

Abstract
As a sensitive signaling system, the mitotic checkpoint ensures faithful chromosome segregation by delaying anaphase onset even when a single kinetochore is unattached to mitotic spindle microtubules. The key signal amplification reaction for the checkpoint is the conformational conversion of “open” mitotic arrest deficient 2 (O-MAD2) into “closed” MAD2 (C-MAD2). The reaction has been suggested to be catalyzed by an unusual catalyst, a MAD1:C-MAD2 tetramer, but how the catalysis is executed and regulated remains elusive. Here, we report that in addition to the well-characterized middle region of MAD1 containing the MAD2-interaction motif (MIM), both N- and C-terminal domains (NTD and CTD) of MAD1 also contribute to mitotic checkpoint signaling. Unlike the MIM, which stably associated only with C-MAD2, the NTD and CTD in MAD1 surprisingly bound both O- and C-MAD2, suggesting that these two domains interact with both substrates and products of the O-to-C conversion. MAD1NTD and MAD1CTD also interacted with each other and with the MPS1 protein kinase, which phosphorylated both NTD and CTD. This phosphorylation decreased the NTD:CTD interaction and also CTD's interaction with MPS1. Of note, mutating the phosphorylation sites in the MAD1CTD, including Thr-716, compromised MAD2 binding and the checkpoint responses. We further noted that Ser-610 and Tyr-634 also contribute to the mitotic checkpoint signaling. Our results have uncovered that the MAD1NTD and MAD1CTD directly interact with each other and with MAD2 conformers and are regulated by MPS1 kinase, providing critical insights into mitotic checkpoint signaling.
Keywords: MAD1, MAD2, mitotic checkpoint, MPS1 kinase, mitosis, signal transduction, kinetochore, mitotic spindle, checkpoint control
Introduction
The mitotic checkpoint is a crucial signal transduction pathway that contributes to faithful chromosome segregation (1–4). A single unattached kinetochore delays anaphase onset, underscoring the importance of signal amplification for the mitotic checkpoint (5). The conversion of mitotic arrest deficient 2 (MAD2)2 domain from open (O-MAD2) to closed (C-MAD2) conformation is a well-recognized signal amplification mechanism for the mitotic checkpoint (6, 7). O-MAD2 is the predominant conformer in interphase cells (8, 9). During prometaphase, intracellular C-MAD2 concentration is increased to promote formation of the mitotic checkpoint complex (MCC), which binds and inhibits the anaphase promoting complex/cyclosome (APC/C) (1–4). In the current model, the MAD2 O–C conversion (i.e. O to C conversion) is catalyzed by a 2:2 MAD1:C-MAD2 tetramer localized at unattached kinetochores. Cytoplasmic O-MAD2 then heterodimerizes with the C-MAD2 moiety in the catalyst and morphs into C-MAD2. The reaction mechanism for the conversion is still unclear, but it may involve some intermediate folding states (I-MAD2) (6, 7, 10).
Two major questions remain unanswered for the model. First, human MAD1 is a mitotic checkpoint protein of 718 amino acid residues, but the formation of a 2:2 heterotetramer with C-MAD2 involves only its MAD2 interaction motif (MIM, 485–584 residues) especially a disordered loop spanning 530–550 residues (Fig. 1a) (11). However, some earlier and recent experiments argued for functional importance of the C-terminal domain of MAD1 (585–718 residues, MAD1CTD) in maintaining the mitotic checkpoint (12–18). Moreover, even the MAD1 fragment encompassing 485–718 residues only exhibits low catalytic activity for MAD2 O–C conversion in vitro (12, 13, 19). The N-terminal domain of MAD1 (1–485 residues, MAD1NTD) was thought to target the protein to nuclear envelope or kinetochores but may also interact with other proteins (20–23). Whether and how MAD1 domains outside MIM contribute to the checkpoint signaling warrants a revisit and careful investigation. Second, MAD1 forms a cell cycle independent complex with C-MAD2; how the complex only becomes an effective catalyst during prometaphase needs to be better defined (18, 24). Several kinetochore-localized mitotic kinases, including MPS1 kinase, were known to elevate C-MAD2 production, but direct biochemical evidence is still incomplete despite exciting recent progress (25–31).
Figure 1.

Both MAD1NTD and MAD1CTD are required for MAD1 activity. a, diagram of human MAD1 showing that multiple segments of MAD1 may form structures (ovals) other than coiled coils (shaded area). b, shown are various MAD1 mutants and truncations used for fusion with mCherry-Mis12. c, images of live cells either transfected with GFP-MAD2L13A alone or together with mCherry-Mis12-WT MAD1. DIC, differential interference contrast. Scale bar, 10 μm. d, mitotic durations of HeLa cells either untransfected (UN) or transfected with mCherry-Mis12 fused with MAD1-WT, AA (defective in MAD2 binding because of mutations in MIM), ΔNTD, ΔCTD, Y634E, or Y634F were shown. Cell numbers imaged for each construct were listed on the right. e, lysates from control (UN) or transfected cells were subjected to anti-Mis12 immunoprecipitation (IP) followed by Western blots for Mis12-MAD1 fusions (probed with anti-Mis12 antibody), MAD2, and endogenous MAD1. Molecular weight markers are marked on the left (in kDa). The arrow indicates the position of endogenous MAD1. Although not always run as a full panel with all the mutants included, any parts of the results have been replicated at least three times by each of three investigators. f, lysates from control (UN) or FLAG-MAD1 transfected cells were subjected to anti-FLAG immunoprecipitation and then probed for MAD1 and MAD2. The asterisk indicates IgG heavy chain.
Here we report our results targeting the above two questions. While our manuscript was prepared, two reports were published indicating that MPS1 phosphorylates MAD1 to enhance MAD2 O–C conversion and MCC assembly (29, 30). Our results support the importance of MAD1 Thr-716 phosphorylation, but we have also uncovered other protein-protein interactions between MAD1, MAD2, and MPS1 and the phosphorylation-dependent regulation of some of the interactions. Our work highlights the coordination of different MAD1 domains in efficient mitotic checkpoint signaling and provides further mechanistic insights into the MAD2 O–C conversion reaction.
Results
MAD1 N-terminal and C-terminal domains (NTD and CTD) are required for efficient mitotic checkpoint signaling
In studying the MAD2 O–C conversion, earlier work has detailed the conformational changes of MAD2 (6, 32). We reasoned that better characterization of the MAD1:C-MAD2 catalyst would provide further mechanistic insights into the conversion reaction and hence the signal amplification step of the mitotic checkpoint. We noted that even though MAD1 is commonly depicted as a rigid coiled-coil protein, parts of its NTD and CTD have been shown or predicted to remain disordered or adopt other structures (Fig. 1a and Fig. S1) (11, 33, 34). We first investigated the possible contribution of MAD1NTD and MAD1CTD to the mitotic checkpoint using a “separation of function” system developed by Maldonado and Kapoor (26). In this system, an mCherry-Mis12-MAD1 fusion construct was exploited to examine catalytic efficiency of the MAD1:C-MAD2 catalyst without concerns over the kinetochore targeting aspect of its regulation (26) (Fig. 1b). Although endogenous MAD1 and MAD2 disappeared from metaphase kinetochores which presumably were occupied by spindle microtubules, expression of wild-type MAD1 (MAD1WT) fused with constitutive kinetochore protein Mis12 retained MAD1 at the metaphase plate and recruited GFP-MAD2L13A to these metaphase kinetochores (26, 28) (Fig. 1c and Fig. S2). MAD2L13A is a MAD2 mutant locked in C conformation (13, 32). The persistence of MAD1 and MAD2 at metaphase attached kinetochores was sufficient to trigger a >12-h mitotic arrest in HeLa cells (26, 28) (Fig. 1d). The arrest was dependent on C-MAD2 binding to MAD1, as cells expressing the fusion with MAD2-binding–deficient MAD1AA mutant (K541A, L543A in MIM) finished mitosis within ∼60 min on average (Fig. 1d and Fig. S2) (26). Note no GFP-MAD2L13A was localized at metaphase kinetochores containing mCherry-Mis12-MAD1AA, although GFP-MAD2L13A did appear at the last few unattached kinetochores, most likely because of presence of endogenous MAD1 there (Fig. S2, compare the second and third columns). Furthermore, co-expression of MAD2ΔC10, an O-conformer locked mutant of MAD2 (6, 7), abolished the mitotic arrest in MAD1WT transfected cells (data not shown), corroborating that the arrest was because of O–C conversion–dependent checkpoint responses (28, 35, 36).
Consistent with previous reports (14–17), MAD1 missing 597–718 residues (MAD1ΔCTD), even as a fusion with Mis12, could not maintain mitotic arrest (98 ± 7 versus 749 ± 22 min for MAD1WT, mean ± S.D., p < 0.0001, Student's t test, there might be an underestimation for MAD1WT transfected cells as the movies lasted only 13 h). Moreover, a specific MAD1Y634E mutant also abolished the mitotic arrest, whereas a MAD1Y634F mutant did not significantly impact mitotic duration (Fig. 1d). Tyr-634 is situated close to the junction between the coiled-coil subdomain (597–637 residues) and the globular subdomain (638–718 residues) of the MAD1CTD (33). We noticed this site during screening potential MAD1 phosphomutants as Tyr-634 was reported to be phosphorylated in vivo (37). Interestingly, MAD1 missing 1–485 residues (MAD1ΔNTD) could not maintain prolonged mitosis either (average duration, 101 ± 22 min) (Fig. 1d).
We then examined the association of different mCherry-Mis12-MAD1 fusion proteins with endogenous MAD1 and MAD2 by Mis12 immunoprecipitation. Little MAD2 was found to associate with MAD1AA as predicted; steady-state levels of MAD2 binding to other MAD1 mutants were also slightly reduced (Fig. 1e). The reduction of MAD2 binding became more obvious for some mutants when FLAG immunoprecipitation was performed using cells transfected with FLAG-tagged MAD1 constructs (Fig. 1f). In addition, MAD1ΔCTD and MAD1ΔNTD did not interact with endogenous MAD1, indicating a dimerization defect (Fig. 1, e and f; note that mCherry-Mis12-MAD1ΔNTD runs at the same position as endogenous MAD1. More on MAD1 dimerization in the last section of “Results”). Taken together, the results shown in Fig. 1 have confirmed the important role of MAD1MIM, but also revealed that both NTD and CTD of MAD1 are required for an efficient mitotic checkpoint.
MAD1NTD and MAD1CTD bind to both O-MAD2 and C-MAD2
We hypothesized that the NTD and CTD of MAD1 facilitate mitotic checkpoint responses by enhancing MAD2 O–C conversion. Based on the analogy to an isomerase which at least transiently interacts with its substrate and product, we prepared recombinant proteins and examined potential interactions between GST-tagged MAD1NTD or MAD1CTD with untagged O- or C-MAD2, supplied as MAD2ΔC10 or MAD2L13A conformation-locked mutants, respectively (6, 7, 36) (Fig. 2a). When proteins were used at roughly endogenous concentrations (see “Experimental Procedures”), GST-pulldown results showed that NTD and CTD bound to both conformers of MAD2 (Fig. 2b, lanes 1, 3 and 5, 7). Under similar conditions, the MAD1MIM only bound to C-MAD2, just as reported previously (compare MAD2 in Fig. 2b, lanes 2 and 6) (11, 38). The conformational status of the O- and C-MAD2 mutants was further verified, because only MAD2L13A bound to GST-CDC20(111–138) or GST-BUBR1(1–371), also as reported before (36, 38) (Fig. S3). The novel interactions were not mediated by tags, as GST alone did not pull down any MAD2 (Fig. 2b, lanes 4 and 8). Importantly, GFP-MAD2ΔC10 was found to be recruited to centromeres in interphase cells expressing mCherry-Mis12-MAD1 fusions, supporting the idea that the interaction between O-MAD2 and MAD1, although surprising, could happen in cells (Fig. 2c, MAD1ΔCTD used here). Maintaining MAD1 fragments at the endogenous concentration of 60 nm, titrating GST pulldown experiments found that binding of MAD2 to NTD, MIM, and CTD could be detected when MAD2 concentrations were as low as 30 nm. Estimations of the half-maximal binding concentrations of MAD1CTD with C-MAD2 and O-MAD2 fell in the range of 100–250 nm (Fig. S3).
Figure 2.

MAD1NTD and MAD1CTD interact with both O-MAD2 and C-MAD2. a, Coomassie Blue stain after SDS-PAGE of purified recombinant proteins. GST-NTD tends to be more labile for degradation. The asterisk marks expected size of GST-NTD. b, GST-tagged MAD1NTD, MIM, CTD, or GST alone were incubated with either MAD2L13A or MAD2ΔC10, and GST pulldown assays were followed by Western blotting of GST and MAD2. The final concentrations of GST or GST-tagged MAD1 fragments were 60 nm, whereas those of MAD2 mutants were 230 nm. Similar results have been replicated for at least five times by two investigators, using different batches of protein preparations. c, immunofluorescence of two interphase cells transfected with GFP-MAD2ΔC10. Note that the GFP signals are recruited to centromeres only in the cell co-expressing mCherry-Mis12-MAD1ΔCTD. Centromeres are stained with anti-CENP-I antibody. Scale bar, 10 μm. d, GST-tagged MAD1 N-terminal truncations were incubated with MAD2ΔC10 or MAD2L13A followed by GST pulldown assays. The result is representative of four replicates. e, GST-tagged MAD1CTD coiled-coil subdomain (597–638 residues) or globular subdomain (638–718 residues) were incubated with MAD2ΔC10 or MAD2L13A followed by GST pulldown and Western blotting. The result is representative of three replicates by two investigators based on different batches of proteins.
We attempted to further define the regions on MAD1NTD or MAD1CTD responsible for association with MAD2. Several MAD1NTD or MAD1CTD truncations produced either insoluble or heavily degraded proteins (data not shown). However, testing with MAD1NTD truncations that we were able to purify, including MAD1(1–327), MAD1(327–423), and MAD1(327–488), revealed dramatically reduced MAD2-binding capability of these fragments (Fig. 2d). The globular subdomain (638–718 residues) of MAD1CTD retained approximately half the MAD2-binding capacity of MAD1CTD whereas the coiled-coil subdomain in the CTD showed only residual binding (Fig. 2e). These combined results suggest that the integrity of MAD1NTD or MAD1CTD is crucial for binding to O-MAD2 or C-MAD2.
A novel interface in MAD2 is employed for its association with NTD or CTD of MAD1
A MAD2 molecule has two well-characterized interfaces for protein-protein interactions: the “safety belt” characteristic of C-MAD2 conformation to which MAD1MIM and CDC20 bind (11, 38–40), and the dimerization domain (primarily αC helix) that allows MAD2 to form O:C or C:C dimers or interact with p31comet or BUBR1 (13, 32, 36, 41, 42). NTD and CTD of MAD1 bound to both MAD2ΔC10 and MAD2L13A, suggesting no discrimination against either MAD2 conformation (Fig. 2b). To further support this notion, wild-type MAD2, which exists as a mixture of both O and C conformers (8), bound to all three MAD1 fragments, whereas another MAD2 mutant MAD2S195D, predominantly in O conformation (43), bound to MAD1NTD or MAD1CTD but not MAD1MIM (Fig. 3a, compare the left and middle panels). On the other hand, MAD2LARQ (L13A/R133E/Q134A), a dimerization defective C-MAD2 mutant (36), associated with MAD1NTD or MAD1CTD at similar levels as MAD2L13A (Fig. 3b). MAD2ΔN10, which might mimic an intermediate conformation during O to C MAD2 conversion (10), bound to all three MAD1 fragments (Fig. 3c). The above results suggest that the safety belt (or C conformation), the dimerization domain (at least the RQ mutant), or the N-terminal 10 amino acids are not essential for MAD2 to interact with MAD1NTD or MAD1CTD. Therefore, a novel interface on MAD2, possibly shared by both O and C conformers, is employed for interactions with MAD1NTD and MAD1CTD.
Figure 3.

A novel interface in MAD2 is employed for its association with NTD or CTD of MAD1. a, GST-pulldown assays using GST-tagged MAD1 fragments after incubation with His6-tagged wild type MAD2 (MAD2WT) or MAD2S195D mutant. b, GST-pulldown assays using GST-tagged MAD1 fragments with MAD2L13A and MAD2LARQ. The asterisk marks expected size of GST-NTD. The result is representative of four replicates. c, GST-pulldown assays using GST-tagged MAD1 fragments with MAD2ΔN10.
MPS1 kinase phosphorylates MAD1NTD and MAD1CTD
It is known that the kinase activity of MPS1 is essential for maintaining mitotic arrest even when MAD1 constitutively localizes at kinetochores as a fusion with mCherry-Mis12 (25–28). We hypothesized that MPS1 might phosphorylate MAD1 or MAD2 to regulate the efficiency of the MAD1:C-MAD2 catalyst. In vitro kinase assays found that MPS1 indeed phosphorylated MAD1NTD and MAD1CTD (Fig. 4a, compare lanes 3 and 5 in both the Coomassie Blue stain panel and the autoradiography panel). The specificity of the kinase assay was validated by the fact that reversine, a previously characterized MPS1 inhibitor (44), reduced in vitro phosphorylation of an artificial MPS1 substrate myelin basic protein (Fig. 4a, compare lanes 1 and 2 in the right panel). GST alone, GST-MAD1MIM, and MAD2 conformers (MAD2L13A and MAD2ΔC10) were not good substrates for MPS1 under the experimental condition (Fig. 4a, lanes 4 and 6-8). Interestingly, recombinant MPS1 kinase binds to MAD1NTD or MAD1CTD but not MIM in the absence of ATP (Fig. 4b, note the presence of MPS1 signal in lanes 1 and 3 but not in lanes 2 and 4).
Figure 4.

MPS1 phosphorylates MAD1 and interacts with MAD1 and MAD2. a, in vitro kinase assays were performed using recombinant GST-MPS1, with myelin basic protein as an artificial substrate of MPS1, or with GST-MAD1NTD, MAD1MIM, MAD1CTD, untagged MAD2L13A, or MAD2ΔC10. The SDS-PAGE gel of the kinase assays were stained with Coomassie Blue (left). Phosphorylation of the proteins by MPS1 was detected by autoradiography after the gel was dried (right). The asterisks indicate the expected sizes of corresponding proteins. The arrow indicates GST-MPS1. Reversine (Rev) was used in lane 2 to validate the kinase specificity. b, GST-pulldown assays using GST or GST-tagged MAD1 fragments (∼80 nm final concentration) after incubation with His-tagged MPS1 (∼40 nm). The binding reactions did not contain ATP. The result is representative of at least three replicates by two investigators. c, GST-pulldown assays using GST or GST-tagged MPS1 (∼60 nm) after incubation with untagged MAD2ΔC10 or MAD2L13A (both at 230 nm final concentrations). The binding reactions did not contain ATP. The result is representative of at least three replicates by two investigators. d and e, immunoprecipitation using anti-His6 antibody (1 μg) to detect His-tagged MAD1CTD interactions with untagged MAD2L13A (d) or MAD2ΔC10) (e) after incubation in the presence/absence of GST-MPS1 kinase, ATP, or MPS1 inhibitor reversine. All recombinant proteins in the incubation reactions were used at final concentrations equivalent to their endogenous levels. The results have been replicated five times.
We then tested whether MPS1 phosphorylation affects the interactions between MAD1CTD and MAD2 conformers. We focused on CTD because it was not only easily purified without apparent degradation but also showed functional significance (see below). We did notice that GST-MPS1 but not GST alone could also bind to MAD2L13A and MAD2ΔC10 (Fig. 4c). In vitro incubation of MPS1, CTD, and MAD2 led to phosphorylation of CTD, as evidenced by the mobility shift detected by anti-His6 antibody, and positive signals of phospho-Thr antibody (Fig. 4, d and e, lane 1). However, the association of either MAD2L13A or MAD2ΔC10 with CTD did not show obvious changes when compared with reactions in the absence of ATP or in the presence of MPS1 inhibitor reversine (Fig. 4, d and e, compare MAD2 signals in lanes 1–3). The interactions between phosphorylated CTD and MPS1, nevertheless, became weaker (Fig. 4, d and e, compare MPS1 signals in lanes 1–3). ATP, reversine, and DMSO (solvent control) did not affect the interactions between CTD and MAD2 conformers (Fig. 4, d and e, lanes 4–6).
Functional characterization of MPS1 phosphorylation sites on MAD1
Realizing some limitations of in vitro binding experiments especially with protein fragments, we decided to directly test the potential effects of MPS1 phosphorylation on MAD1 in cells. Through mass spectrometry we identified eight in vitro MPS1 phosphorylation sites on MAD1NTD and MAD1CTD. The sites are Thr-8, Ser-22, Ser-62, Thr-323, Ser-598, Ser-610, Thr-624, and Thr-716 (Fig. 5a). To test the importance of these sites in cells, a mCherry-Mis12-MAD18A mutant with all the sites mutated into alanine was expressed and found to be defective in maintaining the mitotic arrest (229 ± 23 min mitotic duration) (Fig. 5b). Mutating all four NTD sites into alanine showed no effect, but alanine mutants at the four CTD sites (MAD1CTD4A) shortened the mitotic duration comparable with MAD18A (Fig. 5b). Further tests found that little CTD4A is phosphorylated when compared with wild-type CTD (CTDWT) in vitro, supporting that the CTD four sites, Ser-598, Ser-610, Thr-624 and Thr-716, constituted primary phosphorylation sites of MPS1 kinase under our in vitro experimental conditions (Fig. S4a).
Figure 5.
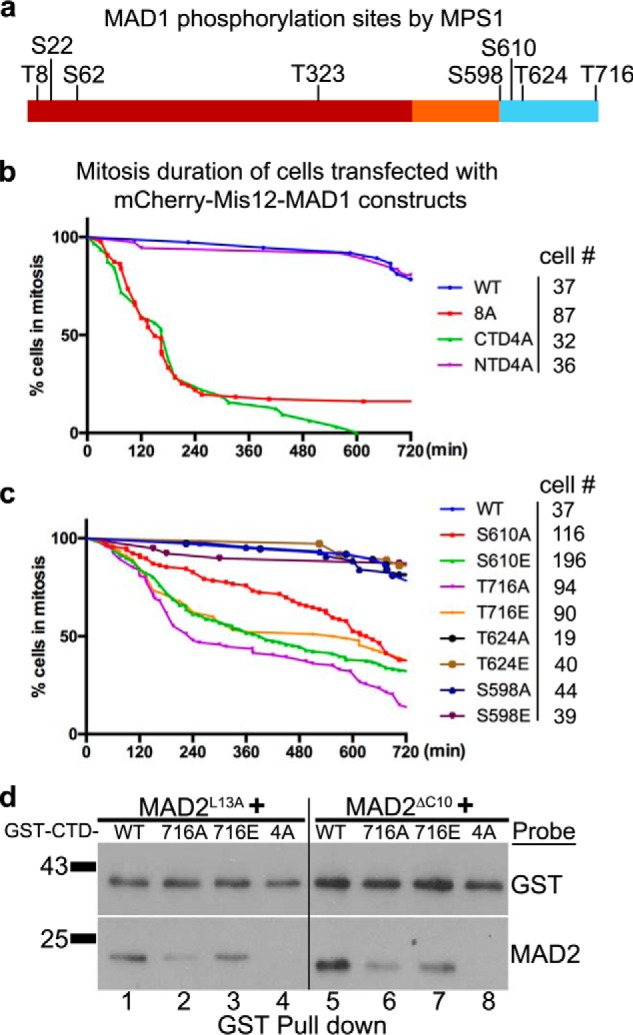
The putative MPS1 phosphorylation sites on MAD1CTD are required for the mitotic checkpoint. a, MAD1 phosphorylation sites by MPS1 kinase in vitro as determined by mass spectrometry. b, mitotic durations of HeLa cells transfected with mCherry-Mis12 fused with MAD1WT, MAD18A (Thr-8, Ser-22, Ser-62, Thr-323, Ser-598, Ser-610, Thr-624, Thr-716 to A), MAD1NTD4A (full length but Thr-8, Ser-22, Ser-62, Thr-323 to A) or MAD1CTD4A (full length but Ser-598, Ser-610, Thr-624, Thr-716 to A). Cell numbers imaged for each construct are listed on the right. c, phosphomimic and phosphoresistant mutants at individual MPS1 phosphorylation sites in the MAD1CTD (Ser-598, Ser-610, Thr-624, Thr-716) were prepared as mCherry-Mis12 fusion constructs and transfected HeLa cells were imaged for mitotic durations as in (b). d, GST-tagged MAD1CTD fragments in WT, or containing T716A or T716E or CTD-4A mutations (∼240 nm) were incubated with either MAD2L13A or MAD2ΔC10 (230 nm), then GST pulldowns were probed with anti-GST and anti-MAD2 antibodies.
Next, a mCherry-Mis12-MAD1CTD4E phosphomimic mutant was prepared and surprisingly found to be also defective in maintaining the mitotic arrest (Fig. S4b). We reasoned that, for a functional mitotic checkpoint in cells, one or more of the four residues identified through in vitro kinase assays could not tolerate mutations. Both phosphoresistant and phosphomimic mutants at individual sites were then prepared. The mutants at Ser-598 or Ser-624 did not show obvious defects in mitotic checkpoint responses when fused with mCherry-Mis12. However, the mutants at Ser-610 and Thr-716 showed significant differences in mitotic arrest durations when compared with the MAD1WT fusion (Fig. 5c). Furthermore, the T716E mutant maintained the checkpoint longer than the T716A mutant (441 ± 38 versus 242 ± 48 min, p < 0.05, Student's t test), indicating that Thr-716 is likely the residue activated by MPS1 for mitotic checkpoint signaling. However, the mCherry-Mis12 fusion of MAD1T716E mutant could not maintain mitotic arrest when transfected cells were challenged by reversine (Fig. S4c), suggesting either the phosphomimic mutant is imperfect or MPS1 has additional key substrates required for a functional mitotic checkpoint.
To further understand the imaging results with different CTD mutants, we examined whether these mutants affected the interaction of MAD1CTD with MAD2. Both CTD4A and CTD4E showed defects in binding to O-MAD2 and C-MAD2 (Fig. S4d). Furthermore, CTDT716A bound less O-MAD2 and C-MAD2 as compared with CTDWT or CTDT716E (Fig. 5d). Similarly, the functional defective MAD1Y634E mutant also bound to less O-MAD2 and C-MAD2 than MAD1Y634F (Fig. S4e). These results correlate mitotic arrest durations with the capability of MAD1CTD mutants to bind to MAD2 (Figs. 1d and 5 and Fig. S4).
MPS1 phosphorylation of MAD1 modulates interdomain and intermolecular interactions
Because MIM, CTD, and NTD of MAD1 are all required for an effective mitotic checkpoint (Fig. 1), we next investigated whether there is coordination among these domains. The presumed non–coiled-coil domains along the length of MAD1 might provide certain bends and turns to the molecule, and an earlier model has suggested that MAD1CTD folds back to the proximity of the catalytic core consisting of MIM and associated C-MAD2 (11). We found that NTD directly interacted with CTD (Fig. 6, a, lane 1 and b, lane 2). CTD interacted with itself (Fig. 6a, lane 3), agreeing with published crystal structure (PDB: 4DZO) and acting as a positive control in this binding assay (33). MIM does not directly interact with CTD or NTD (Fig. 6, a, lane 2 and b, lane 1). Addition of MPS1 kinase reduced the interactions between MAD1NTD and MAD1CTD (Fig. 6c). The effect depended on the MPS1 kinase activity as omitting ATP from the reactions or adding MPS1 inhibitors (reversine or AZ3146) reversed the MPS1 effect on NTD:CTD interaction (Fig. 6c, compare His signals in lane 2 and those in lanes 3–5).
Figure 6.

Interdomain interactions of MAD1 are modulated by MPS1 kinase. a, GST-tagged MAD1NTD, MAD1MIM, MAD1CTD, GST (at 60 nm) or beads alone were incubated with 4 μm His-tagged MAD1CTD. The GST pulldown samples were separated by SDS-PAGE and subjected to Western blotting using anti-GST and anti-His6 antibodies. The asterisks mark the expected bands of GST-MAD1NTD, MAD1MIM, and MAD1CTD. b, GST pulldown assays after GST or GST-MAD1NTD was incubated with His-tagged MAD1MIM and MAD1CTD. c, recombinant GST-MAD1NTD or GST alone was incubated with 9 μm His-MAD1CTD in the absence or presence of MPS1 kinase, reversine (MPS1 inhibitor), AZ3146 (another MPS1 inhibitor), and ATP. GST pulldowns were then probed for anti-GST and anti-His6 antibodies. The asterisk marks expected size of GST-NTD. d, GST-MAD1CTDWT or GST-MAD1CTD4A was incubated with His-tagged MAD1CTDWT or MAD1CTD4A and GST pulldowns were probed. The result has been replicated for at least three times. The asterisk marks expected size of GST-CTD.
MAD1 dimerization has been observed in MAD1MIM:MAD2 and MAD1CTD crystal structures (PDB: 1GO4 and 4DZO, respectively) (11, 33). However, whether MAD1 dimerization is essential for its activity to promote MAD2 O–C conversion is unclear. We noticed that in Fig. 1, e and f, MAD1 truncations missing either NTD or CTD did not co-immunoprecipitate endogenous MAD1 as well as other fusions. Recombinant GST-CTD4A fragment also bound less His-CTDWT and even less His-CTD4A (Fig. 6d, compare lanes 1–3). These results suggest a possible role of CTD in MAD1 dimerization or oligomerization (see “Discussion”).
Discussion
MAD2 O–C conversion is a key signal amplification step for the mitotic checkpoint. The current model suggests that the conversion is catalyzed by an unusual catalyst: the MAD1:C-MAD2 complex localized at unattached kinetochores. How the catalysis is achieved is still unclear. Together with two very recent publications (29, 30), our results support the roles of MAD1CTD and MPS1 kinase in promoting the MAD2 O–C conversion. In particular, our work agrees with the critical role of MAD1 Thr-716, which is phosphorylated by MPS1, but also suggests that Ser-610 and Tyr-634 are potentially key residues for regulating MAD2 O–C conversion (Figs. 1 and 5). Ser-610 is phosphorylated by MPS1 in vitro (Fig. 4), and Tyr-634 was reported to be phosphorylated in vivo, although we have not confirmed this yet in mitotic cells (37). We have also uncovered additional protein-protein interactions between MPS1, MAD1, and MAD2 (Figs. 2–4 and 6). Of particular interest, we found that MAD1NTD and MAD1CTD interact with each other and both bind to O-MAD2 and C-MAD2. These results have been integrated into an updated model for the MAD1:C-MAD2 catalyst that promotes MAD2 O–C conversion (Fig. 7). Many mechanistic details remain to be filled, so we hereby discuss the implications of our results.
Figure 7.
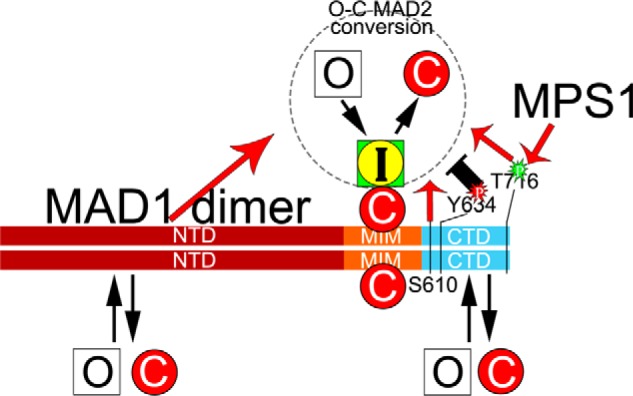
An updated model on the MAD1:C-MAD2 catalyst for MAD2 O–C conversion. In the classical model, a MAD1 dimer tightly binds two C-MAD2 molecules at its MIM region to become the catalyst for MAD2 O–C conversion. Our work has shown that MAD1NTD and MAD1CTD have additional, probably weaker, binding sites for both O-MAD2 and C-MAD2. MAD1NTD and MAD1CTD positively contribute to the MAD2 O–C conversion (red arrows), based on live cell imaging results. Several residues within the CTD domain may have important functional roles. Thr-716 phosphorylation by MPS1 and Ser-610 are required for full MAD1 activity. Tyr-634 might be a residue whose phosphorylation negatively impacts the O–C conversion. Similarly, the NTD:CTD interaction (not shown) may restrain the catalytic efficiency of MAD1, but the interaction can be disrupted by action of MPS1 kinase. It remains unclear whether the two C-MAD2 molecules bound to the MIM regions of the MAD1 dimer are equally engaged in MAD2 O–C conversion.
The functions of MAD1NTD and MAD1CTD in MAD2 O–C conversion
The crystal structure of MAD1MIM in complex with C-MAD2 has solidified the now classical model of the MAD1:C-MAD2 catalyst as a 2:2 heterotetramer (11) (Fig. S1). Each of the two “liganded” C-MAD2s tightly wraps around one MIM monomer through its safety belt loop (11, 18, 38). C-MAD2 then utilizes its dimerization domain to recruit O-MAD2 and converts the latter into C-MAD2, resulting in signal amplification for the mitotic checkpoint (6, 7, 12). The conversion may go through multiple intermediate states (I-MAD2) (6, 7, 10, 13, 32).
Although much of MAD1 has been usually simplified as rather stiff coiled coils, the known structures of MAD1MIM and MAD1CTD and careful analyses of MAD1NTD showed that multiple MAD1 segments may adopt alternative structures or remain disordered (11, 33) (Fig. 1 and Fig. S1). It is likely that liganded C-MAD2 binding to the MIM region of MAD1 still constitutes the catalytic core and represents the MAD2 stably associated with MAD1 in cells (Fig. 1e), but earlier work by others has already lent strong support to possible MAD1CTD involvement in the mitotic checkpoint responses (12–18, 29, 30). At least part of the MAD1NTD contributes to MAD1 nuclear pore or kinetochore localization and interactions with other proteins including Ndc80, Plk1, Nek2A, Tpr, Cep57, and CENP-E (20–23, 33, 45–49), and the regions spanning 400–500 or 420–485 could possibly affect the mitotic checkpoint signaling (14, 29). Nevertheless, the possibility that MAD1NTD and MAD1CTD directly impact the MAD2 O–C conversion was not thoroughly studied until this work.
Our results suggest that a full-length MAD1 molecule may employ NTD and CTD to at least transiently interact with the substrates (O-MAD2), the products (C-MAD2), and even the intermediate states (I-MAD2, represented by MAD2ΔN10 in Fig. 3c) (50) of the MAD2 O–C conversion reaction. The interaction with O-MAD2 might increase local concentration of substrates, driving the conversion reaction. Similarly, enrichment of I-MAD2 by MAD1NTD or MAD1CTD could facilitate its interaction with CDC20, coupling the MAD2 O–C conversion reaction with the energy favorable assembly of the MCC (29, 30). It should be noticed that O-MAD2 conversion to C-MAD2 can certainly be accomplished in the absence of CDC20 (12, 13). Following the law of mass action, the NTD or CTD-retained C-MAD2 could also drive its interaction with BUBR1 as a step of MCC assembly (36). These possible scenarios involving MAD1NTD and MAD1CTD for C-MAD2 production and MCC assembly may underlie the compromised mitotic checkpoint responses when cells were transfected with mCherry-Mis12-MAD1ΔNTD or -MAD1ΔCTD (Fig. 1). Consistent with this idea, our data indicated that MAD1NTD and MAD1CTD bind to a new interface on MAD2. Such a binding mode is postulated to be advantageous, as the I-MAD2 or C-MAD2 products anchored on MAD1 could have two other protein-protein interaction interfaces, the safety belt (for CDC20) and the dimerization domain (for BUBR1), readily available to assemble the MCC. In addition, the MAD2 interactions with NTD or CTD must be weak or transient in cells, as mCherry-Mis12-MAD1ΔNTD and MAD1ΔCTD do not show significant reduction in co-immunoprecipitated MAD2 whereas FLAG-MAD1NTD only shows faint MAD2-binding (Fig. 1, e and f). Such transient interactions are likely also beneficial for stepwise relay mechanisms to facilitate MAD2 O–C conversion and MCC assembly.
In the MAD1MIM:C-MAD2 tetramer structure (11, 32), the dimerization interfaces along the two C-MAD2 molecules to recruit O-MAD2 face toward opposite directions. It is therefore possible that endogenous full-length MAD1, when bound by C-MAD2 at MIM to form a catalyst, contains more than one catalytic center. It remains to be seen whether MAD1NTD and MAD1CTD asymmetrically affect the two presumable MAD1MIM:C-MAD2 catalytic centers or directly contribute to the catalysis through stabilizing some transient I-MAD2 states such as those with unfolded N or C terminus (10, 13). In addition, MAD1CTD and MAD1NTD are important for MAD1 oligomerization (Figs. 1, e and f, and 6d). We noticed an earlier publication suggesting that Ser-214 phosphorylation by ATM affects MAD1 dimerization (51). Whether MAD1 dimerization regulates MAD2 conversion is an interesting topic for future exploration.
The impact of mitotic kinases on MAD2 O–C conversion
Whatever the catalytic mechanism the MAD1:C-MAD2 complexes employ to promote MAD2 O–C conversion, the catalysis is likely initiated or enhanced by mitotic checkpoint kinases whose activities have been shown important for the checkpoint, including MPS1, BUB1, and Aurora B (25, 26, 28, 31). These kinases not only help enrich the MAD1:C-MAD2 catalysts to unattached kinetochores, but also directly impact their catalytic efficiency. In this work we have focused on the potential effect of MPS1 on increasing the catalytic efficiency of the MAD1:C-MAD2 complex.
MPS1 is a key upstream kinase orchestrating the organization of other mitotic checkpoint proteins at unattached kinetochores. It phosphorylates KNL1 to recruit BUB1:BUB3 and BUBR1:BUB3 complexes (reviewed in Refs. 1 and 52). It also phosphorylates BUB1 to recruit MAD1:MAD2 to kinetochores (30, 31, 53). In addition, MPS1 phosphorylating MAD1 at Thr-716 may enhance MAD1 binding to CDC20 (30). Thus MPS1 activity may promote the formation of the MCC by placing all MCC components BUBR1:BUB3, CDC20, and MAD2 in spatial proximity (29, 30). CDC20 binding to BUB1 or BUBR1 might also help the MCC assembly at unattached kinetochores (54–56).
We have demonstrated that MPS1 interacts with and phosphorylates MAD1NTD and MAD1CTD (Fig. 4). Phosphoresistant mutants at in vitro MPS1 phosphorylation sites in MAD1CTD, especially the T716A mutation, compromised the mitotic checkpoint responses in cells and showed reduced interaction with MAD2 in vitro (Fig. 5). No increase in MAD2 interactions with MAD1CTD was detected either using the phosphomimic T716E mutant or after direct in vitro phosphorylation by MPS1 (Figs. 4 and 5). We also expected to see differential bindings of O-MAD2 and C-MAD2 to MAD1NTD or MAD1CTD, especially after MPS1 phosphorylation, but do not have experimental evidence to support the idea yet (Fig. 4). We cannot exclude the possibility that in cells and in the context of full-length MAD1, CTD phosphorylation shows differential binding toward either conformer of MAD2. Synergistic effect with phosphorylation by other kinases such as BUB1 and Aurora B may also be necessary to see such effects. Phosphorylation by MPS1 did reduce its own interaction with CTD and the interaction between NTD and CTD (Figs. 4 and 6).
We propose that the NTD:CTD interaction occurs in interphase cells and represents an inactive state of MAD1 even though its MIM associates with C-MAD2 in a cell cycle–independent manner (18, 24). The interaction between MPS1 and MAD1 might contribute to the MAD1 recruitment to kinetochores. Once concentrated at unattached kinetochores, MPS1 kinase becomes activated and phosphorylates BUB1 to stably anchor MAD1 (30, 31, 57). Activated MPS1 also phosphorylates MAD1CTD, most likely at Thr-716, relieving itself and the NTD from CTD. The now exposed and phosphorylated CTD could bind to O-MAD2 and C-MAD2 and also facilitate the MAD2 O–C conversion either directly or through increased association with CDC20 (30), as discussed in the last section.
Although deleting MAD1NTD compromised the mitotic checkpoint when examined using the mCherry-Mis12 fusions (Fig. 1), mutating presumable MPS1 phosphorylation sites within MAD1NTD had no impact on the mitotic checkpoint (Fig. 5), and others found no requirement for MAD1 N-terminal 400 or 420 residues for MAD2 O–C conversion or checkpoint responses (14, 29). This suggests that a functional critical region lies within 420–485 residues, which happens to contain a likely non–coiled-coil structure (Fig. 1). However, neither of the MAD1 fragments spanning 327–423 or 327–488 residues bind to MAD2 in vitro (Fig. 2), indicating the positive effect of MAD1NTD on the mitotic checkpoint signaling needs further clarification.
Interestingly, Faesen et al. (29) reported that MAD2 can be phosphorylated by MPS1 in vitro at Ser-195. Previously, phosphorylation at this site was proposed to render MAD2 hard to convert and thus more likely to stay in O conformation (43, 58). Whether this particular MAD2 phosphorylation by MPS1 represents a feedback regulatory mechanism can be further explored. In this regard, it might be useful to note that MPS1 interacts with C-MAD2 and O-MAD2 whereas MAD2S195D binds to MAD1 at NTD or CTD but not MIM, behaving indeed like an O-MAD2 (Figs. 3a and 4c).
Although recent work has proposed how BUB1 might help the MAD2 O–C conversion and MCC assembly (29–31), it will still be interesting to further define how other kinases such as Aurora B regulate the MAD1:C-MAD2 catalyst to increase C-MAD2 production. In addition, there have been reports that some tyrosine kinases help silence the mitotic checkpoint to drive anaphase onset (59–63). MAD1 Y634 seems to play important roles in the mitotic checkpoint (Fig. 1 and Fig. S4), and it was once reported to be phosphorylated in vivo (37). Further characterization of this site as well as the apparently essential Ser-610 and Thr-716 residues at the MAD1CTD will provide more mechanistic insights into the functions of MAD1 in MAD2 O–C conversion.
Experimental procedures
DNA constructs
The MAD1 and MAD2 DNA fragments and mutants used in the work are summarized in Table S1. The pMSCV-mCherry-Mis12-MAD1 constructs with wild-type or a mutant MAD1 (K541A, L543A) were gifts from Dr. Tarun Kapoor (Rockefeller University) (26, 28). Other MAD1 mutants or truncations were cloned or mutated in an intermediate pENTR2B vector (Invitrogen), cut out as a NotI-EcoRI fragment, and then used to replace the MAD1 gene in the pMSCV-mCherry-Mis12 backbone also cut with NotI and EcoRI. Mutagenesis was conducted using QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent). Different fragments of MAD1 were also cloned into a pENTR/D-TOPO vector using TOPO Cloning Kit (Invitrogen) and then recombined into pDEST15 (for N-terminal GST tag) or pDEST17 (for N-terminal His6-tag) vectors through LR reactions as instructed for the Gateway cloning system (Invitrogen). MAD2 expression constructs were prepared similarly as before (36), but with a tobacco etch virus (TEV) cleavage site inserted between the tags and second MAD2 codon during pENTR cloning steps. MPS1 was recombined into pDEST10 or pDEST20 and transformed into DH10BAC to prepare recombinant bacmids for Sf9 insect cell transfection. All constructs were confirmed by DNA sequencing.
Cell culture and transfection
HeLaM, a subline of HeLa (35), was maintained in DMEM with 10% fetal bovine serum at 37 °C in 5% CO2. DNA transfection was carried out using TransIT-LT1 reagent (Mirus) following the manufacturer's instructions or using polyethyleneimine (PEI) as described before (64). Sf9 cells were grown at 27 °C in SFX medium (Hyclone) in the presence of antibiotics (streptomycin/penicillin). Cellfectin (Invitrogen) was used to transfect bacmids into Sf9 cells.
Live cell imaging
For determining mitotic durations, HeLa cells grown on No. 1.5 coverslip-bottomed 35-mm dishes (MatTek) were transfected with different mCherry-Mis12-MAD1 constructs. Usually 24 h after transfection, live cell imaging was started on an automated Olympus IX-81 microscope to collect phase contrast and RFP images at 15-min intervals using a 60× objective lens (NA = 1.42) while cells were maintained at 37 °C in a heating chamber. Single-plane images were acquired for up to 13 h at multiple positions using a CoolSNAP HQ2 camera with 2 × 2 binning. Student's t test was used to evaluate the statistical significance between the differences in mitotic durations after different treatment. Nuclear envelope breakdown marks the beginning and appearance of cleavage furrow the end of a mitosis. Some images were collected on a Leica TCS SP8 confocal microscope with a 63× objective (NA = 1.40) as z-stacks of 1.0 μm.
Cell lysates, immunoblotting, and immunoprecipitation
These were performed as described before (28, 64). A list of primary antibodies used in this study is summarized in Table S2.
Recombinant proteins
GST-tagged or His-tagged MAD1 fragments and His-TEV–tagged MAD2L13A or His-TEV–tagged MAD2ΔC10 were expressed in Escherichia coli BL21(DE3)-CodonPlus RIPL (Stratagene), normally at 16 °C. His-MPS1 or GST-MPS1 was expressed in Sf9 cells after infection with recombinant baculoviruses. All expressed proteins were purified using GSH-agarose or Probond nickel beads (Invitrogen). His-tagged TEV(S219P) protease (65) was used to cleave His-tag to make untagged MAD2. Peak fractions of eluted proteins were pooled, buffer-exchanged, and concentrated using Pierce protein concentrators with 10,000 molecular weight cutoff. The storage buffer is 50 mm Hepes pH7.5, 100 mm KCl, 1 mm DTT, 30% glycerol. Concentrations of recombinant proteins were determined by comparing the target band with BSA standards on Coomassie Blue–stained gels using ImageJ software (66).
In vitro binding assays
4 μl of 5× binding buffer (100 mm Tris-HCl, pH 8.0, 750 mm NaCl, 50 mm MgCl2, 2.5% Nonidet P-40, 50% glycerol, 500 μg/ml BSA) was mixed with recombinant GST-tagged MAD1 fragments and MAD2 mutants or His-MAD1CTD or His-MPS1. The proteins (including related fragments or mutants) were used at roughly endogenous intracellular concentrations unless stated otherwise in the figure legends: [MAD2] = 230 nm, [MPS1] = 100 nm, and [MAD1] = 60 nm (8, 67–69). H2O was added to make the final volume 20 μl. The reactions were incubated at 37 °C for 1 h and then mixed with 10 μl GSH agarose beads and shaken at 800 rpm at 4 °C for 40 min (Peqlab Thriller). The beads were pelleted and washed four times with wash buffer (20 mm phosphate buffer, pH 7.4, 250 mm NaCl, 0.5% Nonidet P-40, 10% glycerol). Then 10 μl 2× SDS sample buffer was added to the beads and the samples were heated at 80 °C for 10 min before SDS-PAGE. The gel was transferred to PVDF membranes (Millipore) for immunoblotting. Most of the in vitro binding results have been replicated three times or more.
In vitro kinase assays
GST-tagged MPS1 kinase was purchased from Invitrogen or was purified from Sf9 cell lysates as well as His-tagged MPS1 (64). Myelin basic protein was purchased from Sigma as an artificial substrate for MPS1. In vitro kinase assays were set up similarly as described previously (28, 64): 4 μl of 5× kinase buffer (125 mm Tris-HCl, pH 7.5, 300 mm β-glycerophosphate, 50 mm MgCl2) was mixed with recombinant kinase, substrates, 5μCi 32P ATP and 50 μm cold ATP. In some reactions, MPS1 kinase inhibitors reversine (Calbiochem) or AZ3146 (Selleckchem) was used at 500 nm and 2 μm final concentrations, respectively. DMSO concentration is kept below 0.5%. H2O was added to make the final volume 20 μl. The reactions were incubated at 30 °C for 30 min and then terminated by adding 20 μl 2× SDS sample buffer. Samples were subjected to SDS-PAGE followed by Coomassie Blue staining. After destaining, the SDS-PAGE gel was vacuum dried. Phosphorylation of the substrates was visualized by autoradiography. Samples for mass spectrometry were prepared using only 0.5 mm cold ATP in the kinase reactions and separated on SDS-PAGE. Phosphorylated residues on the excised bands were determined by mass spectrometry (MS Bioworks, Ann Arbor, MI).
For experiments shown in Fig. 4, d and e, in vitro kinase assays were performed by incubating GST-MPS1 and His-MAD1CTD in the presence or absence of 0.5 mm cold ATP and 25 μm reversine at 30 °C for 1 h. Then untagged MAD2L13A or MAD2ΔC10 were directly added into the reaction mixtures and incubated at 37° for another hour before immunoprecipitation with 1 μg anti-His6 antibody.
Author contributions
W. J. and S.-T. L. conceptualization; W. J., Y. L., E. A., and S.-T. L. data curation; W. J., Y. L., E. A., and S.-T. L. formal analysis; W. J., Y. L., E. A., and S.-T. L. investigation; W. J., Y. L., E. A., and S.-T. L. methodology; W. J., Y. L., E. A., and S.-T. L. writing-review and editing; S.-T. L. supervision; S.-T. L. funding acquisition; S.-T. L. writing-original draft; S.-T. L. project administration.
Supplementary Material
Acknowledgments
We thank Drs. Tarun Kapoor and Maria Maldonado for the mCherry-Mis12-MAD1-WT and -AA constructs and Dr. Tim Yen for multiple antibodies. We also thank Dr. William Taylor, Dr. Qian Chen and the members of the Liu lab for stimulating discussions.
This work was supported by National Institutes of Medicine Grant NIH R01CA169500 (to S.-T. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4 and Tables S1 and S2.
- MAD2
- mitotic arrest deficient 2
- O-MAD2
- open MAD2
- C-MAD2
- closed MAD2
- I-MAD2
- intermediate MAD2
- NTD
- N-terminal domain
- CTD
- C-terminal domain
- APC/C
- anaphase promoting complex/cyclosome
- MCC
- mitotic checkpoint complex
- MIM
- MAD2-interaction motif
- TEV
- tobacco etch virus.
References
- 1. Musacchio A. (2015) The molecular biology of spindle assembly checkpoint signaling dynamics. Curr. Biol. 25, R1002–R1018 10.1016/j.cub.2015.08.051 [DOI] [PubMed] [Google Scholar]
- 2. London N., and Biggins S. (2014) Signalling dynamics in the spindle checkpoint response. Nat. Rev. Mol. Cell Biol. 15, 736–747 10.1038/nrm3888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lara-Gonzalez P., Westhorpe F. G., and Taylor S. S. (2012) The spindle assembly checkpoint. Curr. Biol. 22, R966–R980 10.1016/j.cub.2012.10.006 [DOI] [PubMed] [Google Scholar]
- 4. Liu S. T., and Zhang H. (2016) The mitotic checkpoint complex (MCC): looking back and forth after 15 years. AIMS Molecular Science 3, 597–634 10.3934/molsci.2016.4.597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rieder C. L., Cole R. W., Khodjakov A., and Sluder G. (1995) The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J. Cell Biol. 130, 941–948 10.1083/jcb.130.4.941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Luo X., and Yu H. (2008) Protein metamorphosis: The two-state behavior of Mad2. Structure 16, 1616–1625 10.1016/j.str.2008.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mapelli M., and Musacchio A. (2007) MAD contortions: Conformational dimerization boosts spindle checkpoint signaling. Curr. Opin. Struct. Biol. 17, 716–725 10.1016/j.sbi.2007.08.011 [DOI] [PubMed] [Google Scholar]
- 8. Luo X., Tang Z., Xia G., Wassmann K., Matsumoto T., Rizo J., and Yu H. (2004) The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat. Struct. Mol. Biol. 11, 338–345 10.1038/nsmb748 [DOI] [PubMed] [Google Scholar]
- 9. Fava L. L., Kaulich M., Nigg E. A., and Santamaria A. (2011) Probing the in vivo function of Mad1:C-Mad2 in the spindle assembly checkpoint. EMBO J. 30, 3322–3336 10.1038/emboj.2011.239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hara M., Özkan E., Sun H., Yu H., and Luo X. (2015) Structure of an intermediate conformer of the spindle checkpoint protein Mad2. Proc. Natl. Acad. Sci. U.S.A. 112, 11252–11257 10.1073/pnas.1512197112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sironi L., Mapelli M., Knapp S., De Antoni A., Jeang K. T., and Musacchio A. (2002) Crystal structure of the tetrameric Mad1-Mad2 core complex: Implications of a ‘safety belt’ binding mechanism for the spindle checkpoint. EMBO J. 21, 2496–2506 10.1093/emboj/21.10.2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Antoni A., Pearson C. G., Cimini D., Canman J. C., Sala V., Nezi L., Mapelli M., Sironi L., Faretta M., Salmon E. D., and Musacchio A. (2005) The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr. Biol. 15, 214–225 10.1016/j.cub.2005.01.038 [DOI] [PubMed] [Google Scholar]
- 13. Yang M., Li B., Liu C. J., Tomchick D. R., Machius M., Rizo J., Yu H., and Luo X. (2008) Insights into mad2 regulation in the spindle checkpoint revealed by the crystal structure of the symmetric mad2 dimer. PLoS Biol. 6, e50 10.1371/journal.pbio.0060050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kruse T., Larsen M. S., Sedgwick G. G., Sigurdsson J. O., Streicher W., Olsen J. V., and Nilsson J. (2014) A direct role of Mad1 in the spindle assembly checkpoint beyond Mad2 kinetochore recruitment. EMBO Rep. 15, 282–290 10.1002/embr.201338101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heinrich S., Sewart K., Windecker H., Langegger M., Schmidt N., Hustedt N., and Hauf S. (2014) Mad1 contribution to spindle assembly checkpoint signalling goes beyond presenting Mad2 at kinetochores. EMBO Rep. 15, 291–298 10.1002/embr.201338114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ballister E. R., Riegman M., and Lampson M. A. (2014) Recruitment of Mad1 to metaphase kinetochores is sufficient to reactivate the mitotic checkpoint. J. Cell Biol. 204, 901–908 10.1083/jcb.201311113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuijt T. E., Omerzu M., Saurin A. T., and Kops G. J. (2014) Conditional targeting of MAD1 to kinetochores is sufficient to reactivate the spindle assembly checkpoint in metaphase. Chromosoma 123, 471–480 10.1007/s00412-014-0458-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen R. H., Brady D. M., Smith D., Murray A. W., and Hardwick K. G. (1999) The spindle checkpoint of budding yeast depends on a tight complex between the Mad1 and Mad2 proteins. Mol. Biol. Cell 10, 2607–2618 10.1091/mbc.10.8.2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kulukian A., Han J. S., and Cleveland D. W. (2009) Unattached kinetochores catalyze production of an anaphase inhibitor that requires a Mad2 template to prime Cdc20 for BubR1 binding. Dev. Cell 16, 105–117 10.1016/j.devcel.2008.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen R. H., Shevchenko A., Mann M., and Murray A. W. (1998) Spindle checkpoint protein Xmad1 recruits Xmad2 to unattached kinetochores. J. Cell Biol. 143, 283–295 10.1083/jcb.143.2.283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodriguez-Bravo V., Maciejowski J., Corona J., Buch H. K., Collin P., Kanemaki M. T., Shah J. V., and Jallepalli P. V. (2014) Nuclear pores protect genome integrity by assembling a premitotic and mad1-dependent anaphase inhibitor. Cell 156, 1017–1031 10.1016/j.cell.2014.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Akera T., Goto Y., Sato M., Yamamoto M., and Watanabe Y. (2015) Mad1 promotes chromosome congression by anchoring a kinesin motor to the kinetochore. Nat. Cell Biol. 17, 1124–1133 10.1038/ncb3219 [DOI] [PubMed] [Google Scholar]
- 23. Martin-Lluesma S., Stucke V. M., and Nigg E. A. (2002) Role of hec1 in spindle checkpoint signaling and kinetochore recruitment of mad1/mad2. Science 297, 2267–2270 10.1126/science.1075596 [DOI] [PubMed] [Google Scholar]
- 24. Campbell M. S., Chan G. K., and Yen T. J. (2001) Mitotic checkpoint proteins HsMAD1 and HsMAD2 are associated with nuclear pore complexes in interphase. J. Cell Science 114, 953–963 [DOI] [PubMed] [Google Scholar]
- 25. Hewitt L., Tighe A., Santaguida S., White A. M., Jones C. D., Musacchio A., Green S., and Taylor S. S. (2010) Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J. Cell Biol. 190, 25–34 10.1083/jcb.201002133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maldonado M., and Kapoor T. M. (2011) Constitutive Mad1 targeting to kinetochores uncouples checkpoint signalling from chromosome biorientation. Nat. Cell Biol. 13, 475–482 10.1038/ncb2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tighe A., Staples O., and Taylor S. (2008) Mps1 kinase activity restrains anaphase during an unperturbed mitosis and targets Mad2 to kinetochores. J. Cell Biol. 181, 893–901 10.1083/jcb.200712028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tipton A. R., Ji W., Sturt-Gillespie B., Bekier M. E. 2nd, Wang K., Taylor W. R., and Liu S. T. (2013) Monopolar Spindle 1 (MPS1) Kinase Promotes Production of Closed MAD2 (C-MAD2) Conformer and Assembly of the Mitotic Checkpoint Complex. J. Biol. Chem. 288, 35149–35158 10.1074/jbc.M113.522375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Faesen A. C., Thanasoula M., Maffini S., Breit C., Müller F., van Gerwen S., Bange T., and Musacchio A. (2017) Basis of catalytic assembly of the mitotic checkpoint complex. Nature 542, 498–502 10.1038/nature21384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ji Z., Gao H., Jia L., Li B., and Yu H. (2017) A sequential multi-target Mps1 phosphorylation cascade promotes spindle checkpoint signaling. Elife 6, e22513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang G., Kruse T., López-Méndez B., Sylvestersen K. B., Garvanska D. H., Schopper S., Nielsen M. L., and Nilsson J. (2017) Bub1 positions Mad1 close to KNL1 MELT repeats to promote checkpoint signalling. Nat. Commun. 8, 15822 10.1038/ncomms15822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mapelli M., Massimiliano L., Santaguida S., and Musacchio A. (2007) The Mad2 conformational dimer: structure and implications for the spindle assembly checkpoint. Cell 131, 730–743 10.1016/j.cell.2007.08.049 [DOI] [PubMed] [Google Scholar]
- 33. Kim S., Sun H., Tomchick D. R., Yu H., and Luo X. (2012) Structure of human Mad1 C-terminal domain reveals its involvement in kinetochore targeting. Proc. Natl. Acad. Sci. U.S.A. 109, 6549–6554 10.1073/pnas.1118210109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hardwick K. G., and Murray A. W. (1995) Mad1p, a phosphoprotein component of the spindle assembly checkpoint in budding yeast. J. Cell Biol. 131, 709–720 10.1083/jcb.131.3.709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tipton A. R., Tipton M., Yen T., and Liu S. T. (2011) Closed MAD2 (C-MAD2) is selectively incorporated into the mitotic checkpoint complex (MCC). Cell cycle 10, 3740–3750 10.4161/cc.10.21.17919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tipton A. R., Wang K., Link L., Bellizzi J. J., Huang H., Yen T., and Liu S. T. (2011) BUBR1 and closed MAD2 (C-MAD2) interact directly to assemble a functional Mitotic checkpoint complex. J. Biol. Chem. 286, 21173–21179 10.1074/jbc.M111.238543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rikova K., Guo A., Zeng Q., Possemato A., Yu J., Haack H., Nardone J., Lee K., Reeves C., Li Y., Hu Y., Tan Z., Stokes M., Sullivan L., Mitchell J., et al. (2007) Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131, 1190–1203 10.1016/j.cell.2007.11.025 [DOI] [PubMed] [Google Scholar]
- 38. Luo X., Tang Z., Rizo J., and Yu H. (2002) The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol. Cell 9, 59–71 10.1016/S1097-2765(01)00435-X [DOI] [PubMed] [Google Scholar]
- 39. Sironi L., Melixetian M., Faretta M., Prosperini E., Helin K., and Musacchio A. (2001) Mad2 binding to Mad1 and Cdc20, rather than oligomerization, is required for the spindle checkpoint. EMBO J. 20, 6371–6382 10.1093/emboj/20.22.6371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luo X., Fang G., Coldiron M., Lin Y., Yu H., Kirschner M. W., and Wagner G. (2000) Structure of the Mad2 spindle assembly checkpoint protein and its interaction with Cdc20. Nat. Struct. Biol. 7, 224–229 10.1038/73338 [DOI] [PubMed] [Google Scholar]
- 41. Yang M., Li B., Tomchick D. R., Machius M., Rizo J., Yu H., and Luo X. (2007) p31comet blocks Mad2 activation through structural mimicry. Cell 131, 744–755 10.1016/j.cell.2007.08.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chao W. C., Kulkarni K., Zhang Z., Kong E. H., and Barford D. (2012) Structure of the mitotic checkpoint complex. Nature 484, 208–213 10.1038/nature10896 [DOI] [PubMed] [Google Scholar]
- 43. Kim S., Sun H., Ball H. L., Wassmann K., Luo X., and Yu H. (2010) Phosphorylation of the spindle checkpoint protein Mad2 regulates its conformational transition. Proc. Natl. Acad. Sci. U.S.A. 107, 19772–19777 10.1073/pnas.1009000107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Santaguida S., Tighe A., D'Alise A. M., Taylor S. S., and Musacchio A. (2010) Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J. Cell Biol. 190, 73–87 10.1083/jcb.201001036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lince-Faria M., Maffini S., Orr B., Ding Y., Florindo C., Sunkel C. E., Tavares A., Johansen J., Johansen K. M., and Maiato H. (2009) Spatiotemporal control of mitosis by the conserved spindle matrix protein Megator. J. Cell Biol. 184, 647–657 10.1083/jcb.200811012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee S. H., Sterling H., Burlingame A., and McCormick F. (2008) Tpr directly binds to Mad1 and Mad2 and is important for the Mad1-Mad2-mediated mitotic spindle checkpoint. Genes Dev. 22, 2926–2931 10.1101/gad.1677208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chi Y. H., Haller K., Ward M. D., Semmes O. J., Li Y., and Jeang K. T. (2008) Requirements for protein phosphorylation and the kinase activity of polo-like kinase 1 (Plk1) for the kinetochore function of mitotic arrest deficiency protein 1 (Mad1). J. Biol. Chem. 283, 35834–35844 10.1074/jbc.M804967200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lou Y., Yao J., Zereshki A., Dou Z., Ahmed K., Wang H., Hu J., Wang Y., and Yao X. (2004) NEK2A interacts with MAD1 and possibly functions as a novel integrator of the spindle checkpoint signaling. J. Biol. Chem. 279, 20049–20057 10.1074/jbc.M314205200 [DOI] [PubMed] [Google Scholar]
- 49. Zhou H., Wang T., Zheng T., Teng J., and Chen J. (2016) Cep57 is a Mis12-interacting kinetochore protein involved in kinetochore targeting of Mad1-Mad2. Nat. Commun. 7, 10151 10.1038/ncomms10151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Paul A. S., and Pollard T. D. (2008) The role of the FH1 domain and profilin in formin-mediated actin-filament elongation and nucleation. Curr. Biol. 18, 9–19 10.1016/j.cub.2007.11.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang C., Hao J., Kong D., Cui X., Zhang W., Wang H., Guo X., Ma S., Liu X., Pu P., and Xu B. (2014) ATM-mediated Mad1 serine 214 phosphorylation regulates Mad1 dimerization and the spindle assembly checkpoint. Carcinogenesis 35, 2007–2013 10.1093/carcin/bgu087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lischetti T., and Nilsson J. (2015) Regulation of mitotic progression by the spindle assembly checkpoint. Mol. Cell Oncol. 2, e970484 10.4161/23723548.2014.970484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. London N., and Biggins S. (2014) Mad1 kinetochore recruitment by Mps1-mediated phosphorylation of Bub1 signals the spindle checkpoint. Genes Dev. 28, 140–152 10.1101/gad.233700.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Di Fiore B., Davey N. E., Hagting A., Izawa D., Mansfeld J., Gibson T. J., and Pines J. (2015) The ABBA motif binds APC/C activators and is shared by APC/C substrates and regulators. Dev. Cell 32, 358–372 10.1016/j.devcel.2015.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Diaz-Martinez L. A., Tian W., Li B., Warrington R., Jia L., Brautigam C. A., Luo X., and Yu H. (2015) The Cdc20-binding Phe box of the spindle checkpoint protein BubR1 maintains the mitotic checkpoint complex during mitosis. J. Biol. Chem. 290, 2431–2443 10.1074/jbc.M114.616490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lischetti T., Zhang G., Sedgwick G. G., Bolanos-Garcia V. M., and Nilsson J. (2014) The internal Cdc20 binding site in BubR1 facilitates both spindle assembly checkpoint signalling and silencing. Nat. Commun. 5, 5563 10.1038/ncomms6563 [DOI] [PubMed] [Google Scholar]
- 57. Kang J., Chen Y., Zhao Y., and Yu H. (2007) Autophosphorylation-dependent activation of human Mps1 is required for the spindle checkpoint. Proc. Natl. Acad. Sci. U.S.A. 104, 20232–20237 10.1073/pnas.0710519105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wassmann K., Liberal V., and Benezra R. (2003) Mad2 phosphorylation regulates its association with Mad1 and the APC/C. EMBO J. 22, 797–806 10.1093/emboj/cdg071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. St-Denis N., Gupta G. D., Lin Z. Y., Gonzalez-Badillo B., Veri A. O., Knight J. D. R., Rajendran D., Couzens A. L., Currie K. W., Tkach J. M., Cheung S. W. T., Pelletier L., and Gingras A. C. (2016) Phenotypic and interaction profiling of the human phosphatases identifies diverse mitotic regulators. Cell Rep. 17, 2488–2501 10.1016/j.celrep.2016.10.078 [DOI] [PubMed] [Google Scholar]
- 60. Moasser M. M., Srethapakdi M., Sachar K. S., Kraker A. J., and Rosen N. (1999) Inhibition of Src kinases by a selective tyrosine kinase inhibitor causes mitotic arrest. Cancer Res. 59, 6145–6152 [PubMed] [Google Scholar]
- 61. Wolanin K., Magalska A., Kusio-Kobialka M., Podszywalow-Bartnicka P., Vejda S., McKenna S. L., Mosieniak G., Sikora E., and Piwocka K. (2010) Expression of oncogenic kinase Bcr-Abl impairs mitotic checkpoint and promotes aberrant divisions and resistance to microtubule-targeting agents. Mol. Cancer Ther. 9, 1328–1338 10.1158/1535-7163.MCT-09-0936 [DOI] [PubMed] [Google Scholar]
- 62. Caron D., Byrne D. P., Thebault P., Soulet D., Landry C. R., Eyers P. A., and Elowe S. (2016) Mitotic phosphotyrosine network analysis reveals that tyrosine phosphorylation regulates Polo-like kinase 1 (PLK1). Sci. Signal. 9, rs14 10.1126/scisignal.aah3525 [DOI] [PubMed] [Google Scholar]
- 63. Elowe S. (2017) Tyr(less) kinase signaling during mitosis. Cell Cycle 16, 746–748 10.1080/15384101.2017.1302632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ji W., Arnst C., Tipton A. R., Bekier M. E. 2nd, Taylor W. R., Yen T. J., and Liu S. T. (2016) OTSSP167 abrogates mitotic checkpoint through inhibiting multiple mitotic kinases. PLoS One 11, e0153518 10.1371/journal.pone.0153518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kapust R. B., Tözsér J., Fox J. D., Anderson D. E., Cherry S., Copeland T. D., and Waugh D. S. (2001) Tobacco etch virus protease: Mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 14, 993–1000 10.1093/protein/14.12.993 [DOI] [PubMed] [Google Scholar]
- 66. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Meth. 9, 671–675 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fang G. (2002) Checkpoint protein BubR1 acts synergistically with Mad2 to inhibit anaphase-promoting complex. Mol. Biol. Cell 13, 755–766 10.1091/mbc.01-09-0437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tang Z., Bharadwaj R., Li B., and Yu H. (2001) Mad2-Independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev. Cell 1, 227–237 10.1016/S1534-5807(01)00019-3 [DOI] [PubMed] [Google Scholar]
- 69. Howell B. J., Moree B., Farrar E. M., Stewart S., Fang G., and Salmon E. D. (2004) Spindle checkpoint protein dynamics at kinetochores in living cells. Curr. Biol. 14, 953–964 10.1016/j.cub.2004.05.053 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.