

Abstract
Key points
While several studies have investigated the effects of exercise training in human skeletal muscle and the chronic effect of β2‐agonist treatment in rodent muscle, their effects on muscle proteome signature with related functional measures in humans are still incompletely understood.
Herein we show that daily β2‐agonist treatment attenuates training‐induced enhancements in exercise performance and maximal oxygen consumption, and alters muscle proteome signature and phenotype in trained young men.
Daily β2‐agonist treatment abolished several of the training‐induced enhancements in muscle oxidative capacity and caused a repression of muscle metabolic pathways; furthermore, β2‐agonist treatment induced a slow‐to‐fast twitch muscle phenotype transition.
The present study indicates that chronic β2‐agonist treatment confounds the positive effect of high intensity training on exercise performance and oxidative capacity, which is of interest for the large proportion of persons using inhaled β2‐agonists on a daily basis, including athletes.
Abstract
Although the effects of training have been studied for decades, data on muscle proteome signature remodelling induced by high intensity training in relation to functional changes in humans remains incomplete. Likewise, β2‐agonists are frequently used to counteract exercise‐induced bronchoconstriction, but the effects β2‐agonist treatment on muscle remodelling and adaptations to training are unknown. In a placebo‐controlled parallel study, we randomly assigned 21 trained men to 4 weeks of high intensity training with (HIT+β2A) or without (HIT) daily inhalation of β2‐agonist (terbutaline, 4 mg dose−1). Of 486 proteins identified by mass‐spectrometry proteomics of muscle biopsies sampled before and after the intervention, 32 and 85 were changing (false discovery rate (FDR) ≤5%) with the intervention in HIT and HIT+β2A, respectively. Proteome signature changes were different in HIT and HIT+β2A (P = 0.005), wherein β2‐agonist caused a repression of 25 proteins in HIT+β2A compared to HIT, and an upregulation of 7 proteins compared to HIT. β2‐Agonist repressed or even downregulated training‐induced enrichment of pathways related to oxidative phosphorylation and glycogen metabolism, but upregulated pathways related to histone trimethylation and the nucleosome. Muscle contractile phenotype changed differently in HIT and HIT+β2A (P ≤ 0.001), with a fast‐to‐slow twitch transition in HIT and a slow‐to‐fast twitch transition in HIT+β2A. β2‐Agonist attenuated training‐induced enhancements in maximal oxygen consumption (P ≤ 0.01) and exercise performance (6.1 vs. 11.6%, P ≤ 0.05) in HIT+β2A compared to HIT. These findings indicate that daily β2‐agonist treatment attenuates the beneficial effects of high intensity training on exercise performance and oxidative capacity, and causes remodelling of muscle proteome signature towards a fast‐twitch phenotype.
Keywords: physical activity, proteomics, metabolism, beta‐agonists, adrenoceptors, adrenergic, , terbutaline, HIT, athletes
Key points
While several studies have investigated the effects of exercise training in human skeletal muscle and the chronic effect of β2‐agonist treatment in rodent muscle, their effects on muscle proteome signature with related functional measures in humans are still incompletely understood.
Herein we show that daily β2‐agonist treatment attenuates training‐induced enhancements in exercise performance and maximal oxygen consumption, and alters muscle proteome signature and phenotype in trained young men.
Daily β2‐agonist treatment abolished several of the training‐induced enhancements in muscle oxidative capacity and caused a repression of muscle metabolic pathways; furthermore, β2‐agonist treatment induced a slow‐to‐fast twitch muscle phenotype transition.
The present study indicates that chronic β2‐agonist treatment confounds the positive effect of high intensity training on exercise performance and oxidative capacity, which is of interest for the large proportion of persons using inhaled β2‐agonists on a daily basis, including athletes.
Abbreviations
- ATP
adenosine triphosphate
- cAMP
cyclic adenosine monophosphate
- CSA
cross‐sectional area
- FDR
false discovery rate
- HIT
high intensity training + placebo group
- HIT+β2A
high intensity training + β2‐agonist group
- MHC
myosin heavy chain
- PKA
protein kinase A
- TCA
tricarboxylic acid cycle
maximal oxygen consumption
Introduction
Exercise is essential for maintaining physical function and health, and is positively related to quality‐of‐life and life expectancy (Westerterp, 2001; Dhana et al. 2016). Thus, exercise is considered one of the best non‐pharmacological strategies to prevent and even reverse several pathological conditions (Goodyear, 2008). In addition, exercise training is crucial for optimal performance in the vast majority of sport disciplines. High intensity training is widely practised because of its superior efficacy in improving physiological measures compared to low‐ and moderate intensity exercise (Milanovic et al. 2015). Only a few weeks of high intensity training improves cardiovascular fitness and induces several beneficial adaptations in skeletal muscle (Laursen & Jenkins, 2002; Hostrup & Bangsbo, 2017). The adaptations induced by high intensity training are related to major cellular perturbations in skeletal muscle that activate a variety of signalling pathways and gene programmes (Arany et al. 2008; Canto et al. 2009; Powers et al. 2010; Hoffman et al. 2015; Brandt et al. 2016). However, while considerable progress has been made in understanding myocellular adaptations induced by training (Egan et al. 2011; Petriz et al. 2012; Hawley et al. 2014; Murton et al. 2014; Padrão et al. 2016; Powers et al. 2016; Sollanek et al. 2017), only few studies have investigated the effect of high intensity interval training on muscle proteome signature changes (Holloway et al. 2009; Egan et al. 2011; Hussey et al. 2013) and integrated such changes with adaptations in muscle phenotype, contractile function and exercise performance in humans (Petriz et al. 2012; Padrão et al. 2016). Integrated approaches are needed to provide a global perspective on the relation between muscle remodelling and functional changes induced by training from a basic physiological standpoint and for future therapeutic applications.
A large proportion of the population experiences respiratory complications during physically demanding activities (Price et al. 2014). A common cause of this problem is asthma and exercise‐induced bronchoconstriction, with a prevalence of around 10% in Western countries (Cruz, 2007; Kainu et al. 2013). Inhaled β2‐adrenoceptor agonists (β2‐agonists) are used as first‐line treatment of the bronchoconstriction associated with asthma and exercise‐induced bronchoconstriction, and are as such among the most commonly prescribed medications worldwide (Rottenkolber et al. 2015). While the main application of inhaled β2‐agonists is to induce bronchodilatation, a large proportion of the drug enters the systemic circulation and distributes to all organs (Jacobson et al. 2014; Dyreborg et al. 2016). The largest organ of the body, skeletal muscle, contains a high density of β2‐adrenoceptors (Williams et al. 1984; Jensen et al. 2002) that serve an important role in the adrenergic fight‐or‐flight response (Jensen et al. 2008; Emrick et al. 2010; Andersson et al. 2012; Hostrup et al. 2014b). Thus, acute inhalation of β2‐agonist has been shown to affect ion handling and energy production of skeletal muscle in trained young men (Hostrup et al. 2014b; 2016; Kalsen et al. 2016a,b). Furthermore, when administered chronically, β2‐agonists induce hypertrophy and alter metabolic and contractile properties of skeletal muscle (Martineau et al. 1992; Dodd et al. 1996; Rajab et al. 2000; Hostrup et al. 2015). Therefore, β2‐agonists have been proposed as therapeutic agents to combat lifestyle‐related diseases, muscle dysfunction and age‐related muscle atrophy (Lynch & Ryall, 2008; Joassard et al. 2013). However, despite their widespread use, it is unknown what effect daily inhalation of β2‐agonist has on training‐induced adaptations in skeletal muscle and functional capacity in humans. This is important, since inhaled β2‐agonists are often used in conjunction with physical activity in persons with asthma and exercise‐induced bronchoconstriction (Arie, 2012; Price et al. 2014). For instance, in some sports, as many as 50% of athletes use inhaled β2‐agonists in conjunction with training and competition (Parsons & Mastronarde, 2005). A better understanding of these β2‐agonist mediated effects on muscle proteome and phenotype could also lead to improved treatment modalities in a diverse range of diseases such as muscle wasting and obesity.
Thus, the purpose of the present study was to investigate proteome signature and phenotype changes of skeletal muscle induced by high intensity training with and without concomitant daily treatment with β2‐agonist in therapeutic doses, integrating muscle remodelling with relevant functional measures related to maximal oxygen consumption (), exercise performance and muscle contractile properties in trained young men. We hypothesized that daily β2‐agonist treatment would alter training‐induced modulation of muscle proteome signature and cause a shift towards a fast‐twitch muscle phenotype.
Methods
Human subjects and ethics
Twenty‐four healthy trained men were initially screened of which 21 were included in the study. Prior to inclusion, subjects received oral and written information about the aims and contents of the study as well as possible risks involved, including side effects associated with the study drug (terbutaline). Each subject gave his oral and written informed consent. A physician screened each subject for unknown cardiopulmonary disease with lung and heart auscultation and electrocardiography. Inclusion criteria were males, aged 18–36 years, informed consent, a weekly training volume of 2–5 h, a between 40 and 60 mL min kg−1, and a lean mass index between 14 and 22 kg m−2. Exclusion criteria were smoking, allergy towards study drug, and chronic disease. Subjects were recreationally active, engaging in team sports, running, biking and/or light resistance training. Subject characteristics are presented in Table 1. Subjects were told to refrain from competitive events for the entire study and not to change their daily physical activities and nutritional habits, which were recorded. The study was conducted in accordance with the standards set by the 2013 version of the Declaration of Helsinki and was approved by the regional research ethics committee of Copenhagen, Denmark (H‐4‐2014‐002). The study was registered in ClinicalTrials.gov (NCT02557581).
Table 1.
Subject characteristics
| HIT (n = 9) | HIT+β2A (n = 12) | Between‐group difference (P value) | |
|---|---|---|---|
| Age (years) | 24.7 (± 3.0) | 23.5 (± 1.7) | 0.47 |
| Height (cm) | 184 (± 3) | 185 (± 2) | 0.85 |
| Weight (kg) | 78.4 (± 6.7) | 77.6 (± 5.5) | 0.86 |
| Lean body mass (kg) | 61.5 (± 4.4) | 60.6 (± 2.9) | 0.72 |
| (mL min−1) | 3932 (± 233) | 3945 (± 329) | 0.95 |
| MHCI (%) | 49.4 (± 9.6) | 49.2 (± 6.8) | 0.97 |
| MHCIIa (%) | 44.2 (± 9.4) | 45.8 (± 5.9) | 0.74 |
| MHCIIx (%) | 6.5 (± 4.2) | 5.0 (± 2.4) | 0.46 |
, maximal oxygen consumption. MHC, myosin heavy chain. Values are means (±95% CI).
Study design
The study was designed as a block‐randomized controlled parallel study with two groups: Either 4 weeks of high intensity training and daily inhalation of placebo (HIT, n = 9) or 4 weeks of high intensity training and daily inhalation of terbutaline (HIT+β2A, n = 12). Upon inclusion, subjects were randomly allocated in the two groups, stratified for and lean body mass. In HIT, subjects received a placebo inhalator (Turbohaler, AstraZeneca, Cambridge, UK) and in HIT+β2A subjects received an inhalator of the selective β2‐agonist terbutaline (Bricanyl Turbohaler 0.5 mg dose−1, AstraZeneca, Cambridge, UK). Subjects were instructed to inhale eight doses once daily for 28 ± 1 days.
Study drugs
Terbutaline is a commonly prescribed short‐acting β2‐agonist in Northern Europe, which has a high selectivity for the β2‐adrenoceptor (Baker, 2010) and a half‐life of ∼4 h (Krogh et al. 2017). The dosage of inhaled terbutaline administered (8 × 0.5 mg) in the present study is higher than that normally prescribed to asthmatics (Bricanyl Turbohaler; product information, www.astrazeneca.com), but equivalent to the daily upper limit for inhaled salbutamol in competitive sports (2017 list of prohibited substances; www.wada-ama.org). The rationale for the dosage of 4 mg was to ensure an adequate systemic response, while staying within the current upper therapeutic limit in competitive sports. Systemic concentrations of terbutaline after inhalation of 4 mg have been shown to reach their peak within 0.5–1 h with concentrations of ∼5–8 ng mL−1 (Dyreborg et al. 2016; Kreiberg et al. 2017). Such concentrations are equivalent or even higher than those observed after oral administration of 10 mg terbutaline (Elers et al. 2012; Dyreborg et al. 2016). To ensure a drug compliance of 100%, inhalations were monitored by study staff on a daily basis via online video tools (FaceTime/Skype). Because β2‐agonists may affect energy turnover and ion handling in skeletal muscle (Kalsen et al. 2014; Hostrup et al. 2014a,b; Kalsen et al. 2016b), subjects inhaled their daily dose after exercise sessions on training days. During non‐training days, subjects inhaled their daily dose in the time frame of 09.00–18.00. The duration of treatment was based on a previous study showing that 4 weeks of treatment with terbutaline leads to adaptations in skeletal muscle of humans (Hostrup et al. 2015). Both subjects and investigators were blinded to treatment. Terbutaline Turbohalers were delivered by the regional pharmacy of Copenhagen, Denmark. Placebo Turbohalers were kindly delivered by AstraZeneca. Randomization was conducted in SPSS (IBM, Armonk, NY, US) by staff who did not take part in any of the experimental procedures or data analysis.
Experimental protocol
Before the start of the intervention, subjects attended two trials at the laboratory separated by 2 days. At the first trial, subjects’ thigh lean mass was determined by Dual‐energy X‐ray absorptiometry (DXA) (Lunar iDXA, GE Healthcase, Belgium). Subjects were placed in the scanner in supine position undressed. All DXA scans were performed with 10 min of supine rest before scanning to allow distribution of body fluids. To reduce intra‐day variation, two scans were performed. After the scans, subjects rested in a supine position for 20 min and resting metabolic rate was determined by indirect calorimetry breath‐by‐breath with a gas analyser system (Oxycon Pro, CareFusion, San Diego, CA, USA) for 15 min. Subjects then performed a standardized warm‐up on a bike ergometer at 100 W for 10 min (Monark LC4, Monark Exercise AB, Vansbro, Sweden). After warm‐up, subjects’ contractile properties of the quadriceps muscle were assessed by maximal voluntary contraction (MVC). After MVC, subjects’ and performance were determined during incremental cycling to exhaustion on the bike ergometer starting with 4 min of cycling at 100 W, in which substrate utilization was determined by indirect calorimetry as described by Jeukendrup & Wallis (2005), followed by 4 min at 150 W and 200 W, after which workload increased by 30 W every minute until exhaustion.
At the second trial, thigh lean mass and resting metabolic rate were determined similarly to the first trial to minimize day‐to‐day variation. Afterwards, subjects warmed up on the bike ergometer for 4 min, followed by cycling at a workload corresponding to 60% of subjects’ for determination of substrate utilization at same relative intensity of . After the exercise, subjects’ contractile properties of the quadriceps muscle were measured as performed during the first trial to minimize day‐to‐day variation. Subjects then rested for 30 min and had a muscle biopsy taken from the vastus lateralis of the right thigh using a Bergstrøm needle with suction (Bergstrøm, 1975). Prior to biopsy sampling, an incision (3 mm) was made through the skin and fascia at the vastus lateralis belly under local anaesthesia (2 mL lidocaine without epinephrine, 20 mg mL−1 Xylocain, AstraZeneca).
After the second trial, subjects received either placebo or terbutaline according to their group.
Upon conclusion of the 4 week intervention, subjects attended two laboratory trials similar to those conducted before the intervention. To ensure complete washout of terbutaline (half‐life ∼4 h) (Krogh et al. 2017), post‐testing was performed 2–3 days after last inhalation.
Subjects were told to abstain from caffeine, strenuous exercise and alcohol 48 h before each laboratory trial. To minimize variation, subjects ingested a standardized meal and fluid 1.5 h before the laboratory trials.
Training intervention
During the 4 week intervention, subjects performed a high intensity‐training programme of 3 sessions per week on indoor spinning bikes. Each training session was supervized by an instructor and consisted of a standardized 10 min warm‐up followed by three blocks of exercise at ≈85% of maximum heart rate for 10 min with 30 s of all‐out sprinting at the end of each 10 min block. Repeated all‐out sprints of 30 s were chosen, as studies have shown that this type of training effectively induces adaptations in skeletal muscle (Hostrup & Bangsbo, 2017). During each training session, subjects wore heart rate monitors that fed subjects’ heart rate to the instructor's iPad (iPad 4, Apple Inc., CA, USA) using Polar Team app (Polar Electro Denmark, Holte, Denmark), thus ensuring that subjects kept their target heart rate. Training adherence was 100% for all subjects, apart from one subject who missed one training session during the 4 week intervention.
Experimental procedures
Contractile properties of the quadriceps muscle
Subjects’ contractile properties of the quadriceps muscle were determined during MVC with percutaneous electrical muscle stimulation during and immediately after each contraction as described previously (Hostrup et al. 2015). Subjects were familiarized with two 3–4 s submaximal isometric muscle contractions at 40 and 70 % of MVC and with constant current electrostimulation, (Digitimer, model DS7AH, Welwyn Garden City, UK) at 100, 400, 700 and 999 mA with 400 V for 200 μs, before the first MVC. The electrostimulation used could activate 42.6 (±3.3)% of subjects’ peak MVC. Each MVC consisted of a 3–4 s maximal contraction with verbal encouragement. A potentiated twitch was applied when subjects reached their apparent peak MVC, as well as 1 s following relaxation. Peak twitch torque, time‐to‐peak twitch torque, half‐relaxation time and voluntary activation level were calculated as previously described (Bachasson et al. 2013). Rate of force development was calculated as the average slope on the moment–time curve at the time interval 0–200 ms relative to the onset of muscle contraction during MVC (Aagaard et al. 2002). Onset of muscle contraction was defined as the time point at which the moment curve increased to values above 2.5% of the magnitude of the moment achieved during MVC.
For each variable, a grand mean was calculated from the average of the two highest values from both trials. The distance from the base of patella to the middle of the Velcro strap on the ankle was registered and used to calculate torque in newton metres (N m).
Muscle biopsies
Sampled biopsies were divided in two pieces of which the first piece (20–30 mg) was mounted on an embedded medium (OCT Compound Tissue‐Tek; Sakura Finetek, Zoeterwoude, The Netherlands), frozen in isopentane cooled to the freezing point in liquid nitrogen, and stored at −80°C until analysis for fibre type distribution, capillaries and fibre cross‐sectional area by immunohistochemical analysis. The other biopsy piece (50–100 mg) was washed in ice‐cold saline to reduce blood contamination, then dried and frozen in liquid nitrogen, and stored in cryo tubes for later proteomic, enzymatic and Western blot analyses. Prior to these analyses, each biopsy was freeze‐dried and dissected free from apparent non‐muscle tissue. The dissected muscle tissue was then divided in three pieces for proteomics (≈10 mg dry weight), Western blotting (≈1.5 mg dry weight) and enzyme activity assays (≈2 mg dry weight). Because of inadequate muscle tissue yield from one subject in HIT, only proteomic analysis was performed for this subject.
Mass spectrometry proteomics
Proteins were extracted from muscle biopsies using two‐step sequential extraction as described previously (Wilson et al. 2010; Nuez‐Ortin et al. 2016, 2017). Proteins were extracted in progressively denaturing buffers (150 mm NaCl, 50 mm Tris, pH 8.0 followed by 7 m urea, 2 m thiourea, 50 mm Tris, pH 8.0). Protein concentrations were estimated by Bradford assay (Bio‐Rad Laboratories, Hercules, CA, USA), and 100 μg of protein sample was reduced using 10 mm DTT (overnight at 4°C), alkylated using 50 mm iodoacetamide (2 h at ambient temperature in the dark) then trypsin digested by co‐precipitation (Wilson et al. 2010). Briefly, 1 μg trypsin was spotted onto the wall of each sample Eppendorf tube then flushed into the sample with 1 mL 100% methanol chilled to −20°C. Samples were kept at −20°C overnight to allow co‐precipitation of proteins with trypsin, then centrifuged in a benchtop microcentrifuge (5 min at 12,000 g) and pellets allowed to air‐dry for 10 min. After addition of 100 mm ammonium bicarbonate, samples were then incubated for 5 h at 37°C with the further addition of 1 μg trypsin after 3 h. Samples were acidified by the addition of 1% formic acid and non‐digested protein removed by centrifugation in a benchtop microcentrifuge (5 min at 12,000 g). 1 μg aliquots of each peptide sample were analysed using an LTQ‐Orbitrap and Ultimate 3000 nano RSLC system (Thermo Fisher Scientific, MA, USA) using published methods for separation and data‐dependent acquisition (Wilson et al. 2016).
Sequential extraction has been shown to be an effective approach to sample fractionation in complex and challenging tissues rich in structural proteins, such as cartilage (Wilson et al. 2010), whole fish larvae (Nuez‐Ortin et al. 2016) and in white muscle (Nuez‐Ortin et al. 2017) and skeletal muscle (Barbé et al. 2017). Prior to processing the complete set of samples, a subset of six tissue biopsies was used to evaluate the extraction method for human skeletal muscle proteomics. Comparison of the extracted proteins indicated clear separation of the contractile proteins (e.g. actin, myosin and troponin) from readily soluble cytosolic proteins (e.g. glutathione S‐transferases, 14‐3‐3 proteins and carbonic anhydrases) demonstrating the value of this approach to reduce sample complexity at the protein level.
Protein identification and data processing
Data files were imported into MaxQuant version 1.5.1.2 (http://maxquant.org/), where sequential extracts were defined as fractions of the same biological sample, and MS/MS spectra were searched against the Swiss‐Prot Human reference proteome database (downloaded 18/03/2016; 201,015 entries) using the Andromeda search engine. Default settings for protein identification by LTQ‐Orbitrap MS/MS and label‐free quantitation (LFQ) included a maximum of two missed cleavages, mass error tolerances of 20 ppm then 4.5 ppm for initial and main peptide searches, respectively, 0.5 Da tolerance for fragment ions, variable methionine oxidation and fixed cysteine carbamidomethylation. A false discovery rate of 1% was used for both peptide‐spectrum matching and protein identification. The LFQ protein intensity values were extracted from the MaxQuant proteinGroups.txt file into Perseus software (http://perseus-framework.org/), filtered to remove proteins identified by reverse database matching and on the basis of modified peptides only and then log2‐transformed. Proteins detected in fewer than twelve biological samples were also excluded and remaining missing values were imputed using low‐abundance LFQ intensity values drawn from a normal distribution, according to default values in Perseus. In total, 9932 unique peptides were identified matching 783 proteins. Prior to statistical comparative analysis, protein data were filtered for potential non‐skeletal muscle contamination using the Human Protein Atlas (Uhlén et al. 2015; Thul et al. 2017). In accordance with the Human Protein Atlas (June 2017), 297 were determined not to be expressed at a protein level in skeletal muscle and were not included in further statistical analysis. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD005480.
Immunohistochemistry and imaging
Muscle fibre type distribution, capillaries and fibre cross‐sectional area were determined by immunohistochemistry and confocal imaging as described previously (Nyberg et al. 2016). Briefly, the embedded muscle samples were cut in transverse sections of 8 μm in a cryostat. Sections were fixed for 2 min in phosphate buffered saline (PBS, pH 7.2, Gibco 70013‐016, Life Technologies Denmark, Nærum, Denmark) containing 2% formaldehyde and washed in a 1:10 wash buffer (Dako S3006, Glostrup, Denmark), and blocked for 10 min in PBS containing 1% BSA for immunohistochemical staining. Antibodies were diluted in antibody diluent (Dako S0809). First, capillaries and myofibre type IIA were visualized using biotinylated Ulex europaeus agglutinin I lectin (1:100; VECTB‐ 1065, VWR, Bie and Berntsen, Herlev, Denmark) and a monoclonal antibody (1:200; SC‐71, Hybridoma Bank, Iowa City, IA, USA), respectively. Second, myofibre borders were visualized using an antibody against laminin (1:500; Dako Z0097) together with myosin heavy chain (1:1000; Sigma‐Aldrich Denmark M8421, Brøndby, Denmark) added for distinction of myofibre type I. Specific secondary antibodies (order listed: Streptavidin/FITC, (1:200; DAKO F0422), Alexa‐ 555 donkey anti‐mouse (1:1000; Invitrogen A31570, Life Technologies Denmark), Alexa‐350 goat anti‐rabbit (1:1000; Invitrogen P10994) and Alexa‐488 donkey anti‐mouse (1:1000; Invitrogen A21202)) were applied to each primary antibody. Three individual muscle fibre types were identified as type I (green), type IIA (red), and type IIX (unstained/black). Visualization was performed on a computer screen using a light microscope (Carl Zeiss, Germany), and all morphometric analysis were performed using a digital analysis program (ImageJ, NIH ImageJ). Two or more separate sections of a cross‐section were used for analysis, and the cross‐sectional area was assessed by manually drawing the perimeter around each selected section. The number of muscle fibres and capillaries within each section was counted, and capillary supply was subsequently expressed as capillaries per fibre (capillary‐to‐fibre ratio) and capillary density (capillaries mm−2). Mean fibre area was assessed by manual drawing of the perimeter of each muscle fibre. All analyses were carried out manually by the same blinded investigator.
Immunoblotting and SDS‐PAGE
Protein expression by Western blotting were determined as previously described (Thomassen et al. 2010). Protein concentration of each sample was determined with a BSA kit (Thermo Fisher Scientific, MA, USA). Samples were mixed with 6 × Laemmli buffer (7 ml 0.5 m Tris‐base, 3 ml glycerol, 0.93 g DTT, 1 g SDS and 1.2 mg Bromophenol Blue) and double distilled H2O to reach equal protein concentrations. Equal amounts of protein was loaded in each well of pre‐cast gels (Bio‐Rad). Samples from each subject were loaded on the same gel with a mixed human muscle standard lysate loaded in two different wells used for normalization. Bands were visualized with ECL (Millipore, MA, USA) and recorded with a digital camera (ChemiDoc MP Imaging System, Bio‐Rad). Densitometry quantification of band intensity was done using Image Lab version 4.0 (Bio‐Rad) and determined as the total band intensity adjusted for background intensity. Primary antibodies used were citrate synthase (no. ab96600, Abcam, MA, US), GLUT4 (no. PA1‐1065, Thermo Fisher Scientific), glycogen synthase (GS) (no. 3893, Cell Signalling Technology, MA, USA), OXPHOS (no. ab110411, Abcam, MA, USA) and phosphofructokinase (Sc‐166722, Santa Cruz Biotechnology, Inc., Dallas, USA). The secondary antibodies used were HRP conjugated rabbit anti‐sheep (P‐0163), goat anti‐mouse (P‐0447, DAKO, Denmark) and goat anti‐rabbit IgM/IgG (4010‐05 Southern Biotech, AL, USA).
Enzymatic activity
Maximal enzyme activity of citrate synthase, hexokinase, phosphofructokinase, and lactate dehydrogenase was quantified in muscle homogenates using fluorometric methods (Fluoroscan Ascent, Thermo Fisher Scientific) as described by Lowry & Passonneau (1972).
Statistics
Statistical analyses were performed in SPSS version 24. Sample size was based on previous studies of terbutaline (Hostrup et al. 2015; Dyreborg et al. 2016). Data were tested for normality using the Shapiro Wilks test and q–q plots. Data were normally distributed and magnitudes of outcome statistics are presented as means (±95% confidence interval) and P values to represent probability. To estimate within‐ and between‐group changes with the intervention, two‐tailed linear mixed modelling was used with group and trial included as fixed factors and a random factor for subjects for a full factorial design. Age was included as a time‐invariant covariate in the mixed model, since age has been shown to influence the response to β2‐agonists (White et al. 1994). For univariate correlation analyses, the Pearson product‐moment correlation coefficient was used. For the proteomic data, a series of mixed models were performed on the log2 expression for each protein to estimate protein‐specific within‐ and between‐group changes with the intervention. The false discovery rate (FDR) method as described by Storey & Tibshirani (2003) was used to control for multiple testing. Differently expressed proteins (FDR ≤ 5%) were subjected to functional annotation enrichment analysis (David Bioinformatics resources 6.8, NIH, USA) based on gene ontology (GO) terms biological processes (GO:BP) and cellular components (GO:CC) as well as KEGG terms using an EASE score threshold of 0.1 and the Benjamini‐Hochberg procedure to adjust P values. Linear discriminant analysis‐principal component analysis with varimax rotation was used to estimate between‐group separation in the muscle proteome, only including the first eight principal components in the discriminant analysis because of the sample size of nine in HIT.
Results
Muscle proteome remodelling
Of the 486 proteins identified as human skeletal muscle proteins, training upregulated expression of 26 proteins (Table 2) and downregulated expression of 6 proteins (Table 3) in HIT (FDR ≤ 5%)(Fig. 1 A), whereas concomitant daily inhalation of β2‐agonist upregulated expression of 34 proteins (Table 4) and downregulated expression of 51 proteins (Table 5) in HIT+β2A (Fig. 1 B). Inhalation of β2‐agonist caused a repression of 25 proteins in HIT+β2A compared to HIT (group by trial interaction), and an upregulation of 7 proteins compared to HIT (Table 6; Fig. 1 C). Proteome signature changes induced by the intervention in HIT and HIT+β2A were markedly different as indicated by a clear separation of the two groups (Wilks’ Lambda: 0.228, P = 0.005) in a principal component‐discriminant analysis that explained 70% of total proteome variance (Fig. 1 D).
Table 2.
Upregulated proteins in HIT (n = 9)
| Name | Description | Log2‐change | q value |
|---|---|---|---|
| GYS1 | Glycogen [starch] synthase, muscle | 0.46 | ≤ 0.01 |
| FUNDC2 | FUN14 domain‐containing protein 2 | 0.91 | ≤ 0.05 |
| NDUFA2 | NADH dehydrogenase [ubiquinone] 1 α subcomplex subunit 2 | 0.38 | ≤ 0.05 |
| COX6C | Cytochrome c oxidase subunit 6C | 0.63 | ≤ 0.05 |
| S100A13 | Protein S100‐A13 | 1.15 | ≤ 0.05 |
| DNAJA2 | DnaJ homolog subfamily A member 2 | 1.00 | ≤ 0.05 |
| NDUFS6 | NADH dehydrogenase [ubiquinone] iron‐sulfur protein 6, mitochondrial | 0.54 | ≤ 0.05 |
| NDUFB6 | NADH dehydrogenase [ubiquinone] 1 β subcomplex subunit 6 | 0.51 | ≤ 0.05 |
| UQCRH | Cytochrome b‐c1 complex subunit 6, mitochondrial | 0.36 | ≤ 0.05 |
| MRPL12 | 39S ribosomal protein L12, mitochondrial | 1.04 | ≤ 0.05 |
| NDUFB3 | NADH dehydrogenase [ubiquinone] 1 β subcomplex subunit 3 | 0.40 | ≤ 0.05 |
| PHB | Prohibitin | 0.29 | ≤ 0.05 |
| B2M | Β‐2‐microglobulin;Β‐2‐microglobulin form pI 5.3 | 0.97 | ≤ 0.05 |
| ATP5H | ATP synthase subunit d, mitochondrial | 0.28 | ≤ 0.05 |
| SSBP1 | Single‐stranded DNA‐binding protein, mitochondrial | 0.22 | ≤ 0.05 |
| MYOZ2 | Myozenin‐2 | 0.48 | ≤ 0.05 |
| NDUFV2 | NADH dehydrogenase [ubiquinone] flavoprotein 2, mitochondrial | 0.28 | ≤ 0.05 |
| STOML2 | Stomatin‐like protein 2, mitochondrial | 0.43 | ≤ 0.05 |
| ATP5J | ATP synthase‐coupling factor 6, mitochondrial | 0.35 | ≤ 0.05 |
| COX5A | Cytochrome c oxidase subunit 5A, mitochondrial | 0.24 | ≤ 0.05 |
| PLIN2 | Perilipin‐2 | 0.39 | ≤ 0.05 |
| NDUFB8 | NADH dehydrogenase [ubiquinone] 1 β subcomplex subunit 8, mitochondrial | 0.55 | ≤ 0.05 |
| SDHA | Succinate dehydrogenase [ubiquinone] flavoprotein subunit, mitochondrial | 0.14 | ≤ 0.05 |
| CHCHD3 | Coiled‐coil‐helix‐coiled‐coil‐helix domain‐containing protein 3, mitochondrial | 0.30 | ≤ 0.05 |
| AGL | Glycogen debranching enzyme;4‐α‐glucanotransferase;Amylo‐α‐1,6‐glucosidase | 0.23 | ≤ 0.05 |
| IMMT | Mitochondrial inner membrane protein | 0.20 | ≤ 0.05 |
Table 3.
Downregulated proteins in HIT (n = 9)
| Name | Description | Log2‐change | q value |
|---|---|---|---|
| PARK7 | Protein DJ‐1 | −0.25 | ≤ 0.05 |
| MYBPC1 | Myosin‐binding protein C, slow‐type | −0.23 | ≤ 0.05 |
| MYH1 | Myosin‐1 | −0.48 | ≤ 0.05 |
| PNPO | Pyridoxine‐5‐phosphate oxidase | −0.31 | ≤ 0.05 |
| FKBP3 | Peptidyl‐prolyl cis‐trans isomerase FKBP3 | −0.25 | ≤ 0.05 |
| HSPB1 | Heat shock protein β‐1 | −0.19 | ≤ 0.05 |
Figure 1. Proteome signature remodelling in HIT and HIT+β2A.
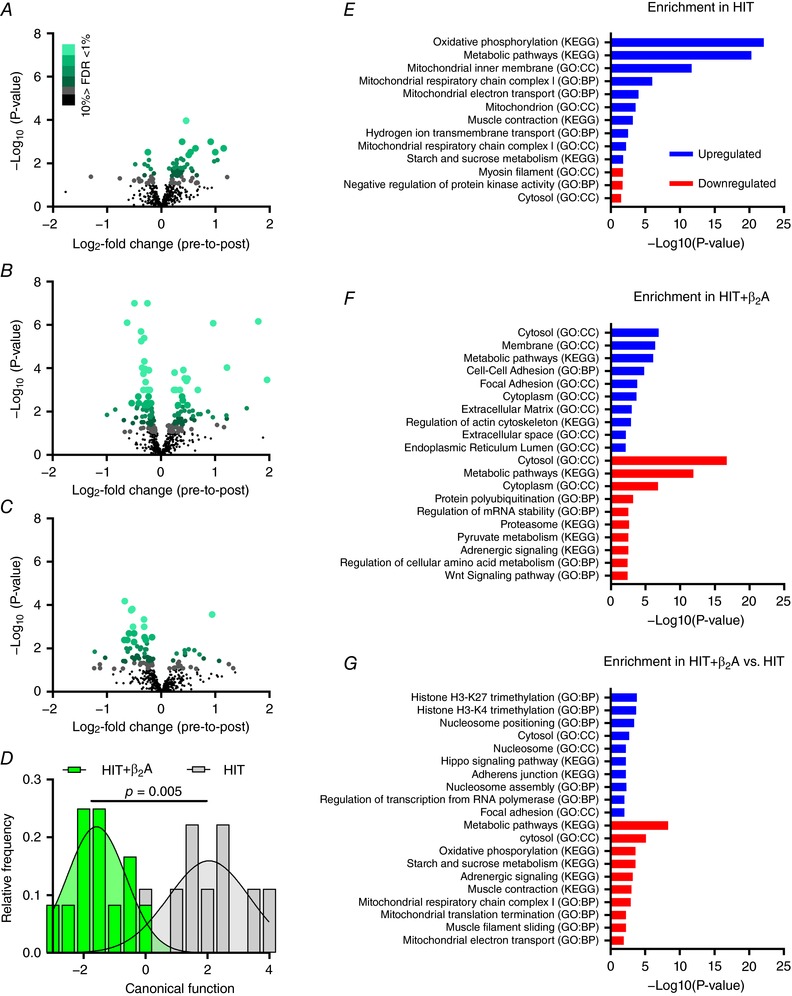
Effect of 4 weeks of high intensity training with (HIT+β2A, n = 12) and without (HIT, n = 9) daily inhalation of terbutaline (4 mg day−1) on proteome signature of the vastus lateralis muscle in trained men. A–C, volcano plots of within‐group changes in skeletal muscle proteins (486 proteins) in HIT (A) and HIT+β2A (B), as well as between‐group interaction (HIT+β2A – HIT) (C). Green scale indicates % false discovery rate (FDR). D, Gaussian fit of frequency distribution of discriminant canonical function score of within‐group changes in proteome signature using principal component‐discriminant analysis (PCA‐DA) of the eight first principal components that explained 70% of total proteome variance. E–G, enrichment analysis of the 10 most up‐ (blue) and downregulated (red) pathways (GO: biological processes, GO: cellular component, and KEGG pathways) in HIT (E) and HIT+β2A (F) as well as between‐group (HIT+β2A – HIT) (G).
Table 4.
Upregulated proteins in HIT+β2A (n = 12)
| Name | Description | Log2‐change | q value |
|---|---|---|---|
| S100A13 | Protein S100‐A13 | 1.80 | ≤ 0.001 |
| ACTG1 | Actin, cytoplasmic 2;Actin, cytoplasmic 2, N‐terminally processed | 0.97 | ≤ 0.001 |
| B2M | Β‐2‐microglobulin;Β‐2‐microglobulin form pI 5.3 | 1.22 | ≤ 0.01 |
| PADI2 | Protein‐arginine deiminase type‐2 | 0.41 | ≤ 0.01 |
| SDHA | Succinate dehydrogenase [ubiquinone] flavoprotein subunit, mitochondrial | 0.25 | ≤ 0.01 |
| AHNAK | Neuroblast differentiation‐associated protein AHNAK | 0.44 | ≤ 0.01 |
| PCBP1 | Poly(rC)‐binding protein 1 | 0.49 | ≤ 0.01 |
| EEF1A1P5;EEF1A1 | Putative elongation factor 1‐α‐like 3;Elongation factor 1‐α 1 | 1.96 | ≤ 0.01 |
| HK1 | Hexokinase‐1 | 0.47 | ≤ 0.01 |
| YWHAZ | 14‐3‐3 protein zeta/delta | 0.27 | ≤ 0.01 |
| ACSL1 | Long‐chain‐fatty‐acid–CoA ligase 1 | 0.35 | ≤ 0.01 |
| PACSIN3 | Protein kinase C and casein kinase substrate in neurons protein 3 | 0.24 | ≤ 0.01 |
| S100A6 | Protein S100‐A6 | 0.68 | ≤ 0.01 |
| DES | Desmin | 0.39 | ≤ 0.05 |
| ACTR3 | Actin‐related protein 3 | 0.40 | ≤ 0.05 |
| RPL5 | 60S ribosomal protein L5 | 0.31 | ≤ 0.05 |
| STOML2 | Stomatin‐like protein 2, mitochondrial | 0.49 | ≤ 0.05 |
| HSPA5 | 78 kDa glucose‐regulated protein | 0.30 | ≤ 0.05 |
| MAPT | Microtubule‐associated protein tau | 0.40 | ≤ 0.05 |
| ANP32A | Acidic leucine‐rich nuclear phosphoprotein 32 family member A | 1.58 | ≤ 0.05 |
| RPSA | 40S ribosomal protein SA | 0.43 | ≤ 0.05 |
| PDIA6 | Protein disulfide‐isomerase A6 | 0.59 | ≤ 0.05 |
| HNRNPK | Heterogeneous nuclear ribonucleoprotein K | 0.37 | ≤ 0.05 |
| MYL9 | Myosin regulatory light polypeptide 9 | 0.87 | ≤ 0.05 |
| PPP1R14B | Protein phosphatase 1 regulatory subunit 14B | 0.52 | ≤ 0.05 |
| TOM1 | Target of Myb protein 1 | 0.31 | ≤ 0.05 |
| HADHA | Trifunctional enzyme subunit α, mitochondrial | 0.32 | ≤ 0.05 |
| NDUFC2 | NADH dehydrogenase [ubiquinone] 1 subunit C2 | 0.73 | ≤ 0.05 |
| TIMM50 | Mitochondrial import inner membrane translocase subunit TIM50 | 0.69 | ≤ 0.05 |
| NDUFS1 | NADH‐ubiquinone oxidoreductase 75 kDa subunit, mitochondrial | 0.33 | ≤ 0.05 |
| ACTC1 | Actin, α cardiac muscle 1 | 1.21 | ≤ 0.05 |
| PFN1 | Profilin‐1 | 0.25 | ≤ 0.05 |
| MAOB | Amine oxidase [flavin‐containing] B | 0.59 | ≤ 0.05 |
| MYL6 | Myosin light polypeptide 6 | 0.96 | ≤ 0.05 |
Table 5.
Downregulated proteins in HIT+β2A (n = 12)
| Name | Description | Log2‐change | q value |
|---|---|---|---|
| HSPB1 | Heat shock protein β‐1 | −0.49 | ≤ 0.001 |
| ALDH9A1 | 4‐trimethylaminobutyraldehyde dehydrogenase | −0.25 | ≤ 0.001 |
| GYG1 | Glycogenin‐1 | −0.62 | ≤ 0.001 |
| CFL2 | Cofilin‐2 | −0.36 | ≤ 0.001 |
| FHL1 | Four and a half LIM domains protein 1 | −0.31 | ≤ 0.001 |
| PTGR2 | Prostaglandin reductase 2 | −0.36 | ≤ 0.001 |
| CRYAB | Α‐crystallin B chain | −0.30 | ≤ 0.01 |
| HSPA2 | Heat shock‐related 70 kDa protein 2 | −0.35 | ≤ 0.01 |
| GSTM2 | Glutathione S‐transferase Mu 2 | −0.30 | ≤ 0.01 |
| ST13 | Hsc70‐interacting protein | −0.22 | ≤ 0.01 |
| CMBL | Carboxymethylenebutenolidase homolog | −0.32 | ≤ 0.01 |
| HSPB7 | Heat shock protein β‐7 | −0.33 | ≤ 0.01 |
| DNPEP | Aspartyl aminopeptidase | −0.28 | ≤ 0.01 |
| ACO1 | Cytoplasmic aconitate hydratase | −0.25 | ≤ 0.01 |
| AGL | Glycogen debranching enzyme | −0.32 | ≤ 0.01 |
| MACROD1 | O‐acetyl‐ADP‐ribose deacetylase MACROD1 | −0.20 | ≤ 0.01 |
| PSMA6 | Proteasome subunit α type‐6 | −0.23 | ≤ 0.01 |
| PSMB3 | Proteasome subunit β type‐3 | −0.32 | ≤ 0.01 |
| NDRG2 | Protein NDRG2 | −0.23 | ≤ 0.01 |
| MDH1 | Malate dehydrogenase, cytoplasmic | −0.23 | ≤ 0.01 |
| CUTC | Copper homeostasis protein cutC homolog | −0.42 | ≤ 0.05 |
| PDLIM5 | PDZ and LIM domain protein 5 | −0.23 | ≤ 0.05 |
| CAB39 | Calcium‐binding protein 39 | −0.26 | ≤ 0.05 |
| NDUFB7 | NADH dehydrogenase [ubiquinone] 1 β subcomplex subunit 7 | −0.54 | ≤ 0.05 |
| PREP | Prolyl endopeptidase | −0.24 | ≤ 0.05 |
| TPM3 | Tropomyosin α‐3 chain | −0.41 | ≤ 0.05 |
| WDR1 | WD repeat‐containing protein 1 | −0.18 | ≤ 0.05 |
| LMCD1 | LIM and cysteine‐rich domains protein 1 | −0.29 | ≤ 0.05 |
| PSMA5 | Proteasome subunit α type‐5 | −0.24 | ≤ 0.05 |
| TNNC1 | Troponin C, slow skeletal and cardiac muscles | −0.30 | ≤ 0.05 |
| MAPRE2 | Microtubule‐associated protein RP/EB family member 2 | −0.44 | ≤ 0.05 |
| YWHAG | 14‐3‐3 protein gamma;14‐3‐3 protein gamma, N‐terminally processed | −0.20 | ≤ 0.05 |
| TXLNB | Β‐taxilin | −0.24 | ≤ 0.05 |
| PYGM | Glycogen phosphorylase, muscle form | −0.31 | ≤ 0.05 |
| LDB3 | LIM domain‐binding protein 3 | −0.17 | ≤ 0.05 |
| TPM2 | Tropomyosin β chain | −0.44 | ≤ 0.05 |
| AMPD1 | AMP deaminase 1 | −0.33 | ≤ 0.05 |
| LMNB2 | Lamin‐B2 | −0.84 | ≤ 0.05 |
| ACADM | Medium‐chain specific acyl‐CoA dehydrogenase, mitochondrial | −0.27 | ≤ 0.05 |
| PRDX1 | Peroxiredoxin‐1 | −0.17 | ≤ 0.05 |
| CRYZ | Quinone oxidoreductase | −0.26 | ≤ 0.05 |
| TPT1 | Translationally‐controlled tumor protein | −0.17 | ≤ 0.05 |
| PHPT1 | 14 kDa phosphohistidine phosphatase | −0.16 | ≤ 0.05 |
| PFN2 | Profilin‐2 | −0.99 | ≤ 0.05 |
| MYOM2 | Myomesin‐2 | −0.21 | ≤ 0.05 |
| NPEPPS | Puromycin‐sensitive aminopeptidase | −0.17 | ≤ 0.05 |
| ATP2A2 | Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 | −0.29 | ≤ 0.05 |
| PCBD2 | Pterin‐4‐α‐carbinolamine dehydratase 2 | −0.34 | ≤ 0.05 |
| LDHA | L‐lactate dehydrogenase A chain | −0.19 | ≤ 0.05 |
| NT5C1A | Cytosolic 5‐nucleotidase 1A | −0.46 | ≤ 0.05 |
| PARK7 | Protein DJ‐1 | −0.15 | ≤ 0.05 |
Table 6.
Between‐group interaction (HIT+β2A – HIT)
| Name | Description | Log2‐change | q value |
|---|---|---|---|
| GYG1 | Glycogenin‐1 | −0.67 | ≤ 0.01 |
| HSPB7 | Heat shock protein β‐7 | −0.53 | ≤ 0.01 |
| AGL | Glycogen debranching enzyme | −0.55 | ≤ 0.01 |
| ACTG1 | Actin, cytoplasmic 2;Actin, cytoplasmic 2, N‐terminally processed | 0.94 | ≤ 0.01 |
| FHL1 | Four and a half LIM domains protein 1 | −0.31 | ≤ 0.01 |
| CFL2 | Cofilin‐2 | −0.31 | ≤ 0.01 |
| GYS1 | Glycogen [starch] synthase, muscle | −0.52 | ≤ 0.01 |
| ATP2A2 | Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 | −0.59 | ≤ 0.05 |
| TNNC1 | Troponin C, slow skeletal and cardiac muscles | −0.50 | ≤ 0.05 |
| CRYAB | Α‐crystallin B chain | −0.31 | ≤ 0.05 |
| ALDH9A1 | 4‐trimethylaminobutyraldehyde dehydrogenase | −0.17 | ≤ 0.05 |
| ACO1 | Cytoplasmic aconitate hydratase | −0.28 | ≤ 0.05 |
| NDUFS6 | NADH dehydrogenase [ubiquinone] iron‐sulfur protein 6, mitochondrial | −0.68 | ≤ 0.05 |
| TPM3 | Tropomyosin α‐3 chain | −0.62 | ≤ 0.05 |
| PYGM | Glycogen phosphorylase, muscle form | −0.50 | ≤ 0.05 |
| LDB3 | LIM domain‐binding protein 3 | −0.26 | ≤ 0.05 |
| MRPS36 | 28S ribosomal protein S36, mitochondrial | −0.44 | ≤ 0.05 |
| PSMB3 | Proteasome subunit β type‐3 | −0.37 | ≤ 0.05 |
| NDUFB6 | NADH dehydrogenase [ubiquinone] 1 β subcomplex subunit 6 | −0.57 | ≤ 0.05 |
| NDUFB7 | NADH dehydrogenase [ubiquinone] 1 β subcomplex subunit 7 | −0.64 | ≤ 0.05 |
| HSPB1 | Heat shock protein β‐1 | −0.29 | ≤ 0.05 |
| HIST1H1C;HIST1H1E | Histone H1.2;Histone H1.4;Histone H1.3 | 0.51 | ≤ 0.05 |
| HIST1H1D | |||
| LMCD1 | LIM and cysteine‐rich domains protein 1 | −0.39 | ≤ 0.05 |
| SSBP1 | Single‐stranded DNA‐binding protein, mitochondrial | −0.27 | ≤ 0.05 |
| ACTN1 | Α‐actinin‐1 | 0.61 | ≤ 0.05 |
| AHNAK | Neuroblast differentiation‐associated protein AHNAK | 0.43 | ≤ 0.05 |
| MRPL12 | 39S ribosomal protein L12, mitochondrial | −1.23 | ≤ 0.05 |
| TPM2 | Tropomyosin β chain | −0.62 | ≤ 0.05 |
| HK1 | Hexokinase‐1 | 0.44 | ≤ 0.05 |
| YWHAZ | 14‐3‐3 protein zeta/delta | 0.28 | ≤ 0.05 |
| NDUFA2 | NADH dehydrogenase [ubiquinone] 1 α subcomplex subunit 2 | −0.30 | ≤ 0.05 |
| KPNB1 | Importin subunit β‐1 | 0.71 | ≤ 0.05 |
Functional annotation enrichment analysis of differently expressed proteins (FDR ≤ 5%) based on gene ontology (GO) terms biological processes (GO:BP), cellular components (GO:CC) and KEGG pathways revealed different enrichments induced by the intervention in HIT and HIT+β2A (Fig. 1 E–G). In HIT, the most dominantly upregulated pathways were related to the mitochondria, in particular the oxidative phosphorylation, as well as metabolic pathways, including glycogen metabolism (Fig. 1 E). In contrast, HIT+β2A predominantly had an upregulation of pathways related to the cytosol, metabolism and cell‐to‐cell adhesion (Fig. 1 F). HIT had a minor downregulation of pathways related to myosin filaments, protein kinases and the cytosol (Fig. 1 E), whereas HIT+β2A had several downregulated pathways, including those related to the cytosol and metabolism, including pyruvate and amino acid metabolism (Fig. 1 F). Between‐group analysis revealed that daily inhalation of β2‐agonist downregulated pathways related to glycogen metabolism and oxidative phosphorylation in HIT+β2A compared to HIT, but upregulated pathways related to histone trimethylation and the nucleosome (Fig. 1 G).
β2‐Agonist blunts training‐induced enhancements in and exercise performance
A key observation was that inhalation of β2‐agonist completely blunted (P ≤ 0.01) training‐induced improvement in and reduced (P ≤ 0.05) enhancement in exercise performance during incremental cycling to exhaustion (Fig. 2 B). While training effectively augmented in HIT (P ≤ 0.01), HIT+β2A showed no relevant changes in with the intervention (Fig. 2 B). Furthermore, HIT had a twofold greater enhancement in exercise performance compared to HIT+β2A (11.6 vs. 6.1%, P ≤ 0.05) (Fig. 2 B). Between‐group changes in were still evident after adjusting for adaptations in resting metabolic rate and thigh lean mass (Fig. 2 C), which indicates that the increase in observed in HIT and the attenuating effect of β2‐agonist observed in HIT+β2A were attributed to factors other than different adaptations in resting metabolic rate and hypertrophy of the exercising muscles. For training‐induced enhancement in exercise performance, the between‐group difference was also evident after adjusting for thigh lean mass (Fig. 2 C). On the other hand, when adjusting for change in , no differences were observed in training‐induced enhancement in exercise performance between the groups, thus indicating that the blunting effect of β2‐agonist on training‐induced enhancement in performance in HIT+β2A was related to the attenuating effect of β2‐agonist on improvement in .
Figure 2. Maximal oxygen consumption, capillary density and muscle oxidative enzymes in HIT and HIT+β2A.
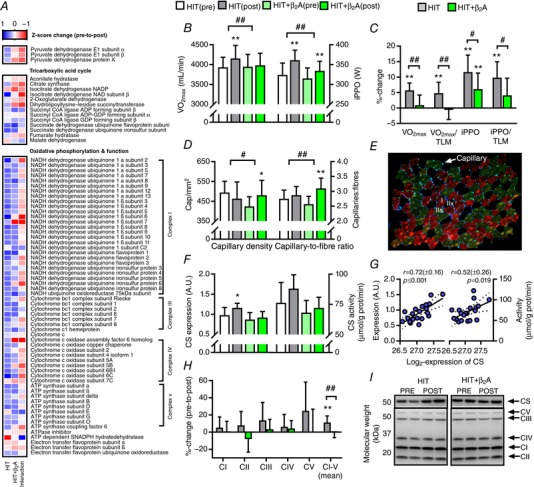
Effect of 4 weeks of high intensity training with (HIT+β2A, n = 12) and without (HIT, n = 9) daily inhalation of terbutaline (4 mg day−1) on outcomes related to exercise performance and oxidative capacity in trained men. A, heat map of mean Z‐score changes of the proteome related to tricarboxylate acid cycle and oxidative phosphorylation in HIT and HIT+β2A, as well as between‐group interaction (HIT+β2A – HIT). B, mean (±95% CI) maximal oxygen consumption (; left y‐axis) and incremental peak power output (iPPO; right y‐axis). C, mean (±95% CI) relative change in and iPPO when unadjusted and adjusted for thigh lean mass (TLM). D, mean (±95% CI) capillary density (left y‐axis) and capillary‐to‐fibre ratio (right y‐axis). E, representative image of immunohistochemical analysis; myosin heavy chain I (red), IIa (green/brown), IIx (black), laminin (blue), capillary (light green). F, mean (±95% CI) expression (left y‐axis) and maximal activity (right y‐axis) of citrate synthase (CS). G, bivariate correlation between intensity (log2‐expression) of CS as determined by proteomics (y‐axis) versus expression (left y‐axis) and maximal activity (right y‐axis) of CS as determined by Western blotting and fluorometrically, respectively. H, mean (±95% CI) relative change in abundance of OXPHOS complexes I–V (CI–V) determined by Western blotting. I, representative blots of CS and OXPHOS CI–V. #Between‐group interaction (P ≤ 0.05). ##Between‐group interaction (P ≤ 0.01). *Within‐group difference from pre (P ≤ 0.05). **Within‐group difference from pre (P ≤ 0.01).
Muscle capillary density has been shown to be related to and associated with endurance performance (Saltin et al. 1977; Coyle et al. 1991). Nonetheless, we observed opposite adaptations in capillarization of the vastus lateralis muscle considering that observed for and exercise performance. Inhalation of β2‐agonist increased capillary density by 13% (P ≤ 0.05) and capillary‐to‐fibre ratio by 21% (P ≤ 0.01) in HIT+β2A, whereas HIT showed no relevant changes with the intervention (Fig. 2 D). Thus, the increased in HIT and the blunted effect in HIT+β2A might be related to an inhibitory effect of β2‐agonist on adaptations of the heart or mitochondrial oxidative capacity of the exercised muscles (Holloszy & Coyle, 1984).
From proteomic analysis, we detected and grouped 13 proteins related to the tricarboxylic acid (TCA) cycle and 58 proteins related to oxidative phosphorylation of the mitochondria (Fig. 2 A). For the TCA cycle, only minor changes were observed, in which succinate dehydrogenase flavoprotein subunit increased in both groups (Tables 2 and 4), whereas aconitate hydratase and malate dehydrogenase decreased in HIT+β2A (Table 5) with a between‐group interaction for aconitate hydratase (Table 6). While proteomic analysis revealed no significant changes in citrate synthase, Western blot analysis showed a minor increase in citrate synthase content in HIT with a tendency towards a higher maximal enzymatic activity (Fig. 2 F), but values were not statistically different from HIT+β2A. There was a good correlation between the proteomic measurements of citrate synthase with that of Western blotting and enzymatic activity (Fig. 2 G). In the oxidative phosphorylation, pronounced modulation was observed with the intervention. In HIT, abundance of a variety of the NADH dehydrogenase subcomplexes and ATP synthase subunits increased with the intervention (Table 2), whereas HIT+β2A only had a few significantly upregulated proteins (Table 4) and even a blunted training‐induced increase in abundance of four NADH dehydrogenase subcomplexes (Table 6). Consistent with this finding, Western blotting revealed an upregulation of overall abundance of OXPHOS complexes I–V in HIT (P ≤ 0.01), whereas this effect was attenuated (P ≤ 0.01) in HIT+β2A (Fig. 2 H).
β2‐Agonist alters contractile phenotype
Some clearly distinct adaptations in muscle contractile properties and phenotype were observed between the groups. As shown in Fig. 3 A and B, muscle proteome signature changed differently in HIT and HIT+β2A as indicated by a between‐group separation (Wilks’ Lambda: 0.127, P ≤ 0.001) (Fig. 3 B) in a principal component‐discriminant analysis that explained 80% of the variance for the contractile proteins listed in Fig. 3 A. These between‐group differences were primarily driven by opposing adaptations in myosin light chains, troponin C and tropomyosin α and β chains (Table 6), all of which may affect contractile properties and myofibrillar Ca2+ sensitivity (Greaser et al. 1988; Lamboley et al. 2014). Muscle fibre phenotype has historically been categorized according to MHC isoforms (Lutz et al. 1979; Harridge et al. 1996). Expression of myosin 1, the most highly expressed myosin isoform in type IIx fibres (Murgia et al. 2015), decreased with the intervention in HIT (log2‐fold change: −0.48, P ≤ 0.05) (Fig. 3 A and Table 2). Consistent with this observation, immunohistochemical analysis of the muscle biopsies revealed significant between‐group changes (P ≤ 0.05) with the intervention, in which HIT had a reduction in distribution of MHCIIx (P ≤ 0.05) (Fig. 3 C), whereas HIT+β2A had an increase in distribution of MHCIIa by 4.1% (P ≤ 0.05) (Fig. 3 C). There was a high degree of consistency between myosin expression data determined by proteomics and MHC isoform distribution determined immunohistochemically (Fig. 3 D). Aside from muscle contractile phenotype changes, HIT and HIT+β2A had some different adaptations in sarcomere architecture and cell‐to‐cell adhesion. Compared to HIT, HIT+β2A had an upregulation of the actin‐ and anchoring‐related proteins actin and α‐actinin‐1 (Table 6).
Figure 3. Muscle contractile phenotype and function in HIT and HIT+β2A.
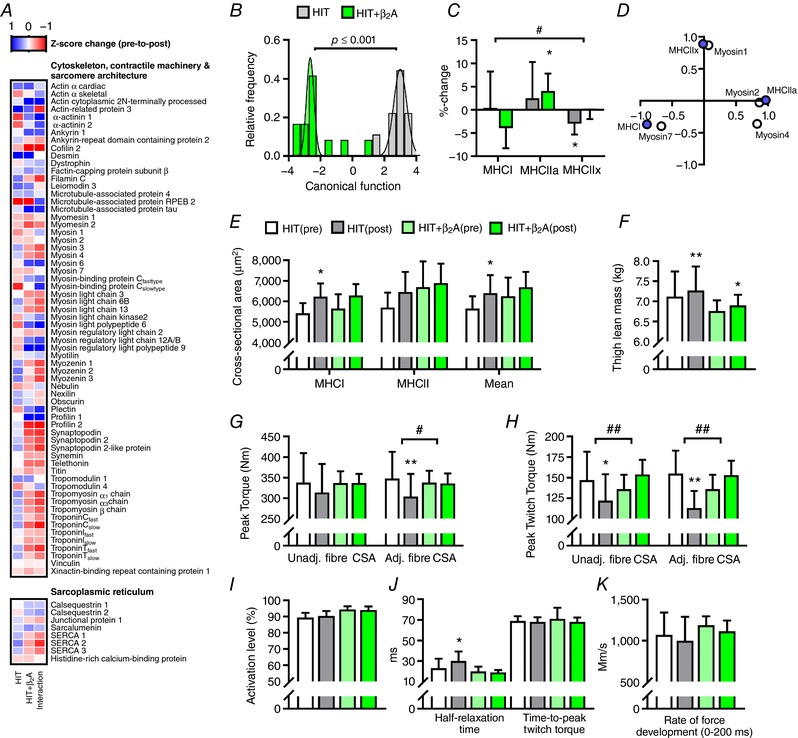
Effect of 4 weeks of high intensity training with (HIT+β2A, n = 12) and without (HIT, n = 9) daily inhalation of terbutaline (4 mg day−1) on outcomes related to contractile phenotype and function in trained men. A, heat map of mean Z‐score changes of the proteome related to the cytoskeleton and contractile phenotype in HIT and HIT+β2A, as well as between‐group interaction (HIT+β2A – HIT). B, Gaussian fit of frequency distribution of discriminant canonical function score of within‐group changes in the proteins listed in A using a principal component‐discriminant analysis (PCA‐DA) of the eight first principal components that explained 80% of the variance. C, mean (±95% CI) change in myosin heavy chain (MHC) isoform distribution. D, PCA of intensity (log2‐expression) of myosin isoforms determined by proteomics (open circles) and MHC isoform distribution determined immunohistochemically (purple circles). E–K, mean (±95% CI) values for muscle mass and contractile properties. CSA, cross‐sectional area. #Between‐group interaction (P ≤ 0.05). ##Between‐group interaction (P ≤ 0.01). *Within‐group difference from pre (P ≤ 0.05). **Within‐group difference from pre (P ≤ 0.01).
Although a common effect of β2‐agonist treatment is muscle growth (Lynch & Ryall, 2008; Joassard et al. 2013; Hostrup et al. 2015), we observed a similar degree of thigh muscle hypertrophy in both groups with the intervention (Fig. 3 F). At the muscle fibre level, however, histochemical analysis revealed that only HIT had an increase in mean cross‐sectional area (CSA) of muscle fibres (P ≤ 0.05), albeit not statistically different from HIT+β2A (Fig. 3 E). Despite the induced hypertrophy of the thigh, maximal isometric muscle strength (peak torque) of the quadriceps did not change in either HIT or HIT+β2A (Fig. 3 G). In fact, when adjusting for adaptations in muscle fibre CSA, peak torque declined by 19% with the intervention in HIT (P ≤ 0.01), whereas no change was observed in HIT+β2A (Fig. 3 G). Likewise, peak twitch torque declined by 17% (P ≤ 0.05) and 27% (P ≤ 0.01) in HIT when unadjusted and adjusted for muscle fibre CSA, respectively, whereas non‐significant increases were observed in HIT+β2A (Fig. 3 H). Furthermore, muscle half‐relaxation time was prolonged by 8 (±6) ms with the intervention in HIT (P ≤ 0.05), but not differently from HIT+β2A which showed no relevant change with the intervention (Fig. 3 J). Degree of voluntary activation level (Fig. 3 I), time‐to‐peak twitch torque (Fig. 3 J) and rate of force development (Fig. 3 K) of the quadriceps did not change in either group with the intervention.
β2‐Agonist represses metabolic pathways
Various myocellular pathways regulate substrate choice during exercise, where glycogenolysis and glycolysis are predominant metabolic pathways for glucose metabolism and β‐oxidation for lipid metabolism. Though only minor changes were observed in proteins related to lipid metabolism, HIT and HIT+β2A had markedly diverse adaptations in the muscle proteome related to glycogenesis and glycogenolysis (Fig. 4 A). Notably, in HIT+β2A, inhalation of β2‐agonist attenuated the training‐induced upregulation of glycogenin 1, glycogen debranching enzyme, glycogen synthase and glycogen phosphorylase observed in HIT (Table 6). HIT+β2A even showed a marked downregulation of glycogen debranching enzyme, glycogen phosphorylase and glycogenin 1 (Fig. 4 A, Table 5). Likewise, glycogen synthase, the rate‐limiting enzyme in glycogenesis, which was the most significantly upregulated protein of the proteome in HIT (Fig. 4 A, Table 2), did not change in HIT+β2A. For further confirmation of this observation, Western blotting of glycogen synthase revealed a 25% increase in glycogen synthase abundance in HIT (P ≤ 0.01), which was blunted (P ≤ 0.01) in HIT+β2A (Fig. 4 D). In the glycolytic pathway, some minor changes were observed. Abundance of hexokinase 1 increased with the intervention in HIT+β2A compared to HIT (Tables 4 and 6) with a concurrent minor repression of lactate dehydrogenase A chain (Table 5). Partly in accordance with these observations, enzymatic analysis showed that maximal activity of hexokinase increased (P ≤ 0.01) (Fig. 4 B) and lactate dehydrogenase decreased in HIT+β2A (P ≤ 0.05) (Fig. 4 C). In contrast, HIT showed a minor increase in abundance and maximal activity of the glycolytic rate‐limiting enzyme, phosphofructokinase (P ≤ 0.05) (Fig. 4 B and D), and a marked increase in lactate dehydrogenase activity (P ≤ 0.01) (Fig. 4 C). In general, principal component analysis revealed a good association between the different assays used to determine changes in glycogen synthase, hexokinase, phosphofructokinase and lactate dehydrogenase (Fig. 4 F).
Figure 4. Substrate choice during exercise and muscle metabolic enzymes in HIT and HIT+β2A.
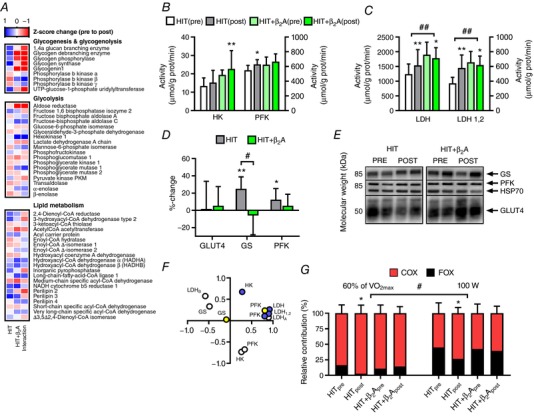
Effect of 4 weeks of high intensity training with (HIT+β2A, n = 12) and without (HIT, n = 9) daily inhalation of terbutaline (4 mg day−1) on outcomes related to metabolism in trained men. A, heat map of mean Z‐score changes of the proteome related to glycogenesis, glycogenolysis, glycolysis and lipid metabolism with the intervention in HIT and HIT+β2A, as well as between‐group interaction (HIT+β2A – HIT). B and C, mean (±95% CI) maximal activity for hexokinase (HK), phosphofructokinase (PFK) and lactate dehydrogenase (LDH). D, mean (±95% CI) relative change in abundance of GLUT4, glycogen synthase (GS) and PFK determined by Western blotting. E, representative blots of GLUT4, GS and PFK. F, principal component analysis of intensity (log2‐expression) of GS, HK, LDHA, LDHB and PFK determined by proteomics (open circles), GS and PFK expression determined by Western blotting (yellow circles), and HK, LDH and PFK activity determined fluorumetrically (purple circles). G, mean (±95% CI) relative contribution of carbohydrate (COX; red bars) and fat oxidation (FOX; black bars) during cycling at 60% of maximal oxygen consumption (; left side) and at 100 W (right side). #Between‐group interaction (P ≤ 0.05). ##Between‐group interaction (P ≤ 0.01). *Within‐group difference from pre (P ≤ 0.05). **Within‐group difference from pre (P ≤ 0.01).
The major sources of substrate for ATP production during endurance exercise are glucose and free fatty acids, whereas the contribution of amino acids is negligible (Jeukendrup & Wallis, 2005). As a measure for changes in substrate choice during exercise, we determined subjects’ glucose and fat oxidation by indirect calorimetry during steady state cycling both at same absolute intensity at 100 W and at an intensity corresponding to 60% of before and after the intervention. While HIT showed an increase in the relative contribution of glucose oxidation by 15.9 (±7.0) percentage‐points and a corresponding decline in fat oxidation with the intervention at both intensities (P ≤ 0.01), concomitant daily inhalation of β2‐agonist blunted (P ≤ 0.05) this training‐induced change in substrate choice in HIT+β2A (Fig. 4 G).
Discussion
Herein we have described adaptations in skeletal muscle proteome signature and phenotype induced by high intensity training with and without concomitant daily inhalation of β2‐agonist, as well as associated physiological adaptations in exercise performance, , muscle contractile properties and substrate utilization during exercise. The most important findings were that daily β2‐agonist treatment altered training‐induced changes in muscle proteome signature and phenotype and attenuated enhancements in and exercise performance.
While several studies have investigated the effect of high intensity interval training in human skeletal muscle (Laursen & Jenkens, 2002; Padrão et al. 2016; Hostrup & Bangsbo, 2017) and the chronic effect of β2‐agonist treatment in rodent muscle (Pearen et al. 2009; Koopman et al. 2010), the present study is the first to describe changes in muscle proteome signature and phenotype with related functional measures after an intervention with high intensity interval training and β2‐agonist treatment in humans. Consistent with previous studies (Laursen & Jenkins, 2002; Milanovic et al. 2015), we observed that high intensity interval training effectively enhanced and exercise performance in trained young men. More surprisingly, however, we observed that daily inhalation of β2‐agonist, terbutaline, completely blunted the training‐induced increase in and reduced enhancement in exercise performance. Although a few studies in rodents have shown that systemic chronic treatment with β2‐agonist clenbuterol blunts the effect of endurance training on exercise capacity (Ingalls et al. 1996; Duncan et al. 2000), the present study is the first to show such effect of β2‐agonists in humans. Furthermore, the present study is the first to show that daily β2‐agonist treatment attenuates the enhancing effect of endurance training on . It is noteworthy that inhalation of β2‐agonist, at close to therapeutic doses, is capable of blunting the enhancing effects of high intensity interval training on and exercise performance. This is possibly related to the relatively high systemic bioavailability of inhaled β2‐agonists (Dyreborg et al. 2016), partitioning into muscle (Jacobson et al. 2014) and inducing a continuous adrenergic fight‐or‐flight response (Emrick et al. 2010; Andersson et al. 2012; Hostrup et al. 2014b). Accordingly, high inhalation of terbutaline has been shown to induce significant β2‐adrenergic stimulation of skeletal muscle in humans (Hostrup et al. 2014b; 2016; Kalsen et al. 2016a,b). The dosage of inhaled terbutaline (4 mg) administered in the present study has also been shown to result in systemic concentrations that exceed those observed after oral administration of 10 mg terbutaline (Elers et al. 2012; Dyreborg et al. 2016).
Our observations indicate that the attenuating effect of β2‐agonist on training‐induced enhancement in exercise performance and , at least in part, may be related to remodelling of muscle proteome signature and phenotype. Indeed, enhancements in exercise performance and observed in HIT were complemented by pronuonced upregulation of mitochondrial proteins and metabolic pathways in skeletal muscle, in particular related to oxidative phosphorylation and glycogen metabolism, being consistent with reports in humans and rodents (Holloway et al. 2009; Egan et al. 2011; Sollanek et al. 2017). In contrast, however, we observed that daily inhalation of β2‐agonist attenuated training‐induced upregulation of NADH dehydrogenase subcomplexes of the oxidative phosphorylation and even caused a repression of carbohydrate metabolic pathways, including downregulation of glycogen debranching enzyme, glycogen phosphorylase and glycogenin 1. The repressing effect of β2‐agonist treatment on oxidative capacity and carbohydrate metabolism is in agreement with observations in rodents (Torgan et al. 1995), in which chronic β2‐agonist treatment has been shown to compromise mitochondrial function and pyruvate oxidation capacity (Hoshino et al. 2012). Given that a high capacity for oxidative metabolism and glycogenolysis is essential for energy and ion homeostasis of skeletal muscle and thus performance during intense exercise (Allen et al. 2008; Ørtenblad et al. 2013; Hostrup & Bangsbo, 2017), the attenuating effect of β2‐agonist on training‐induced upregulation of oxidative and glycogenolytic proteins may explain why training‐induced enhancement in exercise performance was reduced in HIT+β2A. Furthermore, it may be speculated that β2‐agonist treatment compromised glycogen storage in HIT+β2A compared to HIT in the present study, since glycogenin 1 was repressed by β2‐agonist treatment and that training‐induced upregulation of glycogen synthase was blunted in HIT+β2A. Accordingly, expression and activity of glycogenin are proportional to glycogen content in human skeletal muscle (Shearer et al. 2000, 2005) and glycogen synthase is a key enzyme in muscle glycogenesis (Bouskila et al. 2010). Thus, future studies should elucidate whether β2‐agonist‐induced proteome changes in glycogen metabolism are associated with alterations in resting glycogen content and rate of glycogenolysis during exercise.
Aside from the effect of β2‐agonist treatment on training‐induced adaptations in oxidative and metabolic pathways, we observed that terbutaline also modulated adaptations in muscle contractile phenotype and function. While the high intensity interval training regime undertaken induced a shift in muscle contractile phenotype from type IIx towards type IIa, concomitant daily inhalation of β2‐agonist induced a muscle phenotype transition from slow‐ towards a fast‐twitch phenotype. Although slow‐to‐fast‐twitch muscle phenotype transition is a common phenomenon after chronic β2‐agonist treatment in rodents (Dodd et al. 1996; Zhang et al. 1996; Jones et al. 2004; Sirvent et al. 2014), this has not been described previously in humans. The contractile slow‐to‐fast twitch phenotype changes induced by chronic β2‐agonist treatment have been shown to be associated with enhancements in maximal force and acceleration of relaxation time of skeletal muscle in both rodents and humans (Martineau et al. 1992; Dodd et al. 1996; Zhang et al. 1996; Hostrup et al. 2015, 2016). In contrast to these findings, however, β2‐agonist treatment did not increase muscle force and peak twitch force or accelerate relaxation time of the quadriceps in HIT+β2A. This discrepancy is possibly related to the fact that β2‐agonist treatment did not induce muscle hypertrophy or changes in expression of sarcoplasmic reticulum Ca2+ ATPase (SERCA) isoforms in HIT+β2A compared to HIT in the present study. As such, muscle hypertrophy has been shown to be the main mechanism by which chronic β2‐agonist treatment enhances peak twitch and tetanic muscle force (Dodd et al. 1996; Hostrup et al. 2015). Furthermore, studies suggest that the accelerating effect of chronic β2‐agonist treatment on muscle relaxation time is attributed to upregulation of SERCAI (Zhang et al. 1996; Hostrup et al. 2015) and concurrent downregulation of SERCAII and SERCA‐regulatory subunit phospholamban (Zhang et al. 1996). In HIT, on the other hand, peak twitch force declined and relaxation time was prolonged with the intervention, thus indicating that changes in contractile phenotype were associated with some of the adaptations in quadriceps contractile properties. Given that muscle contractile properties are highly dependent on muscle fibre contractile phenotype (Harridge et al. 1996), the phenotype changes in HIT and HIT+β2A may explain why HIT had a slower muscle relaxation time and a decline in peak twitch torque after the intervention.
The observation that β2‐agonist treatment alters training‐induced remodelling of proteome signature and phenotype in relation to oxidative capacity, as well as metabolic and contractile properties of skeletal muscle, coincides with the acute and temporal changes in gene transcription profile observed in rodent skeletal muscle upon β2‐adrenergic stimulation (Spurlock et al. 2006; Pearen et al. 2009). Acute β2‐adrenoceptor activation has profound effects on global gene expression in rodent skeletal muscle, where β2‐agonists formoterol and clenbuterol have been shown to enrich a variety of pathways, including those related to metabolism, cell‐to‐cell communication and transcriptional regulation (Spurlock et al. 2006; Pearen et al. 2009). Apart from the proteome adaptations in metabolism, we also observed that β2‐agonist treatment induced an upregulation of proteome pathways related to transcriptional regulation compared to training alone, including histone trimethylation and the nucleosome. Because β2‐adrenergic stimulation induces an adrenergic fight‐or‐flight response in skeletal muscle (Emrick et al. 2010; Andersson et al. 2012), nucleosome adaptations to chronic β2‐agonist treatment seem to be a logical consequence of the continuous adrenergic myocellular stress and signalling (Pearen et al. 2009). Indeed, non‐selective β‐agonist isoproterenol has been shown to induce expression of histones in cells (Lim & Juhnn, 2016), and Spurlock et al. (2006) observed that chronic β2‐agonist treatment with clenbuterol upregulated transcriptional and translational initiator genes in rodent muscle. In addition, we observed that β2‐agonist treatment upregulated proteome pathways related to adherens junction, cell‐to‐cell communication and cytoskeleton, inducing expression of actin and α‐actinin 1 compared to training alone. It could be speculated that such upregulations may have impacted between‐fibre force transfer positively in HIT+β2A (Hughes et al. 2016; Lambert et al. 2016; Nelson et al. 2016). Thus, a potential modulation of sarcomere architecture and cell‐to‐cell adhesion induced by β2‐agonist treatment may explain why HIT+β2A had no reduction in peak twitch torque and mass‐specific peak torque compared to HIT.
While the acute and chronic response to β2‐adrenoceptor activation may have some similarities with exercise training (Spurlock et al. 2006; Pearen et al. 2009; Hostrup et al. 2015), the present study, as well as studies in rodents (Ingalls et al. 1996; Lynch et al. 1996; Duncan et al. 2000; Mounier et al. 2007), indicate that chronic β2‐agonist treatment confounds the effect of exercise training. For instance, although a common feature of chronic β2‐agonist treatment is muscle hypertrophy in rodents and humans (Lynch & Ryall, 2008; Joassard et al. 2013; Hostrup et al. 2015), we observed no apparent induction of hypertrophy by β2‐agonist treatment compared to training alone. This observation is consistent with observations in rodents, in which exercise training blunts or reduces the hypertrophic effect of chronic clenbuterol treatment (Duncan et al. 2000; Mounier et al. 2007). The mechanisms underlying the attenuating effect of endurance training on the hypertrophic response to β2‐agonist treatment remain to be elucidated, but may be related to the fact that endurance training interferes with the growth‐promoting signalling (Coffey & Hawley, 2017) induced upon β2‐adrenergic stimulation (Koopman et al. 2010).
In summary, the present study showed that daily inhalation of β2‐agonist alters adaptations to 4 weeks of high intensity endurance training in recreationally active young men, both in terms of proteome signature remodelling and phenotype changes of skeletal muscle, but also in relation to functional adaptations in exercise performance, , muscle contractile properties and substrate utilization during exercise. The most notable observation was that β2‐agonist treatment attenuated training‐induced enhancements in exercise performance and , as well as blunted adaptations in oxidative phosphorylation and glycogen metabolism of skeletal muscle. Furthermore, that β2‐agonist treatment induced a slow‐to‐fast twitch muscle fibre type transition. Future studies should elucidate the molecular mechanisms underlying the confounding effect of β2‐agonist treatment on adaptations to endurance training.
Additional information
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD005480.
Competing interests
The authors have no competing interests.
Author contributions
M.H. designed the study, participated in data collection, analysis and interpretation, and drafted the manuscript. J.O. conducted the human experiments and participated in data analysis and interpretation, as well as in drafting of the manuscript. G.A.J. and R.W. conducted the muscle proteomic analysis and assisted in the bioinformatics analysis and interpretation, as well as in revising the manuscript. J.B. participated in designing the study and in the analysis and interpretation data, as well as in drafting of the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The study was supported by a grant from the Danish Ministry of Culture and Team Denmark.
Translational perspective
This study tested the hypothesis that daily inhalation of β2‐agonist alters adaptations in muscle proteome signature incurred from high intensity training. We observed that the β2‐agonist terbutaline blunted training‐induced upregulation of proteome pathways related to oxidative phosphorylation and glycogen metabolism, while concurrently inducing a slow‐to‐fast twitch muscle phenotype transition. Notably, terbutaline also attenuated training‐induced enhancements in and exercise performance. These findings are of interest to the large proportion of persons using inhaled β2‐agonists on a daily basis, including athletes, and indicate that athletes who have a high use of β2‐agonist may benefit from lessening their reliance on β2‐agonist inhalers. This is also consistent with clinical guidelines, where regular reliance on β2‐agonists is indicative of poor asthma control. Furthermore, the present study provides a fingerprint of some of the muscle proteome and phenotype changes induced by high intensity training with and without β2‐agonist treatment, which may be used for future therapeutic applications. Indeed, it has long been proposed that β2‐agonists have potential therapeutic use in the treatment of muscle atrophic conditions (Lynch & Ryall, 2008; Joassard et al. 2013), despite their potential cardiomyotoxic effects when administered in high doses. Accordingly, chronic β2‐agonist treatment may repress oxidative capacity (Léger et al. 2011) and induce collagen infiltration and necrosis in rodent cardiac muscle (Duncan et al. 2000; Burniston et al. 2002, 2005, 2006; Gregorevic et al. 2005). Thus, the attenuating effect of daily β2‐agonist treatment on training‐induced enhancements in exercise performance and may involve modulating effects in both skeletal and cardiac muscle.
Acknowledgements
We express our gratitude for the technical assistance of Anders Krogh Lemminger, Anders Schulze Gad, Christian Narkowicz, Jens Jung Nielsen, Martin Thomassen, Nanna Krogh, Peter Piil and Søren Jessen throughout the study in the collection, analyses, and interpretation of data.
Edited by: Scott Powers & Paul Greenhaff
References
- Aagaard P, Simonsen EB, Andersen JL, Magnusson P & Dyhre‐Poulsen P (2002). Increased rate of force development and neural drive of human skeletal muscle following resistance training. J Appl Physiol 93, 1318–1326. [DOI] [PubMed] [Google Scholar]
- Allen DG, Lamb GD & Westerblad H (2008). Skeletal muscle fatigue: cellular mechanisms. Physiol Rev 88, 287–332. [DOI] [PubMed] [Google Scholar]
- Andersson DC, Betzenhauser MJ, Reiken S, Umanskaya A, Shiomi T & Marks AR (2012). Stress‐induced increase in skeletal muscle force requires protein kinase A phosphorylation of the ryanodine receptor. J Physiol 590, 6381–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z, Foo SY, Ma Y, Ruas JL, Bommi‐Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A & Spiegelman BM (2008). HIF‐independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC‐1α. Nature 451, 1008–1012. [DOI] [PubMed] [Google Scholar]
- Arie S (2012). What can we learn from asthma in elite athletes? BMJ 344, e2556. [DOI] [PubMed] [Google Scholar]
- Bachasson D, Millet GY, Decorte N, Wuyam B, Levy P & Verges S (2013). Quadriceps function assessment using an incremental test and magnetic neurostimulation: a reliability study. J Electromyogr Kinesiol 23, 649–658. [DOI] [PubMed] [Google Scholar]
- Baker JG (2010). The selectivity of β‐adrenoceptor agonists at human β1‐, β2‐ and β3‐adrenoceptors. Br J Pharmacol 160, 1048–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbé C, Bray F, Gueugneau M, Devassine S, Lause P, Tokarski C, Rolando C & Thissen JP (2017). Comparative proteomic and transcriptomic analysis of follistatin‐induced skeletal muscle hypertrophy. J Proteome Res 16, 3477–3490. [DOI] [PubMed] [Google Scholar]
- Bergstrøm J (1975) .Percutaneous needle biopsy of skeletal muscle in physiological and clinical research. Scand J Clin Lab Invest 35, 609–616. [PubMed] [Google Scholar]
- Bouskila M, Hunter RW, Ibrahim AF, Delattre L, Peggie M, van Diepen JA, Voshol PJ, Jensen J & Sakamoto K (2010). Allosteric regulation of glycogen synthase controls glycogen synthesis in muscle. Cell Metab 12, 456–466. [DOI] [PubMed] [Google Scholar]
- Brandt N, Gunnarsson TP, Hostrup M, Tybirk J, Nybo L, Pilegaard H & Bangsbo J (2016). Impact of adrenaline and metabolic stress on exercise‐induced intracellular signaling and PGC‐1α mRNA response in human skeletal muscle. Physiol Rep 4, e12844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burniston JG, Clark WA, Tan LB & Goldspink DF (2006). Dose‐dependent separation of the hypertrophic and myotoxic effects of the β2‐adrenergic receptor agonist clenbuterol in rat striated muscles. Muscle Nerve 33, 655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burniston JG, Chester N, Clark WA, Tan LB & Goldspink DF (2005). Dose‐dependent apoptotic and necrotic myocyte death induced by the β2‐adrenergic receptor agonist, clenbuterol. Muscle Nerve 32, 767–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burniston JG, Ng Y, Clark WA, Colyer J, Tan LB & Goldspink DF (2002). Myotoxic effects of clenbuterol in the rat heart and soleus muscle. J Appl Physiol 93, 1824–1832. [DOI] [PubMed] [Google Scholar]
- Cantó C, Gerhart‐Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P & Auwerx J (2009). AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey VG & Hawley JA (2017). Concurrent exercise training: do opposites distract? J Physiol 595, 2883–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle EF, Feltner ME, Kautz SA, Hamilton MT, Montain SJ, Baylor AM, Abraham LD & Petrek GW (1991). Physiological and biomechanical factors associated with elite endurance cycling performance. Med Sci Sports Exerc 23, 93–107. [PubMed] [Google Scholar]
- Cruz AA (2007). Global Surveillance, Prevention and Control of Chronic Respiratory Diseases: A Comprehensive Approach, eds Bousquet J. & Khaltaev NG. World Health Organization. [Google Scholar]
- Dhana K, Koolhaas CM, Berghout MA, Peeters A, Ikram MA, Tiemeier H, Hofman A, Nusselder W & Franco OH (2016). Physical activity types and life expectancy with and without cardiovascular disease: the Rotterdam Study. J Public Health https://doi.org/10.1093/pubmed/fdw110. [DOI] [PubMed] [Google Scholar]
- Dodd SL, Powers SK, Vrabas IS, Criswell D, Stetson S & Hussain R (1996). Effects of clenbuterol on contractile and biochemical properties of skeletal muscle. Med Sci Sports Exerc 28, 669–676. [DOI] [PubMed] [Google Scholar]
- Duncan ND, Williams DA & Lynch GS (2000). Deleterious effects of chronic clenbuterol treatment on endurance and sprint exercise performance in rats. Clin Sci (Lond) 98, 339–347. [PubMed] [Google Scholar]
- Dyreborg A, Krogh N, Backer V, Rzeppa S, Hemmersbach P & Hostrup M (2016). Pharmacokinetics of oral and inhaled terbutaline after exercise in trained men. Front Pharmacol 7, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan B, Dowling P, O'Connor PL, Henry M, Meleady P, Zierath JR & O'Gorman DJ (2011). 2‐D DIGE analysis of the mitochondrial proteome from human skeletal muscle reveals time course‐dependent remodelling in response to 14 consecutive days of endurance exercise training. Proteomics 11, 1413–1428. [DOI] [PubMed] [Google Scholar]
- Elers J, Hostrup M, Pedersen L, Henninge J, Hemmersbach P, Dalhoff K & Backer V (2012). Urine and serum concentrations of inhaled and oral terbutaline. Int J Sports Med 33, 1026–1033. [DOI] [PubMed] [Google Scholar]
- Emrick MA, Sadilek M, Konoki K & Catterall WA (2010). Β‐adrenergic‐regulated phosphorylation of the skeletal muscle CaV1.1 channel in the fight‐or‐flight response. Proc Natl Acad Sci USA 107, 18712–18717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodyear LJ (2008). The exercise pill – too good to be true? N Engl J Med 359, 1842–1844. [DOI] [PubMed] [Google Scholar]
- Greaser ML, Moss RL & Reiser PJ (1988). Variations in contractile properties of rabbit single muscle fibres in relation to troponin T isoforms and myosin light chains. J Physiol 406, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorevic P, Ryall JG, Plant DR, Sillence MN & Lynch GS (2005). Chronic β‐agonist administration affects cardiac function of adult but not old rats, independent of β‐adrenoceptor density. Am J Physiol Heart Circ Physiol 289, H344–H349. [DOI] [PubMed] [Google Scholar]
- Harridge SD, Bottinelli R, Canepari M, Pellegrino MA, Reggiani C, Esbjörnsson M & Saltin B (1996). Whole‐muscle and single‐fibre contractile properties and myosin heavy chain isoforms in humans. Pflugers Arch 432, 913–920. [DOI] [PubMed] [Google Scholar]
- Hawley JA, Hargreaves M, Joyner MJ & Zierath JR (2014). Integrative biology of exercise. Cell 159, 738–749. [DOI] [PubMed] [Google Scholar]
- Hoffman NJ, Parker BL, Chaudhuri R, Fisher‐Wellman KH, Kleinert M, Humphrey SJ, Yang P, Holliday M, Trefely S, Fazakerley DJ, Stöckli J, Burchfield JG, Jensen TE, Jothi R, Kiens B, Wojtaszewski JF, Richter EA & James DE (2015). Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise‐regulated kinases and AMPK substrates. Cell Metab 22, 922–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloszy JO & Coyle EF (1984). Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J Appl Physiol Respir Environ Exerc Physiol 56, 831–838. [DOI] [PubMed] [Google Scholar]
- Holloway KV, O'Gorman M, Woods P, Morton JP, Evans L, Cable NT, Goldspink DF & Burniston JG (2009). Proteomic investigation of changes in human vastus lateralis muscle in response to interval‐exercise training. Proteomics 9, 5155–5574. [DOI] [PubMed] [Google Scholar]
- Hoshino D, Yoshida Y, Holloway GP, Lally J, Hatta H & Bonen A (2012). Clenbuterol, a β2‐adrenergic agonist, reciprocally alters PGC‐1 α and RIP140 and reduces fatty acid and pyruvate oxidation in rat skeletal muscle. Am J Physiol Regul Integr Comp Physiol 302, R373–R384. [DOI] [PubMed] [Google Scholar]
- Hostrup M & Bangsbo J (2017). Limitations in intense exercise performance of athletes ‐ effect of speed endurance training on ion handling and fatigue development. J Physiol 595, 2897–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostrup M, Kalsen A, Auchenberg M, Bangsbo J & Backer V (2016). Effects of acute and 2‐week administration of oral salbutamol on exercise performance and muscle strength in athletes. Scand J Med Sci Sports 26, 8–16. [DOI] [PubMed] [Google Scholar]
- Hostrup M, Kalsen A, Bangsbo J, Hemmersbach P, Karlsson S & Backer V (2014a). High‐dose inhaled terbutaline increases muscle strength and enhances maximal sprint performance in trained men. Eur J Appl Physiol 114, 2499–2508. [DOI] [PubMed] [Google Scholar]
- Hostrup M, Kalsen A, Onslev J, Jessen S, Haase C, Habib S, Ørtenblad N, Backer V & Bangsbo J (2015). Mechanisms underlying enhancements in muscle force and power output during maximal cycle ergometer exercise induced by chronic β2‐adrenergic stimulation in men. J Appl Physiol 119, 475–486. [DOI] [PubMed] [Google Scholar]
- Hostrup M, Kalsen A, Ortenblad N, Juel C, Mørch K, Rzeppa S, Karlsson S, Backer V & Bangsbo J (2014b). β2‐Adrenergic stimulation enhances Ca2+ release and contractile properties of skeletal muscles, and counteracts exercise‐induced reductions in Na+‐K+‐ATPase in trained men. J Physiol 592, 5445–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DC, Marcotte GR, Marshall AG, West DWD, Baehr LM, Wallace MA, Saleh PM, Bodine SC & Baar K (2016). Age‐related differences in dystrophin: impact on force transfer proteins, membrane integrity, and neuromuscular junction stability. J Gerontol A Biol Sci Med Sci 72, 640–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussey SE, Sharoff CG, Garnham A, Yi Z, Bowen BP, Mandarino LJ & Hargreaves M (2013). Effect of exercise on the skeletal muscle proteome in patients with type 2 diabetes. Med Sci Sports Exerc 45, 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingalls CP, Barnes WS & Smith SB (1996). Interaction between clenbuterol and run training: effects on exercise performance and MLC isoform content. J Appl Physiol 80, 795–801. [DOI] [PubMed] [Google Scholar]
- Jacobson GA, Yee KC, Premilovac D & Rattigan S (2014). Enantioselective disposition of (R/S)‐albuterol in skeletal and cardiac muscle. Drug Test Anal 6, 563–567. [DOI] [PubMed] [Google Scholar]
- Jensen J, Brennesvik EO, Bergersen H, Oseland H, Jebens E & Brørs O (2002). Quantitative determination of cell surface β‐adrenoceptors in different rat skeletal muscles. Pflugers Arch 444, 213–219. [DOI] [PubMed] [Google Scholar]
- Jensen J, Grønning‐Wang LM, Jebens E, Whitehead JP, Zorec R & Shepherd PR (2008). Adrenaline potentiates insulin‐stimulated PKB activation in the rat fast‐twitch epitrochlearis muscle without affecting IRS‐1‐associated PI 3‐kinase activity. Pflugers Arch 456, 969–978. [DOI] [PubMed] [Google Scholar]
- Jeukendrup AE & Wallis GA (2005). Measurement of substrate oxidation during exercise by means of gas exchange measurements. Int J Sports Med 26, 28–37. [DOI] [PubMed] [Google Scholar]
- Joassard OR, Durieux AC & Freyssenet DG (2013). β2‐Adrenergic agonists and the treatment of skeletal muscle wasting disorders. Int J Biochem Cell Biol 45, 2309–2321. [DOI] [PubMed] [Google Scholar]
- Jones SW, Baker DJ, Gardiner SM, Bennett T, Timmons JA & Greenhaff PL (2004). The effect of the β2‐adrenoceptor agonist prodrug BRL‐47672 on cardiovascular function, skeletal muscle myosin heavy chain, and MyoD expression in the rat. J Pharmacol Exp Ther 311, 1225–1131. [DOI] [PubMed] [Google Scholar]
- Kainu A, Pallasaho P, Piirilä P, Lindqvist A, Sovijärvi A & Pietinalho A (2013). Increase in prevalence of physician‐diagnosed asthma in Helsinki during the Finnish Asthma Programme: improved recognition of asthma in primary care? A cross‐sectional cohort study. Prim Care Respir J 22, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsen A, Hostrup M, Backer V & Bangsbo J (2016a). Effect of formoterol, a long‐acting β2‐adrenergic agonist, on muscle strength and power output, metabolism, and fatigue during maximal sprinting in men. Am J Physiol Regul Integr Comp Physiol 310, R1312–R1321. [DOI] [PubMed] [Google Scholar]
- Kalsen A, Hostrup M, Karlsson S, Hemmersbach P, Bangsbo J & Backer V (2014). Effect of inhaled terbutaline on substrate utilization and 300‐kcal time trial performance. J Appl Physiol 117, 1180–1187. [DOI] [PubMed] [Google Scholar]
- Kalsen A, Hostrup M, Söderlund K, Karlsson S, Backer V & Bangsbo J (2016b). Inhaled β2‐agonist increases power output and glycolysis during sprinting in men. Med Sci Sports Exerc 48, 39–48. [DOI] [PubMed] [Google Scholar]
- Koopman R, Gehrig SM, Léger B, Trieu J, Walrand S, Murphy KT & Lynch GS (2010). Cellular mechanisms underlying temporal changes in skeletal muscle protein synthesis and breakdown during chronic β‐adrenoceptor stimulation in mice. J Physiol 588, 4811–4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiberg M, Becker V, Jessen S, Rzeppa S, Hemmersbach P, Backer V & Hostrup M (2017). Influence of exercise in normal and hot ambient conditions on the pharmacokinetics of inhaled terbutaline in trained men. Scand J Med Sci Sports 27, 692–703. [DOI] [PubMed] [Google Scholar]
- Krogh N, Rzeppa S, Dyreborg A, Dehnes Y, Hemmersbach P, Backer V & Hostrup M (2017). Terbutaline accumulates in blood and urine following daily therapeutic inhalation. Med Sci Sports Exerc, 49, 1236–1243. [DOI] [PubMed] [Google Scholar]
- Lambert M, Richard E, Duban‐Deweer S, Krzewinski F, Deracinois B, Dupont E, Bastide B & Cieniewski‐Bernard C (2016). O‐GlcNAcylation is a key modulator of skeletal muscle sarcomeric morphometry associated to modulation of protein‐protein interactions. Biochim Biophys Acta 1860, 2017–2030. [DOI] [PubMed] [Google Scholar]
- Lamboley CR, Murphy RM, McKenna MJ & Lamb GD (2014). Sarcoplasmic reticulum Ca2+ uptake and leak properties, and SERCA isoform expression, in type I and type II fibres of human skeletal muscle. J Physiol 592, 1381–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen PB & Jenkins DG (2002). The scientific basis for high‐intensity interval training: optimising training programmes and maximising performance in highly trained endurance athletes. Sports Med 32, 53–73. [DOI] [PubMed] [Google Scholar]
- Léger B, Koopman R, Walrand S, Gehrig SM, Murphy KT & Lynch GS (2011). Chronic formoterol administration reduces cardiac mitochondrial protein synthesis and oxidative capacity in mice. Int J Cardiol 146, 270–272. [DOI] [PubMed] [Google Scholar]
- Lim JA & Juhnn YS (2016). Isoproterenol increases histone deacetylase 6 expression and cell migration by inhibiting ERK signaling via PKA and Epac pathways in human lung cancer cells. Exp Mol Med 48, e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH & Passonneau JV (1972). A Flexible System of Enzymatic Analysis, pp. 237–249. Academic Press, New York. [Google Scholar]
- Lutz H, Weber H, Billeter R & Jenny E (1979). Fast and slow myosin within single skeletal muscle fibres of adult rabbits. Nature 281, 142–144. [DOI] [PubMed] [Google Scholar]
- Lynch GS, Hayes A, Campbell SP & Williams DA (1996). Effects of β2‐agonist administration and exercise on contractile activation of skeletal muscle fibers. J Appl Physiol 81, 1610–1618. [DOI] [PubMed] [Google Scholar]
- Lynch GS & Ryall JG (2008). Role of β‐adrenoceptor signaling in skeletal muscle: implications for muscle wasting and disease. Physiol Rev 88, 729–767. [DOI] [PubMed] [Google Scholar]
- Martineau L, Horan MA, Rothwell NJ & Little RA (1992). Salbutamol, a β2‐adrenoceptor agonist, increases skeletal muscle strength in young men. Clin Sci (Lond) 83, 615–621. [DOI] [PubMed] [Google Scholar]
- Milanović Z, Sporiš G & Weston M (2015). Effectiveness of high‐intensity interval training (HIT) and continuous endurance training for VO2max improvements: a systematic review and meta‐analysis of controlled trials. Sports Med 45, 1469–1481. [DOI] [PubMed] [Google Scholar]
- Mounier R, Cavalié H, Lac G & Clottes E (2007). Molecular impact of clenbuterol and isometric strength training on rat EDL muscles. Pflugers Arch 453, 497–507. [DOI] [PubMed] [Google Scholar]
- Murgia M, Nagaraj N, Deshmukh AS, Zeiler M, Cancellara P, Moretti I, Reggiani C, Schiaffino S & Mann M (2015). Single muscle fiber proteomics reveals unexpected mitochondrial specialization. EMBO Rep 16, 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murton AJ, Billeter R, Stephens FB, Des Etages SG, Graber F, Hill RJ, Marimuthu K & Greenhaff PL (2014). Transient transcriptional events in human skeletal muscle at the outset of concentric resistance exercise training. J Appl Physiol 116, 113–125. [DOI] [PubMed] [Google Scholar]
- Nelson CE et al (2016). In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351, 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuez‐Ortin WG, Carter CG, Nichols PD & Wilson R (2016). Sequential protein extraction as an efficient method for improved proteome coverage in larvae of Atlantic salmon (Salmo salar). Proteomics 16, 2043–2047. [DOI] [PubMed] [Google Scholar]
- Nuez‐Ortín WG, Carter CG, Wilson R, Cooke IR, Amoroso G, Cobcroft JM & Nichols PD (2017). Triploid Atlantic salmon shows similar performance, fatty acid composition and proteome response to diploids during early freshwater rearing. Comp Biochem Physiol Part D Genomics Proteomics 22, 67–77. [DOI] [PubMed] [Google Scholar]
- Nyberg M, Fiorenza M, Lund A, Christensen M, Rømer T, Piil P, Hostrup M, Christensen PM, Holbek S, Ravnholt T, Gunnarsson TP & Bangsbo J (2016). Adaptations to speed endurance training in highly trained soccer players. Med Sci Sports Exerc 48, 1355–1364. [DOI] [PubMed] [Google Scholar]
- Padrão AI, Ferreira R, Amado F, Vitorino R & Duarte JA (2016). Uncovering the exercise‐related proteome signature in skeletal muscle. Proteomics 16, 816–830. [DOI] [PubMed] [Google Scholar]
- Parsons JP & Mastronarde JG (2005). Exercise‐induced bronchoconstriction in athletes. Chest 128, 3966–3974. [DOI] [PubMed] [Google Scholar]
- Pearen MA, Ryall JG, Lynch GS & Muscat GE (2009). Expression profiling of skeletal muscle following acute and chronic β2‐adrenergic stimulation: implications for hypertrophy, metabolism and circadian rhythm. BMC Genomics 10, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petriz BA, Gomes CP, Rocha LA, Rezende TM & Franco OL (2012). Proteomics applied to exercise physiology: a cutting‐edge technology. J Cell Physiol 227, 885–898. [DOI] [PubMed] [Google Scholar]
- Powers SK, Duarte J, Kavazis AN & Talbert EE (2010). Reactive oxygen species are signalling molecules for skeletal muscle adaptation. Exp Physiol 95, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Radak Z & Ji LL (2016). Exercise‐induced oxidative stress: past, present and future. J Physiol 594, 5081–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price OJ, Hull JH, Backer V, Hostrup M & Ansley L (2014). The impact of exercise‐induced bronchoconstriction on athletic performance: a systematic review. Sports Med 44, 1749–1761. [DOI] [PubMed] [Google Scholar]
- Rajab P, Fox J, Riaz S, Tomlinson D, Ball D & Greenhaff PL (2000). Skeletal muscle myosin heavy chain isoforms and energy metabolism after clenbuterol treatment in the rat. Am J Physiol Regul Integr Comp Physiol 279, R1076–R1081. [DOI] [PubMed] [Google Scholar]
- Rottenkolber M, Voogd E, van Dijk L, Primatesta P, Becker C, Schlienger R, de Groot MC, Alvarez Y, Durand J, Slattery J, Afonso A, Requena G, Gil M, Alvarez A, Hesse U, Gerlach R, Hasford J, Fischer R, Klungel OH & Schmiedl S (2015). Time trends of period prevalence rates of patients with inhaled long‐acting beta‐2‐agonists‐containing prescriptions: a European comparative database study. PLoS One 10, e0117628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltin B, Henriksson J, Nygaard E, Andersen P & Jansson E (1977). Fiber types and metabolic potentials of skeletal muscles in sedentary man and endurance runners. Ann N Y Acad Sci 301, 3–29. [DOI] [PubMed] [Google Scholar]
- Shearer J, Marchand I, Sathasivam P, Tarnopolsky MA & Graham TE (2000). Glycogenin activity in human skeletal muscle is proportional to muscle glycogen concentration. Am J Physiol Endocrinol Metab 278, E177–E180. [DOI] [PubMed] [Google Scholar]
- Shearer J, Wilson RJ, Battram DS, Richter EA, Robinson DL, Bakovic M & Graham TE (2005). Increases in glycogenin and glycogenin mRNA accompany glycogen resynthesis in human skeletal muscle. Am J Physiol Endocrinol Metab 289, E508–E514. [DOI] [PubMed] [Google Scholar]
- Sirvent P, Douillard A, Galbes O, Ramonatxo C, Py G, Candau R & Lacampagne A (2014). Effects of chronic administration of clenbuterol on contractile properties and calcium homeostasis in rat extensor digitorum longus muscle. PLoS One 9, e100281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollanek KJ, Burniston JG, Kavazis AN, Morton AB, Wiggs MP, Ahn B, Smuder AJ & Powers SK (2017). Global proteome changes in the rat diaphragm induced by endurance exercise training. PLoS One 12, e0171007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurlock DM, McDaneld TG & McIntyre LM (2006). Changes in skeletal muscle gene expression following clenbuterol administration. BMC Genomics 7, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD & Tibshirani R (2003). Statistical significance for genomewide studies. Proc Natl Acad Sci USA 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomassen M, Christensen PM, Gunnarsson TP, Nybo L & Bangsbo J (2010). Effect of 2‐wk intensified training and inactivity on muscle Na+‐K+ pump expression, phospholemman (FXYD1) phosphorylation, and performance in soccer players. J Appl Physiol 108, 898–905. [DOI] [PubMed] [Google Scholar]
- Thul PJ et al (2017). A subcellular map of the human proteome. Science 356, eaal3321. [DOI] [PubMed] [Google Scholar]
- Torgan CE, Etgen GJ Jr, Kang HY & Ivy JL (1995). Fiber type‐specific effects of clenbuterol and exercise training on insulin‐resistant muscle. J Appl Physiol 79, 163–167. [DOI] [PubMed] [Google Scholar]
- Uhlén M et al (2015). Proteomics. Tissue‐based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Westerterp KR (2001). Pattern and intensity of physical activity. Nature 410, 539. [DOI] [PubMed] [Google Scholar]
- White M, Roden R, Minobe W, Khan MF, Larrabee P, Wollmering M, Port JD, Anderson F, Campbell D, Feldman AM & Bristow MR (1994). Age‐related changes in β‐adrenergic neuroeffector systems in the human heart. Circulation 90, 1225–1238. [DOI] [PubMed] [Google Scholar]
- Williams RS, Caron MG & Daniel K (1984). Skeletal muscle β‐adrenergic receptors: variations due to fiber type and training. Am J Physiol 246, 160–167. [DOI] [PubMed] [Google Scholar]
- Wilson R, Diseberg AF, Gordon L, Zivkovic S, Tatarczuch L, Mackie EJ, Gorman JJ & Bateman JF (2010). Comprehensive profiling of cartilage extracellular matrix formation and maturation using sequential extraction and label‐free quantitative proteomics. Mol Cell Proteomics 9, 1296–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R, Golub SB, Rowley L, Angelucci C, Karpievitch YV, Bateman JF & Fosang AJ (2016). Novel elements of the chondrocyte stress response identified using an in vitro model of mouse cartilage degradation. J Proteome Res 15, 1033–1050. [DOI] [PubMed] [Google Scholar]
- Zhang KM, Hu P, Wang SW, Feher JJ, Wright LD, Wechsler AS, Spratt JA & Briggs FN (1996). Salbutamol changes the molecular and mechanical properties of canine skeletal muscle. J Physiol 496, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ørtenblad N, Westerblad H & Nielsen J (2013). Muscle glycogen stores and fatigue. J Physiol 591, 4405–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]