

Abstract
Microsatellite alterations within genomic DNA frameshift as a result of defective DNA mismatch repair (MMR). About 15% of sporadic colorectal cancers (CRCs) manifest hypermethylation of the DNA MMR gene MLH1, resulting in mono- and di-nucleotide frameshifts to classify it as microsatellite instability-high (MSI-H) and hypermutated, and due to frameshifts at coding microsatellites generating neo-antigens, produce a robust protective immune response that can be enhanced with immune checkpoint blockade. More commonly, approximately 50% of sporadic non-MSI-H CRCs demonstrate frameshifts at di- and tetra-nucleotide microsatellites to classify it as MSI-low/elevated microsatellite alterations at selected tetranucleotide repeats (EMAST) as a result of functional somatic inactivation of the DNA MMR protein MSH3 via a nuclear-to-cytosolic displacement. The trigger for MSH3 displacement appears to be inflammation and/or oxidative stress, and unlike MSI-H CRC patients, patients with MSI-L/EMAST CRCs show poor prognosis. These inflammatory-associated microsatellite alterations are a consequence of the local tumor microenvironment, and in theory, if the microenvironment is manipulated to lower inflammation, the microsatellite alterations and MSH3 dysfunction should be corrected. Here we describe the mechanisms and significance of inflammatory-associated microsatellite alterations, and propose three areas to deeply explore the consequences and prevention of inflammation’s effect upon the DNA MMR system.
Keywords: Microsatellite instability, Microsatellite stable, Elevated microsatellite alterations at selected tetranucleotide repeats, Colorectal cancer, Mismatch repair, Inflammation, MSH3
Core tip: Inflammation can trigger microsatellite stable colorectal cancers (CRCs) to acquire a nuclear-to-cytoplasm displacement of the DNA mismatch repair protein MSH3, rendering the CRC with di- and tetranucleotide microsatellite instability (MSI-low/elevated microsatellite alterations at selected tetranucleotide repeats) and modifying the biological behavior of the CRC towards metastasis and poor patient survival. We herein discuss the mechanisms and significance of these induced inflammatory-associated microsatellite alterations, and suggest three content areas to further examine interventions that may modify the observed behavior of these CRCs.
INTRODUCTION
The Cancer Genome Atlas (TCGA) for colorectal cancers (CRCs) clarified that there are two types of sporadic CRCs - hypermutated and non-hypermutated. Most hypermutated CRCs have a defect in their mismatch repair (MMR) system due to the loss of MLH1 function by promoter silencing of the MLH1 locus, resulting in high levels of insertion/deletion (I/D) mutations at microsatellite loci (microsatellite instability high: MSI-H)[1]. Most MSI-H CRCs exhibit proximal location, mucinous, undifferentiated histology, abundant CD8+/Th1 T cell infiltrations, and less aggressive clinical behavior, and are susceptible for immune checkpoints blockade[2,3]. Among non-hypermutated CRCs, I/D mutations in microsatellite loci with larger repeat units (di- and tetra-nucleotide repeats) are frequent and have been shown to be caused by tumor cells’ exposure to inflammatory tumor-microenvironments[4,5]. In this review, we describe and discuss the penetrance and causes of inflammation-associated microsatellite alterations (IAMAs), and their significance to patients’ prognoses in CRC. We also raise “Provocative Questions” whose answers could contribute not only to understand the biology of IAMAs but also to treatment of CRC with IAMAs.
MSI-H, MSI-L AND EMAST IN CRC
Microsatellites or simple sequence repeats are composed of 1-6 nucleotide repeats, occupy 3% of the total human genome, and are located in both coding and non-coding regions[6]. MSI is defined as continuous length changes in simple DNA repeat sequences within microsatellite loci[7]. MSI in CRC was first reported by Aaltonen et al[8] and Thibodeau et al[9] followed by Ionov et al[10] in 1993. It was then shown that a subset of sporadic CRC tumors and tumors from hereditary nonpolyposis colon cancer (HNPCC) exhibit MSI and MMR-defects[11]. Subsequently, germline mutations in MSH2, MLH1, PMS2 and MSH6 were found in different HNPCC families[12-18] and tumors from these families exhibited MSI[19,20]. A causal relationship between MMR-defect, MSI and cancer susceptibility was shown by knockout mouse studies[21-24]. Genetic complementation studies using tissue cultured MSI-positive CRC cells also confirmed that MSI is caused by MMR-deficiency in human cells[25-27]. It was also shown that MSI exhibited in 10%-15% of sporadic CRC cases was due to transcriptional down-regulation of MLH1 expression through promoter hyper-methylation[28].
MSI in CRCs was defined at an international workshop meeting sponsored by the National Cancer Institute in 1998[2]. A panel of five microsatellite markers - two markers with mononucleotide repeats and three markers with dinucleotide repeats - were validated to be classified as follows: High-frequency MSI (MSI-H: 2 or more of 5 markers show instability), low-frequency MSI (MSI-L: 1 of 5 markers shows instability), and microsatellite stable (MSS: none of 5 shows instability) CRCs. It was also confirmed that MSI-H in CRC is caused by defective MMR, mainly MSH2 and MLH1, and manifests as sporadic and hereditary forms of CRCs. Both sporadic and inherited MSI-H CRCs have unique clinical and pathological futures compared to MSI-L/MSS sporadic CRCs[2]. At this NIH meeting, the presence of CRCs with MSI-L was appreciated and discussed. However, the etiology of MSI-L and the distinction between MSI-L and MSS CRC remained unclear. Another type of microsatellite alteration, called elevated microsatellite alterations in selected tetra-nucleotide repeats (EMAST), where insertion/deletion mutations in the loci with tri- and/or tetra-nucleotide but not with mono- and/or dinucleotide repeats was recognized as a component of CRC but its etiology and clinic-pathological significance was not determined[2].
Although a consensus on the definition of MSI-L CRC was reached at the NCI meeting, two subsequent studies showed that approximately 80% of non-MSI-H CRC exhibited mutation at < 1 microsatellite locus when a large number of the loci with di-nucleotide repeats were tested for frame-shift mutations, indicating that most of CRC is MSI-L, and that the NCI reference panel was inadequate for detection of MSI-L CRC[29,30]. These studies also showed that there were no genetic or clinic-pathological characteristics of tumors to separate MSI-L from MSS CRC. However, both studies observed that the incidence of MSI was non-randomly distributed among non-MSI-H CRC, suggesting that some tumors were more susceptible than others to slippage mutations at microsatellite loci, especially loci with dinucleotide repeats[29,30]. The reason for the observed variation in instability and its pathological significance in patients’ prognoses remained unclear.
I/D mutations in loci with selected tetra-nucleotide repeats (EMAST), such as (AAAG)n or (ATAG)n, have been reported in non-CRCs including non-small cell lung, bladder, ovary, head and neck, skin and kidney cancers[31]. Haugen et al[32] first described the frequency of EMAST in CRC, its relationship to MSI-L and its possible cause. They used the five NCI-endorsed MSI markers plus 2 additional markers with dinucleotide repeats to identify MSI-H, MSI-L and MSS CRC. They also used 7 EMAST markers and defined EMAST-positive if one or more of the 7 markers showed ID mutations[32]. They found that EMAST is common in sporadic cases of non-MSI-H CRC (approximately 50%) and is associated with decreased nucleus MSH3 expression in tumor cells. Using MSH3-proficient and -deficient colon cancer cell lines, they also showed evidence that EMAST and low levels of instability at dinucleotide loci repeats - but not with mononucleotide repeats - in non-MSI-H CRC cells are caused by loss of MSH3[32]. Frequent incidence of EMAST in CRCs was confirmed by 2 other studies[33,34]. The genetic cause of EMAST due to the loss of MSH3 was also proven by other studies using tissue cultured human cells[35,36].
BIOCHEMICAL BASIS OF MICROSATELLITE ALTERATIONS
Accumulated evidence supports that MSI-H, MSI-L and EMAST are caused by defects in some components of MMR[37]. When DNA polymerase copies template DNA containing microsatellite loci, it mistakenly adds or deletes a repeat unit in the newly synthesized DNA strand (Figure 1A). The DNA polymerase slippage errors create loops between the two strands, which are recognized and repaired by MMR. In vitro experiments using cell extracts and/or purified proteins demonstrate that there are 5 MMR proteins involved in MMR reactions in human cells (Figure 1)[38]. MSH2 plays a major role in recognition of mismatched DNA. MLH1 and PMS2 are the main proteins responsible for down-stream MMR reactions. If MSH2, MLH1 or PMS2 lose their function, slippage errors at microsatellite loci with mono-, di- and tetra-nucleotide repeats are not fixed at all, resulting in MSI-H (Figure 1B). There are 2 pathways for mismatch recognition: (1) MSH2 and MSH6 form a dimer called MutSα that preferentially recognizes mismatched nucleotides and loops containing 1-2 nucleotides; (2) MSH2 and MSH3 form a dimer called MutSβ that recognizes loops containing 2 or more nucleotides generated at di- and tetra-nucleotide repeats, including the EMAST loci (Figure 1A)[39]. Defects in MSH6 result in increased missense mutations and in instability at mononucleotide repeats (Figure 1A)[40]. When only MSH3 is disabled, increases in instability at di-, tri- and tetra-nucleotide repeats (EMAST) but not at mononucleotide repeats are observed (Figure 1)[32]. Biochemical data indicates that loops containing 2 nucleotides are preferentially recognized by MutSβ over MutSα[41]. Thus, when loss of MSH3 leaves many loops containing 2 or more nucleotides unrepaired, MutSα may repair some but not all such loops, resulting in low levels of mutation in di-nucleotide repeat loci and high levels of mutation in loci with tetra-nucleotide repeats (EMAST) loci (Figure 1B and C).
Figure 1.
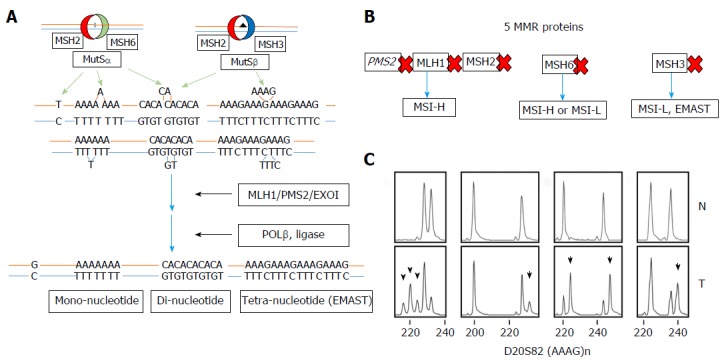
Human DNA mismatch repair. A: Two DNA recognition complexes MutSα, which recognizes insertion-deletion (I/D) loops of 1-2 repeated nucleotides for repair, and MutSβ which recognizes I/D loops of 2 or greater nucleotides for repair, are the key protein complexes of MMR. The MLH1 and PMS2 complex, also known as MutLα, then helps execute the repair with the exonuclease Exo1, polymeraseβ and DNA ligase to fully effect repair; B: Specific efficiency in one of the five DNA MMR proteins yields differing microsatellite instability (MSI) results. Loss of MLH1, MSH2 or PMS2 will yield frameshifts at mono-, di- and tetra-nucleotide microsatellite markers. Loss of MSH6, inactivating MutSα only, will yield mononucleotide mostly but some dinucleotide microsatellite frameshifts, whereas loss of MSH3, inactivating MutSβ, will yield di- and tetranucleotide microsatellite frameshifts, but no mononucleotide microsatellite frameshifts; C: Examples of fragment analysis comparing normal colon tissue (upper panels) with tissue (lower panels) demonstrating frameshifts in the tetranucleotide marker D20S82. MMR: Mismatch repair; MSI: Microsatellite instability; CRC: Colorectal cancer.
MSI-L AND EMAST ARE CAUSED BY MSH3 FUNCTIONAL LOSS IN CRC
The first evidence that loss of MSH3 may result in MSI-L and/or EMAST in CRC was reported by Haugen et al[32] in 2008. They used the colon cancer cell line HCT116 that is deficient in MLH1 due to a hemizygous inactivating mutation in exon 9, and is also deficient in MSH3 due to a homozygous frameshift inactivating mutation in exon 7. Thus, this cell line showed the MSI-H phenotype. Introduction of a normal human chromosome 3 carrying a wild-type MLH1 to HCT116 complemented MLH1-deficiency[25]. The resulting HCT116 with chromosome 3 exhibited stability in loci with mononucleotide repeats but showed low levels of instability at loci with dinucleotide repeats: MSI-L, and high degree of instability at EMAST loci. They further introduced a normal human chromosome 5 carrying wild-type MSH3 into HCT116 + 3 cells. The resulting HCT116 + 3 + 5 cells exhibited complete stability at loci with mono-, dinucleotide repeats and EMAST loci. Finally, they introduced MSH3-shRNA to HCT116 + 3 + 5 cells to knock-down MSH3 and showed that specific knock-down of MSH3 resulted in an MSI-L/EMAST phenotype.
The second evidence that loss of MSH3 results in MSI in loci with di- and tetra-but not mono-nucleotide repeats is from a discovery of two families with bi-allelic MSH3 germ-line mutations, reported by Adam et al[42]. Patients with bi-allelic inactivation mutations of the MSH3 locus suffered from a colorectal adenoma polyposis syndrome and early occurrence of multiple adenoma polyps and tumors in other organs. As expected, the expression of MSH3 was null in normal colon and adenoma polyps from these patients (Table 1). MSI assays showed that instability at di-nucleotide repeat loci and EMAST loci, but not loci with mononucleotide repeats, was detected in adenoma polyps but not in normal colon cells from the same patient. This is because adenoma is monoclonal while the normal colon of these patients consists of mixture of cells with MSI at different loci, masking each alteration that occurred in individual colon cells with the exception of germline alleles. However, there is likely dinucleotide and tetranucleotide instability within normal tissues if they were compared to heterozygous MSH3 germline relatives, or relatives that are homozygous normal for MSH3 mutation. These results support that MSI-L/EMAST in sporadic CRC is caused by loss of MSH3 function.
Table 1.
Expression of MSH3 protein within the epithelium of normal colonic mucosa and adenoma of patients with mono- or bi-allelic germline mutation in MSH3
| Tissue (epithelium) | Monoallelic MSH3 germline mutation | Bi-allelic MSH3 germline mutation |
| Normal colonic mucosa | MSH3 expressed | MSH3 absent |
| Colon adenoma | Not obtained | MSH3 absent |
Extracted from Adam et al[42].
EVIDENCE THAT MSI-L/EMAST IN SPORADIC CRC IS INDUCED BY INFLAMMATION THROUGH DISPLACEMENT OF MSH3 FROM NUCLEUS TO CYTOPLASM
While homogeneous loss of nuclear MSH3 can be detected in adenoma polyp with bi-allelic germline MSH3 mutations, heterogeneous loss of nuclear MSH3 is frequently detected in sporadic CRC exhibiting MSI-L/EMAST (Figure 2A). These results suggest that local loss of MSH3 expression in sporadic MSI-L/EMAST CRC may be not due to genetic loss of MSH3. TCGA data shows that the frequency of MSH3 somatic mutations in CRC is about 6.6%. This does not explain the high incidence of MSI-L/EMAST (approximately 50%) in CRC. Furthermore, most MSH3 mutations are frame-shift mutations in exon 7 that are a resulting target from MLH1 inactivation in sporadic CRC (Table 2)[1].
Figure 2.
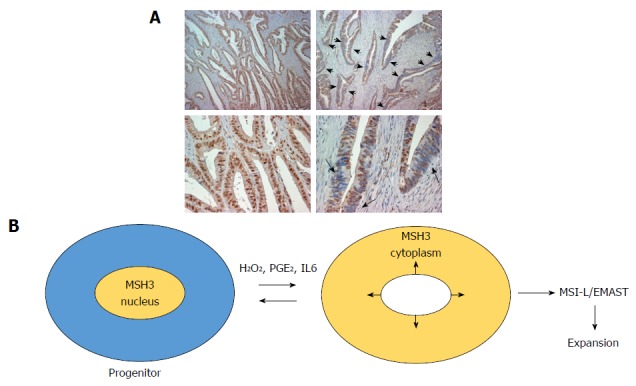
MSH3 expression in sporadic colorectal cancer. A: Immunohistochemistry for MSH3 in sporadic CRC. Arrows show heterogeneous expression of MSH3 in cells and within nuclei in the epithelium; B: Model of MSH3 displacement from the nucleus to the cytosol with inflammatory stimuli to allow accumulation of tetranucleotide frameshift mutations. Progenitor cells could be affected earlier such that subsequent daughter cells amplify the accumulated frameshift mutations. MSI: Microsatellite instability; CRC: Colorectal cancer; EMAST: Elevated microsatellite alterations at selected tetranucleotide repeats.
Table 2.
Comparison of type of mismatch repair gene mutations between sporadic hypermethylated MLH1 colorectal cancers and POLE mutation colorectal cancers from TCGA
| MLH1 promoter hypermethylation | 22/35 (63%) of hypermutated CRCs | 8/22 (36%) with MSH3 frameshift mutation |
| 1/22 (4.5%) with MSH3 missense/nonsense mutation | ||
| 0/22 (0%) with MSH2 mutation | ||
| 5/22 (23%) with MSH6 frameshift mutation | ||
| 4/22 (18%) with MSH6 missense/nonsense mutation | ||
| POLE mutation | 13/35 (37%) of hypermutated CRCs | 3/13 (23%) with MSH3 frameshift mutation |
| 2/13 (15%) with MSH3 missense/nonsense mutation | ||
| 5/13 (38%) with MSH2 missense/nonsense mutation | ||
| 0/13 (0%) with MSH6 frameshift mutation | ||
| 7/13 (54%) with MSH6 missense/nonsense mutation |
Both types of CRCs are hypermutated, containing hundreds of somatic mutations in genomic DNA. Note that the MLH1 hypermethylated CRCs demonstrate higher frequency and consistent frameshift mutations in MSH3 and MSH6 as compared to POLE mutated CRCs, which contain some frameshifts but higher frequency of missense/nonsense mutations in MSH3, MSH2 and MSH6. Extracted from: Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487: 333-337. CRCs: Colorectal cancers.
Lee et al[34] found that EMAST CRC is enriched in in the tumor microenvironment of CD8+ T cells compared to non-EMAST CRC, suggesting that some immunological and inflammatory responses are active in EMAST CRC. They also found that EMAST is significantly high in ulcerated tumors. Devaraj et al[43] further showed that EMAST-positive rectal tumors are associated with the presence of chronic inflammation. These observations led them to hypothesize that inflammation may somehow affect MSH3 function that induces MSI-L/EMAST.
Tseng-Rogenski et al[4,36] demonstrated that several main inflammatory factors, including oxidative stress (hydrogen peroxide), interleukin 6 (IL6) and prostaglandin E2 (PGE2) induce displacement of MSH3 from the nucleus to the cytoplasm in several cancer cell lines. Importantly, other MMR proteins including MLH1, MSH2 and MSH6 do not move from the nucleus to the cytoplasm in response to these stimuli. Repeated treatment of several microsatellite stable colon cancer cell lines with IL6 induced microsatellite instability at EMAST loci. However, other inflammatory cytokines including TNFα, IFNα, IFNβ, and IL1β did not have such an effect. Tseng-Rogenski et al[4] also showed that phosphorylation of STAT3 may be required for displacement of MSH3 when induced by IL6. These studies convincingly show that not all, but some, inflammatory factors induce EMAST through loss of MSH3 from the nucleus (Figure 2B).
Evidence that an inflammatory micro-environment induces MSI-L (low levels of MSI at the loci with dinucleotide repeats) has been shown in regenerated colon tissues from ulcerated colitis (UC) patients. The first study, reported by Brentnall et al[44], showed for the first time the presence of MSI-L but not MSI-H in colon tissues from UC patients. The second study, by Ozaki et al[45], isolated crypts from UC-derived CRC, UC-derived hyperplasia and UC-regenerated colons through laser micro-capture and tested for the presence of microsatellite instability in DNA. Ozaki et al[45] detected MSI-L but not MSI-H in some crypts but not in others, regardless of whether they were from cancer or non-cancer tissues. They also showed that MSI was not detected from stroma cells from these UC patients. Each crypt showed a different MSI-profile, indicating that MSI-L occurs independently at the crypt level. Our recent study showed that regenerated colon cells and CRCs from UC patients have a high frequency of MSH3 displacement from the nucleus to the cytoplasm, and demonstrate MSI-L/EMAST[46]. These results further support the role of inflammation in displacement of MSH3-induced MSI-L/EMAST in human tissues including cancers.
PROGNOSTIC VALUE OF MSI-L/EMAST IN CRC
Several studies have examined the impact of MSI-L and/or EMAST genotypes on patient prognoses in CRC. There have been 4 studies evaluating the prognosis values of MSI-L[47-50]. Kohonen-Corish et al[47] showed that patients with stage C colon cancers defined as MSI-L by the NCI panel plus one tetra-nucleotide marker (MYCL1) showed poor overall survival (OS) compared to patients with MSI-H and/or MSS colon cancers. Similar results were obtained by Wright et al[48]. They showed that stage C CRC patients that are positive for MSI-L as defined by the NCI panel, plus an additional 2 markers with mono-nucleotide repeats, 3 with di-nucleotide repeats and the tetra-nucleotide MYCL1 marker, exhibited poor cancer-specific survival compared to MSS CRC patients[48]. They also observed that most MSI-L CRC exhibited MSI at one di- or tetra-nucleotide but not at mono-nucleotide repeat markers[48]. Lee et al[49] examined 3019 CRC cases for MSI using an NCI microsatellite marker panel and evaluated prognoses of those patients. Similar to other studies, they showed that most MSI-L CRC exhibited MSI at dinucleotide repeats, and patients with MSI-L CRCs was associated with poor OS by Cox regression analysis[49]. Although the previous 2 studies suggested that MSI-L may have a significant prognostic value for stage C CRC patients, Lee et al[49] did not examine the prognostic significance of MSI-L for cancer-specific survival in their large cohort. In contrast to the above three studies, Azzoni et al[50] reported that MSI-L is associated with improved patient survival as compared to MSS CRC. However, the percentage of MSI-H cases in their cohort was unusually high (37%: 68 of 184 cases) compared to other studies (10%-15%), suggesting the presence of some bias in the studied cohort. Lastly, a study reported by Garcia et al[51] did not find any association between MSI-L and disease-free survival (DFS) or OS in stage II and III CRC cohorts.
There are 2 studies examining the relationship between EMAST and OS in CRC; they found no association between the two[33,51]. However, when both MSI-L and EMAST cases were combined, Garcia et al[51] found that MSI-L/EMAST was associated with shorter DFS but not OS compared with non-MSI-L/EMAST CRC. In their cohort, MSI-H CRC patients exhibited the highest survival. Thus, the MSI-L/EMAST genotype in CRC may be associated with recurrence and/or metastasis after surgery. There appears to be heterogeneity even among MSI-L/EMAST CRC patients[52,53]. One group of MSI-L/EMAST CRC exhibited loss of heterozygosity (LOH) at chromosome 9p24.2. and the other did not exhibit 9p24.2 LOH. When the prognoses of these two groups were compared, the one with 9p24.2 LOH at stage III showed improved survival after surgery and OS in Kaplan-Meier analysis and in multi variate analysis over the one without 9p24.2. LOH at stage III[53]. The results also showed that MSI-L/EMAST/9p24.2 LOH is an independent factor that predicts improved OS in stage II/III CRC. Thus, MSI-L/EMAST may be associated with recurrence, but additional genetic or epigenetic changes may modify the behavior of recurrent tumors[53]. Overall, the data presented so far suggest that MSI-L and/or EMAST could be a biomarker for DFS and/or OS of stage II and/or III CRC. However, additional studies using a population-based large cohort are needed to confirm the prognostic value of MSI-L and EMAST.
One concern regarding EMAST is that various studies have not reached a full consensus on the definition of EMAST. As described above, current evidence supports the idea that MSI-L and EMAST in sporadic CRC share the same etiology: both are induced by the absence of nuclear MSH3 in response to exogenous inflammatory factors such as IL6, and oxidative stress[4]. Based on these observations, we propose that EMAST cancer is a non-MSI-H cancer, and MSI at EMAST markers is not caused by loss of other MMR proteins including MLH1, MSH2, PMS2[51,53]. The next question should be whether or not non-EMAST CRC really exits. Similar to MSI-L in CRC[29,30], almost all CRC could be EMAST-positive if a large number of EMAST markers are used. A recent study by Cortes-Ciriano et al[54] showed that all non-MSI-H cancers contain various levels of frame-shift mutations in microsatellite loci with mono-, di-, tri- and tetra-nucleotide repeats. Considering that all tumor tissues contain some degree of inflammatory elements, many of those mutations could be induced by the loss of MSH3 triggered by inflammation in the tumor-microenvironment. Furthermore, a study for UC suggested that frequent exposure to inflammation increased the incidence of MSI-L and EMAST[46]. Thus, while the purpose of the MSI assay is primarily to detect MMR-deficient CRC, the purpose of an EMAST assay could be to distinguish CRCs whose precursors were exposed to high levels of inflammation to CRCs whose precursors were exposed to lower levels of inflammation. Therefore, the results of the studies by Kohonen-Corish, Wright, Lee and Garcia could be re-interpreted according to the idea that high levels of inflammatory tumor-microenvironments not only induce MSI-L/EMAST in cancer cells at the primary site but also include some property that promotes recurrence and/or metastasis when they disseminate. Additional studies will be required to determine whether the numbers and kinds of EMAST markers and cut-off levels for determining EMAST-positive/negative used so far are adequate to distinguish CRCs with different prognoses[31].
PROVOCATIVE QUESTIONS
Here, we have raised three questions whose answers can be important for not only clinical but also basic aspects of MSI-L/EMAST in CRC (Figure 3).
Figure 3.
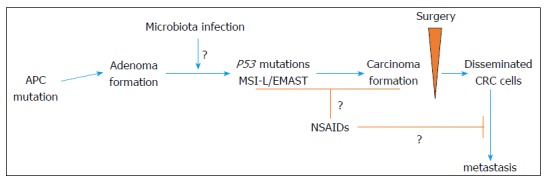
Model of adenoma-to-carcinoma formation in the human colon, with actual and potential sites of interventions to improve survival. MSI: Microsatellite instability; CRC: Colorectal cancer; EMAST: Elevated microsatellite alterations at selected tetranucleotide repeats.
Question 1: Does treatment of CRC with non-steroidal anti-inflammatory drugs reduce not only recurrence/metastasis but also the incidence of MSI-L/EMAST?
The idea that inflammation is associated with recurrence and/or metastasis is indirectly supported by observations that an intake of the anti-inflammatory drug, aspirin, may not only prevent adenomas[55] and CRC formations[56], but also prevent recurrence and metastasis of CRC following surgery[57]. Other non-steroidal anti-inflammatory drugs (NSAID) including celecoxib and rofecoxib, specific inhibitors of cyclooxygenase 2 (COX-2), have been shown to reduce the incidence of adenomas[58-60]. But it was also found that COX-2 inhibitors suppressed colorectal tumor growth and metastasis in mouse models[61,62]. Furthermore, Chan et al[63] reported that the regular intake of aspirin after curative surgery reduced cancer-specific mortality in a sub-group of CRC cells expressing a high level of COX-2 protein. In addition, CRCs expressing HLA class I compared to those not expressing HLA class I are susceptible for aspirin treatment after diagnosis[64]. Also, CRCs with PIK3CA mutations responded better to aspirin treatment after diagnosis than did CRCs with wild-type PIK3CA[65]. However, a recent study by Gray et al[66] showed that the efficacy of aspirin on cancer-specific survival, and OS was associated with levels of COX-2 expression but not with mutational status of PIK3CA in CRCs. Ng et al[67] showed that aspirin and COX-2 inhibitors improved recurrence-free survival, DFS and OS of stage III CRC patients who either received fluorouracil (FU) plus leucovorin (LV) or FU plus LV with irinotecan. These studies support the idea that NSAIDs can be used as part of adjuvant therapy for stage I-III CRC, however, the efficacy of NSAIDs on the recurrence/metastasis of CRC are still under investigation through several randomized controlled trials[57].
Ma et al[68] showed that PGE2 and its receptor, the prostaglandin E receptor 2 (EP2), are necessary for colon cancer formation in inflammatory tissue environments. Compared to wild-type mice treated with azoxymethan (AOM) followed by dextran sodium sulfate (DSS), AOM/DSS-treated EP2-knockout and prostaglandin E synthase (Ptges)-knockout mice bore a significantly reduced number of colon tumors. They identified neutrophil, probably myeloid-derived suppressor cells (MDSC), and cancer-associated fibroblast (CAF) as the main cell components recruited in tumor-microenvironments, expressing EP2, responding to PEG2, and contributing to tumor formation. These cells form a positive-feedback loop of COX-2-PGE2-EP2-NF-κB-COX-2 cycles, and produce TNF-α and IL6[68]. The presence of MDSC and CAF in the tumor-microenvironment are also significantly associated with stage progression and a poor prognosis for CRC, while activation of Th1 helper and cytotoxic memory T cells play a key role in anti-tumor activities preventing recurrence and/or metastasis in CRC[69,70]. Interestingly, Zelenay et al[71] showed that, depending on the level of COX-activity in cancer, the immunological landscape of tumor-microenvironments can be switched between anti-tumor and inflammatory pro-tumor. Therefore, the level of PEG2 and of COX-2 may be major factors in controlling immunological responses to cancer cells, and thereby a patient’s prognosis. Regarding the relationship between MSI-L/EMAST and PEG2, we have observed that the exposure of colon cancer cells in tissue cultures to PGE2 triggers movement of MSH3 from the nucleus to the cytoplasm, which may induce MSI-L/EMAST. Therefore, it is reasonable to speculate that MSI-L/EMAST in CRC may be associated with high levels of COX-2 expression in cancer cells and/or in tumor-microenvironments. This could be the reason why patients with MSI-L/EMAST CRCs exhibit a shorter RFS[51,53]. Thus, reduction of PGE2 by NSAIDs may reduce the incidence and recurrence/metastasis of MSI-EMAST. If this is the case, MSI-L/EMAST could be a biomarker for susceptibility to the NSAIDs treatment.
Question 2: Do microbiota play a role in MSI-L/EMAST formation, adenoma/carcinoma transition and recurrence/metastasis?
Lee et al[34] discovered that EMAST is less frequent in colorectal adenomas and well-differentiated adenocarcinomas than in moderately differentiated and poorly differentiated adenocarcinomas, suggesting that EMAST is progressively acquired during the histological adenoma-carcinoma sequence, from adenoma to well-differentiated carcinomas to moderately and poorly differentiated carcinomas. Because a key gene alteration responsible for adenoma-carcinoma sequence in CRC is p53 mutation[72], MSI-L/EMAST formation may be associated with p53 mutation. In fact, Ahrendt et al[73] reported that EMAST is associated with p53 mutations in non-small cell lung cancer. Li et al[74] observed an association between LOH at TP53 and EMAST in CRC. Interestingly, p53 mutations are the most frequently found in inflammatory bowel disease (IBD)-associated CRC among other gene mutations (60%-90%)[75,76]. One half of the p53 mutations are C:G>T:G transitions, thought to be caused by nitric oxide exposure due to increased inducible nitric oxide synthase expression in IBD[75]. Our preliminary data showed that IBD-associated CRC exhibit a higher frequency of MSI-L/EMAST than do sporadic CRC (unpublished data). Taken together, these results suggest that the inflammatory tissue environment may enhance p53 mutations and MSI-L/EMAST formation in sporadic adenomas, leading to carcinoma transition. As mentioned earlier, MSI-L/EMAST in stage II CRC patients is associated with shorter RFS, suggesting that the inflammatory tumor-environment in primary tumor tissues somehow promotes recurrence or metastasis. These observations lead to the next question: What establishes an inflammatory environment in colorectal adenoma and carcinoma?
Microbiota in the colon and rectum create an inflammatory microenvironment and promote CRC formation[77]. Several bacterial organisms including Fusobacterium nucleatum (F. nucleatum), Enterotoxigenic Bacteroides fragilis (ETBF), and colibactin-producing Escherichia coli (E. coli) are epidemiologically associated with CRC, and have been found to be enriched in CRC[77,78]. The enrichment of F. nucleatum was also found in colorectal adenoma relative to non-adenoma or surrounding tissues[79-81]. McCoy et al[79] showed that F. nucleatum abundance in colorectal adenoma is associated with local inflammatory cytokine gene expression including IL-10 and TNF-α. Kostic et al[80] investigated the effect of F. nucleatum infection on the development of intestinal tumors in APCMin/+, IL10-/- and T-bet-/- X Rag2-/- mice. There was an increase in the number of tumors in APCMin/+ mice. Importantly, infection with F. nucleatum accelerated adenocarcinoma formation in the small intestines of APCMin/+ mice compared to sham-treated control mice. In contrast, infection with F. nucleatum did not induce any tumor formation in IL10-/- and T-bet-/- X Rag2-/- mice. These results suggest that the effects of F. nucleatum may manifest on existing adenomas, and may stimulate adenoma-carcinoma transition by creating an oxidative stress-rich, carcinogenic environment[80]. It would be interesting to determine whether F. nucleatum -induced adenocarcinomas in APCMin/+ mice gain p53 mutations. Kostic et al[80] further showed that infection of tumor tissues with F. nucleatum results in recruitment of MDSCs, tumor-associated macrophages, and dendritic cells in tumor tissues, and modulate the tumor immune micro-environment that promote tumor progression. In addition, they found the up-regulation of genes that are down-stream of NF-κB including PTGS2 (COX-2), IL6, IL1β, and TNF in both human and mouse CRC infected with F. nucleatum[80]. It is tempting to speculate that F. nucleatum-induced adenocarcinoma may gain MSI-L/EMAST in response to oxidative stress, PEG2 and/or IL6 that cause displacement of MSH3 from the nucleus to the cytoplasm. Recently, a heavy load of F. nucleatum has been associated with MSI-H CRC, proximal colon cancer and a poor prognosis[81-84]. Yu et al[85] showed that F. nucleatum infection in primary CRC is associated with recurrence after surgery followed by adjuvant chemotherapy. They showed that F. nucleatum induces chemo-resistance in infected cells through autophagy[85]. One of the reasons why 5-FU-based adjuvant therapy does not have benefit for a sub-group of MSI-H CRC[86,87] could be partly explained by the infection of F. nucleatum[85]. It is also possible that the CpG Island Methylator Phenotype (CIMP) including promoter methylation of the MLH1 locus could be induced by chronic inflammation due to a heavy load of F. nucleatum infection[82]. Considering that infection of F. nucleatum is associated with recurrence of CRC after surgery, a group of such CRCs may exhibit MSI-L/EMAST CRC[51,53].
Another bacterium, ETBF, is also associated with CRC[88-90] and can target colorectal cells to promote an adenoma and/or adenoma-carcinoma transition in APCMin/+ mice[91]. ETBF produces a metalloprotease toxin called BFT. BFT binds to the surface of colorectal epithelial cells and induces E-cadherin cleavage, resulting in an increase in barrier permeability and inducing an inflammatory micro-environment with Th-17/IL-17 predominance[91,92]. Th-17/IL-17 plays a major role in ETBF tumorigenesis because the depletion of CD4+ T cells and blockade of IL-17 inhibited it. IL-17 attracts neutrophils, MDSCs and macrophages, and induces carcinogenic and immunosuppressive factors including nitric oxide, ROS, and Arg1 in mouse models[92]. Colibactin-producing E. coli is also associated with CRC[93,94] and initiates inflammation and promotes adenoma formation in APCMin/+mice[94] and in APCMin/+, IL10-/- mice[95]. Taken together, infection with all three bacterial organisms, that are found to be associated with CRC, induces an inflammatory environment in adenoma tissue and promotes adenoma and/or a transition from adenoma to carcinoma in mouse models. It would be interesting to determine whether MSI-L/EMAST and p53 mutations coincide with bacterial-induced transitions to adenoma/carcinoma. Recently, Scott et al[96] showed that the efficacy of 5-FU treatment maybe largely influenced by microbiota in the gut.
Question 3: Is MSH3 a component of DNA damage signaling?
The MutSβ hetero-duplex between MSH3 and MSH2 not only functions in MMR but may also play a role in double strand break (DSB) repair via homologous recombination (HR)[97-100]. DNA double strand breaks (DSB) induce cell death if not repaired. Cells have evolved two pathways to re-connect the broken DNA ends: Non-homologous end joining (NHEJ) and homologous recombination (HR). If one of these pathways is disabled when DSB is created, cells use the other pathway for survival. The HR reaction starts with a nuclease-mediated resection of broken DNA ends to be coated by the single stranded (ss) DNA-binding protein, replication protein A (RPA). Then, Ataxia telangiectasia and Rad3-related (ATR) kinase is recruited to the RPA-coated ssDNA via an ATR-interacting partner (ATRIP). The topoisomerase IIβ-binding protein 1 (TOPBP1), which is recruited to the DSB site, interacts with ATRIP and activates ATR. Activated ATR phosphorylates CHEK2 that regulate cell cycle progression. TOPBP1 also interacts with polo-like kinase (PLK) that phosphorylate RAD51 for its loading on resected ssDNA[101]. Burdova et al[99] showed that recruitment of ATR/ATRIP to RPA-coated ssDNA is mediated by MutSβ which binds to the loop structure formed within the ssDNA. Therefore, MutSβ is required in the early stage of HR-DSB repair and its loss due to an MSH2 or MSH3 defect forces a cell to use NHEJ for survival under the presence of DSBs[98,100]. Thus, when oxidative stress causes DSBs, it may induce elimination of MSH3 from the nucleus, resulting in activation of Ataxia-telangiectasia mutated (ATM)[102] but not ATR, and dependence of NHEJ for survival.
An intriguing question is why and how nuclear MSH3 proteins translocate in response to oxidative stress or exposure to IL6 or PGE2 (Figure 2B)[4,36]. H2O2 and oxidative stress causes DSBs, resulting in activation of NF-κB[103,104]. IL6 and PGE2 are mediators that possibly form a loop associated with activation of NF-κB through STAT3[105-107]. IL6 activates STAT3, which directly interacts with the NF-κB family member RELA, contributing to constitutive NF-κB activation[105], and COX2/PGE2 also activates STAT3, leading to NF-κB activation[106]. We found that MSH3 itself is a shuttling protein. It contains a bona fide bipartite nuclear localization signal (NLS) that directs its nuclear import to perform DNA repair (unpublished data). It also contains two functional nuclear export signals (NESs) that allow it to exit the nucleus upon the treatment of a pro-inflammatory cytokine, IL-6 (unpublished data). Among the other main MMR proteins including MSH2, MSH3 is the only MMR protein that shifts into the cytoplasm upon oxidative stress or IL6 treatment, suggesting that MSH3 moves alone or does so with other unknown partner proteins. Recent data indicate that the NF-κB Essential Modulator (NEMO), when used as a bait, can pull down MSH3, suggesting physical interaction between these two proteins[108]. As one of the three components of the IKK complex, NEMO’s role in regulating the NF-κB pathway is well documented[104]. It is possible that simultaneous or sequential movement of MSH3, NEMO and ATM in the cell may transmit a DNA damage signal to NF-κB, depending on the degree of DNA damage. Further studies are necessary for clarify these possibilities.
MSI-L/EMAST IS COMMON IN HUMAN CANCERS
Since the discovery of MSI in CRC, MSI-L and EMAST have been examined in cancers from other organs and tissues. MSI-L has been found in stomach[109], cervical[110], pancreatic[111], ovarian[112], skin[113], nerve[114], breast, endometrial[115], liver[116], esophageal[117], eye[118], soft tissue[119], gallbladder[120], head and neck[121], prostate[122], lung[123] and cancers of the urinary tract[124]. EMAST has also been widely detected in other various human cancers[31]. A recent study by Cortes-Ciriano et al[54] showed that there are MSI-H prone cancers including colorectal, esophageal, stomach and endometrial cancers, and non-MSI-H prone cancers that include ovarian, kidney, liver, breast, head and neck, cervical, lung, pancreatic, bladder, prostate, skin, adrenal, cortical and thyroid cancers. They also showed that most non-MSI-H cancers exhibit different degrees of MSI at not only loci with mono- but also loci with di-, tri- and tetra-nucleotide repeats, suggesting that inflammation-induced MSH3 replacement from the nucleus to the cytoplasm is probably common in human cancers[54,125,126]. Thus, the answers to the provocative questions raised above may also apply to many human cancers.
CONCLUSION
MSI-L/EMAST is common in human cancers. MSI-L/EMAST is caused by displacement of MSH3 from the nucleus to the cytoplasm in replicating cells triggered by inflammatory stimuli, and can be termed Inflammatory-Associated Microsatellite Alterations (IAMAs). MSI-L/EMAST is associated with recurrence and/or metastasis in CRC patients. MSI-L/EMAST CRC is a heterogeneous group and consists of sub-groups with different genetic changes and prognoses.
Footnotes
Supported by United States Public Health Service, Nos. DK067287, CA162147 and CA206010; and the A. Alfred Taubman Medical Research Institute of the University of Michigan. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict-of-interest statement: Authors declare no conflict of interests for this article.
Peer-review started: October 29, 2017
First decision: November 23, 2017
Article in press: December 6, 2017
Manuscript source: Invited manuscript
Specialty type: Oncology
Country of origin: United States
Peer-review report classification
Grade A (Excellent): A, A
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
P- Reviewer: Fan RY, Li C, Wani IA S- Editor: Ji FF L- Editor: A E- Editor: Wang CH
Contributor Information
Minoru Koi, Division of Gastroenterology, Department of Internal Medicine and Comprehensive Cancer Center, University of Michigan, Ann Arbor, MI 48109-5368, United States.
Stephanie S Tseng-Rogenski, Division of Gastroenterology, Department of Internal Medicine and Comprehensive Cancer Center, University of Michigan, Ann Arbor, MI 48109-5368, United States.
John M Carethers, Division of Gastroenterology, Department of Internal Medicine and Comprehensive Cancer Center, University of Michigan, Ann Arbor, MI 48109-5368, United States.
References
- 1.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 3.Overman MJ, Kopetz S, Lonardi S, McDermott R, Leone F, Leach J, Lenz H, Hendlisz A, Morse M, Garcia-Alfonso P, et al. Nivolumab ± ipilimumab in treatment (tx) of patients (pts) with metastatic colorectal cancer (mCRC) with and without high microsatellite instability (MSI-H): CheckMate-142 interim results. J Clin Oncol. 2016;34:3501. [Google Scholar]
- 4.Tseng-Rogenski SS, Hamaya Y, Choi DY, Carethers JM. Interleukin 6 alters localization of hMSH3, leading to DNA mismatch repair defects in colorectal cancer cells. Gastroenterology. 2015;148:579–589. doi: 10.1053/j.gastro.2014.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carethers JM. Microsatellite Instability Pathway and EMAST in Colorectal Cancer. Curr Colorectal Cancer Rep. 2017;13:73–80. doi: 10.1007/s11888-017-0352-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Subramanian S, Madgula VM, George R, Mishra RK, Pandit MW, Kumar CS, Singh L. Triplet repeats in human genome: distribution and their association with genes and other genomic regions. Bioinformatics. 2003;19:549–552. doi: 10.1093/bioinformatics/btg029. [DOI] [PubMed] [Google Scholar]
- 7.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 8.Aaltonen LA, Peltomäki P, Leach FS, Sistonen P, Pylkkänen L, Mecklin JP, Järvinen H, Powell SM, Jen J, Hamilton SR. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812–816. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 9.Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993;260:816–819. doi: 10.1126/science.8484122. [DOI] [PubMed] [Google Scholar]
- 10.Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–561. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- 11.Parsons R, Li GM, Longley MJ, Fang WH, Papadopoulos N, Jen J, de la Chapelle A, Kinzler KW, Vogelstein B, Modrich P. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell. 1993;75:1227–1236. doi: 10.1016/0092-8674(93)90331-j. [DOI] [PubMed] [Google Scholar]
- 12.Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1994;77:1 p following 166. [PubMed] [Google Scholar]
- 13.Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, Peltomäki P, Sistonen P, Aaltonen LA, Nyström-Lahti M. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- 14.Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, Kane M, Earabino C, Lipford J, Lindblom A. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258–261. doi: 10.1038/368258a0. [DOI] [PubMed] [Google Scholar]
- 15.Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM, Adams MD. Mutation of a mutL homolog in hereditary colon cancer. Science. 1994;263:1625–1629. doi: 10.1126/science.8128251. [DOI] [PubMed] [Google Scholar]
- 16.Nicolaides NC, Papadopoulos N, Liu B, Wei YF, Carter KC, Ruben SM, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature. 1994;371:75–80. doi: 10.1038/371075a0. [DOI] [PubMed] [Google Scholar]
- 17.Palombo F, Gallinari P, Iaccarino I, Lettieri T, Hughes M, D’Arrigo A, Truong O, Hsuan JJ, Jiricny J. GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science. 1995;268:1912–1914. doi: 10.1126/science.7604265. [DOI] [PubMed] [Google Scholar]
- 18.Miyaki M, Konishi M, Tanaka K, Kikuchi-Yanoshita R, Muraoka M, Yasuno M, Igari T, Koike M, Chiba M, Mori T. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet. 1997;17:271–272. doi: 10.1038/ng1197-271. [DOI] [PubMed] [Google Scholar]
- 19.Eshleman JR, Markowitz SD. Microsatellite instability in inherited and sporadic neoplasms. Curr Opin Oncol. 1995;7:83–89. [PubMed] [Google Scholar]
- 20.Kolodner RD, Tytell JD, Schmeits JL, Kane MF, Gupta RD, Weger J, Wahlberg S, Fox EA, Peel D, Ziogas A, et al. Germ-line msh6 mutations in colorectal cancer families. Cancer Res. 1999;59:5068–5074. [PubMed] [Google Scholar]
- 21.de Wind N, Dekker M, Berns A, Radman M, te Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82:321–330. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- 22.Reitmair AH, Schmits R, Ewel A, Bapat B, Redston M, Mitri A, Waterhouse P, Mittrücker HW, Wakeham A, Liu B. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat Genet. 1995;11:64–70. doi: 10.1038/ng0995-64. [DOI] [PubMed] [Google Scholar]
- 23.Baker SM, Bronner CE, Zhang L, Plug AW, Robatzek M, Warren G, Elliott EA, Yu J, Ashley T, Arnheim N, et al. Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell. 1995;82:309–319. doi: 10.1016/0092-8674(95)90318-6. [DOI] [PubMed] [Google Scholar]
- 24.Edelmann W, Yang K, Umar A, Heyer J, Lau K, Fan K, Liedtke W, Cohen PE, Kane MF, Lipford JR, et al. Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell. 1997;91:467–477. doi: 10.1016/s0092-8674(00)80433-x. [DOI] [PubMed] [Google Scholar]
- 25.Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, Boland CR. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N’-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 1994;54:4308–4312. [PubMed] [Google Scholar]
- 26.Umar A, Koi M, Risinger JI, Glaab WE, Tindall KR, Kolodner RD, Boland CR, Barrett JC, Kunkel TA. Correction of hypermutability, N-methyl-N’-nitro-N-nitrosoguanidine resistance, and defective DNA mismatch repair by introducing chromosome 2 into human tumor cells with mutations in MSH2 and MSH6. Cancer Res. 1997;57:3949–3955. [PubMed] [Google Scholar]
- 27.Watanabe Y, Haugen-Strano A, Umar A, Yamada K, Hemmi H, Kikuchi Y, Takano S, Shibata Y, Barrett JC, Kunkel TA, et al. Complementation of an hMSH2 defect in human colorectal carcinoma cells by human chromosome 2 transfer. Mol Carcinog. 2000;29:37–49. [PubMed] [Google Scholar]
- 28.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Halford S, Sasieni P, Rowan A, Wasan H, Bodmer W, Talbot I, Hawkins N, Ward R, Tomlinson I. Low-level microsatellite instability occurs in most colorectal cancers and is a nonrandomly distributed quantitative trait. Cancer Res. 2002;62:53–57. [PubMed] [Google Scholar]
- 30.Laiho P, Launonen V, Lahermo P, Esteller M, Guo M, Herman JG, Mecklin JP, Järvinen H, Sistonen P, Kim KM, et al. Low-level microsatellite instability in most colorectal carcinomas. Cancer Res. 2002;62:1166–1170. [PubMed] [Google Scholar]
- 31.Watson MM, Berg M, Søreide K. Prevalence and implications of elevated microsatellite alterations at selected tetranucleotides in cancer. Br J Cancer. 2014;111:823–827. doi: 10.1038/bjc.2014.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haugen AC, Goel A, Yamada K, Marra G, Nguyen TP, Nagasaka T, Kanazawa S, Koike J, Kikuchi Y, Zhong X, et al. Genetic instability caused by loss of MutS homologue 3 in human colorectal cancer. Cancer Res. 2008;68:8465–8472. doi: 10.1158/0008-5472.CAN-08-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamada K, Kanazawa S, Koike J, Sugiyama H, Xu C, Funahashi K, Boland CR, Koi M, Hemmi H. Microsatellite instability at tetranucleotide repeats in sporadic colorectal cancer in Japan. Oncol Rep. 2010;23:551–561. [PMC free article] [PubMed] [Google Scholar]
- 34.Lee SY, Chung H, Devaraj B, Iwaizumi M, Han HS, Hwang DY, Seong MK, Jung BH, Carethers JM. Microsatellite alterations at selected tetranucleotide repeats are associated with morphologies of colorectal neoplasias. Gastroenterology. 2010;139:1519–1525. doi: 10.1053/j.gastro.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campregher C, Schmid G, Ferk F, Knasmüller S, Khare V, Kortüm B, Dammann K, Lang M, Scharl T, Spittler A, et al. MSH3-deficiency initiates EMAST without oncogenic transformation of human colon epithelial cells. PLoS One. 2012;7:e50541. doi: 10.1371/journal.pone.0050541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tseng-Rogenski SS, Chung H, Wilk MB, Zhang S, Iwaizumi M, Carethers JM. Oxidative stress induces nuclear-to-cytosol shift of hMSH3, a potential mechanism for EMAST in colorectal cancer cells. PLoS One. 2012;7:e50616. doi: 10.1371/journal.pone.0050616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carethers JM. Hereditary, sporadic and metastatic colorectal cancer are commonly driven by specific spectrums of defective DNA mismatch repair components. Trans Am Clin Climatol Assoc. 2016;127:81–97. [PMC free article] [PubMed] [Google Scholar]
- 38.Hsieh P, Zhang Y. The Devil is in the details for DNA mismatch repair. Proc Natl Acad Sci USA. 2017;114:3552–3554. doi: 10.1073/pnas.1702747114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gupta S, Gellert M, Yang W. Mechanism of mismatch recognition revealed by human MutSβ bound to unpaired DNA loops. Nat Struct Mol Biol. 2011;19:72–78. doi: 10.1038/nsmb.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.You JF, Buhard O, Ligtenberg MJ, Kets CM, Niessen RC, Hofstra RM, Wagner A, Dinjens WN, Colas C, Lascols O, et al. Tumours with loss of MSH6 expression are MSI-H when screened with a pentaplex of five mononucleotide repeats. Br J Cancer. 2010;103:1840–1845. doi: 10.1038/sj.bjc.6605988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kantelinen J, Kansikas M, Korhonen MK, Ollila S, Heinimann K, Kariola R, Nyström M. MutSbeta exceeds MutSalpha in dinucleotide loop repair. Br J Cancer. 2010;102:1068–1073. doi: 10.1038/sj.bjc.6605531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adam R, Spier I, Zhao B, Kloth M, Marquez J, Hinrichsen I, Kirfel J, Tafazzoli A, Horpaopan S, Uhlhaas S, et al. Exome Sequencing Identifies Biallelic MSH3 Germline Mutations as a Recessive Subtype of Colorectal Adenomatous Polyposis. Am J Hum Genet. 2016;99:337–351. doi: 10.1016/j.ajhg.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Devaraj B, Lee A, Cabrera BL, Miyai K, Luo L, Ramamoorthy S, Keku T, Sandler RS, McGuire KL, Carethers JM. Relationship of EMAST and microsatellite instability among patients with rectal cancer. J Gastrointest Surg. 2010;14:1521–1528. doi: 10.1007/s11605-010-1340-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brentnall TA, Crispin DA, Bronner MP, Cherian SP, Hueffed M, Rabinovitch PS, Rubin CE, Haggitt RC, Boland CR. Microsatellite instability in nonneoplastic mucosa from patients with chronic ulcerative colitis. Cancer Res. 1996;56:1237–1240. [PubMed] [Google Scholar]
- 45.Ozaki K, Nagasaka T, Notohara K, Kambara T, Takeda M, Sasamoto H, Jass JR, Tanaka N, Matsubara N. Heterogeneous microsatellite instability observed within epithelium of ulcerative colitis. Int J Cancer. 2006;119:2513–2519. doi: 10.1002/ijc.22095. [DOI] [PubMed] [Google Scholar]
- 46.Munakata K, Koi M, Leconte P, Kitajima T, Tseng-Rogenski S, Uemura M, Mizushima T, Carethers JM. Loss of MSH3 and Subsequent EMAST Determines the Pathological Significance of MSI-L in Ulcerative Colitis. Gastroenterology. 2016;150:S962. [Google Scholar]
- 47.Kohonen-Corish MR, Daniel JJ, Chan C, Lin BP, Kwun SY, Dent OF, Dhillon VS, Trent RJ, Chapuis PH, Bokey EL. Low microsatellite instability is associated with poor prognosis in stage C colon cancer. J Clin Oncol. 2005;23:2318–2324. doi: 10.1200/JCO.2005.00.109. [DOI] [PubMed] [Google Scholar]
- 48.Wright CM, Dent OF, Newland RC, Barker M, Chapuis PH, Bokey EL, Young JP, Leggett BA, Jass JR, Macdonald GA. Low level microsatellite instability may be associated with reduced cancer specific survival in sporadic stage C colorectal carcinoma. Gut. 2005;54:103–108. doi: 10.1136/gut.2003.034579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee SY, Kim DW, Lee HS, Ihn MH, Oh HK, Min BS, Kim WR, Huh JW, Yun JA, Lee KY, et al. Low-Level Microsatellite Instability as a Potential Prognostic Factor in Sporadic Colorectal Cancer. Medicine (Baltimore) 2015;94:e2260. doi: 10.1097/MD.0000000000002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Azzoni C, Bottarelli L, Cecchini S, Silini EM, Bordi C, Sarli L. Sporadic colorectal carcinomas with low-level microsatellite instability: a distinct subgroup with specific clinicopathological and molecular features. Int J Colorectal Dis. 2011;26:445–453. doi: 10.1007/s00384-011-1133-8. [DOI] [PubMed] [Google Scholar]
- 51.Garcia M, Choi C, Kim HR, Daoud Y, Toiyama Y, Takahashi M, Goel A, Boland CR, Koi M. Association between recurrent metastasis from stage II and III primary colorectal tumors and moderate microsatellite instability. Gastroenterology. 2012;143:48–50.e1. doi: 10.1053/j.gastro.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carethers JM, Koi M, Tseng-Rogenski SS. EMAST is a Form of Microsatellite Instability That is Initiated by Inflammation and Modulates Colorectal Cancer Progression. Genes (Basel) 2015;6:185–205. doi: 10.3390/genes6020185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koi M, Garcia M, Choi C, Kim HR, Koike J, Hemmi H, Nagasaka T, Okugawa Y, Toiyama Y, Kitajima T, et al. Microsatellite Alterations With Allelic Loss at 9p24.2 Signify Less-Aggressive Colorectal Cancer Metastasis. Gastroenterology. 2016;150:944–955. doi: 10.1053/j.gastro.2015.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cortes-Ciriano I, Lee S, Park WY, Kim TM, Park PJ. A molecular portrait of microsatellite instability across multiple cancers. Nat Commun. 2017;8:15180. doi: 10.1038/ncomms15180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cole BF, Logan RF, Halabi S, Benamouzig R, Sandler RS, Grainge MJ, Chaussade S, Baron JA. Aspirin for the chemoprevention of colorectal adenomas: meta-analysis of the randomized trials. J Natl Cancer Inst. 2009;101:256–266. doi: 10.1093/jnci/djn485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cook NR, Lee IM, Zhang SM, Moorthy MV, Buring JE. Alternate-day, low-dose aspirin and cancer risk: long-term observational follow-up of a randomized trial. Ann Intern Med. 2013;159:77–85. doi: 10.7326/0003-4819-159-2-201307160-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frouws MA, van Herk-Sukel MPP, Maas HA, Van de Velde CJH, Portielje JEA, Liefers GJ, Bastiaannet E. The mortality reducing effect of aspirin in colorectal cancer patients: Interpreting the evidence. Cancer Treat Rev. 2017;55:120–127. doi: 10.1016/j.ctrv.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 58.Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, Tang J, Rosenstein RB, Wittes J, Corle D, et al. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355:873–884. doi: 10.1056/NEJMoa061355. [DOI] [PubMed] [Google Scholar]
- 59.Arber N, Eagle CJ, Spicak J, Rácz I, Dite P, Hajer J, Zavoral M, Lechuga MJ, Gerletti P, Tang J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006;355:885–895. doi: 10.1056/NEJMoa061652. [DOI] [PubMed] [Google Scholar]
- 60.Baron JA, Sandler RS, Bresalier RS, Quan H, Riddell R, Lanas A, Bolognese JA, Oxenius B, Horgan K, Loftus S, et al. A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology. 2006;131:1674–1682. doi: 10.1053/j.gastro.2006.08.079. [DOI] [PubMed] [Google Scholar]
- 61.Yamauchi T, Watanabe M, Hasegawa H, Nishibori H, Ishii Y, Tatematsu H, Yamamoto K, Kubota T, Kitajima M. The potential for a selective cyclooxygenase-2 inhibitor in the prevention of liver metastasis in human colorectal cancer. Anticancer Res. 2003;23:245–249. [PubMed] [Google Scholar]
- 62.Yao M, Kargman S, Lam EC, Kelly CR, Zheng Y, Luk P, Kwong E, Evans JF, Wolfe MM. Inhibition of cyclooxygenase-2 by rofecoxib attenuates the growth and metastatic potential of colorectal carcinoma in mice. Cancer Res. 2003;63:586–592. [PubMed] [Google Scholar]
- 63.Chan AT, Ogino S, Fuchs CS. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N Engl J Med. 2007;356:2131–2142. doi: 10.1056/NEJMoa067208. [DOI] [PubMed] [Google Scholar]
- 64.Reimers MS, Bastiaannet E, Langley RE, van Eijk R, van Vlierberghe RL, Lemmens VE, van Herk-Sukel MP, van Wezel T, Fodde R, Kuppen PJ, et al. Expression of HLA class I antigen, aspirin use, and survival after a diagnosis of colon cancer. JAMA Intern Med. 2014;174:732–739. doi: 10.1001/jamainternmed.2014.511. [DOI] [PubMed] [Google Scholar]
- 65.Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, Imamura Y, Qian ZR, Baba Y, Shima K, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med. 2012;367:1596–1606. doi: 10.1056/NEJMoa1207756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gray RT, Cantwell MM, Coleman HG, Loughrey MB, Bankhead P, McQuaid S, O’Neill RF, Arthur K, Bingham V, McGready C, et al. Evaluation of PTGS2 Expression, PIK3CA Mutation, Aspirin Use and Colon Cancer Survival in a Population-Based Cohort Study. Clin Transl Gastroenterol. 2017;8:e91. doi: 10.1038/ctg.2017.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ng K, Meyerhardt JA, Chan AT, Sato K, Chan JA, Niedzwiecki D, Saltz LB, Mayer RJ, Benson AB 3rd, Schaefer PL, Whittom R, Hantel A, Goldberg RM, Venook AP, Ogino S, Giovannucci EL, Fuchs CS. Aspirin and COX-2 inhibitor use in patients with stage III colon cancer. J Natl Cancer Inst. 2014;107:345. doi: 10.1093/jnci/dju345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ma X, Aoki T, Tsuruyama T, Narumiya S. Definition of Prostaglandin E2-EP2 Signals in the Colon Tumor Microenvironment That Amplify Inflammation and Tumor Growth. Cancer Res. 2015;75:2822–2832. doi: 10.1158/0008-5472.CAN-15-0125. [DOI] [PubMed] [Google Scholar]
- 69.Angelova M, Charoentong P, Hackl H, Fischer ML, Snajder R, Krogsdam AM, Waldner MJ, Bindea G, Mlecnik B, Galon J, et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015;16:64. doi: 10.1186/s13059-015-0620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Becht E, de Reyniès A, Giraldo NA, Pilati C, Buttard B, Lacroix L, Selves J, Sautès-Fridman C, Laurent-Puig P, Fridman WH. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin Cancer Res. 2016;22:4057–4066. doi: 10.1158/1078-0432.CCR-15-2879. [DOI] [PubMed] [Google Scholar]
- 71.Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell. 2015;162:1257–1270. doi: 10.1016/j.cell.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 73.Ahrendt SA, Decker PA, Doffek K, Wang B, Xu L, Demeure MJ, Jen J, Sidransky D. Microsatellite instability at selected tetranucleotide repeats is associated with p53 mutations in non-small cell lung cancer. Cancer Res. 2000;60:2488–2491. [PubMed] [Google Scholar]
- 74.Li J, Koike J, Kugoh H, Arita M, Ohhira T, Kikuchi Y, Funahashi K, Takamatsu K, Boland CR, Koi M, et al. Down-regulation of MutS homolog 3 by hypoxia in human colorectal cancer. Biochim Biophys Acta. 2012;1823:889–899. doi: 10.1016/j.bbamcr.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Robles AI, Traverso G, Zhang M, Roberts NJ, Khan MA, Joseph C, Lauwers GY, Selaru FM, Popoli M, Pittman ME, et al. Whole-Exome Sequencing Analyses of Inflammatory Bowel Disease-Associated Colorectal Cancers. Gastroenterology. 2016;150:931–943. doi: 10.1053/j.gastro.2015.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yaeger R, Shah MA, Miller VA, Kelsen JR, Wang K, Heins ZJ, Ross JS, He Y, Sanford E, Yantiss RK, et al. Genomic Alterations Observed in Colitis-Associated Cancers Are Distinct From Those Found in Sporadic Colorectal Cancers and Vary by Type of Inflammatory Bowel Disease. Gastroenterology. 2016;151:278–287.e6. doi: 10.1053/j.gastro.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brennan CA, Garrett WS. Gut Microbiota, Inflammation, and Colorectal Cancer. Annu Rev Microbiol. 2016;70:395–411. doi: 10.1146/annurev-micro-102215-095513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe. 2014;15:317–328. doi: 10.1016/j.chom.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McCoy AN, Araújo-Pérez F, Azcárate-Peril A, Yeh JJ, Sandler RS, Keku TO. Fusobacterium is associated with colorectal adenomas. PLoS One. 2013;8:e53653. doi: 10.1371/journal.pone.0053653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207–215. doi: 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ito M, Kanno S, Nosho K, Sukawa Y, Mitsuhashi K, Kurihara H, Igarashi H, Takahashi T, Tachibana M, Takahashi H, et al. Association of Fusobacterium nucleatum with clinical and molecular features in colorectal serrated pathway. Int J Cancer. 2015;137:1258–1268. doi: 10.1002/ijc.29488. [DOI] [PubMed] [Google Scholar]
- 82.Tahara T, Yamamoto E, Suzuki H, Maruyama R, Chung W, Garriga J, Jelinek J, Yamano HO, Sugai T, An B, et al. Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res. 2014;74:1311–1318. doi: 10.1158/0008-5472.CAN-13-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nosho K, Sukawa Y, Adachi Y, Ito M, Mitsuhashi K, Kurihara H, Kanno S, Yamamoto I, Ishigami K, Igarashi H, et al. Association of Fusobacterium nucleatum with immunity and molecular alterations in colorectal cancer. World J Gastroenterol. 2016;22:557–566. doi: 10.3748/wjg.v22.i2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mima K, Nishihara R, Qian ZR, Cao Y, Sukawa Y, Nowak JA, Yang J, Dou R, Masugi Y, Song M, et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut. 2016;65:1973–1980. doi: 10.1136/gutjnl-2015-310101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, Qian Y, Kryczek I, Sun D, Nagarsheth N, et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell. 2017;170:548–563.e16. doi: 10.1016/j.cell.2017.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kawakami H, Zaanan A, Sinicrope FA. Implications of mismatch repair-deficient status on management of early stage colorectal cancer. J Gastrointest Oncol. 2015;6:676–684. doi: 10.3978/j.issn.2078-6891.2015.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Suzuki S, Iwaizumi M, Tseng-Rogenski S, Hamaya Y, Miyajima H, Kanaoka S, Sugimoto K, Carethers JM. Production of truncated MBD4 protein by frameshift mutation in DNA mismatch repair-deficient cells enhances 5-fluorouracil sensitivity that is independent of hMLH1 status. Cancer Biol Ther. 2016;17:760–768. doi: 10.1080/15384047.2016.1178430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Toprak NU, Yagci A, Gulluoglu BM, Akin ML, Demirkalem P, Celenk T, Soyletir G. A possible role of Bacteroides fragilis enterotoxin in the aetiology of colorectal cancer. Clin Microbiol Infect. 2006;12:782–786. doi: 10.1111/j.1469-0691.2006.01494.x. [DOI] [PubMed] [Google Scholar]
- 89.Boleij A, Hechenbleikner EM, Goodwin AC, Badani R, Stein EM, Lazarev MG, Ellis B, Carroll KC, Albesiano E, Wick EC, et al. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin Infect Dis. 2015;60:208–215. doi: 10.1093/cid/ciu787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Purcell RV, Pearson J, Aitchison A, Dixon L, Frizelle FA, Keenan JI. Colonization with enterotoxigenic Bacteroides fragilis is associated with early-stage colorectal neoplasia. PLoS One. 2017;12:e0171602. doi: 10.1371/journal.pone.0171602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sears CL, Geis AL, Housseau F. Bacteroides fragilis subverts mucosal biology: from symbiont to colon carcinogenesis. J Clin Invest. 2014;124:4166–4172. doi: 10.1172/JCI72334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bonnet M, Buc E, Sauvanet P, Darcha C, Dubois D, Pereira B, Déchelotte P, Bonnet R, Pezet D, Darfeuille-Michaud A. Colonization of the human gut by E. coli and colorectal cancer risk. Clin Cancer Res. 2014;20:859–867. doi: 10.1158/1078-0432.CCR-13-1343. [DOI] [PubMed] [Google Scholar]
- 95.Tomkovich S, Yang Y, Winglee K, Gauthier J, Mühlbauer M, Sun X, Mohamadzadeh M, Liu X, Martin P, Wang GP, et al. Locoregional Effects of Microbiota in a Preclinical Model of Colon Carcinogenesis. Cancer Res. 2017;77:2620–2632. doi: 10.1158/0008-5472.CAN-16-3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Scott TA, Quintaneiro LM, Norvaisas P, Lui PP, Wilson MP, Leung KY, Herrera-Dominguez L, Sudiwala S, Pessia A, Clayton PT, et al. Host-Microbe Co-metabolism Dictates Cancer Drug Efficacy in C. elegans. Cell. 2017;169:442–456.e18. doi: 10.1016/j.cell.2017.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sugawara N, Pâques F, Colaiácovo M, Haber JE. Role of Saccharomyces cerevisiae Msh2 and Msh3 repair proteins in double-strand break-induced recombination. Proc Natl Acad Sci USA. 1997;94:9214–9219. doi: 10.1073/pnas.94.17.9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dietlein F, Thelen L, Jokic M, Jachimowicz RD, Ivan L, Knittel G, Leeser U, van Oers J, Edelmann W, Heukamp LC, et al. A functional cancer genomics screen identifies a druggable synthetic lethal interaction between MSH3 and PRKDC. Cancer Discov. 2014;4:592–605. doi: 10.1158/2159-8290.CD-13-0907. [DOI] [PubMed] [Google Scholar]
- 99.Burdova K, Mihaljevic B, Sturzenegger A, Chappidi N, Janscak P. The Mismatch-Binding Factor MutSβ Can Mediate ATR Activation in Response to DNA Double-Strand Breaks. Mol Cell. 2015;59:603–614. doi: 10.1016/j.molcel.2015.06.026. [DOI] [PubMed] [Google Scholar]
- 100.Dietlein F, Thelen L, Reinhardt HC. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 2014;30:326–339. doi: 10.1016/j.tig.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 101.Moudry P, Watanabe K, Wolanin KM, Bartkova J, Wassing IE, Watanabe S, Strauss R, Troelsgaard Pedersen R, Oestergaard VH, Lisby M, et al. TOPBP1 regulates RAD51 phosphorylation and chromatin loading and determines PARP inhibitor sensitivity. J Cell Biol. 2016;212:281–288. doi: 10.1083/jcb.201507042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304:93–96. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- 103.Sedelnikova OA, Redon CE, Dickey JS, Nakamura AJ, Georgakilas AG, Bonner WM. Role of oxidatively induced DNA lesions in human pathogenesis. Mutat Res. 2010;704:152–159. doi: 10.1016/j.mrrev.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.McCool KW, Miyamoto S. DNA damage-dependent NF-κB activation: NEMO turns nuclear signaling inside out. Immunol Rev. 2012;246:311–326. doi: 10.1111/j.1600-065X.2012.01101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, Forman S, Jove R, Pardoll DM, Yu H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009;15:283–293. doi: 10.1016/j.ccr.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu X, Ji Q, Ye N, Sui H, Zhou L, Zhu H, Fan Z, Cai J, Li Q. Berberine Inhibits Invasion and Metastasis of Colorectal Cancer Cells via COX-2/PGE2 Mediated JAK2/STAT3 Signaling Pathway. PLoS One. 2015;10:e0123478. doi: 10.1371/journal.pone.0123478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xiong H, Du W, Sun TT, Lin YW, Wang JL, Hong J, Fang JY. A positive feedback loop between STAT3 and cyclooxygenase-2 gene may contribute to Helicobacter pylori-associated human gastric tumorigenesis. Int J Cancer. 2014;134:2030–2040. doi: 10.1002/ijc.28539. [DOI] [PubMed] [Google Scholar]
- 108.Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 2015;163:712–723. doi: 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- 109.Leung SY, Yuen ST, Chung LP, Chu KM, Chan AS, Ho JC. hMLH1 promoter methylation and lack of hMLH1 expression in sporadic gastric carcinomas with high-frequency microsatellite instability. Cancer Res. 1999;59:159–164. [PubMed] [Google Scholar]
- 110.Chung TK, Cheung TH, Wang VW, Yu MY, Wong YF. Microsatellite instability, expression of hMSH2 and hMLH1 and HPV infection in cervical cancer and their clinico-pathological association. Gynecol Obstet Invest. 2001;52:98–103. doi: 10.1159/000052951. [DOI] [PubMed] [Google Scholar]
- 111.Yamamoto H, Itoh F, Nakamura H, Fukushima H, Sasaki S, Perucho M, Imai K. Genetic and clinical features of human pancreatic ductal adenocarcinomas with widespread microsatellite instability. Cancer Res. 2001;61:3139–3144. [PubMed] [Google Scholar]
- 112.Watanabe Y, Koi M, Hemmi H, Hoshai H, Noda K. A change in microsatellite instability caused by cisplatin-based chemotherapy of ovarian cancer. Br J Cancer. 2001;85:1064–1069. doi: 10.1054/bjoc.2001.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hussein MR, Sun M, Tuthill RJ, Roggero E, Monti JA, Sudilovsky EC, Wood GS, Sudilovsky O. Comprehensive analysis of 112 melanocytic skin lesions demonstrates microsatellite instability in melanomas and dysplastic nevi, but not in benign nevi. J Cutan Pathol. 2001;28:343–350. doi: 10.1034/j.1600-0560.2001.280702.x. [DOI] [PubMed] [Google Scholar]
- 114.Szybka M, Bartkowiak J, Zakrzewski K, Polis L, Liberski P, Kordek R. Microsatellite instability and expression of DNA mismatch repair genes in malignant astrocytic tumors from adult and pediatric patients. Clin Neuropathol. 2003;22:180–186. [PubMed] [Google Scholar]
- 115.Halford SE, Sawyer EJ, Lambros MB, Gorman P, Macdonald ND, Talbot IC, Foulkes WD, Gillett CE, Barnes DM, Akslen LA, et al. MSI-low, a real phenomenon which varies in frequency among cancer types. J Pathol. 2003;201:389–394. doi: 10.1002/path.1453. [DOI] [PubMed] [Google Scholar]
- 116.Chiappini F, Gross-Goupil M, Saffroy R, Azoulay D, Emile JF, Veillhan LA, Delvart V, Chevalier S, Bismuth H, Debuire B, et al. Microsatellite instability mutator phenotype in hepatocellular carcinoma in non-alcoholic and non-virally infected normal livers. Carcinogenesis. 2004;25:541–547. doi: 10.1093/carcin/bgh035. [DOI] [PubMed] [Google Scholar]
- 117.Evans SC, Gillis A, Geldenhuys L, Vaninetti NM, Malatjalian DA, Porter GA, Guernsey DL, Casson AG. Microsatellite instability in esophageal adenocarcinoma. Cancer Lett. 2004;212:241–251. doi: 10.1016/j.canlet.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 118.Choy KW, Pang CP, Fan DS, Lee TC, Wang JH, Abramson DH, Lo KW, To KF, Yu CB, Beaverson KL, et al. Microsatellite instability and MLH1 promoter methylation in human retinoblastoma. Invest Ophthalmol Vis Sci. 2004;45:3404–3409. doi: 10.1167/iovs.03-1273. [DOI] [PubMed] [Google Scholar]
- 119.Kawaguchi K, Oda Y, Takahira T, Saito T, Yamamoto H, Kobayashi C, Tamiya S, Oda S, Iwamoto Y, Tsuneyoshi M. Microsatellite instability and hMLH1 and hMSH2 expression analysis in soft tissue sarcomas. Oncol Rep. 2005;13:241–246. [PubMed] [Google Scholar]
- 120.Saetta AA, Gigelou F, Papanastasiou PI, Koilakou SV, Kalekou-Greca H, Miliaras D, Michalopoulos NV, Patsouris E. High-level microsatellite instability is not involved in gallbladder carcinogenesis. Exp Mol Pathol. 2006;80:67–71. doi: 10.1016/j.yexmp.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 121.Koy S, Plaschke J, Luksch H, Friedrich K, Kuhlisch E, Eckelt U, Martinez R. Microsatellite instability and loss of heterozygosity in squamous cell carcinoma of the head and neck. Head Neck. 2008;30:1105–1113. doi: 10.1002/hed.20857. [DOI] [PubMed] [Google Scholar]
- 122.Perinchery G, Nojima D, Goharderakhshan R, Tanaka Y, Alonzo J, Dahiya R. Microsatellite instability of dinucleotide tandem repeat sequences is higher than trinucleotide, tetranucleotide and pentanucleotide repeat sequences in prostate cancer. Int J Oncol. 2000;16:1203–1209. doi: 10.3892/ijo.16.6.1203. [DOI] [PubMed] [Google Scholar]
- 123.Arai H, Okudela K, Oshiro H, Komitsu N, Mitsui H, Nishii T, Tsuboi M, Nozawa A, Noishiki Y, Ohashi K, et al. Elevated microsatellite alterations at selected tetra-nucleotide (EMAST) in non-small cell lung cancers--a potential determinant of susceptibility to multiple malignancies. Int J Clin Exp Pathol. 2013;6:395–410. [PMC free article] [PubMed] [Google Scholar]
- 124.Catto JW, Azzouzi AR, Amira N, Rehman I, Feeley KM, Cross SS, Fromont G, Sibony M, Hamdy FC, Cussenot O, et al. Distinct patterns of microsatellite instability are seen in tumours of the urinary tract. Oncogene. 2003;22:8699–8706. doi: 10.1038/sj.onc.1206964. [DOI] [PubMed] [Google Scholar]
- 125.Carethers JM, Jung BH. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology. 2015;149:1177–1190.e3. doi: 10.1053/j.gastro.2015.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ashktorab H, Kupfer SS, Brim H, Carethers JM. Racial Disparity in Gastrointestinal Cancer Risk. Gastroenterology. 2017;153:910–923. doi: 10.1053/j.gastro.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]