‐specific re‐entry morphology may be a reliable approach for identifying the optimal illumination target in each individual (i.e. the critical AT isthmus).
The above‐described approach yields very high success rates (up to 100%) and requires dramatically less input power than distributed illumination
We conclude that simulations in patient‐specific models show that targeted light pulses lasting longer than the AT cycle length can efficiently and reliably terminate AT if the human atria can be successfully light‐sensitized via gene delivery of ChR2.
Abstract
Optogenetics has emerged as a potential alternative to electrotherapy for treating arrhythmia, but feasibility studies have been limited to ventricular defibrillation via epicardial light application. Here, we assess the efficacy of optogenetic atrial tachycardia (AT) termination in human hearts using a strategy that targets for illumination specific regions identified in an automated manner. In three patient‐specific models reconstructed from late gadolinium‐enhanced MRI scans, we simulated channelrhodopsin‐2 (ChR2) expression via gene delivery. In all three models, we attempted to terminate re‐entrant AT (induced via rapid pacing) via optogenetic stimulation. We compared two strategies: (1) distributed illumination of the endocardium by multi‐optrode grids (number of optrodes, N opt = 64, 128, 256) and (2) targeted illumination of the critical isthmus, which was identified via analysis of simulated activation patterns using an algorithm based on flow networks. The illuminated area and input power were smaller for the targeted approach (19–57.8 mm2; 0.6–1.8 W) compared to the sparsest distributed arrays (N opt = 64; 124.9 ± 6.3 mm2; 3.9 ± 0.2 W). AT termination rates for distributed illumination were low, ranging from <5% for short pulses (1/10 ms long) to ∼20% for longer stimuli (100/1000 ms). When we attempted to terminate the same AT episodes with targeted illumination, outcomes were similar for short pulses (1/10 ms long: 0% success) but improved for longer stimuli (100 ms: 54% success; 1000 ms: 90% success). We conclude that simulations in patient‐specific models show that light pulses lasting longer than the AT cycle length can efficiently and reliably terminate AT in atria light‐sensitized via gene delivery. We show that targeted optogenetic stimulation based on analysis of AT morphology may be a reliable approach for defibrillation and requires less power than distributed illumination.
Keywords: cardiac optogenetics, atrial tachycardia, patient‐specific computational models, anti‐arrhythmia, fibrotic remodelling
Key points
Optogenetics has emerged as a potential alternative to electrotherapy for treating heart rhythm disorders, but its applicability for terminating atrial arrhythmias remains largely unexplored.
We used computational models reconstructed from clinical MRI scans of fibrotic patient atria to explore the feasibility of optogenetic termination of atrial tachycardia (AT), comparing two different illumination strategies: distributed vs. targeted.
We show that targeted optogenetic stimulation based on automated, non‐invasive flow‐network analysis of patient‐specific re‐entry morphology may be a reliable approach for identifying the optimal illumination target in each individual (i.e. the critical AT isthmus).
The above‐described approach yields very high success rates (up to 100%) and requires dramatically less input power than distributed illumination
We conclude that simulations in patient‐specific models show that targeted light pulses lasting longer than the AT cycle length can efficiently and reliably terminate AT if the human atria can be successfully light‐sensitized via gene delivery of ChR2.
Introduction
Cardiac optogenetics is an emerging technology that involves expression of light‐sensitive ion channels (opsins) in heart tissue to enable fine‐grain spatiotemporal control of membrane voltage (V m) via illumination (Entcheva, 2013; Boyle et al. 2015b). A major potential application of this technology is the development of novel light‐based approaches to modulate electrophysiological properties of the beating human heart in clinical settings (Knollmann, 2010; Ambrosi & Entcheva, 2014; Boyle et al. 2014a,b; Karathanos et al. 2016b; Crocini et al. 2017). Optogenetic pacing has been demonstrated in cardiac tissue and in mouse hearts rendered light‐sensitive by viral gene delivery of channelrhodopsin‐2 (ChR2) (Bruegmann et al. 2010; Nussinovitch & Gepstein, 2015; Vogt et al. 2015) or by injection of ChR2‐rich donor cells (Jia et al. 2011; Nussinovitch et al. 2014). Moreover, experiments conducted in ChR2‐expressing cell monolayers (Bingen et al. 2014; Feola et al. 2016) and rodent hearts (Bruegmann et al. 2016; Crocini et al. 2016; Nyns et al. 2017) have shown that optogenetic disruption of re‐entrant arrhythmia is possible. The potential translational feasibility of the latter approach has been confirmed via simulations conducted in computational models of the human ventricles (Bruegmann et al. 2016; Karathanos et al. 2016a). In terms of atrial applications, optogenetic modulation of action potential duration (APD) has been explored via computational modelling (Karathanos et al. 2014). However, the feasibility of light‐based atrial arrhythmia termination at the organ scale remains completely unknown. The atria are an attractive target for optogenetic arrhythmia termination because they have thinner walls than the ventricles, meaning that complete transmural depolarization can be achieved with weaker light stimuli.
Previous studies concerning anti‐arrhythmic applications of optogenetics have largely focused on illuminating the entire ventricular surface with light sources (i.e. optrodes) arranged in uniform (Bingen et al. 2014; Bruegmann et al. 2016; Nyns et al. 2017) or punctate (Karathanos et al. 2016a) spatial patterns. In contrast, experiments in cardiac cell monolayers have highlighted the intricate spatiotemporal control of electrophysiology enabled by optogenetics (e.g. reversing spiral wave chirality; Burton et al. 2015) by using more targeted illumination patterns. Most recently, application of illumination patterns designed to disrupt spiral wave propagation was shown to be highly effective for terminating ventricular arrhythmias in mouse hearts (up to ∼100% success rate with the brightest light pulses attempted) (Crocini et al. 2016). However, it remains unclear whether the ability to achieve fine spatiotemporal control of cardiac activation with light could be leveraged to develop effective and reliable approaches for arrhythmia termination involving targeted depolarization of specific tissue regions based on re‐entrant circuit morphology.
The aim of this study was to assess the feasibility of optogenetic termination of atrial tachyardia (AT) perpetuated by macro‐re‐entry involving a critical isthmus (i.e. a region of tissue that can be ablated to destroy the re‐entrant circuit, rendering AT non‐inducible). To achieve this goal, we conducted simulations in multiscale atrial models incorporating fibrotic remodelling, which were reconstructed from cardiac late gadolinium‐enhanced magnetic resonance imaging (LGE‐MRI) scans of patients with AT. We simulated light‐sensitization of the tissue and induced AT in each model. Then, we compared AT termination success rates for two spatial illumination strategies: (1) distributed illumination of the atrial endocardium by multi‐optrode grids and (2) targeted illumination of the critical isthmus only. Relevant parameters associated with illumination (e.g. optrode density, light pulse duration and onset time) were systematically varied to assess their effect on the success rate of AT termination for the distributed and targeted strategies. Mechanisms of light‐induced termination were quantitatively assessed by calculating the safety factor (SF) of wavefront propagation. We present proof‐of‐concept for optogenetic termination of AT in patient‐specific models of fibrotic human atria and we show that targeted illumination of the critical isthmus tissue region leads to more robust termination of arrhythmia and requires less energy compared to distributed illumination.
Methods
Computational modelling of the fibrotic atria
Patient‐specific electrophysiological models of fibrotic human atria were reconstructed from the LGE‐MRI scans of three individuals. The patients were from a cohort described in a previous study (Zahid et al. 2016b). The Johns Hopkins Institutional Review Board approved the use of imaging data in this retrospective study and all patients provided informed consent. We chose to use three different models to explore the effects of inter‐patient variability in cardiac geometry (e.g. differences in wall thickness). A detailed description of the methodology used to reconstruct atrial models and represent electrophysiology can be found in previous work (Zahid et al. 2016a, b). Briefly, we first extracted each individual's unique atrial geometry and spatial distribution of fibrotic and non‐fibrotic tissue (Fig. 1 A), which were differentiated via image intensity ratio, as described previously (Khurram et al. 2014). Then, fibre orientations were mapped from an atrial atlas geometry into each patient‐specific model using an image‐based estimation approach (McDowell et al. 2012, 2013, 2015; Vadakkumpadan et al. 2012). Finally, model parameters controlling membrane kinetics (Courtemanche et al. 1998; Krummen et al. 2012) and anisotropic conduction velocities (Konings et al. 1994; Li et al. 1999; Burstein et al. 2009) were calibrated to match relevant experimental recordings from fibrotic and non‐fibrotic atrial tissue. Fibrotic tissue was represented by modifying cell‐ and tissue‐scale electrophysiological parameters rather than dense scar. The rationale underlying this approach is outlined in our previous papers (Roney et al. 2016, Zahid et al. 2016a). Our representation assumes that tissue in regions of late gadolinium enhancement comprises an interdigitated mixture of fibroblasts, collagen and myocytes with modified electrophysiology due to fibrogenic signalling. This method is consistent with available data from experimental measurements of atrial tissue subjected to fibrotic remodelling. Although it is possible that inexcitable dense scars might be present in the diseased atria of some individuals, the prevalence of such regions prior to ablation is low (Squara et al. 2014).
Figure 1. Simulation methodology.
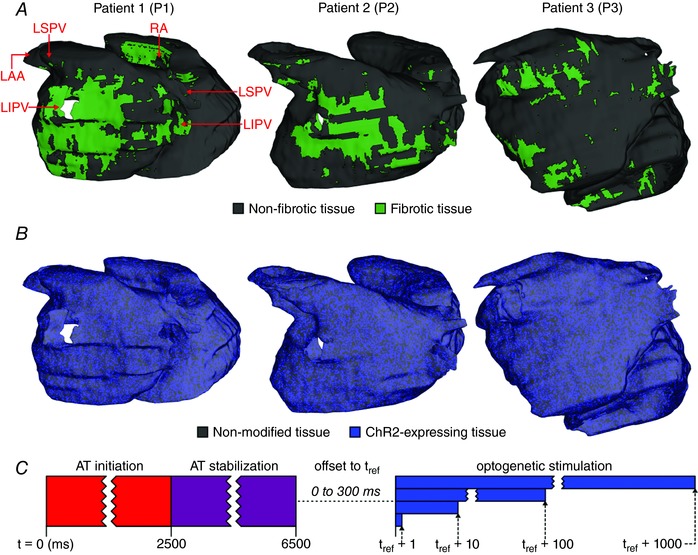
A, 3D renderings of models reconstructed from LGE‐MRI scans including patient‐specific distribution of fibrotic and non‐fibrotic tissue. Models are shown by a posterior view of the LA; key anatomical landmarks are indicated. B, maps showing distribution of ChR2‐expressing tissue. C, schematic diagram summarizing protocol used to assess feasibility of optogenetic AT termination in each model. See Methods for full details. [Color figure can be viewed at wileyonlinelibrary.com]
The validity of AT simulations conducted in patient‐specific models reconstructed according to this method was established in our previous paper (Zahid et al. 2016b), in which we compared critical isthmus locations predicted in silico with locations targeted for catheter ablation during clinical procedures in the corresponding patients. We found that there was a high degree of similarity between the locations predicted by our methodology and those ablated clinically.
Modelling ChR2 expression and light attenuation
Optogenetic transduction of the human atria was simulated using our previously validated multiscale modelling framework (Boyle et al. 2013, 2015a; Ambrosi et al. 2015). ChR2 expression characteristics were modelled as in a recent study (Bruegmann et al. 2016). Briefly, a model of light‐sensitive ChR2 current (Williams et al. 2013) was incorporated in 58.2% of all myocardial tissue with a diffuse spatial pattern (Fig. 1 B), consistent with experimental measurements of ChR2 expression rates in wild‐type mouse hearts after gene transfer (Vogt et al. 2015). Although the latter study did not consider the effects of disease‐related structural changes as we do in the present work, our approach represents fibrotic tissue as myocardial cells with intrinsic electrophysiology and coupling affected by the presence of fibroblasts and other fibrogenic factors (i.e. not as dense scar), so it is reasonable to expect a homogeneous expression level. ChR2 conductance was set to 0.17 mS cm−2 to match photocurrent properties observed in vitro (Vogt et al. 2015). Light attenuation due to scattering and energy absorption in myocardial tissue was modelled using the steady‐state photon diffusion equation, an approach used in numerous cardiac modelling studies (Bishop et al. 2006, 2007; Boyle et al. 2013; Ambrosi et al. 2015; Bruegmann et al. 2016; Karathanos et al. 2016a). For all simulations, we used published values for optical properties of cardiac tissue illuminated by blue light (wavelength, λ = 488 nm, diffusivity, D = 0.18 mm, absorptivity, μa = 0.52 mm–1), resulting in an approximate exponential decay space constant of ∼0.588 mm. Unless noted otherwise, all optogenetic stimuli used a light intensity (irradiance E e = 1.5 mW mm−2) consistent with the maximum value tested in our previous experimental study (Bruegmann et al. 2016).
Modelling different illumination strategies
Distributed light sources were modelled as grids with different numbers (number of optrodes, N opt = 64, 128, or 256) of 1 mm‐radius optrodes on the endocardial surface of the left atrium (LA). Optrode locations were generated by applying the principle of random point relaxation (Turk, 1991). For each grid density in each model, we generated five different sets of optrodes to rule out the possibility of any particular configuration performing unusually well because optrodes were serendipitously placed in locations that favoured arrhythmia termination. To achieve this, we first found a set of 5 × N opt evenly spaced points on the LA endocardium. The latter points were then subdivided into five disjoint subsets of N opt points each, which were also evenly spaced. We calculated the distance between each point and its nearest neighbour for each generated set to ensure that optrodes were always evenly spaced.
The targeted illumination strategy involved optogenetic stimulation of the critical isthmus of the re‐entrant circuit sustaining AT. In each patient model, the critical isthmus was identified by applying a minimum cut (i.e. min‐cut) algorithm, as described and validated in our previous study (Zahid et al. 2016b). Briefly, we recast wavefront propagation during a single AT cycle as a flow network. We then used an algorithm from graph theory (Boykov & Kolmogorov, 2004) to identify the smallest group of nodes that could be removed to separate this network into two discontinuous components. For each model, the region subjected to light in simulations of targeted illumination was defined by the subset of min‐cut points on the LA endocardial surface.
To calculate the input power for each optical stimulus, we assumed light source efficiency of 4.8% to estimate that the required input power (P in) per unit area for illumination stimuli used in this study (E e = 1.5 mW mm−2) would be approximately 31.25 mW mm−2. This was based on a previously described approach (Boyle et al. 2015b), which used reported measurements of blue light stimuli (Kim et al. 2013).
Characterizing robustness of conduction
To better understand mechanisms of light‐induced arrhythmia termination, we calculated the SF of wavefront propagation (Boyle & Vigmond, 2010; Boyle et al. 2014b). This analysis enables quantitative evaluation of the dynamic relationship between electrotonic sources and sinks for wavefront propagation in tissue with complex geometry. Briefly, SF is the ratio between the net charge delivered by the propagating wavefront and the approximate threshold charge required to elicit an action potential in an isolated membrane patch in the same electrophysiological state.
Simulation protocol
The protocol used to induce AT and assess the feasibility of optogenetic arrhythmia termination, via the two illumination strategies described above, is illustrated in Fig. 1 C. As in previous studies (Zahid et al. 2016a, b), sustained arrhythmia was initiated using a clinically relevant burst pacing sequence (Narayan et al. 2012). In each patient model, the stability of the induced AT was verified by confirming that there was no cycle‐to‐cycle variation in re‐entrant wavefront pathway. Then, for each illumination pattern (multi‐optrode grids of different density vs. targeted), we attempted to terminate AT using light pulses with different durations (1, 10, 100 or 1000 ms). To determine whether the likelihood of AT termination was sensitive to the timing of illumination onset, we tested each light pulse with multiple offset times between 0 and 300 ms. Since we previously showed that AT induced in such models has a cycle length <300 ms (Zahid et al. 2016b), this range of offsets ensured that stimuli were delivered at different timings spanning all phases of the re‐entrant cycle. In each patient, six different offsets (60 ms increments) were tested for each distributed illumination scheme and 16 different offsets (20 ms increments) were tested for the targeted illumination scheme. We tested different numbers of offsets for the two illumination strategies due to constraints imposed by limited computing resources. Specifically, it was possible to test more offsets for targeted illumination because there was only one light stimulus pattern per model, as opposed to three optrode densities (N = 64, 128, or 256) per patient for distributed illumination.
All simulations were conducted using the CARP software package (Vigmond et al. 2002; Vigmond et al. 2003). Each second of simulated activity required 40–50 min of computing time on 24 CPU cores (2.80 GHz) in parallel. The total computational burden for simulations discussed in this paper was ∼120,000 CPU hours.
Statistics
Because of the non‐normal distribution and the unequal variances in the data sets involved, the following non‐parametric tests were used: the Kruskal–Wallis test to compare inter‐optrode spacing and the efficacy of illumination approaches; Friedman's test to compare the efficacy of different light pulse durations; and the Mann–Whitney U test to compare LA wall thickness at illuminated sites in different patient models. Statistics were calculated with Prism (GraphPad Software, La Jolla, CA, USA). Data are shown as means ± SEM.
Results
In each patient‐specific atrial model, the re‐entrant circuit sustaining AT was located completely in the LA (Fig. 2 A) and was stable (same location for >7.5 s following the final pacing stimulus). For patients 1 and 3 (i.e. P1 and P3), re‐entry had a figure‐of‐eight morphology (Fig. 2 B, left/right); for P2, arrhythmia was sustained by a single re‐entrant wavefront (Fig. 2 B, middle).
Figure 2. AT in patient‐specific models.
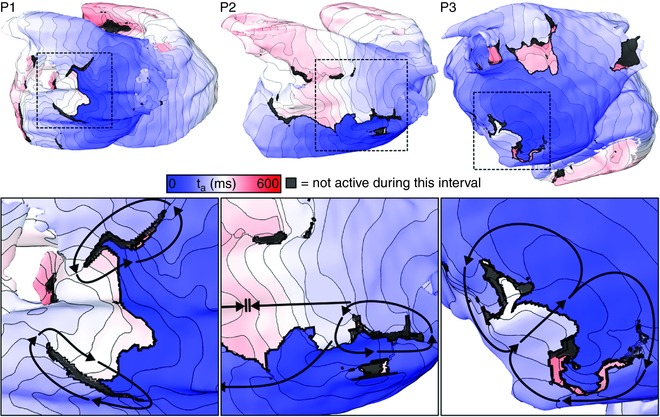
3D maps of activation time (t a) to highlight locations of re‐entrant activity during simulated episodes of AT induced. Arrows indicate directions of wavefront propagation. Double lines indicate conduction block. Dashed boxes indicate inset panel locations in bottom row. Isochronal lines are separated by 10 ms in this figure and all subsequent t a maps. Supplementary Videos 1–3 show the spatiotemporal evolution of V m on the endocardium and epicardium for simulations corresponding to the three columns of this figure. [Color figure can be viewed at wileyonlinelibrary.com]
Distributed LA endocardial arrays with different optrode grid densities are shown in Fig. 3 A. For each density in each patient‐specific atrial model, statistical analysis indicated that there was no significant difference in inter‐optrode spacing (Fig. 4). For the sparsest arrays (64 optrodes distributed uniformly on the LA endocardium), the total illuminated surface area (A i) was 124.9 ± 6.3 mm2, corresponding to an approximate P in of 3.9 ± 0.2 W; for the 128‐ and 256‐optrode arrays, the illuminated area values were 248.5 ± 11.3 mm2 (P in = 7.8 ± 0.4 W) and 499.3 ± 0.2 mm2 (P in = 15.6 ± 0.6 W). Illumination patterns for targeted optogenetic stimulation determined by the min‐cut algorithm are shown in Fig. 3 B. A i and P in values varied between models (P1: A i = 57.8 mm2, P in = 1.8 W; P2: A i = 37.3 mm2, P in = 1.2 W; P3: A i = 19.0 mm2, P in = 0.6 W). Notably, in all cases, the A i and P in values associated with targeted optogenetic stimulation were ≤50% of those corresponding to the sparsest optrode grids.
Figure 3. Distributed and targeted illumination schemes.
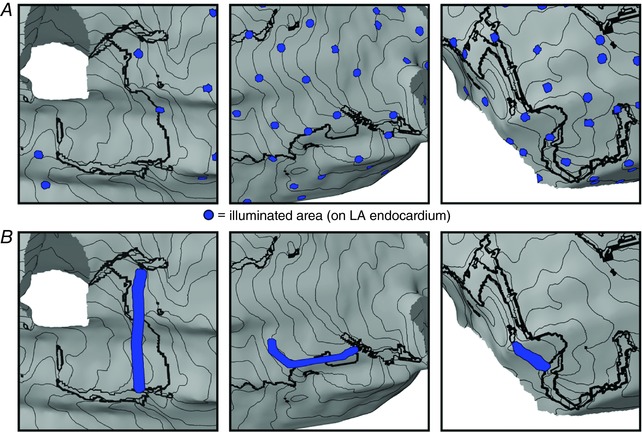
A, maps showing directly illuminated LA endocardial areas for examples of distributed optogenetic stimuli with 64, 128 and 256 optrodes (left, middle and right, respectively). B, same as A but for targeted optogenetic stimuli customized to target regions identified by min‐cut analysis, as described in Methods. In both panels, views and isochrones lines are the same as in inset panels of Fig. 2. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 4. Spatially distributed optrode distributions.
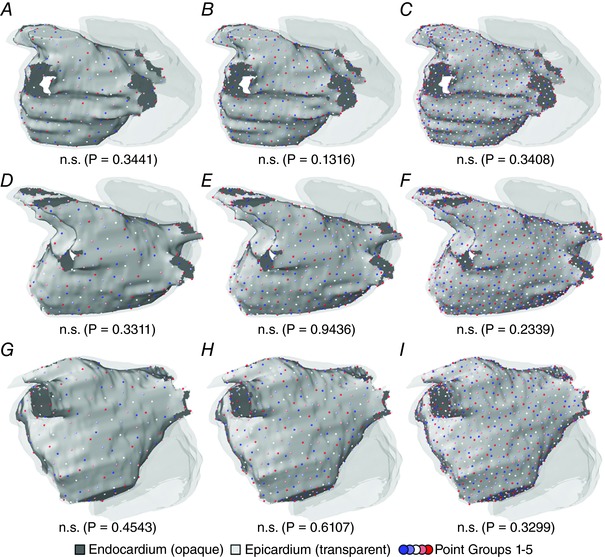
LA endocardium is rendered in opaque grey; other cardiac surfaces are translucent. Each panel shows the five disjoint spatial patterns used for each optrode density in one patient model; different sets are represented by different colours. The value shown beneath each panel is the result of a statistical test used to ascertain whether inter‐point spacing was uneven (Kruskal–Wallis). A–C, distributions with 64, 128 and 256 evenly spaced optrodes, respectively, for the P1 model. D–F, same as A–C but for P2. G–I, same as A–C but for P3. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 5 shows activation maps and snapshots of transmembrane voltage (V m) distribution illustrating the three potential outcomes of stimulation delivered via distributed endocardial arrays. For all examples, the activation sequence in the absence of the applied stimulus (leftmost panel of each row) is provided to facilitate comparison with the transient response to illumination. For the case highlighted in Fig. 5 A, the counter‐clockwise‐rotating wavefront interacted with optogenetically depolarized sites (indicated by asterisk) near the min‐cut location for that patient model (see Fig. 3 B, panel 2), resulting in dramatically slowed conduction. On the subsequent excitation cycle, the wavefront failed to excite this region altogether due to changes in refractoriness. Subsequently, all remaining activity terminated spontaneously. Since the last excitation was observed prior to the end of light stimulation, this outcome was classified as rapid termination.
Figure 5. Response to illumination via three distributed optrode configurations.
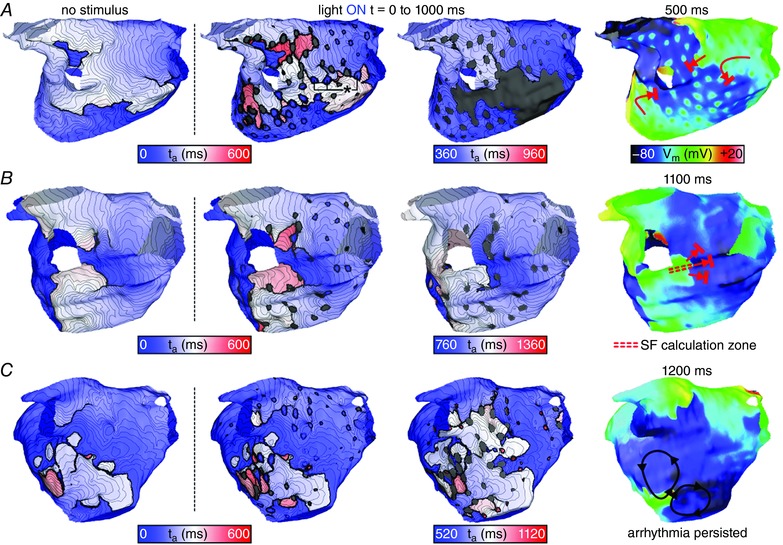
Maps are rendered exclusively on the LA endocardial surface (epicardial surfaces not shown). t a = 0 corresponds to the instant of stimulus onset (regardless of pulse timing). Column 1 shows the excitation sequence that would have occurred in the absence of stimulation; columns 2–4 show the response to illumination and subsequent propagating activity via maps of both ta and V m. Red arrows show wavefront propagation and conduction block for cases where stimulation terminated AT; black arrows show persistent re‐entrant activity following light pulses that failed to terminate AT. A, result of a 1000 ms‐long pulse applied by a 256‐optrode array in the P2 model; classified as rapid termination (see text). *indicates new excitations induced directly by the optogenetic stimulus that were located near the target identified by min‐cut analysis. B, same as A but for illumination with a 128‐optrode array in P1, leading to delayed termination (see text). Double‐dashed red lines indicate pathway along which SF was calculated (see text and Fig. 6). C, same as A but for illumination with a 256‐optrode array in the P3 model, which failed to disrupt AT. Supplementary Videos 4–6 show the spatiotemporal evolution of V m on the endocardium and epicardium for simulations corresponding to panels A–C, respectively. [Color figure can be viewed at wileyonlinelibrary.com]
For some other combinations of distributed light source density and light pulse duration/offset, re‐entrant AT did not terminate until after the end of light stimulation; such cases were classified as delayed termination. An example of this outcome is illustrated in Fig. 5 B. Shortly after illumination onset, light‐induced depolarization caused conduction block near the left inferior pulmonary vein. This resulted in spontaneous conversion from figure‐of‐eight re‐entry into AT perpetuated by a single clockwise‐rotating wavefront, which persisted throughout the 1 s‐long optogenetic stimulus. Then, the AT terminated during the first post‐illumination cycle when the re‐entrant wavefront underwent conduction block as it attempted to excite tissue that had recently recovered from light‐induced depolarization. To pinpoint the underlying mechanism of conduction failure in this case, we calculated SF along the critical pathway (double‐dashed lines in Fig. 5 B). This analysis revealed a drop in SF as the re‐entrant wavefront approached the site of conduction block on the first post‐illumination cycle (Fig. 6). Tissue in this region had recently been depolarized by optogenetic stimulation and subsequently remained refractory, thus acting as an electrotonic sink that leeched excitatory current from the re‐entrant wavefront, ultimately leading to AT termination.
Figure 6. Safety factor (SF) of cardiac conduction.
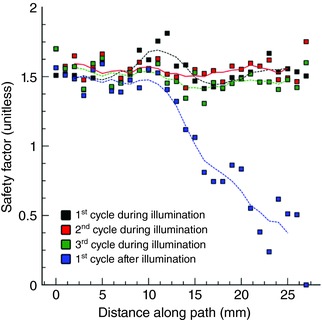
Different‐coloured squares indicate SF values at nodes along the red‐highlighted pathway in Fig. 5 B for three consecutive cycles of AT that persisted during application of a 1000 ms‐long optogenetic stimulus and the subsequent cycle, during which AT terminated via conduction block that occurred in the analysed region. Propagation during AT was robust (i.e. no SF values <1.25 were observed) but quickly deteriorated towards critical values (SF ≤ 1) on the first cycle after the light stimulus was turned off. Dashed lines show the result of applying a five‐point moving average filter to the SF values. [Color figure can be viewed at wileyonlinelibrary.com]
The third (and most common) outcome of distributed illumination was failure to terminate AT, an example of which is shown in Fig. 5 C. For such cases, the re‐entry morphology was unaffected by the presence of multiple small regions of optogenetically depolarized tissue, even when illuminated sites were located near the critical isthmus identified by min‐cut analysis.
A comprehensive summary of all 1080 simulations of distributed illumination (3 patient models × 3 optrode grid densities × 5 distributions per density × 4 pulse durations × 6 pulse timings) is presented in Table 1. The success rate was extremely low for shorter light pulses (<5% for duration of 1 or 10 ms). Longer light pulses modestly improved outcomes (up to ∼20% success rate for duration = 100 or 1000 ms). The incidence of rapid termination was extremely low for the patient models in which AT was driven by figure‐of‐eight re‐entry (2 of 720 cases for P1 and P3) but relatively common in the case where AT was perpetuated by a single re‐entrant source (∼80% of all successes observed for 1000 ms light pulses applied in P2).
Table 1.
Rates of arrhythmia termination (%) for distributed (i.e. optrode grid) illumination configurations in all three patient models
| Patient 1 | Patient 2 | Patient 3 | Average | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of optrodes | Light pulse duration (ms) | Rapid | Delayed | Overall | Rapid | Delayed | Overall | Rapid | Delayed | Overall | Rapid | Delayed | Overall |
| 64 | 1 | ||||||||||||
| 10 | |||||||||||||
| 100 | 20 | 20 | 7 | 7 | |||||||||
| 1000 | 3 | 3 | 23 | 40 | 63 | 8 | 14 | 22 | |||||
| 128 | 1 | ||||||||||||
| 10 | 10 | 10 | 3 | 3 | |||||||||
| 100 | 3 | 14 | 17 | 23 | 23 | 1 | 12 | 13 | |||||
| 1000 | 3 | 3 | 47 | 16 | 63 | 16 | 6 | 22 | |||||
| 256 | 1 | ||||||||||||
| 10 | 10 | 10 | 3 | 3 | |||||||||
| 100 | 53 | 53 | 18 | 18 | |||||||||
| 1000 | 3 | 14 | 17 | 40 | 10 | 50 | 14 | 8 | 22 | ||||
n = 6 pulse timings per entry. See text for explanation of rapid vs. delayed termination.
The activation maps in Fig. 7 show examples of the response to targeted optogenetic stimulation based on min‐cut analysis. In each case, the boundary of the illuminated LA endocardial area is highlighted by a dashed yellow line. For the example classified as rapid termination (Fig. 7 A), the two re‐entrant wavefronts (clockwise and counter‐clockwise) were largely unperturbed by a new wave of propagating excitation that was directly triggered by the light pulse (shown by an asterisk). However, immediately after pivoting back towards the optogenetically depolarized tissue, both re‐entrant wavefronts were blocked and AT terminated abruptly. Similar behaviour was seen for the second example, highlighted in Fig. 7 B. Both wavefronts in the figure‐of‐eight re‐entry pattern failed to propagate into the illuminated region defined by the min‐cut analysis. Because the last excitations spontaneously extinguished ∼200 ms after the end of the 100 ms‐long light pulse, this case was classified as delayed termination. Finally, Fig. 7 C showcases a targeted optogenetic stimulus that failed to terminate AT. For this pulse duration/timing configuration, optogenetic depolarization completely disrupted re‐entrant wavefront propagation near the endocardium but excitation along a similar pathway persisted in the epicardial layer of atrial tissue (shown by pink arrow; see also Supplementary Video 9). Quantitative analysis of atrial geometry (Fig. 8) showed that LA wall thickness in the illuminated region of this model (P2) was significantly larger (median = 3.133 mm) than the stimulated regions in either P1 (median = 2.359 mm; P < 0.0001) or P3 (median = 1.829 mm; P < 0.0001).
Figure 7. Response to illumination via three targeted configurations.
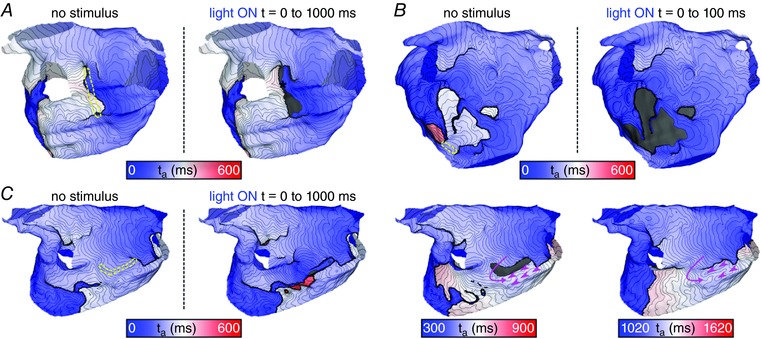
See Fig. 5 for conventions regarding t a, column headings, etc. Yellow dashed lines indicate LA illumination targets identified by min‐cut analysis (see text and Fig. 3 B). A, result of a 1000 ms‐long targeted light pulse in the P1 model; classified as rapid termination (see text). B, same as A but for a 100 ms pulse in P3; classified as delayed termination (see text). C, same as A but for a 1000 ms pulse in P2 that failed to terminate AT. Pink arrows indicate pathways along which wavefronts propagated in the epicardial tissue layer (despite conduction block occurring on the endocardial surface, as rendered here). Pink arrowheads show sites where activity propagating from the epicardial layer broke through and excited the endocardium. Supplementary Videos 7–9 show the spatiotemporal evolution of V m on the endocardium and epicardium for simulations corresponding to panels A–C, respectively. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 8. Analysis of wall thickness.
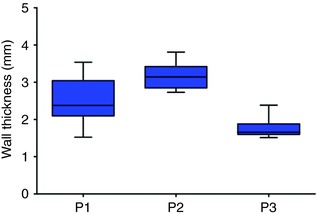
Endocardial to epicardial distances for regions of targeted optogenetic stimulation in all three patient models are shown. Wall thickness was significantly greater in P2 than in either P1 (P < 0.0001, Kruskal–Wallis test with Dunn's multiple comparison post‐test) or P3 (P < 0.0001). Number of endocardial points used for calculation: P1: n = 120, P2: n = 47, P3: n = 35. [Color figure can be viewed at wileyonlinelibrary.com]
Based on the latter analysis, we surmised that the behaviour observed in Fig. 7 C was due to limited light penetration in the vicinity of the particularly thick region of the atria identified by min‐cut analysis for P2. To confirm that this was the case, we repeated all five simulations in the P2 model in which targeted 1000 ms‐duration illumination failed to terminate AT with a more powerful (i.e. deeper penetrating) light source (E e = 10 mW mm−2, consistent with optical stimuli previously used to optogenetically stimulate ChR2‐expressing human iPSC‐CM cells in vitro; Abilez et al. 2011). In all five cases, increased irradiance changed the outcome from failure to rapid AT termination.
Outcomes for all 192 simulations of targeted optogenetic stimulation (3 patient models × 4 pulse durations × 16 pulse timings) are presented in Table 2. Although short light pulses were ineffective (no AT terminations for duration of 1 or 10 ms), the use of intermediate stimulus duration (100 ms) resulted in dramatically increased efficacy (54%). Moreover, for the longest pulse durations tested (1000 ms), the targeted illumination approach was highly effective (90%). Notably, as indicated above, for the small number of failures that were observed for this stimulus type, increasing optical stimulus strength E e from 1.5 to 10 mW mm−2 resulted in successful AT termination. Consistent with findings from distributed light source simulations, rapid termination of AT was much more likely to occur in response to 1000 ms‐long light stimuli (87% of all terminations were rapid) compared to 100 ms pulses (25% rapid).
Table 2.
Rates of arrhythmia termination (%) for targeted (i.e. min‐cut‐based) illumination configurations in all three patient models
| Patient 1 | Patient 2 | Patient 3 | Average | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Light pulse duration (ms) | Rapid | Delayed | Overall | Rapid | Delayed | Overall | Rapid | Delayed | Overall | Rapid | Delayed | Overall |
| 1 | ||||||||||||
| 10 | ||||||||||||
| 100 | 31 | 25 | 56 | 50 | 50 | 12 | 44 | 56 | 14 | 40 | 54 | |
| 1000 | 87 | 13 | 100 | 44 | 25 | 69 | 100 | 100 | 77 | 13 | 90 | |
n = 16 pulse timings per entry. See text for explanation of rapid vs. delayed termination.
Light pulse duration had a significant influence on optogenetic AT termination success rates, regardless of the illumination strategy used (distributed or targeted), with extremely low efficacy values (i.e. overall success rates) for light pulses <100 ms (Fig. 9 A). Finally, the choice of illumination strategy significantly influenced the likelihood of successful AT termination for both 100 ms‐ and 1000 ms‐long light pulses (Fig. 9 B and C, respectively), with the highest success rates observed for targeted illumination in both cases.
Figure 9. Determinants of success in simulations of optogenetic AT termination.
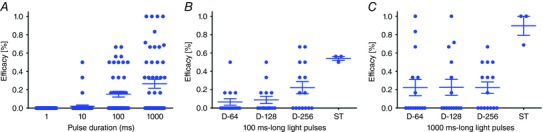
A, effect of light pulse duration on overall efficacy of the optogenetic approach (n = 48 points per pulse duration, P < 0.0001, Friedman's test). Each point shows the total percentage of successful optogenetic AT termination attempts (i.e. rapid + delayed success rates) for one optrode configuration in one patient model. B and C, effect of illumination strategy (i.e. distributed optrode grids of different density (n = 15): D‐64, D‐128, D‐128; spatially targeted (n = 3): ST) on efficacy of the optogenetic approach for 100 ms‐long light pulses (B: P = 0.01, Kruskal–Wallis test) and 1000 ms‐long light pulses (C: P = 0.04). [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
In this study, we used computational models reconstructed from LGE‐MRI scans of the fibrotic atria of individuals with AT to explore the feasibility of optogenetic defibrillation based on viral gene delivery of ChR2 and distributed or targeted illumination strategies. This work provides a rigorous assessment of the potential for organ‐scale translational applications of cardiac optogenetics and presents a novel anti‐AT optogenetic stimulation strategy based on targeting the critical AT isthmus, identified via fully automated, non‐invasive flow‐network analysis of activation patterns in patient‐specific simulations. We showed that (1) light‐based termination of re‐entrant AT in human hearts is theoretically feasible with standard optogenetic constructs (i.e. ChR2‐H134R); (2) targeted optogenetic stimulation based on analysis of patient‐specific re‐entry morphology is a highly reliable approach for terminating AT, with very high success rates (up to 100%), and requires dramatically less input power than distributed illumination; and (3), importantly, we show that each patient's optimal target for optogenetic stimulation could be identified offline via non‐invasive, patient‐specific clinical MRI‐based modelling.
Since optogenetics emerged as a means of exercising fine‐grain control of V m in the heart, researchers in the field have speculated that it might be possible to devise spatially targeted, low energy, light‐based alternatives to cardiac defibrillation (Knollmann, 2010; Ambrosi & Entcheva, 2014; Boyle et al. 2014a, 2015b; Karathanos et al. 2016b; Crocini et al. 2017). Our findings demonstrate that optogenetic depolarization of specific ChR2‐sensitive tissue regions could indeed robustly terminate re‐entrant arrhythmias if the illuminated area is carefully chosen based on the specific pattern of wavefront propagation. Most prior explorations of organ‐scale optogenetic defibrillation have focused on non‐targeted illumination strategies (Bruegmann et al. 2016; Karathanos et al. 2016a; Nyns et al. 2017). Although our study is a departure in terms of optrode spatial distribution, we reach a similar conclusion to the latter papers in terms of light pulse duration. Namely, regardless of species (mouse, rat, human) or arrhythmia type (VT or VF), optical stimuli lasting longer than the arrhythmia cycle length are a baseline requirement for light‐based defibrillation success.
A noteworthy point of contrast is a recent study (Crocini et al. 2016) that used sequences of 10 short light pulses and compared full epicardial illumination to a generalized targeting strategy where light was applied at three rod‐shaped regions (i.e. ‘triple barrier’) on different parts of the ventricular surface. The rationale of this approach was to increase the likelihood that the re‐entrant wavefront would collide with refractory tissue. The two illumination patterns had similar efficacies in terms of terminating VT in mouse hearts (∼50% success for the cases compared), even though the triple‐barrier approach required only the delivery of ∼25 times less energy. When further triple‐barrier experiments were performed with longer light pulses (10 × 10 ms) and stronger stimuli (40 mW mm−2), the success rate increased to 100%. Here, in the context of optogenetic AT termination examined in silico, we show that much weaker light sources can be used (1.5 mW mm−2) and high success rates can be achieved, but only if the light stimulus is considerably longer (i.e. pulse duration exceeds the arrhythmia cycle length) and carefully targeted to depolarize the critical isthmus of the re‐entrant circuit. This is relevant to potential translational applications, since it suggests that optogenetic stimuli may be able to robustly terminate arrhythmia under a wide range of illumination parameters (which may be constrained by technical factors, including light source properties), as long as they are appropriately calibrated to ensure effectiveness and safety (e.g. a 1000 ms light pulse at 40 mW mm−2 might be ruled out due to tissue heating effects).
Our study furthers understanding of the mechanisms by which optogenetic stimulation could extinguish AT (or fail to do so). While light stimuli produce new excitatory wavefronts and/or regions of conduction block proximal to the illuminated tissue, these effects are often not directly responsible for AT termination. As shown by our SF analysis, re‐entrant circuits underlying AT can persist for several cycles spanning the entire interval of illumination, but changes in source–sink mismatch following the end of optogenetic stimulation can then reduce the robustness of wavefront conduction, ultimately leading to conduction block. The high incidence of delayed termination observed in our simulations (see Tables 1 and 2) suggests that this termination mode occurred relatively frequently. Second, our wall thickness analysis shows that some failed attempts to terminate AT can be attributed to incomplete transmural depolarization by optogenetic stimuli applied endocardially. Despite the fact that the atrial walls are thin compared to ventricular tissue (2–4 mm), it is still possible for the endocardium to be robustly depolarized while wavefront propagation continues undeterred in the epicardial layer. This is attributed to the limited penetration depth of blue light (exponential decay with δ ≈ 0.57 mm) and the relatively short space constant of electrotonic coupling (<1 mm). Our results suggest that this potential shortcoming could be overcome by adjusting light pulse intensity on a case‐by‐case basis, which is analogous to the standard shock strength calibration process for implantable electrotherapy devices. Finally, we note that successful AT termination via the mechanism observed in this study depends fundamentally on optogenetic modulation of V m and could not be achieved via conventional electrical stimuli. This is because chronic or prolonged current injection elicits Faradaic reactions, which have deleterious effects in cardiac tissue, meaning that only the shortest duration light pulses used in this study (t = 1–10 ms) could theoretically be mimicked via electric stimuli. Given that most attempts to optogenetically terminate AT with short light pulses failed, it is clear that replacing optrodes with stimulating electrodes would be ineffective.
Taken as a whole, our results provide strong proof‐of‐concept that an optogenetics‐based approach may be able to provide a safe, effective and efficient means of terminating episodes of AT in patients. This is an important finding, since there is a need for better clinical solutions in patients with this type of arrhythmia. One type of AT, atypical LA flutter, occurs in up to 31% of patients following ablation of atrial fibrillation, carries a major risk of stroke, and is difficult to manage pharmacologically (Gerstenfeld et al. 2004; Daoud et al. 2006; Weerasooriya et al. 2009). Potential treatments include cardioversion and catheter ablation but both options have shortcomings. In addition to the fact that it provides only temporary relief from symptoms, external cardioversion is painful (Adlam & Azeem, 2003) and necessitates anaesthesia, which is expensive, time consuming, and can lead to complications (Boodhoo et al. 2004). Conventional identification of ablation targets via intracardiac mapping is tedious and requires invasive electrophysiological study (Patel et al. 2008; Miyazaki et al. 2012). Previously, we validated an automated, non‐invasive solution to determine AT ablation targets by applying the min‐cut algorithm to AT activation sequences predicted by patient‐specific models of the fibrotic atria (Zahid et al. 2016b). The present study combines that technique with the emergent technology of optogenetics to envision a completely novel light‐based approach for robust AT termination.
Our calculation of input power for each optrode configuration also provides a basis for comparison to existing intracardiac electrotherapy approaches for terminating atrial arrhythmia. The threshold energy for biphasic intracardiac atrial cardioversion shocks lasting ∼5 ms is 6.5 ± 3.0 J (Schmitt et al. 1996). This is well above the reported pain threshold of 0.4–1 J (Mitchell et al. 2004). By comparison, we report successful AT termination for light stimuli involving the delivery of less energy over longer times (P in = 0.6–1.8 J, duration = 100–1000 ms). Thus, our results show that targeted optogenetic stimuli could be associated with modest reduction of total delivered energy (∼4–10×) and dramatic reduction of power requirements (∼1000×). As such, should light‐based devices become a reality, the proposed approach could lead to considerable prolongation of battery life for implantable atrial defibrillators (IADs), especially in patients requiring frequent shocks. Our findings are also noteworthy in the context of limited device‐industry interest in IAD development, which is largely due to concerns regarding patient discomfort from shocks (Santini & Santini, 2015). Since optogenetic treatment would involve the delivery of less energy over longer time intervals and excitatory current would be induced only in light‐sensitized myocardium (and not surrounding skeletal muscle), it would theoretically be totally pain‐free. As such, its development could potentially overcome this long‐standing problem associated with IADs.
Of course, as with all translational applications of optogenetics, it is important to recognize that before such an approach could be tested in humans or even live animals, two major technical issues must be overcome: (1) light‐sensitization of the beating heart and (2) intra‐cardiac illumination. Our simulations were designed specifically to probe the feasibility of clearing these hurdles. Whereas our prior work has shown that optogenetic defibrillation of the human ventricles would require the use of theoretical opsins with both increased light sensitivity and red‐shifted absorption spectrum (Bruegmann et al. 2016; Karathanos et al. 2016a), here we show that the widely available ChR2‐H134R variant is sufficient in the context of terminating AT. Moreover, the rate of opsin expression in our models (58.2% of atrial cardiomyocytes) was calibrated to match the reported value one full year after a single AAV9‐ChR2 injection (Vogt et al. 2015). Since there are no known safety concerns associated with adeno‐associated virus‐based treatment in humans (Greenberg et al. 2016), this suggests that it should be feasible to express ChR2 in the heart in vivo. With regard to intracardiac illumination, it is not difficult to envision practical solutions using currently available technologies to achieve the different types of light sources simulated in this paper. Distributed optrode grids could be constructed by extending and adapting basket mapping catheters routinely deployed in patients during AF ablation procedures (Narayan et al. 2014) and replacing electrodes with light emitting diodes. Localized light stimuli such as those used in simulations of targeted illumination could be fabricated using flexible, biocompatible optoelectronics (Kim et al. 2010, 2013) and then implanted in the atria. Due to the low energy requirements for the proposed approach, it might even be possible to control and power such devices wirelessly (Park et al. 2015).
Finally, we note that our study considered a small number of patient‐specific models due to the large computational burden associated with conducting each set of simulations to assess the feasibility of optogenetic AT termination. It is also noteworthy that the proposed approach was only tested for AT involving a macro‐re‐entrant circuit related to a fibrotic substrate. Nonetheless, the fact that stimulation with blue light at the endocardium successfully disrupted re‐entrant arrhythmias in our study does suggest that this type of approach is generally more promising for atrial applications compared to optogenetic defibrillation of the human ventricles, which we have previously shown to be impractical without the use of channels that open in response to deeper‐penetrating red light (Bruegmann et al. 2016; Karathanos et al. 2016a).
In conclusion, we used computational modelling to present the proof‐of‐concept for a novel, patient‐specific approach involving the use of very low‐energy, targeted light pulses to terminate AT in the human atria. In all three atrial models tested, targeted illumination of the critical isthmus of the re‐entrant circuit, which was identified via an automated, non‐invasive approach, terminated AT more effectively and more efficiently (i.e. less energy delivered) compared to distributed illumination via optrode grids. Impressively, we observed near‐perfect efficacy (≥90%) in simulations with long‐duration (1000 ms) targeted light pulses, which suggests that the proposed approach is quite robust and may warrant further investigation (e.g. in a large animal model of atrial arrhythmia).
Additional information
Competing interests
None.
Author contributions
P.M.B.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing. M.J.M.: data analysis and interpretation, manuscript writing. T.V.K.: data analysis and interpretation. S.Z.: conception and design. R.C.B.: conception and design. N.A.T.: conception and design, manuscript writing. All co‐authors approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons listed qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by American Heart Association 16‐SDG‐30440006 (to P.M.B.); by an NSF Graduate Research Fellowship and an ARCS Foundation Award (to S.Z.); and by NIH DP1‐HL123271 (to N.A.T.).
Supporting information
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Video 1. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 2 (Patient 1).
Video 2. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 2 (Patient 2).
Video 3. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 2 (Patient 3).
Video 4. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 5A.
Video 5. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 5B.
Video 6. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 5C.
Video 7. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 7A.
Video 8. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 7B.
Video 9. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 7C.
Acknowledgements
None.
Edited by: Don Bers & Colleen Clancy
References
- Abilez OJ, Wong J, Prakash R, Deisseroth K, Zarins CK & Kuhl E (2011). Multiscale computational models for optogenetic control of cardiac function. Biophys J 101, 1326–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adlam D & Azeem T (2003). Ventricular fibrillation during electrical cardioversion of pre‐excited atrial fibrillation. Postgrad Med J 79, 297–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosi CM, Boyle PM, Chen K, Trayanova NA & Entcheva E (2015). Optogenetics‐enabled assessment of viral gene and cell therapy for restoration of cardiac excitability. Sci Rep 5, 17350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosi CM & Entcheva E (2014). Optogenetics' promise: pacing and cardioversion by light? Future Cardiol 10, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingen BO, Engels MC, Schalij MJ, Jangsangthong W, Neshati Z, Feola I, Ypey DL, Askar SF, Panfilov AV, Pijnappels DA & de Vries AA (2014). Light‐induced termination of spiral wave arrhythmias by optogenetic engineering of atrial cardiomyocytes. Cardiovasc Res 104, 194–205. [DOI] [PubMed] [Google Scholar]
- Bishop MJ, Rodriguez B, Eason J, Whiteley JP, Trayanova N & Gavaghan DJ (2006). Synthesis of voltage‐sensitive optical signals: application to panoramic optical mapping. Biophys J 90, 2938–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop MJ, Rodriguez B, Qu F, Efimov IR, Gavaghan DJ & Trayanova NA (2007). The role of photon scattering in optical signal distortion during arrhythmia and defibrillation. Biophys J 93, 3714–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boodhoo L, Bordoli G, Mitchell AR, Lloyd G, Sulke N & Patel N (2004). The safety and effectiveness of a nurse led cardioversion service under sedation. Heart 90, 1443–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boykov Y & Kolmogorov V (2004). An experimental comparison of min‐cut/max‐flow algorithms for energy minimization in vision. IEEE Trans Pattern Anal Mach Intell 26, 1124–1137. [DOI] [PubMed] [Google Scholar]
- Boyle PM, Entcheva E & Trayanova NA (2014a). See the light: can optogenetics restore healthy heartbeats? And, if it can, is it really worth the effort? Expert Rev Cardiovasc Ther 12, 17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PM, Karathanos TV, Entcheva E & Trayanova NA (2015a). Computational modeling of cardiac optogenetics: Methodology overview & review of findings from simulations. Comput Biol Med 65, 200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PM, Karathanos TV & Trayanova NA (2015b). “Beauty is a light in the heart”: the transformative potential of optogenetics for clinical applications in cardiovascular medicine. Trends Cardiovasc Med 25, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PM, Park CJ, Arevalo HJ, Vigmond EJ & Trayanova NA (2014b). Sodium current reduction unmasks a structure‐dependent substrate for arrhythmogenesis in the normal ventricles. PLoS One 9, e86947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PM & Vigmond EJ (2010). An intuitive safety factor for cardiac propagation. Biophys J 98, L57–L59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PM, Williams JC, Ambrosi CM, Entcheva E & Trayanova NA (2013). A comprehensive multiscale framework for simulating optogenetics in the heart. Nat Commun 4, 2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruegmann T, Boyle PM, Vogt CC, Karathanos TV, Arevalo HJ, Fleischmann BK, Trayanova NA & Sasse P (2016). Optogenetic defibrillation terminates ventricular arrhythmia in mouse hearts and human simulations. J Clin Invest 126, 3894–3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruegmann T, Malan D, Hesse M, Beiert T, Fuegemann CJ, Fleischmann BK & Sasse P (2010). Optogenetic control of heart muscle in vitro and in vivo. Nat Methods 7, 897–900. [DOI] [PubMed] [Google Scholar]
- Burstein B, Comtois P, Michael G, Nishida K, Villeneuve L, Yeh YH & Nattel S (2009). Changes in connexin expression and the atrial fibrillation substrate in congestive heart failure. Circ Res 105, 1213–1222. [DOI] [PubMed] [Google Scholar]
- Burton RAB, Klimas A, Ambrosi CM, Tomek J, Corbett A, Entcheva E & Bub G (2015). Optical control of excitation waves in cardiac tissue. Nat Photon 9, 813–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtemanche M, Ramirez RJ & Nattel S (1998). Ionic mechanisms underlying human atrial action potential properties: insights from a mathematical model. Am J Physiol Heart Circ Physiol 275, H301–H321. [DOI] [PubMed] [Google Scholar]
- Crocini C, Ferrantini C, Coppini R, Scardigli M, Yan P, Loew LM, Smith G, Cerbai E, Poggesi C, Pavone FS & Sacconi L (2016). Optogenetics design of mechanistically‐based stimulation patterns for cardiac defibrillation. Sci Rep 6, 35628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocini C, Ferrantini C, Pavone FS & Sacconi L (2017). Optogenetics gets to the heart: A guiding light beyond defibrillation. Prog Biophys Mol Biol 130, 132–139. [DOI] [PubMed] [Google Scholar]
- Daoud EG, Weiss R, Augostini R, Hummel JD, Kalbfleisch SJ, Van Deren JM, Dawson G & Bowman K (2006). Proarrhythmia of circumferential left atrial lesions for management of atrial fibrillation. J Cardiovasc Electrophysiol 17, 157–165. [DOI] [PubMed] [Google Scholar]
- Entcheva E (2013). Cardiac optogenetics. Am J Physiol Heart Circ Physiol 304, H1179–H1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feola I, Teplenin A, de Vries AA & Pijnappels DA (2016). Optogenetic engineering of atrial cardiomyocytes. Methods Mol Biol 1408, 319–331. [DOI] [PubMed] [Google Scholar]
- Gerstenfeld EP, Callans DJ, Dixit S, Russo AM, Nayak H, Lin D, Pulliam W, Siddique S & Marchlinski FE (2004). Mechanisms of organized left atrial tachycardias occurring after pulmonary vein isolation. Circulation 110, 1351–1357. [DOI] [PubMed] [Google Scholar]
- Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS, Barnard D, Bouchard A, Jaski B, Lyon AR, Pogoda JM, Rudy JJ & Zsebo KM (2016). Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double‐blind, placebo‐controlled, phase 2b trial. Lancet 387, 1178–1186. [DOI] [PubMed] [Google Scholar]
- Jia Z, Valiunas V, Lu Z, Bien H, Liu H, Wang H‐Z, Rosati B, Brink PR, Cohen IS & Entcheva E (2011). Stimulating cardiac muscle by light: cardiac optogenetics by cell delivery. Circ Arrhythm Electrophysiol 4, 753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karathanos TV, Bayer JD, Wang D, Boyle PM & Trayanova NA (2016a). Opsin spectral sensitivity determines the effectiveness of optogenetic termination of ventricular fibrillation in the human heart: a simulation study. J Physiol 594, 6879–6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karathanos TV, Boyle PM & Trayanova NA (2014). Optogenetics‐enabled dynamic modulation of action potential duration in atrial tissue: feasibility of a novel therapeutic approach. Europace 16(Suppl 4), iv69–iv76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karathanos TV, Boyle PM & Trayanova NA (2016b). Light‐based approaches to cardiac arrhythmia research: from basic science to translational applications. Clin Med Insights Cardiol 10, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurram IM, Beinart R, Zipunnikov V, Dewire J, Yarmohammadi H, Sasaki T, Spragg DD, Marine JE, Berger RD, Halperin HR, Calkins H, Zimmerman SL & Nazarian S (2014). Magnetic resonance image intensity ratio, a normalized measure to enable interpatient comparability of left atrial fibrosis. Heart Rhythm 11, 85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim RH, Kim DH, Xiao J, Kim BH, Park SI, Panilaitis B, Ghaffari R, Yao J, Li M, Liu Z, Malyarchuk V, Kim DG, Le AP, Nuzzo RG, Kaplan DL, Omenetto FG, Huang Y, Kang Z & Rogers JA (2010). Waterproof AlInGaP optoelectronics on stretchable substrates with applications in biomedicine and robotics. Nat Mater 9, 929–937. [DOI] [PubMed] [Google Scholar]
- Kim TI, McCall JG, Jung YH, Huang X, Siuda ER, Li Y, Song J, Song YM, Pao HA, Kim RH, Lu C, Lee SD, Song IS, Shin G, Al‐Hasani R, Kim S, Tan MP, Huang Y, Omenetto FG, Rogers JA & Bruchas MR (2013). Injectable, cellular‐scale optoelectronics with applications for wireless optogenetics. Science 340, 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knollmann BC (2010). Pacing lightly: optogenetics gets to the heart. Nat Methods 7, 889–891. [DOI] [PubMed] [Google Scholar]
- Konings KT, Kirchhof CJ, Smeets JR, Wellens HJ, Penn OC & Allessie MA (1994). High‐density mapping of electrically induced atrial fibrillation in humans. Circulation 89, 1665–1680. [DOI] [PubMed] [Google Scholar]
- Krummen DE, Bayer JD, Ho J, Ho G, Smetak MR, Clopton P, Trayanova NA & Narayan SM (2012). Mechanisms of human atrial fibrillation initiation: clinical and computational studies of repolarization restitution and activation latency. Circ Arrhythm Electrophysiol 5, 1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Fareh S, Leung TK & Nattel S (1999). Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation 100, 87–95. [DOI] [PubMed] [Google Scholar]
- McDowell KS, Vadakkumpadan F, Blake R, Blauer J, Plank G, MacLeod RS & Trayanova NA (2012). Methodology for patient‐specific modeling of atrial fibrosis as a substrate for atrial fibrillation. J Electrocardiol 45, 640–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell KS, Vadakkumpadan F, Blake R, Blauer J, Plank G, Macleod RS & Trayanova NA (2013). Mechanistic inquiry into the role of tissue remodeling in fibrotic lesions in human atrial fibrillation. Biophys J 104, 2764–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell KS, Zahid S, Vadakkumpadan F, Blauer J, MacLeod RS & Trayanova NA (2015). Virtual electrophysiological study of atrial fibrillation in fibrotic remodeling. PLoS One 10, e0117110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AR, Spurrell PA, Boodhoo LE & Sulke N (2004). Long‐term care of the patient with the atrial defibrillator. Am Heart J 147, 210–217. [DOI] [PubMed] [Google Scholar]
- Miyazaki S, Shah AJ, Kobori A, Kuwahara T & Takahashi A (2012). How to approach reentrant atrial tachycardia after atrial fibrillation ablation. Circ Arrhythm Electrophysiol 5, e1–e7. [DOI] [PubMed] [Google Scholar]
- Narayan SM, Baykaner T, Clopton P, Schricker A, Lalani GG, Krummen DE, Shivkumar K & Miller JM (2014). Ablation of rotor and focal sources reduces late recurrence of atrial fibrillation compared with trigger ablation alone: extended follow‐up of the CONFIRM trial (Conventional Ablation for Atrial Fibrillation With or Without Focal Impulse and Rotor Modulation). J Am Coll Cardiol 63, 1761–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan SM, Krummen DE, Shivkumar K, Clopton P, Rappel WJ & Miller JM (2012). Treatment of atrial fibrillation by the ablation of localized sources: CONFIRM (Conventional Ablation for Atrial Fibrillation With or Without Focal Impulse and Rotor Modulation) trial. J Am Coll Cardiol 60, 628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinovitch U & Gepstein L (2015). Optogenetics for in vivo cardiac pacing and resynchronization therapies. Nat Biotechnol 33, 750–754. [DOI] [PubMed] [Google Scholar]
- Nussinovitch U, Shinnawi R & Gepstein L (2014). Modulation of cardiac tissue electrophysiological properties with light‐sensitive proteins. Cardiovasc Res 102, 176–187. [DOI] [PubMed] [Google Scholar]
- Nyns EC, Kip A, Bart CI, Plomp JJ, Zeppenfeld K, Schalij MJ, de Vries AA & Pijnappels DA (2017). Optogenetic termination of ventricular arrhythmias in the whole heart: towards biological cardiac rhythm management. Eur Heart J 38, 2132–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SI, Brenner DS, Shin G, Morgan CD, Copits BA, Chung HU, Pullen MY, Noh KN, Davidson S, Oh SJ, Yoon J, Jang KI, Samineni VK, Norman M, Grajales‐Reyes JG, Vogt SK, Sundaram SS, Wilson KM, Ha JS, Xu R, Pan T, Kim TI, Huang Y, Montana MC, Golden JP, Bruchas MR, Gereau RW 4th & Rogers JA (2015). Soft, stretchable, fully implantable miniaturized optoelectronic systems for wireless optogenetics. Nat Biotechnol 33, 1280–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AM, d'Avila A, Neuzil P, Kim SJ, Mela T, Singh JP, Ruskin JN & Reddy VY (2008). Atrial tachycardia after ablation of persistent atrial fibrillation: identification of the critical isthmus with a combination of multielectrode activation mapping and targeted entrainment mapping. Circ Arrhythm Electrophysiol 1, 14–22. [DOI] [PubMed] [Google Scholar]
- Roney CH, Bayer JD, Zahid S, Meo M, Boyle PMJ, Trayanova NA, Haïssaguerre M, Dubois R, Cochet H & Vigmond EJ (2016). Modelling methodology of atrial fibrosis affects rotor dynamics and electrograms. Europace 18, iv146–iv155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini L & Santini M (2015). The role of implantable devices to treat atrial fibrillation. Future Cardiol 11, 689–695. [DOI] [PubMed] [Google Scholar]
- Schmitt C, Alt E, Plewan A, Ammer R, Leibig M, Karch M & Schomig A (1996). Low energy intracardiac cardioversion after failed conventional external cardioversion of atrial fibrillation. J Am Coll Cardiol 28, 994–999. [DOI] [PubMed] [Google Scholar]
- Squara F, Frankel DS, Schaller R, Kapa S, Chik WW, Callans DJ, Marchlinski FE & Dixit S (2014). Voltage mapping for delineating inexcitable dense scar in patients undergoing atrial fibrillation ablation: a new end point for enhancing pulmonary vein isolation. Heart Rhythm 11, 1904–1911. [DOI] [PubMed] [Google Scholar]
- Turk G (1991). Generating textures on arbitrary surfaces using reaction‐diffusion. In SIGGRAPH ′91. Proceedings of the 18th Annual Conference on Computer Graphics and Interactive Techniques, pp. 289–298. ACM, New York.
- Vadakkumpadan F, Arevalo H, Ceritoglu C, Miller M & Trayanova N (2012). Image‐based estimation of ventricular fiber orientations for personalized modeling of cardiac electrophysiology. IEEE Trans Med Imaging 31, 1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigmond EJ, Aguel F & Trayanova NA (2002). Computational techniques for solving the bidomain equations in three dimensions. IEEE Trans Biomed Eng 49, 1260–1269. [DOI] [PubMed] [Google Scholar]
- Vigmond EJ, Hughes M, Plank G & Leon LJ (2003). Computational tools for modeling electrical activity in cardiac tissue. J Electrocardiol 36(Suppl), 69–74. [DOI] [PubMed] [Google Scholar]
- Vogt CC, Bruegmann T, Malan D, Ottersbach A, Roell W, Fleischmann BK & Sasse P (2015). Systemic gene transfer enables optogenetic pacing of mouse hearts. Cardiovasc Res 106, 338–343. [DOI] [PubMed] [Google Scholar]
- Weerasooriya R, Jais P, Wright M, Matsuo S, Knecht S, Nault I, Sacher F, Deplagne A, Bordachar P, Hocini M & Haissaguerre M (2009). Catheter ablation of atrial tachycardia following atrial fibrillation ablation. J Cardiovasc Electrophysiol 20, 833–838. [DOI] [PubMed] [Google Scholar]
- Williams JC, Xu J, Lu Z, Klimas A, Chen X, Ambrosi CM, Cohen IS & Entcheva E (2013). Computational optogenetics: empirically‐derived voltage‐ and light‐sensitive channelrhodopsin‐2 model. PLoS Comput Biol 9, e1003220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahid S, Cochet H, Boyle PM, Schwarz EL, Whyte KN, Vigmond EJ, Dubois R, Hocini M, Haissaguerre M, Jais P & Trayanova NA (2016a). Patient‐derived models link re‐entrant driver localization in atrial fibrillation to fibrosis spatial pattern. Cardiovasc Res 110, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahid S, Whyte KN, Schwarz EL, Blake RC 3rd, Boyle PM, Chrispin J, Prakosa A, Ipek EG, Pashakhanloo F, Halperin HR, Calkins H, Berger RD, Nazarian S & Trayanova NA (2016b). Feasibility of using patient‐specific models and the “minimum cut” algorithm to predict optimal ablation targets for left atrial flutter. Heart Rhythm 13, 1687–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supporting information has been peer‐reviewed but not copyedited.
Video 1. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 2 (Patient 1).
Video 2. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 2 (Patient 2).
Video 3. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 2 (Patient 3).
Video 4. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 5A.
Video 5. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 5B.
Video 6. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 5C.
Video 7. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 7A.
Video 8. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 7B.
Video 9. Epicardial and endocardial renderings of voltage sequence corresponding to Fig. 7C.