

Abstract
IL-36 cytokines signal through the IL-36 receptor (IL-36R) and a shared subunit, IL-1RAcP (IL-1 receptor accessory protein). The activation mechanism for the IL-36 pathway is proposed to be similar to that of IL-1 in that an IL-36R agonist (IL-36α, IL-36β, or IL-36γ) forms a binary complex with IL-36R, which then recruits IL-1RAcP. Recent studies have shown that IL-36R interacts with IL-1RAcP even in the absence of an agonist. To elucidate the IL-36 activation mechanism, we considered all possible binding events for IL-36 ligands/receptors and examined these events in direct binding assays. Our results indicated that the agonists bind the IL-36R extracellular domain with micromolar affinity but do not detectably bind IL-1RAcP. Using surface plasmon resonance (SPR), we found that IL-1RAcP also does not bind IL-36R when no agonist is present. In the presence of IL-36α, however, IL-1RAcP bound IL-36R strongly. These results suggested that the main pathway to the IL-36R·IL-36α·IL-1RAcP ternary complex is through the IL-36R·IL-36α binary complex, which recruits IL-1RAcP. We could not measure the binding affinity of IL-36R to IL-1RAcP directly, so we engineered a fragment crystallizable–linked construct to induce IL-36R·IL-1RAcP heterodimerization and predicted the binding affinity during a complete thermodynamic cycle to be 74 μm. The SPR analysis also indicated that the IL-36R antagonist IL-36Ra binds IL-36R with higher affinity and a much slower off rate than the IL-36R agonists, shedding light on IL-36 pathway inhibition. Our results reveal the landscape of IL-36 ligand and receptor interactions, improving our understanding of IL-36 pathway activation and inhibition.
Keywords: immunology, kinetics, protein–protein interaction, signaling, surface plasmon resonance (SPR), IL-1Rrp2, IL-36R
Introduction
IL-36 cytokines, including three agonists (IL-36α, IL-36β, and IL-36γ) and one antagonist (IL-36Ra),2 are members of IL-1 family that activate and inhibit MYD88 (myeloid differentiation primary response gene 88)–mediated pathways leading to nuclear factor κ-light-chain enhancer of activated B cells (NF-κB) and MAPKs (1–3). It has been shown that IL-36 agonists do not activate NF-κB unless both IL-36R and IL-1RAcP are present (1). A natural antagonist, IL-36Ra, exists to inhibit IL-36 (4).
Mutations and aberrant expression of IL-36 cytokines have been associated with inflammation and skin diseases. Recent studies have shown that loss of function mutations in IL-36Ra leads to a severe and rare form of skin inflammation: generalized pustular psoriasis (5). Importantly, studies have also shown a potential link of IL-36 signaling to psoriasis vulgaris. It has been shown that the levels of IL-36γ are elevated in skin and serum of patients with psoriasis, and the level is correlated with the disease severity. Furthermore, elevated levels of IL-36γ can be normalized with anti-TNFα treatment (6).
Because IL-36, similar to other IL-1 members, requires both receptor and co-receptor being present for activation, it has been speculated that the activation mode for IL-36 is the same as for IL-1 (7, 8). IL-1β activates signaling by binding to membrane bound IL-1 receptor (IL-1R) and subsequent recruitment of the co-receptor IL-1RAcP (9, 10). Formation of the IL-1R·IL-1β·IL-1RAcP ternary complex brings together intracellular Toll-IL-1 receptor domains, which is necessary for activating downstream signaling (11). However, some attempts to measure the first event of IL-36 activation, which is the direct binding of cytokines and IL-36R, have not been successful (1). Furthermore, a recent study shows IL-36R interacts with IL-1RAcP even in the absence of the agonist (12). Knowledge of how IL-36R becomes activated and inhibited is important to understand a potential drug target that has implications in multiple inflammatory diseases including psoriasis.
In this study, we examined three theoretical paths for IL-36 activation using an SPR binding assay. In addition, an orthogonal time-resolved (TR)–FRET assay was also applied to evaluate direct binding. Through these measurements, we were able to establish the temporal order of binding events associated with IL-36 ligands and receptors. In addition, we also measured direct binding of IL-36Ra·IL-36R, which was much stronger than the agonist·IL-36R. Collectively, these results offer a biochemical basis for elucidating IL-36 activation and inhibition.
Results
To achieve the IL-36R·IL-36α·IL-1RAcP ternary complex, a binary complex must form first. Theoretically, there may be three ways to form binary complexes, for example, IL-36α·IL-36R, IL-36α·IL-1RAcP, and IL-36R·IL-1RAcP (Fig. 1). To help elucidate which path is preferred for IL-36 activation from a binding affinity perspective, we expressed and purified each component and measured their direct binding in an SPR assay.
Figure 1.

A schematic diagram of binding events involved in IL-36 activation and inhibition. This illustration shows that among the three possible activating binary complexes, IL-36α·IL-36R is the most likely to form because of its stronger interactions when comparing to the other two. IL-1RAcP is recruited to IL-36α·IL-36R at a KD of 42 nm for ternary complex formation. IL-36Ra, when present, competes with IL-36α for IL-36R binding. The KD values are averages of at least two independent SPR binding results. Solid boxes represent likely bound or unbound states of receptors, co-receptors, and cytokines in cells. Dashed boxes represent either the unlikely state or a transient state. Activation paths are illustrated with black arrows and boxes, whereas the inhibition path is shown in red.
IL-36 cytokines and receptors were purified to more than 95% homogeneity
It has been reported that N-terminal processing of IL-36 cytokines is required for full agonistic and antagonistic activity (13). Therefore, we expressed and purified truncated cytokines, IL-36α (6–158), IL-36γ (18–169), and IL-36Ra (2–155). Extracellular domains (ECDs) of IL-36R (1–335) and IL-1RAcP (1–367) were expressed with a FLAG-His-AVI tag on their C terminus for purification and SPR chip capturing purpose. We also purified the non-AVI-tagged version of both IL-36R and IL-1RAcP to study them as analytes by SPR. Finally, an fragment crystallizable (Fc)–linked IL-36R·IL-1RAcP heterodimer with mutations to enhance heterodimerization during expression was purified (14). All proteins were purified to more than 95% purity estimated by SDS-PAGE (Fig. 2). Prior to binding studies, samples were further purified by SEC and quantified by UV280.
Figure 2.

SDS-PAGE gels showing purified soluble forms of receptors, co-receptors, and cytokines. All soluble proteins were purified to more than 95% purity as estimated from the single band on SDS-PAGE. IL-36α, IL-36γ, and IL-36Ra migrate as a single band with a molecular mass of ∼17 kDa, which is consistent with theoretical molecular masses. IL-36R and IL-1RAcP show up as single band with a molecular mass of ∼49 kDa, which is higher than theoretical molecular mass of 37 kDa. Each half of the heterodimer is showing up as single band with a molecular mass of ∼70 kDa, which is higher than theoretical molecular mass of 63 kDa. Receptors showing higher molecular mass by SDS-PAGE are likely due to glycosylation (12).
Purified IL-36 cytokines are biologically active
To test binding properties of expressed and purified IL-36 cytokines, we first needed to confirm their biological activity. It was previously demonstrated that IL-36 cytokines can induce a robust expression and secretion of chemokines, including CXCL1, from keratinocytes in culture (15, 16). Using a HaCaT keratinocyte cell line, we demonstrated a robust, dose-dependent production of CXCL1 being induced by IL-36α and IL-36γ treatment (Fig. 3). Moreover, IL-36γ-induced production of CXCL1 was inhibited in a dose-dependent fashion by treatment with IL-36Ra (Fig. 3). Measurements of secreted CXCL1 demonstrate that the IL-36Ra potency is consistent with previously reported values (15, 16).
Figure 3.

Purified IL-36 cytokines retain their biological activity as measured in secreted CXCL1 assay. Human HaCaT keratinocyte cell line was treated for 24 h with IL-36α (left panel) or IL-36γ (middle panel) at the indicated concentrations. Measurement of secreted CXCL1 shows similar potencies between the two cytokines. HaCaT keratinocyte cells were pretreated with IL-36Ra (right panel) at the indicated concentrations for 15 min and then overlaid with 60 ng/ml of IL-36γ for 24 h. Measurement of secreted CXCL1 shows IL-36Ra potency is consistent with reported values (15, 16). Averages of triplicate measurements are shown in the plot. Standard deviations are represented as error bars. The data are fit using Equation 1 to obtain EC50.
IL-36α and IL-36γ showed direct binding to IL-36R
Multiple attempts were made by other groups to detect direct binding between cytokines and IL-36R (1, 4). To the best of our knowledge, this is the first study to provide quantitative measurements of this critical binding event. IL-36R (1–335)-FLAG-His-AVI was biotinylated and captured onto a NeutrAvidin chip surface (Fig. 4A). Various concentrations of IL-36α (6–158) or IL-36γ (18–169) were injected as analytes in solution. IL-36α bound to IL-36R with a KD = 480 ± 90 nm, whereas IL-36γ showed weaker binding affinity (KD = 1800 ± 200 nm) (Table 1). Both IL-36α and IL-36γ showed fast-on and fast-off kinetics to IL-36R (Fig. 5A and Table 1). The on-rate constant and off-rate constant of IL-36γ could not be obtained accurately because of instrument limitations. Only the equilibrium dissociation constant was obtained for IL-36γ·IL-36R binding.
Figure 4.
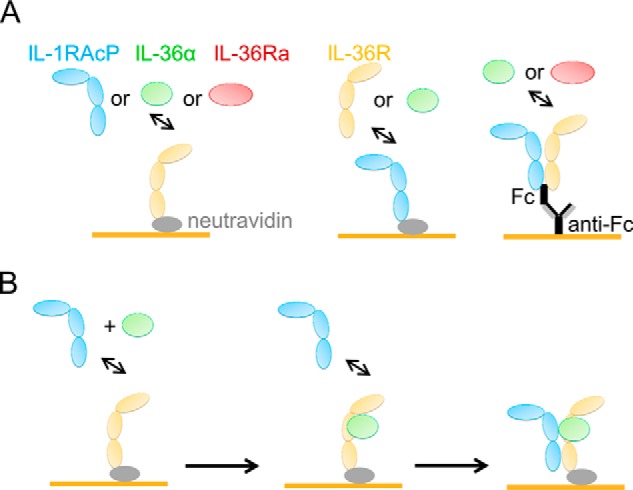
Illustrations of SPR binding study design. A, experimental designs for cytokines binding to IL-36R, IL-1RAcP, or Fc-linked IL-36R·IL-1RAcP heterodimer. Receptors are captured onto the chip surface (yellow bar) either though biotin–NeutrAvidin interactions or through Fc–anti-Fc interactions. Cytokines or receptors are injected in solution at various concentrations. B, binding events associated with IL-1RAcP binding to IL-36R in the presence of IL-36α. Various concentrations of IL-1RAcP and a saturating concentration of IL-36α (4 μm) are added to the chip surface captured with IL-36R. Fast-on and fast-off kinetics allows IL-36α to quickly bind to IL-36R, followed by IL-1RAcP recruitment.
Table 1.
Summary of SPR binding results for IL-36 receptor, co-receptor, and cytokines
ka (× 105 m−1 s−1) and kd (× 10−3 s−1) are determined from fitting of SPR sensorgram with 1:1 binding model. KD (× 10−9 m) is calculated from the ratio of kd and ka in each independent experiment. The values displayed are the averages ± S.D. from two or more independent experiments. Two significant figures are reported for the averages.
| Analyte | IL-36R |
IL-1RAcP |
Heterodimer |
||||
|---|---|---|---|---|---|---|---|
| ka | kd | KD | KD | ka | kd | KD | |
| IL-36R | NDa | >1000b | ND | ||||
| IL-1RAcP | >1000b | ND | ND | ||||
| IL-1RAcP + IL-36α | 1.5 ± 0.3 | 6.3 ± 0.3 | 42 ± 10 | ND | ND | ||
| IL-36α | 3.3 ± 0.6 | 160 ± 10 | 480 ± 90 | >60,000b | 9.9 ± 7.0 | 0.20 ± 0.05 | 0.27 ± 0.13 |
| IL-36γ | 1800 ± 200c | >60,000b | 0.25 ± 0.01 | 0.14 ± 0.01 | 5.6 ± 0.6 | ||
| IL-36Ra | 8.3 ± 0.5 | 4.8 ± 0.1 | 5.8 ± 0.3 | ND | 7.8 ± 0.2 | 4.3 ± 0.1 | 5.5 ± 0.2 |
a Upper limit of affinity estimated from the highest concentration of analyte tested.
b ND, not determined.
c Affinity determined from fitting of sensorgram at steady state.
Figure 5.

IL-36 agonists and the antagonist bind to receptor IL-36R and heterodimer with distinct kinetics. A, left panels, agonists IL-36α/γ bind to IL-36R with fast-on and fast-off rates, whereas antagonist IL-36Ra dissociates from bound IL-36Ra·IL-36R complex at a slower off rate. Right panels, agonists IL-36α/γ bind to Fc-linked IL-36R·IL-1RAcP heterodimer and dissociate from it at a much slower rate than IL-36Ra. B, top panels, little or no IL-36α/γ binding to IL-1RAcP. Bottom panels, IL-1RAcP binds to IL-36R only when IL-36α is present. One representative sensorgram from at least two independent experiments is shown. Sensorgrams are colored lines, whereas fits are in black. Sensorgrams are globally fit using a 1:1 kinetics model with a mass transfer term included. Binding kinetics of IL-36γ·IL-36R is out of instrument limit. Affinity values for IL-36γ·IL-36R are obtained using steady-state fit.
IL-36α showed no detectable binding to IL-1RAcP
To examine the possibility of another binary complex, the IL-36α·IL-1RAcP, we biotinylated IL-1RAcP-FLAG-His-AVI and captured it onto a NeutrAvidin chip. Up to ∼60 μm of IL-36α was injected onto the chip. No IL-1RAcP binding was detected by SPR (Fig. 5B). We concluded that IL-36α and IL-1RAcP were not likely to exist as a stable binary complex in cells because of undetectable binding at high concentrations.
IL-36R showed no detectable binding to IL-1RAcP up to 1 μm
Interestingly, it was reported that IL-36R and IL-1RAcP co-immunoprecipitated in the absence of a cytokine such as IL-36γ (12). To study interactions between IL-36R and IL-1RAcP, we coupled each one of them separately on a chip and injected the other component. However, no binding was detected up to 1 μm of analytes (Fig. 5B). We did not use analyte concentrations higher than 1 μm because it would produce nonspecific binding in the SPR assay. Reconciling our negative binding results with reported co-immunoprecipitation results, we believe that the interactions between IL-36R and IL-1RAcP are rather weak. Therefore, the KD value for receptor and co-receptor interaction was estimated to be larger than 1 μm based on the highest concentration tested in this study.
IL-1RAcP showed strong binding to IL-36R in the presence of IL-36α
To bring Toll-IL-1 receptor domain domains in proximity, IL-1RAcP needs to join the IL-36α·IL-36R binary complex. It has been reported that interactions between IL-36R and IL-1RAcP are increased in the presence of a IL-36 cytokine (12). We measured the binding kinetics and affinity of the second step for activation, IL-1RAcP joining IL-36α·IL-36R. A chip surface with IL-36R was injected with various concentrations of IL-1RAcP in the presence of 4 μm (a saturating concentration) of IL-36α. We have already determined that IL-1RAcP does not bind IL-36R at 1 μm and that IL-36α binds to IL-36R with fast-on and fast-off kinetics. At 4 μm, IL-36α would occupy nearly all of the IL-36R on chip surface almost immediately because of the fast-on rate. Therefore any additional binding observed in a mix of IL-36α and IL-1RAcP should reflect interactions between IL-1RAcP and IL-36α·IL-36R (Fig. 4B). Notably, we observed strong binding of IL-1RAcP (KD = 42 ± 10 nm) that did not exist in the absence of IL-36α (Fig. 5B and Table 1).
IL-36α or IL-36γ showed strong binding to Fc-linked IL-36R·IL-1RAcP heterodimer
Although we could not detect the interactions between IL-1RAcP and IL-36R up to 1 μm in SPR studies, it has been shown that the interactions do exist by co-immunoprecipitation (12). Because we could not quantify the weak interactions between IL-36R and IL-1RAcP directly through binding measurements, we sought to predict the binding affinity through thermodynamic analysis. To complete the thermodynamic cycle, we needed to measure the affinity of IL-36α binding to a preassociated IL-36R·IL-1RAcP heterodimer (Fig. 1). An IL-36R·IL-1RAcP heterodimer that was dimerized through Fc “knob and hole” mutations was purified as described (4). In the SPR assay, the heterodimer was captured onto an anti-human Fc chip at a low ligand density. IL-36α or IL-36γ was injected in solution. Both cytokines showed much stronger binding to the heterodimer than to the IL-36R monomer. IL-36α had a binding affinity of 0.27 ± 0.13 nm, whereas IL-36γ bound to heterodimer at a KD of 5.6 ± 0.6 nm (Table 1). Both IL-36α and IL-36γ dissociated from heterodimer with very slow off-rates (Fig. 5A and Table 1). Such slow dissociation was likely caused by stabilization of IL-1RAcP because both cytokines dissociated from IL-36R alone rather quickly (Fig. 5A). Notably, IL-36α displayed higher affinity than IL-36γ to both IL-36R and IL-36R·IL-1RAcP heterodimer.
Antagonist IL-36Ra binds IL-36R with a higher affinity than agonists
Finally, we examined the binding between the antagonist IL-36Ra and the IL-36R. IL-36R was captured onto a NeutrAvidin chip, and IL-36Ra was injected in solution. Antagonist IL-36Ra showed stronger binding (KD = 5.8 ± 0.3 nm) to IL-36R than agonist IL-36α (KD = 480 ± 90 nm) or IL-36γ (KD = 1800 ± 200 nm) (Table 1). In contrast to the fast-off kinetics observed for agonist, IL-36Ra showed a much slower off-rate (4.8 ± 1.1 × 10−3 s−1) (Fig. 5A and Table 1). To date, this is the first quantitative measurement of IL-36Ra binding to IL-36R. It has been reported that IL-36Ra bound to the Fc-linked IL-36R·IL-1RAcP heterodimer at KD values ranging from 10 to 81 nm (4, 13). We measured IL-36Ra bound to heterodimer at a KD value of 5.5 ± 0.2 nm, which was identical to the affinity of IL-36Ra·IL-36R interactions.
IL-36 family cytokines bind to the receptor in solution by TR–FRET
To test for the ability of IL-36 cytokines to bind to IL-36R monomer and IL-36R·IL-1RAcP heterodimer in solution; we developed a TR–FRET assay using the biotinylated AVI-tagged version of IL-36α and IL-36Ra. The cytokines were coupled with streptavidin-Alexa Fluor 488 and tested for binding to IL-36R monomer or IL-36R·IL-1RAcP. IL-36α binds to IL-36R·IL-1RAcP with a KD of 0.52 nm (Fig. 6), very close to results from SPR assay. Concomitantly, IL-36Ra binds to IL-36R and the heterodimer with the same affinity, 33 ± 5 and 38 ± 5 nm, respectively (Fig. 6).
Figure 6.

IL-36α and IL-36Ra bind to IL-36R and heterodimer in solution by TR–FRET. IL-36α (left panel) binds to Fc-linked IL-36R·IL-1RAcP heterodimer strongly in solution. IL-36Ra binds to heterodimer (middle panel) and IL-36R (right panel) with similar affinities. The ratio of fluorescent signal at 520 and 495 nm is normalized to the maximal signal ratio. Averages of triplicate measurements are shown in the plot. Standard deviations are represented as error bars. The data are fit using Equation 3 to obtain KD.
IL-36γ binding to the receptor in physiological conditions by glyco-FRET
The limitations of testing protein–protein interactions by SPR and in solution lay in the absence of membrane, transmembrane, and intercellular domains that may alter those interactions. Other binding assays such as flow cytometry suffer from the limitation that binding is typically not measured under equilibrium conditions because washing steps are required prior to measurement. To test for the more native interactions, we used a novel glyco-FRET binding assay, which is a live-cell binding assay that allows the characterization of ligand–receptor binding in equilibrium (17). Briefly, cell-surface receptors are labeled with a chelated lanthanide reporter for time-resolved FRET. Importantly, receptor labeling does not interfere with receptor dimerization or ligand binding, because the reporter is installed on the glycans of the receptor. Next, the cells are incubated with a fluorescently labeled ligand, followed by FRET measurement without removal of ligand. We stably overexpressed either IL-36R alone or together with IL-1RAcP in HEK293 cells that do not express the receptor (4). The surface levels of IL-36R in both transfected populations were similar (Fig. 7A). All glycosylated cell-surface receptors were labeled with the TR–FRET donor terbium. IL-36γ Alexa Fluor 488–labeled cytokine was assessed for binding to cell surface–expressed full length IL-36R or IL-36R·IL-1RAcP using TR–FRET. Similarly to SPR and TR–FRET assays, IL-36R alone shows almost no binding up to 60 nm of labeled ligand, whereas IL-36R·IL-1RAcP heterodimer binds with an EC50 of 1 nm (Fig. 7B).
Figure 7.

IL-36γ binds to IL-36R and heterodimer expressed on HEK293 cells by glyco-FRET. A, IL-36R surface expression analysis on expanded IL-36R and IL-36R·IL-1RAcP HEK 293H cell lines with FACS using anti-HA antibody described under “Experimental Procedures.” The Geo mean values of fluorescence are 2.3 × 104, 1.5 × 104, and 2.2 × 102 for IL-36R (blue), IL-36R·IL-1RAcP (orange), and parental HEK293H (red), respectively. B, glycol-FRET analysis on HEK293 cells overexpressing IL-36R or IL-36R·IL-1RAcP shows strong binding of IL-36γ to receptor dimer but not the monomer. The ratio of fluorescent signal at 520 and 495 nm is shown (TR–FRET ratio). Averages of triplicate measurements are shown in the plot. Standard deviations are represented as error bars.
Discussion
With the evidence linking the IL-36 pathway to pustular psoriasis, understanding how IL-36 ligand and receptors interact is important to gain insights into IL-36 activation and inhibition and is useful in developing a strategy for targeting IL-36. To date, limited quantitative evidence has been reported for the temporal order of association and dissociation of IL-36R, IL-1RAcP, and cytokines. Little direct binding has been shown for any of the three possible paths for activation (Fig. 1). We have explored and confirmed that the dominant path to IL-36R·agonist·IL-1RAcP is through agonist·IL-36R complex formation and subsequent recruitment of IL-1RAcP.
We have also studied a less prominent path to ternary complex, through IL-36R·IL-1RAcP. Semiquantitative evidence from co-immunoprecipitation has suggested that the IL-36R interacts with the IL-1RAcP even in the absence of cytokines (12). Interestingly, others have reported a high basal activation level in a cell line co-transfected with IL-36R and IL-1RAcP (1). We believe that this high basal activation level could be due to heterodimerization of IL-36R and IL-1RAcP induced by high expression. Nevertheless, both co-immunoprecipitation and co-transfection studies suggest that IL-36R and IL-1RAcP interact with each other reversibly. However, detection of the binary complex under our SPR experimental conditions is difficult, because these interactions are rather weak. Our measurements provide an estimation of KD that is larger than 1 μm for IL-36R·IL-1RAcP interactions. Binding of IL-1RAcP to IL-36R in the presence of IL-36α can be measured accurately. The affinity is 42 ± 10 nm, which is at least a 24-fold increase from IL-36R·IL-1RAcP interactions without an agonist. IL-36α binds to IL-36R·IL-1RAcP heterodimer with a KD of 0.27 nm. The affinity of IL-36α·IL-36R is 480 nm. Through a complete thermodynamic cycle, we predict the affinity of IL-36R and IL-1RAcP to be 74 μm, which explains why we could not detect the binding at 1 μm. Enhancement of IL-1RAcP binding in the presence of IL-36α also suggests there is interaction between IL-1RAcP and IL-36α, although it is too weak to measure (Fig. 5B). Therefore, we estimated the KD for the interactions between IL-1RAcP and IL-36α to be larger than 60 μm based on the highest concentration tested in our SPR study (Table 1).
When both agonists and antagonist are present, IL-36Ra competes with agonists for IL-36R occupancy. We have found that IL-36Ra (KD = 5.8 ± 0.3 nm) binds to IL-36R much stronger than either IL-36α (KD = 480 ± 90 nm) or IL-36γ (KD = 1800 ± 200 nm). Higher affinity makes this natural antagonist an efficient inhibitor for IL-36R. The half-life of inhibited IL-36R is estimated to be 144 s based on dissociation rate of IL-36Ra (Equation 3). In cells, antagonistic signals are often generated after activation. When IL-36Ra is induced after formation of a stable activation ternary complex, IL-36Ra only binds to IL-36Rs that are either newly synthesized or dissociated from the ternary complex. Agonist IL-36α dissociates from heterodimer at a kd of 0.20 × 10−3 s−1, whereas IL-1RAcP dissociates from IL-36α·IL-36R at a kd that is 32-fold faster (6.3 × 10−3 s−1). It is likely that IL-1RAcP is the first component to dissociate from ternary complex. Therefore, the half-life of the activating ternary complex is estimated to be 110 s based on this dissociation rate constant of IL-1RAcP (Equation 3).
IL-36Ra has been reported to bind to Fc-linked IL-36R·IL-1RAcP heterodimer at KD values ranging from 10 to 84 nm (4, 13). We measured IL-36Ra bound to either heterodimer or IL-36R at the same KD (5.8 nm by SPR and ∼35 nm by TR–FRET). The 6-fold loss of affinity in TR–FRET may be due to insertion of the AVI tag for fluorescence labeling. Nevertheless, IL-36Ra binds to both monomer and heterodimer with similar affinity in both assays. Although it is expected that there would be no enhancement for interactions between IL-36Ra and IL-36R when IL-1RAcP is present, it is interesting to note that there was no loss of binding either. In a published IL-36R·IL-36Ra·IL-1RAcP model, steric hindrance between IL-1RAcP and loop β 11/12 on IL-36Ra is said to be the structural basis for the antagonism of IL-36Ra (4). It seems plausible that the Fc-linked IL-36R·IL-1RAcP still contains enough flexibility to allow for IL-36Ra binding.
Both IL-36R and IL-1RAcP contain ECD and transmembrane and intracellular Toll-IL-1 receptor domains. Based on what has been reported, other domains likely do not play an important role in ligand recognition (4). However, soluble ECD of receptor in solution may behave differently than membrane bound form. Membrane-bound receptor also has one less dimension for rotation than receptors in solution (18). This reduction of dimension in membrane bound receptors may also contribute to an increase in binding affinity. In SPR studies, receptors are tethered onto a chip surface, which may have helped to bridge the gap between membrane-bound measurement and solution-based measurement. Indeed, agonist IL-36γ binds strongly to heterodimer but not the monomer in either soluble ECD or membrane-bound form (Figs. 5A and 7B). In this study, receptor ECD was used consistently to compare agonist and antagonist binding. This interpretation of activation and inhibition should be comparable with studies using membrane-bound receptors.
An interesting observation we made from literature is that it takes much less IL-1β than IL-36α to activate reporter genes. Even more IL-36γ is needed to activate reporter signal when compared with IL-36α (1). We believe the observed difference in activation potency may be due to differences in binding affinity between cytokines and their corresponding cognate receptors. For example, IL-1β binds to IL-1 receptor at a KD of ∼1 nm (19), whereas IL-36α binds to IL-36R at a KD of 480 ± 90 nm, and IL-36γ binds to IL-36R at an even lower affinity of 1800 ± 200 nm.
This study provides kinetic insight into binding events associated with IL-36. Future studies can be directed to characterize other members of the IL-1 family. IL-1RAcP is a shared subunit among IL-1, IL-33, and IL-36 (2, 3). With a finite number of IL-1RAcP, cells may also regulate the IL-36 pathway through sequestering and releasing the IL-1RAcP from the other IL-1 receptors. Kinetics for binding and spatial distribution of IL-1RAcP could help us to understand the signaling dynamics of IL-1 family members, which carry out important biological functions such as innate immunity and inflammation (20).
Experimental procedures
Protein expression and purification
ECDs of both IL-36R (1–335; UniProt Q9HB29) and IL- 1RAcP (1–367; UniProt Q9NPH3) were engineered with a FLAG-His6-Avi tag at the C terminus and a TEV cleavage site for tag removal. Clones pIL-36R-Fc (hole) and pIL-1RAcP-Fc (knob)-His-Avi were created for expression of IL-36R·IL-1RAcP heterodimer. In these constructs, both ECDs were N-terminally fused to human IgG1 Fc. To promote heterodimerization, the Fc regions were mutated to create a knob-in-hole interaction (14). Specifically, the Fc for IL-1RAcP component had a T150W mutation, creating the knob, whereas the Fc for IL-36R contained mutations T150S, L152A, and Y191V, creating the hole. Protein encoding sequences were cloned into pFastBac1 for expression in Sf9 cells. Protein expression for IL-36R-FLAG-His-AVI and IL-1RAcP-FLAG-His-AVI, as well as corresponding Fc fusion clones, was done in Sf9 cells. Baculovirus was generated according to the Bac-to-Bac procedure (Life Technologies). Fermentation was performed by infecting Sf9 cells at 2.5 million/ml cell density in Sf900IISFM medium (Life Technologies). The culture was incubated for 72 h at 27 °C. The cells were removed by centrifugation, and supernatant containing the secreted protein was filtered through a Pall AcroPak 500 filtration device before being concentrated. Filtered sample was incubated with anti-FLAG M2 affinity gel (Sigma) overnight at 4 °C. Protein was eluted in PBS plus 0.2 mg/ml FLAG peptide (Sigma) and further purified by gel filtration in PBS, pH 7.4 buffer. Heterodimer was purified by nickel column followed by an IL-36α affinity column (4). Supernatant was mixed with 5 ml of nickel Sepharose resin (GE) overnight at 4 °C. The resin was washed with 30 column volumes of PBS, pH 7.4, plus 20 mm imidazole and eluted with PBS plus 300 mm imidazole. After overnight dialysis into PBS to remove imidazole, the eluted material was mixed with 10 ml of IL-36α affinity resin overnight at 4 °C. The resin was washed with 20 column volumes of PBS, and the heterodimer was eluted with successive applications of 10 ml of 0.1 m glycine, pH 3.7, followed by neutralization by adding 1 ml of 0.5 m Tris, pH 8.0, 1.5 m NaCl. Fractions containing the IL-36R heterodimer were pooled, dialyzed into PBS, pH 7.4, and concentrated to the desired concentration.
Amino acids 6–158 of IL-36α (UniProt Q9UHA7), 18–169 of human IL-36γ (NCBI accession no. NP_062564), and 2–155 of human IL-36Ra (UniProt Q9UBH0) were reverse translated, codon-optimized for Escherichia coli expression, and synthesized with an appropriate N-terminal sequence encoding a His6-SUMO tag. For FRET assay, Avi tag was inserted into IL-36α at residue 79 and into IL-36Ra at residue 73. Synthetic DNA was cloned into pET28a(+) or pET29b(+), and protein was overexpressed in E. coli BL21(DE3) (Life Technologies) by isopropyl β-d-thiogalactopyranoside induction overnight at 18–20 °C.
Columns and instruments used for purification were from GE Healthcare, and all purification steps were performed at 4 °C. Cell paste from expression of IL-36α was lysed on an EmulsiFlex C-50 homogenizer (Avestin) in 25 mm HEPES, pH 7.5, 300 mm NaCl, 5 mm MgCl2, 0.5 mm TCEP. The protein was first purified on a nickel affinity column, followed by dialysis into 25 mm HEPES, pH 7.5, 150 mm NaCl, 0.5 mm TCEP to remove the imidazole. To cleave off the His-SUMO tag, the protein was mixed with ULP1 protease overnight at 4 °C, and the IL-36α protein was separated from the cleaved tag by nickel column. IL-36α was further purified by gel filtration in 25 mm HEPES, pH 7.5, 150 mm NaCl, 2 mm DTT. Cell pastes from expression of IL-36γ and IL-36Ra were suspended in PBS, pH 7.4, and disrupted by three passages through a Microfluidizer (Microfluidics). After centrifugation, the supernatant was loaded onto a HisTrap FF column equilibrated with PBS, pH 7.4. The column was sequentially washed with PBS, PBS containing detergent, and PBS containing 25 mm imidazole. Bound protein was eluted with PBS containing 300 mm imidazole. The His6-SUMO tag was cleaved off the eluted protein with ULP1 protease while dialyzing overnight into PBS. The dialyzed sample was reapplied to HisTrap FF to separate the His-tagged ULP1 protease and the liberated His6-SUMO tag. hIL-36γ(18–169) and hIL-36Ra(2–155) proteins were further purified by SEC on Superdex 200 equilibrated and run with PBS, pH 7.4.
IL-36R and IL-36R·IL-1RAcP cell line generation and characterization
HA tag sequence was inserted into human IL-36R at N-terminal following its signal peptide and was cloned into pCMV6 vector. Full-length of human IL-1RAcP was cloned into pLVX-IRES-Puro vector. The cell lines were generated with transient transfection method followed by cell sorting. Briefly, HEK293H was maintained in hybridoma SFM (Invitrogen catalog no. 12045) with 0.1 mm HEPES, 1% FBS at 37 °C. The day before transfection, 1 million HEK293H cells were seeded into a 6-well plate. Then cells were transfected with IL-36R plasmid alone or IL-36R and IL-1RAcP plasmids 1:2 ratio with Lipofectamine 2000 according to the manufacturer's directions. Two days after transfection, the cells were put into antibiotic selection for 2 weeks: 1 mg/ml G418 for IL-36R cell line, 1 mg/ml G418, and 2 μg/ml puromycin for IL-36R·IL-1RAcP respectively. The cells were then stained with anti-HA-PE (Miltenyl Biotech catalog no. 130-092-257), and high expressors were selected for expansion, used for ligand binding assays.
Keratinocyte culture, treatment, and CXCL1 measurement
Human keratinocyte cell line HaCaT was cultured in DMEM with 10% fetal bovine serum and 1% penicillin/streptomycin added (Sigma). For IL-36–induced chemokine release experiments, the cells were seeded in 96-well tissue culture plates (Corning) at 25,000/well and allowed to adhere and recover for 24 h. The cells were then rinsed with PBS (Sigma) and treated with cytokines diluted in 200/well μl of DMEM supplemented with 0.5% FBS at concentrations indicated. After 24 h of treatment, supernatants were collected and tested for the presence of CXCL1 using a custom immunoassay (Meso Scale Discovery). EC50 were estimated from a four-parameter logistic fit (Equation 1).
SPR binding assay
Binding kinetics of cytokines, receptor IL-36R, and co-receptor IL-1RAcP were determined by surface plasmon resonance-based measurements made on SPR T100 or T200 instruments (GE Healthcare) at 25 °C using a NeutrAvidin-biotin capturing approach. Measurements were made in HBS-EP+ assay buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm EDTA, 0.05% Tween 20). For example, ∼10,000 RU of NeutrAvidin (Thermo Fisher Scientific) diluted at 25 μg/ml in 10 mm sodium acetate, pH 5.5, was directly immobilized across a CM5 chip using a standard amine coupling kit according to the manufacturer's instructions. Unreacted moieties were blocked with ethanolamine. IL-36R and IL-1RAcP were biotinylated through AVI tag according to manufacturer's protocol (Avidity). Labeled samples were further purified by SEC in HBS-N buffer (10 mm HEPES, pH 7.4, 150 mm NaCl) before being captured onto a NeutrAvidin chip at 6000–8000 RU. Ligands were captured at such high density because active chip surface was low. Various concentrations of analyte in 3-fold dilutions were injected onto chip surface at a flow rate of 80 μl/min, for example, IL-36α (900 - 4 nm), IL-36γ (2700 - 33 nm), and IL-36Ra (900 - 4 nm). In some studies where we did not observe binding, higher concentrations of analytes were tested. For example, ∼60 μm of IL-36α/γ were tested to bind IL-1RAcP; ∼1 μm of IL-36R and IL-1RAcP were tested to probe interactions with each other. Various concentrations of IL-1RAcP (653 - 41 nm, 3× dilutions) and a fixed concentration of IL-36α (4 μm) were injected simultaneously onto a chip with IL-36R captured for binary complex binding study. Analytes were injected for 90 s, and dissociation was measured for 600 s. No regeneration was needed. During the assay, all measurements were double referenced against surface alone and buffer-only injections. Association and dissociation rate constants, ka (m−1 s−1) and kd (s−1) were derived by fitting SPR sensorgram simultaneously and globally to an equation, which was derived from the 1:1 Langmuir binding model with a mass transfer term included. Equilibrium dissociation constant KD (m) was either calculated from ratio of kd and ka or through steady-state fitting using 1:1 model.
To measure heterodimer binding, ∼400 RU of heterodimer was captured onto an anti-hu Fc chip. Approximately 2000 RU of anti-hu Fc (Thermo Fisher Scientific) diluted at 15 μg/ml in 10 mm sodium acetate, pH 4.5, was directly immobilized through amine-coupling onto a CM5 chip. Assay buffer was HBS-EP+. Various concentrations of analyte in 3-fold dilutions were injected onto chip surface at a flow rate of 80 μl/min, for example, IL-36α (44 - 1 nm), IL-36γ (900 - 1 nm), and IL-36Ra (900 - 4 nm). After each cycle the chip surface was regenerated using 10 mm glycine-HCl, pH 1.5.
TR–FRET assay
Biotinylated IL-36α and IL-36Ra were conjugated overnight at 4 °C to streptavidin-Alexa Fluor 488 according to the manufacturers' instructions (Life Technologies). Impurities were removed, and conjugated protein was transferred to 1× PBS, pH 7.4 (Sigma), using Zeba Spin 7K desalting columns (Thermo Scientific). TR–FRET assay was run in low-volume 384-well Proxy Plates (PerkinElmer Life Sciences). 5 μl of assay buffer (1% BSA in 1× PBS), 5 μl of either IL-36R·IL-1RAcP knob-in-hole or IL-36R monomer His6-tagged protein (1.5 μg/ml final concentration) in assay buffer and 5 ml of detection solution (6 nm final concentration of LanthaScreen Elite Tb-anti-His Antibody (Thermo Scientific) together with serial dilutions of streptavidin-Alexa Fluor 488–conjugated IL-36α or IL-36Ra at concentrations indicated) were added in individual wells. The plates were sealed and incubated overnight at 4 °C. After incubation, the plates were brought back to room temperature, and the ratio between fluorescent signals at 495 and 520 nm (TR–FRET ratio) was read using the TR–FRET enabled EnVision plate reader (PerkinElmer Life Sciences). KD was obtained by fitting normalized TR–FRET ratio to titrant concentration using Equation 2.
Glyco-FRET assay
The assay was performed as described elsewhere (17). Briefly, IL-36γ ligand was labeled using Alexa Fluor 488 N-hydroxysuccinimidyl ester (Molecular Probes) and purified into PBS, pH 7.4, using Zeba Spin 7K desalting columns (Thermo Scientific). IL-36R and IL-36R·IL-1RAcP stably transfected HEK293 cells were incubated for 48 h in medium containing 50 μm Ac4ManNAz. The cells were washed with Dulbecco's PBS (Invitrogen), dissociated and suspended in labeling buffer (1% FBS in Dulbecco's PBS) at 1 × 107 cells/ml. The cells were then incubated with the ADIBO-Terbium chelate at a concentration of 5 μm for 1 h on ice. Following wash, the cells were suspended in labeling buffer at 1 × 107 cells/ml. Next, the cells were aliquoted into a 384-well Opti plates (PerkinElmer Life Sciences) at 5 × 104 cells/well. 5 μl of ligand was added, and the plate was incubated on ice for 2 h.
The FRET signal was measured with an EnVision plate reader (PerkinElmer Life Sciences) as described above for the TR–FRET assay. EC50 values were calculated using Equation 1 in Prism software (GraphPad),
| (Eq. 1) |
where Max and Min indicate the maximum and minimum CXCL1 concentrations respectively; X is the titrant concentration; and Y is the measured CXCL1 concentration.
| (Eq. 2) |
In Equation 2, Bmax is the maximum magnitude of normalized FRET ratio, Kd is the dissociation constant, NS is the linear slope of nonspecific binding, X is the titrant concentration, and Y is the measured normalized FRET ratio.
| (Eq. 3) |
In Equation 3, t½ is the half-life, and kd is the dissociation rate constant.
Author contributions
L. Z. conceived the study, conducted SPR binding measurements, analyzed the results, and drafted the paper. V. T. validated IL-36 cytokines in CXCL1 release assay, developed TR–FRET assay with S. K., H. S., and P. L. R., and participated in manuscript writing. S. K. designed BEV constructs and purified receptors and IL-36α. L. W. and R. S. were responsible for designing and expression of E. coli. IL-36γ and IL-36Ra and cell line generation for IL-36R and IL-36R·IL-1RAcP. B. S. and L. M. purified IL-36γ and IL-36Ra. E. D. and C. S. coordinated resources and participated in construct designs. V. S. coordinated resources and participated in manuscript writing.
Acknowledgment
We thank Dr. W. Blaine Stine for reviewing the manuscript.
This work was supported by AbbVie Inc. All authors are employees of AbbVie. The design, study conduct, and financial support for this research were provided by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the publication.
- IL-36Ra
- IL-36 receptor antagonist
- IL-36R
- IL-36 receptor
- SPR
- surface plasmon resonance
- Fc
- fragment crystallizable
- IL-1R
- IL-1 receptor
- TR
- time-resolved
- ECD
- extracellular domain
- SEC
- size exclusion chromotography
- RU
- response units.
References
- 1. Towne J. E., Garka K. E., Renshaw B. R., Virca G. D., and Sims J. E. (2004) Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-κB and MAPKs. J. Biol. Chem. 279, 13677–13688 10.1074/jbc.M400117200 [DOI] [PubMed] [Google Scholar]
- 2. Garlanda C., Dinarello C. A., and Mantovani A. (2013) The interleukin-1 family: back to the future. Immunity 39, 1003–1018 10.1016/j.immuni.2013.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sims J. E., and Smith D. E. (2010) The IL-1 family: regulators of immunity. Nat. Rev. Immunol 10, 89–102 [DOI] [PubMed] [Google Scholar]
- 4. Günther S., and Sundberg E. J. (2014) Molecular determinants of agonist and antagonist signaling through the IL-36 receptor. J. Immunol. 193, 921–930 10.4049/jimmunol.1400538 [DOI] [PubMed] [Google Scholar]
- 5. Marrakchi S., Guigue P., Renshaw B. R., Puel A., Pei X.-Y., Fraitag S., Zribi J., Bal E., Cluzeau C., Chrabieh M., Towne J. E., Douangpanya J., Pons C., Mansour S., Serre V., et al. (2011) Interleukin-36–receptor antagonist deficiency and generalized pustular psoriasis. N. Engl. J. Med. 365, 620–628 10.1056/NEJMoa1013068 [DOI] [PubMed] [Google Scholar]
- 6. D'Erme A. M., Wilsmann-Theis D., Wagenpfeil J., Hölzel M., Ferring-Schmitt S., Sternberg S., Wittmann M., Peters B., Bosio A., Bieber T., and Wenzel J. (2015) IL-36γ (IL-1F9) is a biomarker for psoriasis skin lesions. J. Invest. Dermatol. 135, 1025–1032 10.1038/jid.2014.532 [DOI] [PubMed] [Google Scholar]
- 7. Gabay C., and Towne J. E. (2015) Regulation and function of interleukin-36 cytokines in homeostasis and pathological conditions. J. Leukoc. Biol. 97, 645–652 10.1189/jlb.3RI1014-495R [DOI] [PubMed] [Google Scholar]
- 8. Dietrich D., and Gabay C. (2014) Inflammation: IL-36 has proinflammatory effects in skin but not in joints. Nat. Rev. Rheumatol. 10, 639–640 10.1038/nrrheum.2014.156 [DOI] [PubMed] [Google Scholar]
- 9. Wang D., Zhang S., Li L., Liu X., Mei K., and Wang X. (2010) Structural insights into the assembly and activation of IL-1β with its receptors. Nat. Immunol. 11, 905–911 10.1038/ni.1925 [DOI] [PubMed] [Google Scholar]
- 10. Sims J. E. (2010) Accessory to inflammation. Nat. Immunol. 11, 883–885 10.1038/ni1010-883 [DOI] [PubMed] [Google Scholar]
- 11. O'Neill L. A. (2008) The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 226, 10–18 10.1111/j.1600-065X.2008.00701.x [DOI] [PubMed] [Google Scholar]
- 12. Yi G., Ybe J. A., Saha S. S., Caviness G., Raymond E., Ganesan R., Mbow M. L., and Kao C. C. (2016) Structural and functional attributes of the interleukin-36 receptor. J. Biol. Chem. 291, 16597–16609 10.1074/jbc.M116.723064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Towne J. E., Renshaw B. R., Douangpanya J., Lipsky B. P., Shen M., Gabel C. A., and Sims J. E. (2011) Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J. Biol. Chem. 286, 42594–42602 10.1074/jbc.M111.267922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gunasekaran K., Pentony M., Shen M., Garrett L., Forte C., Woodward A., Ng S. B., Born T., Retter M., Manchulenko K., Sweet H., Foltz I. N., Wittekind M., and Yan W. (2010) Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J. Biol. Chem. 285, 19637–19646 10.1074/jbc.M110.117382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Foster A. M., Baliwag J., Chen C. S., Guzman A. M., Stoll S. W., Gudjonsson J. E., Ward N. L., and Johnston A. (2014) IL-36 promotes myeloid cell infiltration, activation, and inflammatory activity in skin. J. Immunol. 192, 6053–6061 10.4049/jimmunol.1301481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carrier Y., Ma H.-L., Ramon H. E., Napierata L., Small C., O'Toole M., Young D. A., Fouser L. A., Nickerson-Nutter C., Collins M., Dunussi-Joannopoulos K., and Medley Q. G. (2011) Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J. Invest. Dermatol. 131, 2428–2437 10.1038/jid.2011.234 [DOI] [PubMed] [Google Scholar]
- 17. Stockmann H., Todorovic V., Richardson P. L., Marin V., Scott V., Gerstein C., Lake M., Wang L., Sadhukhan R., and Vasudevan A. (2017) Cell-surface receptor-ligand interaction analysis with homogenous time-resolved FRET and metabolic glycan engineering: application to transmembrane and GPI-anchored receptors. J. Am. Chem. Soc. 139, 16822–16829 10.1021/jacs.7b09359 [DOI] [PubMed] [Google Scholar]
- 18. Wu Y., Vendome J., Shapiro L., Ben-Shaul A., and Honig B. (2011) Transforming binding affinities from three dimensions to two with application to cadherin clustering. Nature 475, 510–513 10.1038/nature10183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jobling S. A., Auron P. E., Gurka G., Webb A. C., McDonald B., Rosenwasser L. J., and Gehrke L. (1988) Biological activity and receptor binding of human prointerleukin-1β and subpeptides. J. Biol. Chem. 263, 16372–16378 [PubMed] [Google Scholar]
- 20. O'Neill L. A. J. (2000) The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Science's STKE 2000, re1 [DOI] [PubMed] [Google Scholar]