

Abstract
The calcium‐dependent antibiotics (CDAs) are an important emerging class of antibiotics. The crystal structure of the CDA laspartomycin C in complex with calcium and the ligand geranyl‐phosphate at a resolution of 1.28 Å is reported. This is the first crystal structure of a CDA bound to its bacterial target. The structure is also the first to be reported for an antibiotic that binds the essential bacterial phospholipid undecaprenyl phosphate (C55‐P). These structural insights are of great value in the design of antibiotics capable of exploiting this unique bacterial target.
Keywords: bacterial cell wall, C55-P, calcium-dependent antibiotics, crystal structure, laspartomycin c
Key to addressing the growing threat of drug‐resistant bacteria is the identification and characterization of antibiotics that operate by unexploited mechanisms.1, 2 Notable in this regard are the calcium‐dependent antibiotics (CDAs), which have gained clinical prominence owing to their activity against multi‐drug‐resistant pathogens.3, 4, 5 While a variety of antibacterial mechanisms have been ascribed to the various CDAs, an atomic‐level understanding of the recognition of their bacterial target(s) remains elusive. At present, daptomycin is the only clinically used CDA, and its mode of action is the topic of ongoing investigation.6, 7, 8, 9 By comparison, the structurally similar CDAs laspartomycin C, friulimycin B, tsushimycin, and amphomycin have more clearly understood antibacterial mechanisms.10, 11, 12, 13, 14 It was recently reported that laspartomycin C forms a high‐affinity complex (K d=7.3±3.8 nm) with the bacterial cell wall precursor undecaprenyl phosphate (C55‐P) and in doing so inhibits peptidoglycan biosynthesis, ultimately leading to cell death.14 Notably, the sequestration of C55‐P is a mechanism not exploited by any current clinically used antibiotic making it an attractive target for further study. To date, the only structural insights available for the C55‐P binding CDAs are provided by the unliganded structure of tsushimycin.15 To achieve a deeper understanding of the high‐affinity C55‐P binding and specificity, herein we report the crystal structure of laspartomycin C in complex with calcium and geranyl phosphate (C10‐P, a soluble C55‐P analogue) at a resolution of 1.28 Å. The target specificity of laspartomycin C was also studied by examining its interaction with other common phospholipids. Furthermore, the stereochemical implications of laspartomcyin C target binding revealed by the crystal structure were probed by synthesizing and testing the enantiomeric form of the antibiotic. Not only is the structure here reported the first for a CDA in complex with both calcium and its biomolecular target, it is also the first for any antibiotic that targets C55‐P.
While laspartomycin C can be isolated from fermentation of S. viridochromogenes,16, 17 it was found to be more convenient to prepare the compound by synthetic means as previously described (see the Supporting Information for synthetic details).14 The calcium‐dependent antibiotic activity of the synthetic laspartomycin C was confirmed against a number of bacterial strains, including drug‐resistant pathogens (Supporting Information, Table S1). Crystals of the laspartomycin C/Ca2+/geranyl phosphate (C10‐P) complex were grown and diffracted to 1.28 Å resolution (Supporting Information, Table S3). The refined structure reveals a stoichiometry for the complex of a single laspartomycin C molecule bound to one geranyl phosphate ligand along with two calcium ions playing key roles in mediating recognition (Figure 1).
Figure 1.
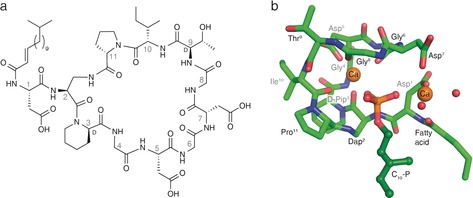
Laspartomycin C forms a 1:1:2 complex with C10‐P and Ca2+. a) Structure of laspartomycin C. b) Structure of the ternary complex with laspartomycin C (green stick representation), two bound Ca2+ ions (orange spheres), bound water molecules (red spheres), and the C10‐P ligand (dark green ball‐and‐stick representation). The Ca2+ ion on the left is referred to as the central Ca2+ ion and the Ca2+ ion on the right as the peripheral Ca2+ ion.
Laspartomycin C adopts a saddle‐shaped amphipathic fold with polar residues (Asp5, Asp7, d‐allo‐Thr9) aligning the top plain and aliphatic residues (d‐Pip3 and Pro11) forming the hydrophobic bottom plane from which the fatty acid side chain protrudes downwards (Figure 2). The cavity created within the laspartomycin C macrocycle envelops the phosphate head group of the C10‐P, which is held in place by hydrogen bonding to the peptide backbone and chelation of both calcium ions. The C10‐P isoprenyl tail appears to be stabilized by hydrophobic interactions with the laspartomycin C fatty acid side chain as it exits the binding pocket. The complex is well‐resolved except for the outermost parts of the C10‐P isoprenyl tail and the laspartomycin C fatty acid side chain. In the crystal lattice, the C10‐P isoprenyl units and fatty acid side chains of multiple complexes cluster together to form continuous hydrophobic sheets (Supporting Information, Figure S13). This may also hint at the biologically relevant orientation that laspartomycin C adopts on the bacterial cell surface in its sequestration of C55‐P at the membrane interface.
Figure 2.
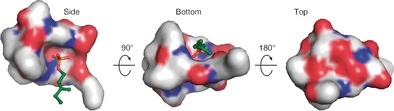
Surface distribution of polar regions of the Laspartomycin C/Ca2+/C10‐P complex. The “top” and the sides of the complex are hydrophilic whereas the bottom face is much more hydrophobic. The C10‐P ligand (green) is bound within the cavity of the laspartomycin C macrocycle. The atoms in laspartomycin C are colored as follows: O red, N blue, C white.
The structure of the complex provides an explanation for the high‐affinity binding of C55‐P by laspartomycin C. Target recognition takes place via direct interactions of the phospholipid head group with the laspartomycin C backbone and both calcium ions (Figure 3). The backbone and side chain amides of Dap2 as well as the backbone amide of Gly8 form hydrogen bonds with three of the four phosphate oxygen atoms (Supporting Information, Table S4). Of key importance are the two calcium ions that interact with the phosphate moiety as part of an octahedral coordination symmetry. The central Ca2+ is coordinated by 5 interactions with laspartomycin C involving four backbone carbonyls (Dap2, Gly6, Gly8, Ile10) and one aspartic acid side chain (Asp5) with all distances (2.3 Å) in agreement with expected Ca−O coordination distances (Figure 3 b; Supporting Information, Table S5).18 The peripheral Ca2+ is bound via interactions provided by two aspartic acid side chains (Asp1, Asp7) and the N‐terminal fatty acid carbonyl group along with two H2O molecules that complete the octahedral coordination (Figure 3 c).
Figure 3.
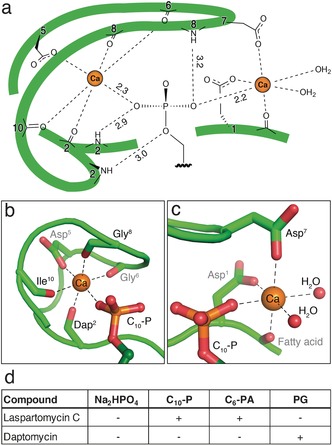
Target recognition by laspartomycin C. a) The C10‐P phosphate headgroup interacts via the coordinated Ca2+ ions and three laspartomycin C backbone amides. b) The central Ca2+ ion is coordinated by backbone carbonyl and side chain interactions as well as the C10‐P head group. c) The peripheral Ca2+ ion is coordinated by two side chain interactions, the fatty acid carbonyl, and the C10‐P head group with octahedral coordination completed by two water molecules. d) Antagonization of the antibiotic activity of laspartomycin C and daptomycin. Antibiotic activity was antagonized (+) or unaffected (−) by the presence of ≤2.0 mol equiv antagonist. Antagonization by Na2HPO4 was assessed employing a fixed 1.25 mm concentration corresponding to 24 and 500 mol equiv for laspartomycin C and daptomycin, respectively. Full data given in the Supporting Information, Table S2 and Figures S7–S9.
The binding of C10‐P by laspartomycin C as revealed by the crystal structure is not intrinsically dependent on a chiral interaction. To confirm this, we synthesized the enantiomeric form of laspartomycin C. As expected, circular dichroism (CD) analysis of laspartomycin C and its enantiomer yielded identical ellipticities of opposite sign with clear effects observed in the absence and presence of Ca2+ (Supporting Information, Figures S5,S6). Additionally, the CD spectra of both peptides indicate a second significant conformational change upon addition of 1.0 equiv of C10‐P. Addition of excess C10‐P did not result in further elliptic changes, in line with a 1:1 peptide:C10‐P binding stoichiometry. Finally, antibiotic testing of ent‐laspartomycin C in parallel with laspartomycin C showed identical antibiotic minimum inhibitory concentrations (MICs) for both compounds against a panel of five Gram‐positive pathogens (Supporting Information, Table S1).
The specificity of laspartomycin C binding to phosphate monoesters was evaluated using an antagonization assay wherein the antibiotic peptide was pre‐mixed with various phospholipids and the effect on activity assessed (Figure 3 d; Supporting Information, Figure S7–S9). Mixing laspartomycin C with inorganic phosphate (HPO4 2−) at concentrations corresponding to normal serum levels resulted in no observable antagonization. As expected, treatment of laspartomycin C with 1.0 equivalent of C10‐P led to loss of antibiotic activity while the activity of daptomycin was not inhibited by C10‐P addition even at higher concentrations. We also assessed the antagonization potential of a C6 truncated variant of the aliphatic mammalian lipid phosphatidic acid (C6‐PA). These studies revealed that C6‐PA is also capable of blocking the antibiotic action of laspartomycin C, with 1.0 mol equiv eliciting complete antagonization. This effect appears to be specific for phosphate monoesters. When the common lipid phosphodiester phosphatidyl glycerol (PG) was added to laspartomycin C in equimolar amount, no antagonization was observed. In fact only after adding a large excess of PG (8.0 molar equivalents) was the activity of laspartomycin C diminished. By comparison, the activity of daptomycin is much more readily antagonized by PG with 2.0 mol equiv, leading to complete loss of antibiotic action. Interestingly, PG is found in high concentrations in mammalian lung surfactant and is the likely cause of the ineffectiveness of daptomycin in treating lung infections.19 The effect of various biologically relevant phosphate monoesters on the activity of laspartomycin C was also investigated. No appreciable antagonization of activity was observed when laspartomycin C was treated with adenosine 5′‐monophosphate, rac‐glycerol‐1‐phosphate, dihydroxyacetone phosphate, glucose 6‐phosphate, O‐phospho‐l‐serine, or γ,γ‐dimethylallyl phosphate (Supporting Information, Table S2). Furthermore, to examine the role played by the length of the alkyl substituent, a series of saturated linear C1, C3, C6, and C10 phosphate monoesters where prepared and tested for their ability to impact the activity of laspartomycin C. These investigations revealed that only the C10 saturated linear phospholipid caused antagonization, suggesting that shorter unbranched aliphatic lipid tails do not support high affinity phosphate monoester binding by laspartomycin C.
The laspartomycin C crystal structure provides key insights into the structural feature that differentiate the antibiotic mechanisms of the lipopeptide and lipodepsipeptide CDA subclasses. In the case of laspartomycin C the side chain of Dap2 closes the peptide macrocycle via an amide bond with the C‐terminal proline. This newly formed amide linkage plays a role in target recognition as it contributes a hydrogen bonding interaction with the phosphate group in the binding pocket (Figure 3 a). Conversely, for lipodepsipeptide CDAs such as daptomycin an ester linkage is found at this position, resulting from cyclization of the C‐terminus with a threonine side chain. The ester linkage found in daptomycin is unable to serve as a H‐bond donor and instead a repulsive electrostatic oxygen‐oxygen interaction is expected to arise when encountering a phosphate monoester.
The laspartomycin C/Ca2+/C10‐P ternary complex exists as a dimer in the asymmetric unit related by a two‐fold rotation and is stabilized by direct and indirect interactions between the two ternary units (Figure 4). We also found evidence for the formation of the dimeric species in solution based on mass spectrometry studies (Supporting Information, Figures S10–S12). The two ternary units in the dimer are nearly identical to each other with only a substantial difference in the side chain rotamer for d‐allo‐Thr9 (Supporting Information, Figure S14). As illustrated in Figure 4, there are a number of interactions unique to the dimer. A hydrogen bond is observed between the d‐allo‐Thr9 backbone amide of one laspartomycin C molecule and the Asp7 side chain carboxylate of the other. Additional indirect hydrogen bonding interactions are mediated by the d‐allo‐Thr9 backbone carbonyl with the water molecules coordinated by the peripheral Ca2+ of the other ternary unit (Supporting Information, Figure S15). The fatty acid side chain of one laspartomycin C molecule has hydrophobic interactions with the Pro11 side chain of the other molecule in the dimer. Furthermore, the absence of a sidechain in Gly8 prevents steric hindrance from occurring at the dimer interface.
Figure 4.
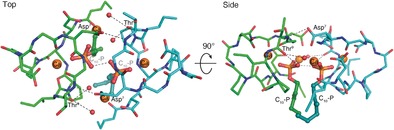
The laspartomycin C/Ca2+/C10‐P complex is a dimer in the crystal. Two views of the dimer interface, with cross‐dimer interactions shown as dotted lines. The two laspartomycin C chains of the two ternary complexes that make up the dimer are colored green and cyan. For clarity, in the side view only the interactors closet to the viewer are labeled.
In the dimer complex, the two C10‐P phosphate head groups are fully coordinated and completely sequestered from the solvent. The two C10‐P head groups also directly interact through a hydrogen bond (Figure 4; Supporting Information, Table S6). Furthermore, the isoprenyl tails of the two C10‐P ligands have hydrophobic interactions with each other and with the Pro11 side chains in the opposing laspartomycin C monomers. The more distal part of the isoprenyl tails are not stabilized in the complex and disordered in the crystals. The full coordination of the C10‐P headgroup and the interactions of laspartomycin C with the headgroup‐proximal part of the isoprenyl tails in the dimer complex is consistent with the high‐affinity laspartomycin C‐undecaprenyl phosphate interaction and explains the specificity of laspartomycin C for phosphate monoesters with both long and short hydrocarbon tails.
A straightforward model of how laspartomycin C is positioned on the bacterial membrane follows from the C10‐P bound dimer structure. The two laspartomycin C fatty acid side chains and the two C10‐P isoprenyl tails are all orientated perpendicular to a hydrophobic plane formed by the bottom sides of two laspartomycin C molecules in the dimer (Supporting Information, Figure S16). This plane is likely oriented parallel to the cell surface. The C10‐P head groups are sequestered above the hydrophobic bottom plane within the core of the laspartomycin C dimer. This suggests that when bound to C55‐P, laspartomycin C is slightly embedded in the bacterial membrane and that the hydrophobic side chains of d‐Pip3 and Pro11 contribute to interactions with the hydrophobic part of the lipid bilayer. Structural features that are important for dimer formation and Ca2+ binding are predominantly conserved within the CDA family. Notably, the structure of the unliganded tsushimycin/Ca2+ complex is also dimeric15 and superimposes closely to our laspartomycin C dimer in complex with Ca2+ and C10‐P (Supporting Information, Table S7, Figure S17). Whether this indicates that the structures of all CDAs are similar and whether they all form dimers awaits further experimental verification.
In summary, the laspartomycin C structure reported herein is the first of a CDA bound to its bacterial target and provides a clear explanation for the two conserved calcium‐binding sites common to all CDAs. Furthermore, it is also is the first structure to be reported for an antibiotic that binds the essential bacterial phospholipid C55‐P. At present, no clinically used antibiotics operate via C55‐P binding. Our findings provide a structural blueprint for the design of new antibiotics capable of exploiting this unique bacterial target.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Johan Kemmink for assistance in recording 2D NMR spectra. Financial support provided by Utrecht University and the Netherlands Organization for Scientific Research (NWO) is acknowledged. L.H.J.K. and T.M.W. were supported by NWO PhD student grants, H.C.V. by NWO grant no 01.80.104.00, B.J.C.J. by NWO‐VIDI grant no 723.012.002, and N.I.M. by an NWO‐VIDI grant no. 016.102.338.
L. H. J. Kleijn, H. C. Vlieg, T. M. Wood, J. Sastre Toraño, B. J. C. Janssen, N. I. Martin, Angew. Chem. Int. Ed. 2017, 56, 16546.
Contributor Information
Bert J. C. Janssen, Email: b.j.c.janssen@uu.nl
Dr. Nathaniel I. Martin, Email: n.i.martin@uu.nl.
References
- 1. Chellat M. F., Raguz L., Riedl R., Angew. Chem. Int. Ed. 2016, 55, 6600–6626; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6710–6738. [Google Scholar]
- 2. Tommasi R., Brown D. G., Walkup G. K., Manchester J. I., Miller A. A., Nat. Rev. Drug Discovery 2015, 14, 529–542. [DOI] [PubMed] [Google Scholar]
- 3. Schneider T., Mullera A., Miess H., Gross H., Int. J. Med. Microbiol. 2014, 304, 37–43. [DOI] [PubMed] [Google Scholar]
- 4. Strieker M., Marahiel M. A., ChemBioChem 2009, 10, 607–616. [DOI] [PubMed] [Google Scholar]
- 5.Book chapter “The Cyclic Lipopeptide Antibiotics”: Kleijn L. H. J., Martin N. I., Topics in Medicinal Chemistry, Vol. 47, Springer, Berlin, 2017, pp. 1–27. [Google Scholar]; https://doi.org/10.1007/7355_2017_9.
- 6. Zhang J., Scott W. R. P., Gabel F., Wu M., Desmond R., Bae J., Zaccai G., Russ Algar W., Straus S. K., Biochim. Biophys. Acta 2017, 1865, 1490–1499. [DOI] [PubMed] [Google Scholar]
- 7. Taylor S. D., Palmer M., Bioorg. Med. Chem. 2016, 24, 6253–6268. [DOI] [PubMed] [Google Scholar]
- 8. Muller A., Wenzel M., Strahl H., Grein F., Saaki T. N., Kohl B., Siersma T., Bandow J. E., Sahl H. G., Schneider T., Hamoen L. W., Proc. Natl. Acad. Sci. USA 2016, 113, E7077–E7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pogliano J., Pogliano N., Silverman J. A., J. Bacteriol. 2012, 194, 4494–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schneider T., Gries K., Josten M., Wiedemann I., Pelzer S., Labischinski H., Sahl H. G., Antimicrob. Agents Chemother. 2009, 53, 1610–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rubinchik E., Schneider T., Elliott M., Scott W. R., Pan J., Anklin C., Yang H., Dugourd D., Muller A., Gries K., Straus S. K., Sahl H. G., Hancock R. E., Antimicrob. Agents Chemother. 2011, 55, 2743–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dugourd D., Yang H., Elliott M., Siu R., Clement J. J., Straus S. K., Hancock R. E., Rubinchik E., Antimicrob. Agents Chemother. 2011, 55, 3720–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. 't Hart P., Kleijn L. H. J., de Bruin G., Oppedijk S. F., Kemmink J., Martin N. I., Org. Biomol. Chem. 2014, 12, 913–918. [DOI] [PubMed] [Google Scholar]
- 14. Kleijn L. H. J., Oppedijk S. F., 't Hart P., van Harten R. M., Martin-Visscher L. A., Kemmink J., Breukink E., Martin N. I., J. Med. Chem. 2016, 59, 3569–3574. [DOI] [PubMed] [Google Scholar]
- 15. Bunkóczi G., Vertesy L., Sheldrick G. M., Acta Crystallogr. Sect. D 2005, 61, 1160–1164. [DOI] [PubMed] [Google Scholar]
- 16. Naganawa H., Hamada M., Maeda K., Okami Y., Takeushi T., J. Antibiot. 1968, 21, 55–62. [DOI] [PubMed] [Google Scholar]
- 17. Borders D. B., Leese R. A., Jarolmen H., Francis N. D., Fantini A. A., Falla T., Fiddes J. C., Aumelas A., J. Nat. Prod. 2007, 70, 443–446. [DOI] [PubMed] [Google Scholar]
- 18. Harding M. M., Acta Crystallogr. Sect. D 2006, 62, 678–682. [DOI] [PubMed] [Google Scholar]
- 19. Silverman J. A., Mortin L. I., Vanpraagh A. D., Li T., Alder J., J. Infect. Dis. 2005, 191, 2149–2152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary