

Abstract
Cytochrome P450 (P450) 17A1 catalyzes the oxidations of progesterone and pregnenolone and is the major source of androgens. The enzyme catalyzes both 17α-hydroxylation and a subsequent 17α,20-lyase reaction, and several mechanisms have been proposed for the latter step. Zebrafish P450 17A2 catalyzes only the 17α-hydroxylations. We previously reported high similarity of the crystal structures of zebrafish P450 17A1 and 17A2 and human P450 17A1. Five residues near the heme, which differed, were changed. We also crystallized this five-residue zebrafish P450 17A1 mutant, and the active site still resembled the structure in the other proteins, with some important differences. These P450 17A1 and 17A2 mutants had catalytic profiles more similar to each other than did the wildtype proteins. Docking with these structures can explain several minor products, which require multiple enzyme conformations. The 17α-hydroperoxy (OOH) derivatives of the steroids were used as oxygen surrogates. Human P450 17A1 and zebrafish P450s 17A1 and P450 17A2 readily converted these to the lyase products in the absence of other proteins or cofactors (with catalytically competent kinetics) plus hydroxylated 17α-hydroxysteroids. The 17α-OOH results indicate that a “Compound I” (FeO3+) intermediate is capable of formation and can be used to rationalize the products. We conclude that zebrafish P450 17A2 is capable of lyase activity with the 17α-OOH steroids because it can achieve an appropriate conformation for lyase catalysis in this system that is precluded in the conventional reaction.
Keywords: biomimetic models
Introduction
The human genome contains 57 genes coding for cytochrome P450 (P450,6 CYP) enzymes. These are involved in the metabolism of a great variety of chemicals (1), including drugs, chemical carcinogens, fatty acids, eicosanoids, and fat-soluble vitamins (2). P450s also dominate the metabolism of steroids (2, 3), with P450s 11A1, 17A1, 21A2, 11B1, 11B2, and 19A1 plus four dehydrogenases accounting for most of the steps between cholesterol and the androgens, estrogens, glucocorticoids, and mineralocorticoids (3). P450 17A1 plays a critical role in steroid metabolism, converting both progesterone and pregnenolone to their 17α-OH products (Fig. 1), which have several roles. P450 17A1 also catalyzes 17α,20-lyase (cleavage, or desmolase) reactions, with the two 17α-OH steroids as substrates, yielding the androgen androstenedione (Andro, from 17α-OH progesterone) and dehydroepiandrosterone (DHEA, from 17α-OH pregnenolone), which is converted to Andro by a steroid dehydrogenase (3). At least 50 natural mutations in the enzyme are known to cause clinical deficiencies (4, 5). On the other hand, prostate cancer can be exacerbated by androgens, and one therapeutic approach is to target P450 17A1 to lower androgen production (6, 7).
Figure 1.

Major reactions catalyzed by P450 17A1 enzymes.
A number of questions in the field of P450 17A1 research have not been resolved. One problem is that the catalytic specificity of P450 17A1 varies among species, in terms of the balance between utilization of progesterone and pregnenolone and between 17α-hydroxylation and lyase activity (3, 8, 9). Cytochrome b5 (b5) stimulates P450 17A1 activity, and the extent to which the lyase reaction is stimulated varies among species, as well as the concentration of b5 in steroidogenic tissues and their individual zones (3, 10, 11). Another controversy is the chemical mechanism of the lyase reaction, for which both electrophilic (Compound I, FeO3+) and nucleophilic (Compound 0, ferric peroxide, Fe2+O2−) mechanisms have been proposed (12–17). A major question that we are trying to address is the molecular basis for the selective loss of the lyase activity, which is an inherent clinical problem (3, 4, 18–20) but a goal in prostate cancer therapy (21–24).
We have utilized a fish model discovered by Zhou et al. (25). Teleost (bony) fish have two genes, CYP17A1 and CYP17A2, which both code P450 subfamily 17A enzymes (∼50% identical) (Fig. S1). P450 17A2 catalyzes 17α-hydroxylation of both progesterone and pregnenolone but does not catalyze the lyase reaction; zebrafish P450 17A1 catalyzes both 17α-hydroxylation and the lyase activities, as in the case of the mammalian enzymes (26). Understanding the molecular basis of the lack of lyase activity of zebrafish P450 17A2 may reveal the basis of the lyase deficiency in humans. We crystallized both zebrafish P450s 17A1 and 17A2, in the presence of the inhibitor abiraterone (26), but the structures of the active sites were very similar to each other and to that of human P450 17A1 bound to the same ligand (5). We attributed the difference in lyase activity of P450s 17A1 and 17A2 to either of two possibilities: either (i) four amino acid differences in the heme binding region or a difference at residue 369 (Arg-369 in zebrafish P450 17A1, corresponding to Arg-358 in human P450 17A1 and Cys-358 in zebrafish P450 17A2) are responsible for a change in the mechanism, ablating lyase activity, or (ii) the X-ray crystal structures are not revealing alternate conformational states of P450 17A1 that are needed for lyase activity (and are not available for P450 17A2) (26). The zebrafish P450 17A1 Arg-369/human P450 17A1 Arg-358 site is particularly interesting in that the human R358Q variant has been known to lead to selective loss of the lyase activity in patients for >20 years (18). This phenomenon has been suggested to be due to a selective loss of b5 binding, in that some lyase activity could be restored by the addition of high concentrations of purified b5 to human P450 17A1 expressed in a heterologous yeast system (19). The results of both covalent cross-linking (27) and NMR (28) studies also suggest an interaction of human P450 17A1 Arg-358 with b5. However, the same region has been implicated in binding of NADPH-P450 reductase in NMR studies, and this protein and b5 compete (28, 29).
We now report zebrafish P450 17A1 and 17A2 mutants in which we have made the corresponding Arg/Cys substitution and the other four residue changes based on the above information (26) (Fig. S1). The X-ray crystal structure of the five-residue P450 17A1 mutant was determined and was also utilized in docking studies. We had previously shown that human P450 17A1 could exhibit lyase activity in a biomimetic model with the established “oxygen surrogate” iodosylbenzene, which is considered to generate either Compound I or a related species (17). We have re-examined reactions with 17α-OOH steroids, proposed early by Tan et al. (30–32) to be intermediates in the lyase mechanism. The 17α-OOH steroids were converted to lyase products and hydroxylation products by human P450 17A1 at very catalytically competent rates. The 17α-OOH steroids were also converted to lyase products by zebrafish P450s 17A1 and 17A2 as well as a human P450 R358C mutant that is devoid of lyase activity in the usual assay system. We interpret the 17α-OOH reactions in terms of a Compound I or related mechanism. P450 17A2 and other normally lyase-inactive P450 17A1 mutants appear to have inherent features capable of lyase activity, but they are not manifested in the normal catalytic reaction, even in the presence of b5.
Results
Identification of more reaction products of zebrafish P450 17A1 and 17A2
In our previous work with zebrafish P450 17A2 (33), we reported only the 17α-OH steroids formed from progesterone and pregnenolone and concluded that they were final products. Zebrafish P450 17A1 formed the lyase products Andro and DHEA from 17α-OH progesterone and 17α-OH pregnenolone, respectively. P450 17A2 did not form any lyase products with either 17α-OH steroid (33).
We increased the enzyme concentrations and reaction time and identified more products, including 17α,21-(OH)2 progesterone (11-deoxycortisol), 16,17α-(OH)2 progesterone (algestone), 6β,16,17α-(OH)3 progesterone, 17α,21-(OH)2 pregnenolone, and 16,17α-(OH)2 pregnenolone from both zebrafish P450 17A1 and 17A2 (Fig. 2). These were identified by their mass spectra and by co-chromatography with the products we characterized in oxidations catalyzed by human P450 17A1 (17) plus a commercial sample of 11-deoxycortisol (17,21-(OH)2 progesterone) (Fig. S9).
Figure 2.

Oxidations catalyzed by human and zebrafish P450 enzymes (17). Products of the zebrafish P450s identified in this work include 17α,21-(OH)2 progesterone (11-deoxycortisol), 16,17α-(OH)2 progesterone (algestone), 6β,16,17α-(OH)3 progesterone, 17α,21-(OH)2 pregnenolone, and 16,17α-(OH)2 pregnenolone.
Two products derived from 17α-OH progesterone correspond to hydroxylation at the 21- and 16-positions. The 21-hydroxy product had not been identified in our previous work with human P450 17A1, but 17α,21-(OH)2 progesterone (11-deoxycortisol) co-chromatographed with a commercial standard and had a very similar mass spectrum (Fig. S9). In particular, the m/z 317 peak corresponds to the loss of -CH2OH, and the loss of the elements of H2O (probably 17α-OH) from this yields m/z 299 (positive-ion atmospheric pressure chemical ionization MS) (34). P450 17A1–catalyzed 21-hydroxylation activity of a steroid possessing a 3-keto-Δ4,5 skeleton has been observed previously in the 21-hydroxylation of progesterone by human P450 17A1 (Fig. 2) (35). A similar approach was used in the identification of the 16-hydroxylation product of 17α-OH progesterone (algestone; Fig. S10). The corresponding site of hydroxylation (carbon 16) for 17α-OH pregnenolone was identified by oxidizing the pregnenolone products with cholesterol oxidase to form the 17α-OH products mentioned above. In neither case could we ascertain the stereochemistry at the 16-position. Another product was identified as 6β,16,17α-(OH)3 progesterone by comparison (HPLC, MS) with the product formed by human P450 17A1 (17).
Site-directed mutagenesis
Although zebrafish P450 17A1 and 17A2 are only ∼50% identical (Fig. 2), their three-dimensional structures are very similar, particularly in the substrate-binding region (33). Thus, superimposition of the zebrafish P450 17A1 and 17A2 complexes with abiraterone results in a root mean square deviation (r.m.s.d.) between the two models of 1.49 Å (see Fig. 14 (C and D) in Ref. 33), with only minimal differences in the active sites.
We considered the residue Arg-358 (numbering system of human P450 17A1). This amino acid is Arg in both zebrafish (Arg-369) and human P450 17A1 (Arg-358), but it is Cys (Cys-358) in zebrafish P450 17A2 (33) (Fig. S1). Because the human P450 17A1 mutation R358Q results in preferential loss of lyase activity compared with 17α-hydroxylation (3, 18–20), we tested the hypothesis that changing this single residue to arginine could generate lyase activity in zebrafish P450 17A2.
Human P450 17A1-R358C had similar kcat and Km values and catalytic efficiency (kcat/Km) for progesterone 17α-hydroxylation compared with the wildtype enzyme (Table 1). Human P450 17A1-R358C had no detectable lyase activity with either 17α-OH progesterone or 17α-OH pregnenolone in the absence or presence of b5. The zebrafish P450 17A1-R369C mutant had only 3% of the 17α-OH progesterone lyase activity of the wildtype enzyme (Fig. S2) but retained about one-third of its progesterone 17α-hydroxylation catalytic efficiency (Table 1). Zebrafish P450 17A2-C358R also had reduced 17α-hydroxylation efficiency (∼⅓) (Table 1) but no detectable lyase activity with 17α-OH progesterone or 17α-OH pregnenolone.
Table 1.
Progesterone 17α-hydroxylation activities of P450 17A Arg/Cys mutant proteins
All rates were measured in the presence of an equimolar concentration of b5 relative to P450 (human b5 was used with human P450 17A1, and zebrafish b5 was used with zebrafish P450s), with a 2-fold molar excess of NADPH-P450 reductase and equimolar concentration of b5 when indicated.
| Human P450 17A1 |
Zebrafish P450 17A1 |
Zebrafish P450 17A2 |
||||
|---|---|---|---|---|---|---|
| WTa | R358C | WTb | R369C | WTb | C358R | |
| kcat, min−1 | 10.2 ± 0.6 | 8.3 ± 0.3 | 0.8 ± 0.1 | 0.37 ± 0.01 | 42 ± 1 | 23 ± 2 |
| Km, μm | 4.3 ± 0.4 | 3.6 ± 0.5 | 0.64 ± 0.06 | 0.90 ± 0.15 | 4.4 ± 0.6 | 7.7 ± 1.3 |
| kcat/Km, μm−1 min−1 | 2.4 ± 0.2 | 2.3 ± 0.3 | 1.2 ± 0.2 | 0.41 ± 0.07 | 9.4 ± 1.3 | 3.0 ± 0.6 |
Previous X-ray crystallography studies showed no major differences between residues in the substrate-binding sites of wildtype zebrafish P450s 17A1 and 17A2 (33). However, four differences were observed in the heme-binding region (Fig. S1). In addition to switching the Arg/Cys pair at positions (Arg) 369 (17A1) and (Cys) 358 (17A2) (Table 1), we added these four substitutions (zebrafish P450 17A1: D309E, V321T, L372M, and L407M; 17A2: E298D, T310D, T310V, M361L, and M396L), with the hypothesis that making the changes could collectively convert the activity of P450 17A1 to that of P450 17A2 and vice versa (Fig. 3, Table S1, and Figs. S3–S5). As reported previously (33), wildtype P450 17A2, in the presence of zebrafish b5, has higher catalytic efficiency for 17α-hydroxylation than P450 17A1 with both the substrates progesterone and pregnenolone (Fig. 3 (A and B), Table S1, and Fig. S3). Making the five substitutions (ETCMM) increased the 17α-hydroxylation catalytic efficiency of zebrafish P450 17A1 4–6-fold with both progesterone and pregnenolone as substrates (Fig. 3B). This was the case with or without b5; the presence of b5 reduced the 17α-hydroxylation catalytic efficiencies severalfold (Table S1). Conversely, making the P450 17A1-like mutations in zebrafish P450 17A2 (DVRLL) decreased the 17α-hydroxylation catalytic efficiencies severalfold toward both progesterone and pregnenolone (Fig. 3 (A and B) and Table S1). The catalytic efficiencies for the lyase activity were measured for the P450 17A1 mutant ETCMM (Fig. 3B, Table S1, and Fig. S4) with and without b5: 0.06 (without b5) and 1.1 (with b5) μm−1 min−1 and 0.17 (without b5) and 0.47 (with b5) μm−1 min−1 for 17α-OH progesterone and 17α-OH pregnenolone, respectively. The effects of b5 stimulation of lyase activity were ∼18- and ∼2.8-fold, respectively, for 17α-OH progesterone and 17α-OH pregnenolone, which contrasts with the case of wildtype zebrafish P450 17A1 (Fig. 3B and Table S1). In the wildtype P450 17A1, the b5 stimulation of lyase activities was consistent between 17α-OH progesterone and 17α-OH pregnenolone, with 3.5-fold stimulation for both substrates (0.43–1.5 and 0.78–2.7 μm−1 min−1 for 17α-OH progesterone and 17α-OH pregnenolone, respectively). These differences in the b5 effect indicate the complex nature of how b5 affects the lyase activity of P450 17A1 (i.e. b5 interaction with P450 17A1 is not just related to a single amino acid residue).
Figure 3.

Summary of steady-state kinetic parameters of zebrafish P450s 17A1 and 17A2 and the mutants. Parameters are shown for the reactions done in the absence and presence of zebrafish b5. No lyase activity was detected for zebrafish P450 17A2 or the mutant derived from it (DVRLL). A, 17α-hydroxylation with progesterone or pregnenolone as susbstrate; B, 17α,20-lyase activity with 17α-OH progesterone or 17α-OH pregnenolone as susbstrate. C, the substrate was 17α-OH progesterone, and the indicated three minor products were measured; D, the substrate was 17α-OH pregnenolone, and the indicated two minor products were measured. All values are catalytic efficiencies (kcat/Km ± S.D. (error bars)); see Tables S1 and S2 and Figs. S3–S8 for details regarding the substrate concentrations used and the error analysis for kcat and Km values.
Wildtype zebrafish P450 17A2 and its five-residue mutant (DVRLL) were also analyzed for the three hydroxylated products of 17α-OH progesterone and 17α-OH pregnenolone (Fig. 3 (C and D), Table S2, and Figs. S6–S8) (i.e. the 16- and 21-hydroxy and 6β,16-dihydroxy products; see above). The five mutations all increased the catalytic efficiencies for all three hydroxylations (Fig. 3 (C and D), Table S2, and Figs. S6–S8). The catalytic efficiencies of the hydroxylations were not so strongly affected by the addition of b5 (Fig. 3, C and D).
X-ray structure of the zebrafish P450 17A1-ETCMM mutant
To allow comparisons with the wildtype zebrafish P450 17A1 enzyme (33), we determined the crystal structure of the zebrafish P450 17A1-ETCMM mutant protein in the presence of the drug abiraterone at 3.03 Å resolution (Fig. 4 and Table 2). As in the case of the wildtype protein complex with inhibitor, crystals of the ETCMM mutant complex with abiraterone were grown using in situ proteolysis with subtilisin (37). However, unlike the crystallization experiments with wildtype zebrafish P450 17A1 that were conducted at room temperature, crystals for the mutant were grown at 4 °C, and their space (P4122) differs from that of the wildtype protein (P3121). The structure of the mutant protein complex was solved by molecular replacement using the coordinates of one of the two wildtype zebrafish P450 17A1 protein molecules minus water and ions as a search model. As in the case of the wildtype protein crystals, the tetragonal zebrafish P450 17A1-ETCMM mutant crystals contained two complexes per asymmetric unit. Molecule B could be completely traced with no interruptions in the chain between residues 39 and 508, and in molecule A, only four amino acids (residues 176–179) in the loop region between helices D and E are missing in the final model (Fig. 4A). Thus, a total of 935 residues from two complexes are included in the final model of the zebrafish P450 17A1-ETCMM structure (Table 2) as compared with 828 residues in the crystal structure of the wildtype protein (33). Whereas the C-terminal region is resolved up to residue 508 in both the wildtype and mutant crystals, the N-terminal region features an additional 44 residues in the latter (Fig. 4A). A shared property of the two crystals is the formation of a dimer that is stabilized by two disulfide bridges involving Cys-146 and Cys-437. The two independent molecules A and B in the zebrafish P450 17A1-ETCMM mutant crystal structure exhibit rather similar conformations with a root mean square deviation (r.m.s.d.) of only 0.67 Å for 465 Cα pairs (Fig. 4C). Conversely, the r.m.s.d. between the wildtype and mutant P450 17A1-abiraterone complexes is significantly higher (molecules B) and amounts to 2.16 Å for 415 Cα pairs. Therefore, the structures of the zebrafish P450 17A1 and 17A2 complexes with abiraterone exhibit a closer similarity (r.m.s.d. of 1.49 Å; see above) (33) than those of the zebrafish P450 17A1 wildtype and ETCMM mutant complexes.
Figure 4.

Crystal structure of zebrafish P450 17A1-ETCMM, bound to abiraterone. A, overall view of the complex with the P450 protein colored in rainbow style, from the N terminus (blue) to the C terminus (red). Individual α-helices are labeled, and every tenth residue is numbered. Heme carbon, nitrogen, and oxygen atoms are colored in black, blue, and red, respectively, and Fe3+ is shown as an orange sphere. Carbon and oxygen atoms of the abiraterone inhibitor are colored in magenta and red, respectively. B, quality of the omit 2Fo − Fc Fourier electron density (1.2σ level) around abiraterone. C, an overlay of the two complexes per crystallographic asymmetric unit reveals highly similar conformations of molecules A (beige ribbon) and B (light blue ribbon).
Table 2.
Selected crystal data, data collection, and refinement parameters for the P450 z17A1-ETCMM mutant abiraterone complex
| Data collection | |
| Wavelength (Å) | 0.9787 |
| Space group | P4122 |
| Resolution (outer shell) (Å) | 51.26–3.03 (3.11–3.03)a |
| Unit cell constants | |
| a = b, c (Å) | 104.92, 235.00 |
| α = β = γ(degrees) | 90 |
| Unique reflections | 26,303 (1,896) |
| Completeness (%) | 99.8 (99.7) |
| I/σ(I) | 14.0 (2.0) |
| Rmerge | 0.119 (0.959) |
| Rpim | 0.047 (0.371) |
| Redundancy | 7.2 (7.4) |
| Refinement | |
| Phasing method | Molecular replacement |
| Rwork | 0.192 (0.301)b |
| Rfree | 0.256 (0.395)b |
| No. of amino acids | 935c |
| No. of protein atoms | 7,390 |
| No. of heme/ligand atoms | 86/52 |
| Average B-factor, protein atoms (Å2) | 78.3 |
| Average B-factor, heme/ligand atoms (Å2) | 58.5/60.7 |
| Average B-factor, water/ions (Å2) | 57.8/89.6 |
| r.m.s.d. bond lengths (Å) | 0.011 |
| r.m.s.d. bond angles (degrees) | 1.6 |
| Ramachandran plot: favored/allowed/outlier residues | 859/59/11 |
| PDB code | 6B82 |
a Numbers in parentheses refer to the outer shell.
b Outer shell for refinement is 3.108–3.03 Å.
c For two independent molecules per asymmetric unit: molecule A, residues 39–175/180–507; molecule B, residues 39–508.
Four of the five mutated residues are ∼10 Å away from the iron center in the heme prosthetic group (Glu-309, Val-321, Met-372, Met-407), and Cys-369 is farther removed (∼15 Å) (Fig. 5A). The overlay of the wildtype and mutant complex structures demonstrates that the active site regions exhibit closely similar main and side chain conformations in the majority of cases. The side chain of Glu-309 is more extended as a consequence of the additional methylene group relative to aspartate in the P450 17A1 wildtype protein. As a result, the side chain of the adjacent Asp-124 shifts and rotates slightly, and the shortest distance between carboxylate oxygens of the two is 2.6 Å in the mutant complex (Fig. 5B, dashed line). The closest distance between the side chains of Asp-309 and Asp-124 in the wildtype complex is identical, and thus the mutation does not appear to create a clash. No basic residue is situated within a close range from these two residues, and it is possible that one of the carboxylates is protonated to prevent a Coulombic repulsion.
Figure 5.

Environments of the five mutated residues in the structure of the zebrafish P450 17A1-abiraterone complexes. A, overlay of wildtype zebrafish P450 17A1 (lilac ribbon, side chains shown in stick mode) (33) and zebrafish P450 17A1-ETCMM (light blue ribbon, side chains of mutated residues shown in ball and stick mode), with individual mutants labeled and the closest distance between a side chain atom from a mutated residue and the Fe3+ atom shown in Å. B–E, close-up views: D309E (B), V321T and L372M (C), R369C and L372M (D), and L407M (E). In B–E, side chains of mutated residues are highlighted in a ball and stick mode. Carbon atoms of side chains are colored lilac (wildtype) and light blue (mutant), and heme and inhibitor carbon atoms in the two structures are colored in an analogous fashion. Amino acids in the vicinity of mutated residues along with the mutant residue are labeled, and contacts with distances of <3.3 Å in the zebrafish P450 17A1-ETCMM mutant complex are indicated by thin solid lines.
The Val-321 in wildtype P450 17A1 and Thr-321 in the mutant protein superimpose neatly (Fig. 5C), indicating that the two residues are interchangeable, and insertion of a hydroxyl in place of the native methyl group in close vicinity of the vinyl moiety of heme (3.6 Å distance between the Oγ1 of threonine and the heme methylene carbon) is well tolerated. Val-377, adjacent to Thr-321, is slightly displaced relative to its position in the wildtype structure next to Val-321 (Fig. 5C). The distance between the Oγ1 of Thr-321 and Cγ2 of Val-377 in the mutant structure (4.2 Å) is slightly longer than the corresponding distance between the Cγ2 of Val-321 and Cγ2 of Val-377 in the structure of the wildtype protein (3.7 Å). The closest distance between Thr-321 (Cγ2) and Met-372 (Cγ; mutated from leucine) is 4.3 Å (Fig. 5C). The distance from the methyl carbon of the threonine to the methionine sulfur is 5.1 Å. The closest distance between the side chains of Val-321 and Leu-372 in the wildtype structure is rather similar, 4.2 Å, again indicative of only minor adjustments of residues that line the active site as a consequence of the mutations.
Compared with the above D309E, V321T, and L372M mutations, the R369C mutation is more drastic in that it replaces a longer, polar side chain with a short hydrophobic one and removes a positive charge. Remarkably, this mutation alone nearly abolished the lyase activity of zebrafish P450 17A1 (Fig. S2). In the structure of the abiraterone complex, the R369C mutation results in significant conformational adjustments of two neighboring side chains and the insertion of a water molecule that occupies a portion of the space vacated by the arginine side chain (Fig. 5D; in complex A the position of this water is occupied by a chloride ion). Mutation of Arg-369 flips the side chains of Glu-365 and Arg-358 around (the latter of which corresponds to the conserved Arg-347 of human P450 17A1, known to also be critical to lyase function (20)), such that the latter two engage in a salt bridge in the mutant structure. In addition, the guanidino moiety of Arg-358 also forms a hydrogen bond to the water newly present at this site. In the structure of the wildtype protein complex, the headgroup of Glu-365 is inserted between the guanidino moieties of Arg-369 and Arg-358 (Fig. 5D, dashed lines), but the distances are too long (>4 Å) to allow formation of salt bridges. Instead, the particular arrangement of the three amino acids produces electrostatically favorable interactions that tie together helices J′ and K (Fig. 5D). Thus, the R369C mutation may affect the relative mobility of these helices, one of which cradles heme (K). The side chain of Lys-460 from a third helix (L) that juts into the region affected by the R369C mutation also alters its conformation slightly in the mutant structure relative to the situation in the wildtype complex. However, a salt bridge between Lys-460 and Glu-456 is conserved in both structures (Fig. 5D). Residues of the K and L helices also interact with helix I, which spans one side of the active site (Fig. 4, A and B). Fig. 5D also illustrates, together with Fig. 5C, how the L372M mutation slightly pushes away the Phe-446 phenyl ring (the methionine sulfur stacks onto the edge of the aromatic moiety).
The L407M mutation is more benign compared with the above replacement of arginine by cysteine. Consistent with the similar spatial requirements and hydrophobic nature of leucine and methionine, the mostly hydrophobic residues (Leu, Ile, Val, Pro, and His) surrounding residue 407 show only minor adjustments in their side chain conformations (Fig. 5E). The closest distance between Met-407 and His-411 is 4 Å (S … Nϵ2). Importantly, in terms of the vicinity of the amino acid at position 407 and heme, the closest distances between Leu-407 and heme in the wildtype structure and Met-407 and heme in the mutant structure are very similar (4.7 and 4.5 Å, respectively).
Lack of conversion of progesterone to 17-O-acetyl testosterone
One piece of evidence offered for a ferric peroxide (Compound 0) mechanism is the reported conversion of progesterone to 17-O-acetyl testosterone by porcine testicular P450 17A1 (Fig. 6) (38, 39). Such a Baeyer–Villiger reaction is a derivative of the proposed FeO2− mechanism (12) and has also been proposed for the P450 51A1 lanosterol 14α-demethylation reaction (40). We conducted UPLC-UV assays with human P450 17A1 and progesterone (under the usual conditions with NADPH-P450 reductase, b5, NADPH, and O2) and could not detect any 17-O-acetyl testosterone (separable by HPLC), at a level of <0.15% of the 17α-OH progesterone formed (rate of formation <0.0013 min−1, corresponding to less than one turnover/13 h). Porcine P450 17A1 was not available for direct comparison, but this reaction cannot be universal in its occurrence among P450 17A1 enzymes. Thus, the previous studies with 17-O-acetyl testosterone (38, 39) cannot be used to support a general FeO2− Baeyer–Villiger mechanism (Fig. 6).
Figure 6.

Baeyer–Villiger reaction proposed for conversion of progesterone to testosterone 17-O-acetate (38, 39). This is a modification of a ferric peroxide mechanism (12).
Reaction of 17α-hydroperoxy steroids with P450 17A enzymes
17α-OOH pregnenolone and 17α-OOH progesterone have both been synthesized previously (41–43) and considered as possible intermediates in the lyase reactions (30–32), although these proposals can no longer be considered viable in light of current information about the catalytic mechanism (16, 17, 44). However, Tan et al. (30–32) had reported the rearrangement of the 17α-OOH steroids to lyase products using crude tissue extracts. We synthesized both 17α-OOH pregnenolone and 17α-OOH progesterone (Figs. S11 and S12) and added these biomimetic reagents to P450 17A enzymes, without NADPH or reductase, to test the rearrangement hypothesis proposed by Tan et al. (32) (Fig. 7).
Figure 7.

Hypothesized conversion of 17α-OOH steroids to lyase products (b) and proposed relationship to normal catalysis (a) (32).
The same products were observed with human P450 17A1 (Fig. 8, A and B) and with zebrafish P450s 17A1 and 17A2 (Fig. S15). Major products included the 17α-OH products, the lyase products (Andro and DHEA), and the 16,17-(OH)2 steroid products (Fig. S15). The identities of DHEA and Andro (Fig. 8B) were confirmed by LC-MS (Figs. S13 and S14).
Figure 8.

Conversion of 17α-OOH pregnenolone to DHEA by human P450 17A1. A, HPLC of standard 17α-OOH pregnenolone, 17α-OH pregnenolone, and DHEA, showing tR for elution. B, chromatograms of reaction of 25 μm 17α-OOH pregnenolone without (green) or with (blue) 60 nm human P450 17A1 after 150 s. Absorbance (relative units) was integrated over the UV range (200–350 nm). See “Experimental procedures” for other details regarding chromatography. C, rate of conversion of 17α-OOH pregnenolone to DHEA, with or without human b5. D, rate of conversion of 17α-OOH progesterone to Andro. The points indicate single assays done at each time.
The lyase reactions were rapid with human P450 17A1 (without NADPH or reductase), with initial rates of > 120 min−1 (Fig. 8, C and D), more than fast enough to be catalytically competent (cf. kcat = 4.3 and 1.1 min−1 for 17α-OH progesterone and 17α-OH pregnenolone in the normal NADPH-supported reactions, respectively) (36). No further acceleration of the reactions was seen with b5 (Fig. 8C), which is very stimulatory in the normal NADPH-P450 reductase-supported reaction (36, 45) but apparently without the involvement of electron transfer (12, 46).
With both 17α-OOH pregnenolone and 17α-OOH progesterone, the lyase reactions were observed with zebrafish P450 17A2 (Figs. S15 and S16). These had never been shown in the NADPH-P450 reductase-supported reaction (33), even in the presence of b5. In other experiments (data not shown), the human P450 17A1-R358C and zebrafish P450 17A1-R369C mutants, devoid of lyase activities in the usual NADPH-supported reactions, showed lyase activity with both 17α-OOH pregnenolone and 17α-OOH progesterone. This was not stimulated by the addition of b5. The rates of formation were about one-half of those observed for the wildtype enzymes. We also saw similar lyase activity with the zebrafish P450 17A2-C358R mutant.
None of the observed products corresponded to 21-hydroxylation (i.e. 17α-,21-(OH)2 progesterone (11-deoxycortisol) or pregnenolone (11-deoxycorticosterone), the former of which had been reported in the reaction of 17α-OOH progesterone with bovine adrenal cortex microsomes (31) (and was a product of 17α-OH steroids in the usual NADPH-dependent assay (Fig. 3C, Table S2))). The major unidentified products (Fig. S15) all had mass spectra with the same parent ions as the 17α-OOH steroids that they were derived from and are proposed to be hydroxylated 17α-OH steroids. Attempts to define the structures of the three major peaks have not been successful yet due to the limited amount of material; we obtained NMR spectra of three peaks, and in two of these, the chemical shift of the 18-CH3 protons was shifted downfield, but we were unable to make an unambiguous assignment. Conceivably, epoxides could also be derived from the unsaturated moieties in the steroids, or alternatively, the structures of these products could be rationalized as hemiketals (which would form if the 17-hydroperoxide bond cleaved and either the C17–C16 bond or the C17–C13 bond were cleaved to generate a C17-radical that could oxygen-rebound to the Compound II iron hydroxide intermediate).
Discussion
One of the most interesting questions about P450 17A1 is the basis of the 17α,20-lyase activity, and a major issue is why some single base variants selectively lose this activity (3, 18, 20). We have been studying zebrafish P450 17A2 as a means of addressing the problem. We now show that zebrafish P450s 17A1 and 17A2 have multiple hydroxylation activities and, in addition to 17α-hydroxylation, are able to form the 16- and 21-hydroxylated products from 17α-OH progesterone and 17α-OH pregnenolone plus 6β,16,17α-(OH)3 progesterone.
The five mutations we made based on the crystal structures (26) rendered zebrafish P450 17A1 and 17A2 more similar to each other. The mutations increased the 17α-hydroxylation activity of zebrafish P450 17A1 and reduced the 17α-hydroxylation activity of P450 17A2 (Fig. 3). The changes also decreased the lyase activity of P450 17A1 (Fig. 3 (A and B), Fig. S4, and Table S1). The R369C mutation nearly ablated the lyase activity of zebrafish P450 17A1 (Fig. S2), but the inverse change (C358R) in P450 17A2 (Fig. S1) did not result in any detectable lyase activity. Further, the five amino acid changes (P450 17A2-DVRLL) did not impart lyase activity.
The structure of zebrafish P450 17A1-ETCMM determined with crystals grown at 4 °C and in the presence of subtilisin yielded a model with more than 40 additional amino acids on the N-terminal side compared with the structure of wildtype zebrafish P450 17A1 (26). The region between residues 39 and 82 entails a 22-residue loop, an α-helix with three full turns, and a six-residue β-strand. The latter forms an antiparallel sheet with amino acids 84–90, which were present in the previous model (Fig. 4A). The likely reason for the different length of the protein trapped in crystals of zebrafish P450 17A1-ETCMM is that in situ proteolysis with subtilisin progressed more slowly at the lower temperature (wildtype crystals had been grown at room temperature). Loop regions that were absent in the model of the wildtype zebrafish P450 17A1 protein could be completely built in the case of complex B in the structure of the mutant. Missing loops in the former were not a consequence of subtilisin action but probably could not be traced in the electron density due to locally increased mobility of the chain (the resolution of the wildtype zebrafish P450 17A1 structure was also somewhat lower (26)). In addition, altered packing interactions resulting from the difference in space group symmetry between the zebrafish wildtype and mutant P4540 17A1 may have resulted in some loop regions becoming more ordered. However, the formation of two disulfide bridges between the two independent abiraterone complexes per crystallographic asymmetric unit is a feature that is conserved in the zebrafish wildtype P450 17A1 and P450 17A1-ETCMM mutant structures. Overlays of the structures of the abiraterone complexes of zebrafish wildtype P450s 17A1 and 17A2 (26) and wildtype P450 17A1 and P450 17A1-ETCMM (this work) reveal a smaller deviation between the former pair.
Despite these conformational differences in the overall structures of zebrafish wildtype P450 17A1 and the ETCMM quintuple mutant (r.m.s.d. of 2.16 Å), the active site regions exhibit only minor differences (Fig. 5A). Two of the mutations are rather conservative (L372M and L407M), and inspection of the structural adjustments in the immediate vicinities of the newly introduced methionines confirms that these are indeed rather minor (Fig. 5, C–E). Even in the cases of the D309E (Fig. 5B) and V321T (Fig. 5C) mutations, comparison of the zebrafish P450 17A1 and ETCMM mutant structures reveals no significant movements of these residues or formation of novel interactions. The longer glutamate side chain at position 309 is accommodated by a slight repositioning of the carboxylate moiety from Asp-124, without loss of the close approach between the two. Similarly, the orientation of the Thr-321 side chain barely changes relative to that of Val-321 in the wildtype structure. Nevertheless, introduction of these four residues that are all some 10 Å or less away from the Fe3+ center (shortest contact involving a side chain atom; Fig. 5A) from the zebrafish P450 17A2 into the P450 17A1 protein improves the hydroxylation activity of the latter. In turn, transplanting the four 17A1 residues at positions 309, 321, 372, and 407 into the P450 17A2 protein lowers the hydroxylation activity of the latter. Clearly, the proximity of these amino acids to the heme group affects the relative activity that is shared between the P450 17A1 and 17A2 enzymes. The static image provided by the structures may not allow us to pinpoint the actual reason(s) for the observed changes in hydroxylation activity. More subtle differences in direct contacts to heme atoms, caused by the four above mutations that may affect the iron center and possibly have consequences also at the level of the dynamic behavior of the two enzymes, obviously have a significant effect on their hydroxylation activities.
By comparison, residue 369 (zebrafish P450 17A1-R369C mutant) has no direct interaction with heme, yet it causes loss of lyase activity with zebrafish P450 17A1 (Fig. S2) (also true with human P450 17A1 Arg-358). The arginine sits adjacent to the junction of helices J′, K, and L (Fig. 5D), whereby the two latter stabilize heme and interact with the long I helix that forms one wall of the active site. Lys-460, from helix L, is directly adjacent to Arg-369 and is just six residues downstream from Cys-453, which is coordinated to the iron center. Although the arginine is farther removed from heme than the other four mutated residues, it appears to nevertheless occupy a key position, and its mutation to cysteine changes the electrostatics at a three-helix junction. Thus, the mutation may affect the relative mobility of these helices and most likely also alter the dynamics of the interaction of helices K and L with helix I, which forms a backdrop for the substrates. Importantly, the corresponding arginine in human 17A1 (Arg-358) was mapped to the binding region with b5 (47). Indeed, inspection of a computational model of the human 17A1-b5 complex based on a cross-linking/mass spectrometric analysis (27, 48) reveals that Arg-358 is in proximity to Ser-43, Glu-68, and Glu-69 of b5 (Fig. S17). Therefore, beyond stabilizing internal interactions in 17A1, Arg-369 (human 17A1–Arg-358) sits at the interface with b5 that enhances the lyase activity. The Arg → Cys mutation would be expected to significantly alter the 17A1-b5 binding interface.
In our earlier study on zebrafish P450 17A enzymes (26), we did not find additional products, other than those depicted in Fig. 1. Secondary products were seen here, similar to those observed with human P450 17A1 (17, 36), after raising the enzyme concentrations and reaction time (Fig. 2). These are minor products, as seen by the lower rates of formation (Fig. 3 (C and D) and Table S2), and we do not know whether any have physiological activities. 16α-OH progesterone has been detected in vivo in human serum (49). We used molecular mechanics to calculate models of the complexes between zebrafish P450 17A1-ETCMM and mono-, di-, and trihydroxyprogesterones as well as the mono- and dihydroxypregnenolones (Fig. 9). In all cases, these substrates can easily be accommodated in the active site, using as guides the orientations of abiraterone in zebrafish P450 17A1 (wildtype and ETCMM mutant; this work, Fig. 9A) and progesterone in the wildtype zebrafish P450 17A2 complex structures (26) as well as the structures of human P450 17A1-substrate complexes (50). Taking into consideration sterics and hydrogen bond interactions, all manually built models could be quickly relaxed with the Amber force field in the UCSF Chimera suite (51) (see “Experimental procedures”) and relying on repeated rounds of steepest descent and conjugate gradient energy minimization approaches. In particular, the 16α-OH oxygen in the dihydroxy products assumes a position that lies only moderately farther way from Fe3+ than the 17α-OH oxygen: 4.5 versus 4.0 Å (progesterone) and 4.9 versus 4.4 Å (pregnenolone), respectively. As well, in the case of the trihydroxyprogesterone product, the 6β-OH oxygen is situated 5 Å from the metal ion. Therefore, these models are qualitatively consistent with the experimentally established formation of additional products catalyzed by the zebrafish P450 17A1 wildtype and ETCMM mutant enzymes.
Figure 9.
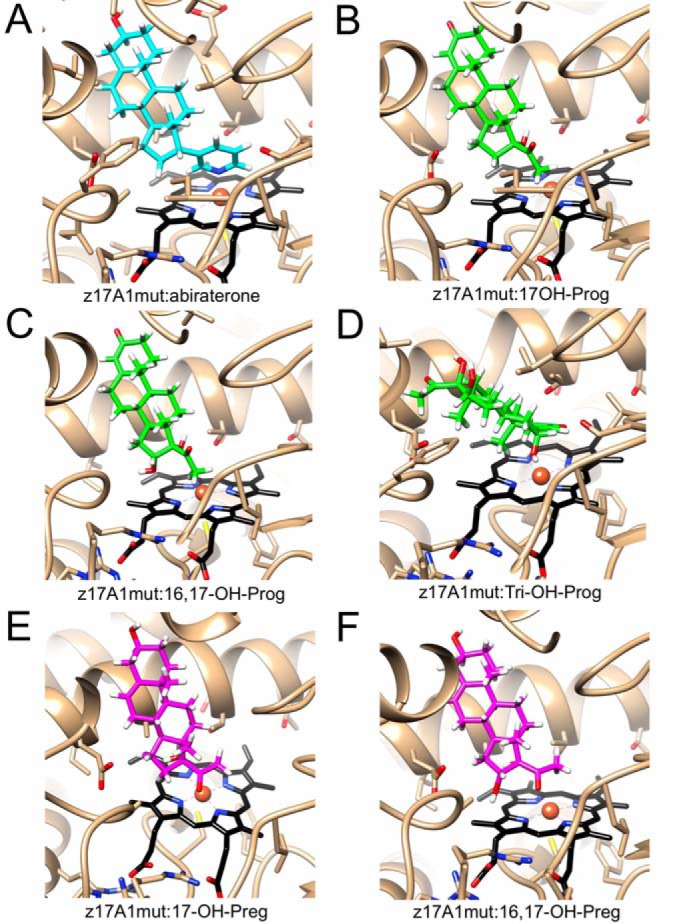
Docking of mono-, di-, and trihydroxyprogesterone and mono- and dihydroxypregnenolone molecules in zebrafish P450 17A1-ETCMM to rationalize the observed products. A, crystal structure of zebrafish P450 17A1-ETCMM (z17A1mut) with abiraterone (cyan carbon atoms). B–F, models docked (in z17A1mut): 17α-OH progesterone (B), 16α,17α-(OH)2 progesterone (C), 6β,16α,17α-(OH)3 progesterone (D), 17α-OH pregnenolone (E), and 16α,17α-(OH)2 pregnenolone (F). The active site is shown in roughly the same orientation in all six panels, and hydroxylated progesterones and pregnenolones are depicted with carbon atoms colored in green and magenta, respectively. Hydrogen atoms (calculated positions) of inhibitor and substrate/product molecules are colored in white.
17α-OOH steroids have previously been considered as intermediates in androgen synthesis (30–32), but there is no evidence that these are formed, and a dioxygenase reaction to generate them has never been reported. Nevertheless, the original conclusions reached about P450-catalyzed rearrangements of the synthetic hydroperoxides (done with crude beef adrenal and rat testis preparations) appear to be valid (30–32). The rates of formation of the lyase products (Fig. 8 (C and D) and Fig. S16) are more than fast enough to be consistent with mechanistic interpretations, which can be raised as a criticism of our earlier biomimetic work with iodosylbenzene (17).
17α-OH steroids and several hydroxylated 17α-OH steroids were generated in the 17α-OOH reactions (Fig. S15). These are relevant to the mechanism of the 17α-OOH reactions and are not explained by mechanism A or C in Fig. S15. In mechanism A (Fig. 10), the only products would result from lyase cleavage, and neither 17α-OH nor other hydroxylated steroids are explained. Mechanism C, involving homolytic cleavage of the hydroperoxide, can explain the lyase reactions. However, like mechanism A, it does not explain the 17α-OH products or the hydroxylated 17α-OH steroids (e.g. 16-OH and others; Fig. S15). Compound II would not be expected to do methylene hydroxylations. Mechanism B involves heterolytic cleavage of the hydroperoxide to form Compound I. Mechanism Bai readily explains 16-hydroxylation. Mechanism Baii can explain the lyase reaction. A test for this hypothesis would involve using 16-d2 17α-OOH steroids and checking for loss of deuterium label in the DHEA/Andro formed, but we have not prepared this material (a similar experiment could be done with 16-d2 steroids in the normal NADPH-supported reaction). Mechanism Bb involves abstraction of a hydrogen atom from the alcohol of the 17α-OH steroid. Bonomo et al. (52) have argued against such a mechanism based on density functional theory calculations (with human P450 17A1). Mechanism Bc involves nucleophilic attack of the 17α-OH oxygen on Compound I (not considered in Bonomo et al. (52)), as proposed in Fig. 7, plus a dioxetane intermediate originally proposed by Tan and Rousseau (32) (mechanism A, also Fig. 7). In principle, the dioxetane mechanism should yield chemiluminescence (53, 54). We have not looked for chemiluminescence; the expected energy yield would be expected to be both low and transient.
Figure 10.

Possible pathways for decomposition of 17α-OOH steroids. A, dioxetane mechanism. B, formation of Compound I (FeO3+) by heterolytic cleavage of 17α-OOH steroids. The a reactions are the result of C–H hydrogen abstraction reactions, and reactions similar to ai are proposed to result in the observed 6β- and other hydroxylations (Fig. 2, Table S2, and Figs. S5–S7). Reaction b involves abstraction of a hydrogen atom from the 17α-OH group and leads to the lyase product. Reaction c results from the reaction of the nucleophilic 17α-alcohol with Compound I and parallels the dioxetane mechanism in A. C, homolytic scission of the 17α-OOH steroid yields Compound II. The mechanism can explain the lyase products but not the hydroxylations.
We propose that the products are dependent upon Compound I (FeO3+) chemistry, and our overall results imply that Compound I (or an equivalent) is formed and has the capability of producing the lyase reactions. Thus, these results, coupled with our earlier work with the oxygen surrogate iodosylbenzene (17), provide evidence that P450 17A1 Compound I (which must be made, to rationalize the many hydroxylation reactions (Fig. 11)) is capable of lyase reaction chemistry. However, our work, as mentioned earlier (17), does not indicate the extent to which Compound I (FeO3+) and Compound 0 (Fe2+O2−) reactions contribute to the normal enzymatic lyase reaction.
Figure 11.

Possible catalytic mechanisms for P450 17A1 17α,20-lyase reactions. See Fig. 10 and text for details.
We conclude that P450 17A2 has a conformation such that it cannot position the 17α-OH group for proper attack by Compound I or Compound 0 (i.e. a conformation to facilitate the reactions is not available to P450 17A2). However, a possible conformation(s) must exist for the productive interaction of the heme ferric iron with the 17α-OOH steroids to generate Compound I (Fig. 9). b5 is not needed to facilitate this latter biomimetic reaction, but in the normal 17α-OH steroid/NADPH-based reaction, even the presence of b5 is not sufficient to yield lyase activity for P450 17A2 or the P450 17A2-DVRLL mutant.
Overall, we conclude that zebrafish P450s 17A1 and 17A2, like human P450 17A1 (17), form a number of minor hydroxylation products (Fig. 2). Productive juxtapositions in the active site can be rationalized in the crystal structure (Fig. 9). Site-directed mutations of zebrafish P450s 17A1 and 17A2, based on their crystal structures (26), alter the catalytic selectivity of the enzymes to be more like each other (Fig. 3, Fig. S1, and Table S1). However, none of the mutations (Fig. S1) yielded lyase activity with P450 17A2 (Table S1). The effects of the four substitutions near the heme periphery were not as dramatic as those resulting from mutation of a more distant arginine (Arg-358 in human P450 17A2, Arg-369 in zebrafish P450 17A1). However, changing Cys-358 in zebrafish P450 17A2 to arginine did not impart lyase activity. The interpretation of these site-directed mutagenesis studies is complex in that the Arg → Cys mutation attenuated progesterone 17α-hydroxylation activity in zebrafish P450 17A1 by about two-thirds, but so did the opposite mutation in zebrafish P450 17A2; the analogous mutation had no effect on progesterone 17α-hydroxylation activity catalytic efficiency in human P450 17A1 (Table 1).
Work with the biomimetic model 17α-OOH steroids showed facile conversion to multiple products, especially the lyase products, with both 17α-OOH progesterone and 17α-OOH pregnenolone (Fig. 8 and Figs. S15 and S16). The lyase products were formed even with P450s that did not have this capability in the normal NADPH-supported reactions. What does this mean? We interpret the results in the context of formation of Compound I (FeO3+), which can proceed to yield all of the observed products (Fig. 10, B and C). There are important caveats in the use of alkyl hydroperoxides as oxygen surrogates (55), in that some of the reactions observed with these can be attributed to reactions involving alkoxy radicals (56). However, the generation of hydroxylated steroid products argues against the involvement of alkoxy radicals, which would be unlikely to use such chemistry. We conclude (Fig. 11), in support of our earlier studies with the oxygen surrogate iodosylbenzene (17), that P450 17A enzymes can use Compound I chemistry (Fig. 11, B and C) to perform lyase reactions. However, our results cannot preclude involvement of Compound 0 (Fig. S11A) in the normal NADPH-based reaction.
Why do zebrafish P450 17A2 and human P450 Arg-358 mutants (Table 1) (18–20) fail to catalyze lyase reactions? At least with P450 17A2, the reason is more than one residue not being arginine (Cys-358) (Fig. S1). Substitution at that position has a more general effect on the structure, as discussed earlier (Figs. 4 and 5), and substituting one residue is not able to overcome a basic defect in the enzyme. However, the lyase-defunct enzymes are able to utilize the 17α-OOH steroids in a manner somewhat distinct from the normal reaction (Fig. 7). We have already concluded (36) that multiple conformations of P450 17A1 are involved in catalysis, in agreement with Scott and associates (22, 47), who have defined structural variants found both in the absence and presence of ligands in NMR and X-ray crystallography studies. Zebrafish P450 17A2 is capable of lyase activity with the 17α-OOH steroids in that it can achieve an appropriate conformation that appears to be precluded in the conventional reaction.
Experimental procedures
Chemicals
Synthesis of 17α-OOH pregnenolone
Preparation of 17α-OOH pregnenolone was done as described previously (43). Pregnenolone (316 mg, 1.0 mmol) was dissolved in anhydrous benzene (50 ml) and tetrahydrofuran (6 ml) in a Parr pressure reaction vessel. Potassium superoxide (284 mg, 4.0 mmol) and 18-crown-6 (449 mg, 1.7 mmol) were added, and the vessel was closed securely. The vessel was pressurized with 8 atmospheres of O2 gas (∼130 p.s.i., from a cylinder) and the mixture was stirred at 6 °C for 4 h. The reaction was quenched with cold 10% HCl (10 ml, aqueous). The product was extracted with diethyl ether, the organic phase was washed once with H2O and dried over anhydrous MgSO4, and the solvent was removed using a rotary evaporator. The crude product (17α-OOH pregnenolone) was purified by semipreparative HPLC; a 250 mm × 10 mm Phenomenex octadecylsilane C18 (5-μm) column was used at a flow rate of 3 ml min−1 with a linear gradient of 40–70% B (v/v) over 40 min (solvent A was H2O, and solvent B was CH3CN). A portion of the product (tR = 21 min) was collected (in ∼4% overall yield). 1H NMR (400 MHz, d6-DMSO, Fig. S11) δ 5.27 (d, 1H), 4.59 (d, 1H) 3.29–3.22 (m, 1H), 2.45–2.38 (1H), 2.17 (s, 3H), 2.14–2.08 (overlapped, 2H), 1.99–1.88 (overlapped, 2H), 1.77–1.16 (overlapped, 13H), 1.02–0.84 (overlapped, 2H), 0.92 (s, 3H), 0.54 (s, 3H) (57) (see also (58–60)). ESI HRMS: calculated for C21H33O4 [MH+-18] m/z 331.2268; found 331.2275 (Δ 2.1 ppm).
Treatment of the product with triphenylphosphine converted most of the product to 17α-OH pregnenolone, as judged by co-chromatography on UPLC (data not shown).
Synthesis of 17α-OOH progesterone
Preparation of 17α-OOH progesterone was done by adapting a previous procedure (41). Progesterone (1.5 g, 4.0 mmol) was dissolved in dioxane (150 ml) and H2SO4 (1.5 ml of 2% concentrated (v/v) H2SO4 in dioxane) was added followed by ethyl orthoformate (4.5 ml, 27 mmol). The mixture was stirred at room temperature for 1.5 h, monitoring development of a (single) product spot by TLC. The reaction was quenched by the addition of pyridine (3 ml) and poured into 500 ml of H2O. The product (3-O-ethoxypregnenolone) was extracted with ethyl acetate, washed once with water and brine, and dried over MgSO4; the solvent was removed using a rotary evaporator. The crude product was purified by silica gel chromatography (20% (v/v) ethyl acetate in hexanes). After removal of the solvent, the resulting product (322 mg, 0.90 mmol) was added to a mixture of potassium tert-butoxide (1.0 g, 9.0 mmol) in tert-butanol (20 ml). The mixture was stirred at room temperature while bubbling O2 gas directly in the solvent. After about 40 min, the tert-butanol began to freeze; 8 ml of tetrahydrofuran was added, and the reaction was continued with stirring for a total of 4 h. The reaction was diluted with H2O (500 ml) and extracted with ethyl acetate. The organic phase was washed twice with H2O, followed by brine, and dried over anhydrous MgSO4. The solvent was removed using a rotary evaporator. The crude solid was dissolved in warm CH3OH (12 ml), and glacial acetic acid was added (12 ml) to cleave the 3-O-ethoxy group. The mixture was stirred at room temperature for 3 h, concentrated using a rotary evaporator, and poured into H2O to form an off-white precipitate. The crude product (17α-OOH progesterone) was purified by silica gel chromatography (20% (v/v) ethyl acetate in hexanes). The product was further purified with a semipreparative 250 mm × 10 mm Phenomenex octadecylsilane C18 column (5 μm, linear gradient of 40–70% (v/v) over 40 min; solvent A was H2O, and solvent B was CH3CN). The product (tR = 22 min) was collected in ∼12% overall yield. 1H NMR (400 MHz, d6-DMSO, Fig. S12) δ 5.62 (s, 1H), 2.45–2.35 (m, 3H) 2.25–2.13 (m, 2H), 2.17 (s, 3H), 1.99–1.88 (overlapped, 2H), 1.81–1.55 (overlapped, 9H), 1.44–1.34 (overlapped, 2H), 1.13 (s, 3H), 1.01–0.78 (overlapped, 2H), 0.57 (s, 3H) (57) (see also Refs. 58–60). ESI HRMS: calculated for C21H33O4 [MH+] m/z 347.2217; found 347.2222 (Δ 1.4 ppm).
Enzymes
Recombinant human P450 17A1 (17), rat NADPH-P450 reductase (61), zebrafish P450 17A1 and 17A2 (26), human b5 (62), and zebrafish b5 (26) were expressed in Escherichia coli and purified as reported previously in detail.
Reactions
Incubations with P450 17A1
Generally, incubations were done at 37 °C in a shaking water bath. Time courses were performed using recombinant P450 17A1 in 100 mm potassium phosphate buffer (pH 7.4) in the presence of 30 μm l-α-1,2-dilauroyl-sn-glycero-3-phosphocholine (with or without b5), using general conditions described elsewhere (17, 26, 36). Reconstituted enzyme systems contained P450 17A1 or 17A2, NADPH-P450 reductase, and b5 (when indicated) added in 1:2:1 molar ratios (17, 26). Human b5 was used with human P450 17A1, and zebrafish b5 was used with zebrafish P450s 17A1 and 17A2. An NADPH-generating system was used, consisting of 10 mm glucose 6-phosphate, 0.5 mm NADP+, and 1 IU of yeast glucose-6-phosphate dehydrogenase/ml (63). In most of the catalytic assays done with NADPH-supported systems, the P450 concentration was 0.1–0.5 μm. Typically, the reaction time was 1–5 min.
In the experiments described with a search for conversion of progesterone to 17-O-acetyl testosterone (Fig. 6), the human P450 17A1 concentration was 1 μm (with 2 μm NADPH-P450 reductase, 1 μm b5, and 20 or 200 μm progesterone), and the incubations were run for as long as 60 min at 37 °C.
Alternatively, incubations were initiated by the addition of 17α-OOH pregnenolone or 17α-OOH progesterone to a final concentration of 25 μm in 0.5% C2H5OH, 0.5% (CH3)2SO (v/v). No NADPH-reductase or NADPH was used in these experiments.
Aliquots (500 μl) were removed and quenched by the addition of 2 ml of CH2Cl2, followed by centrifugation (3 × 103 × g, 10 min) to separate the layers. Aliquots (1.7 ml) of the CH2Cl2 phase were dried and resuspended in 100 μl of CH3CN and diluted with 150 μl of water for HPLC separation. Δ5-Steroids (from pregnenolone series) were oxidized to Δ4-steroids (with increased UV absorbance) for quantitation (HPLC, A244) using cholesterol oxidase, as described in more detail previously (26, 35).
For steady-state kinetic assays (reconstituted system with NADPH and NADPH-P450 reductase), plots of reaction velocity versus substrate concentration were fit to hyperbolic plots to estimate kcat and Km ± S.D., using Prism software (GraphPad, La Jolla, CA). The calculated S.D. values from Prism were used to estimate the S.D. values for the ratio kcat/Km (square root of the sum of the squares of S.D. values for the two parameters).
Chromatography
UPLC, with UV-visible absorbance detection, was routinely used for measurement of product formation. Products were generally separated using a Waters Acquity UPLC system and an Acquity UPLC-BEH octadecylsilane (C18) column (50 mm × 2.1 mm, 1.7 μm) with the following solvent system: Solvent A, 4.9% CH3CN, 0.1% HCO2H, 95% H2O; Solvent B, 95% CH3CN, 0.1% HCO2H, 4.9% H2O (v/v). Samples were injected with a 20-μl loop and separated with the following linear gradient for detection of lyase products: 20–80% B (with 17α-OOH pregnenolone) or 20–50% B (all v/v) (with 17α-OOH progesterone) over 10 min at a flow rate of 0.2 ml min−1. Absorbance (diode array, UV, 200–350 nm) was monitored and used for quantitation with external standards. The same UPLC systems were used to perform separations for mass spectrometry. A244 measurements were used for quantitation of Δ4-steroids.
For the analysis of any 17-O-acetyl testosterone formed (Fig. 6), the UPLC system utilized the same column but a gradient of increasing CH3OH in aqueous 0.1% (v/v) CF3CO2H: 0–5 min, 5% CH3OH; 5–10 min, 5–80% CH3OH; 10–11 min, 80–100% CH3OH; hold (all v/v).
MS and HRMS
LC-MS analyses of 17α-OOH-steroid transformation was done with samples by UPLC as described above. A Thermo LTQ-Orbitrap mass spectrometer was generally used in the FTMS mode at 7500 resolution. The mass spectrometer was operated in the ESI positive ion mode and was tuned with authentic pregnenolone. The tune settings were as follows: sheath gas flow rate, 30; auxiliary gas flow rate, 10; sweep gas flow rate, 0; capillary temperature, 275 °C; source voltage set to 5 kV; source current, 100 μA; capillary voltage, 29 V; tube lens, 80 V.
Crystallization of the zebrafish P450 17A1 ETCMM mutant protein in complex with abiraterone
To generate the P450 complex, the inhibitor abiraterone (dissolved in DMSO) was added to purified zebrafish P450 17A1-ETCMM mutant protein in a protein/arbiraterone molar ratio of 1:10. The complex was concentrated to 30 mg of protein/ml in 50 mm potassium phosphate buffer (pH 7.4) containing 10% glycerol (v/v), 0.1 mm dithiothreitol, 1 mm EDTA, 0.004% C12E9 detergent (w/v), and 125 mm NaCl. Before setting up crystallization droplets, subtilisin was added to the complex, such that the protease/P450 17A1 ratio (w/w) was 1:1000. Crystals were grown using the sitting-drop vapor diffusion technique by mixing equal volumes of complex solution and Crystal Screen 1 (Hampton Research, Aliso Viejo, CA) condition 21 (0.2 m magnesium acetate, 0.1 m sodium cacodylate (pH 6.5), and 30% (v/v) (±)-2-methyl-2,4-pentanediol) and equilibrating the droplets against 60 μl of reservoir solution at 4 °C. Crystals of the zebrafish P450 17A1-ETCMM-abiraterone complex appeared within a few days. One round of microseeding was performed to optimize the size of crystals. Wells containing microcrystals were washed with 10 μl of reservoir buffer, and the mixture was then transferred to a microcentrifuge tube with 40 μl of mother liquor and a PTFE Seed Bead (Hampton Research, Aliso Viejo, CA). After mixing the contents of the tube with a vortex device and diluting 1:1000 with mother liquor (v/v), 200 nl of the solution was mixed with 200 nl of protein for each drop. Larger crystals of the complex (with typical sizes of 0.2–0.25 mm) grew from microseeded droplets. Crystals were mounted in nylon loops and flash-frozen in liquid nitrogen.
Structure determination and refinement
X-ray diffraction data were collected on the 21-ID-F beam line of the Life Sciences Collaborative Access Team, located at Sector 21 of the Advanced Photon Source, Argonne National Laboratory (Argonne, IL), at a wavelength of 0.9787 Å and using a Mar225 CCD detector. Data were processed with the programs xia2/XDS (64) and Pointless/Aimless (64, 65). Selected crystal data and data collection statistics are summarized in Table 2. The structure of the zebrafish P450 17A1-ETCMM mutant in complex with abiraterone was solved by the molecular replacement technique with the program Phaser (66), using the native zebrafish P450 17A1 structure as the starting model (PDB code 4R1Z; protein alone) (33). The N- and C-terminal regions as well as several loops in the two independent molecules per asymmetric unit were manually built into the electron density maps using the program Coot (67). The structure was refined with the program Refmac 5 (68) in the CCP4 suite (70). After about 10 rounds of refinement, heme and abiraterone were built into regions of positive Fo − Fc difference Fourier electron density (2.5σ level), and the refinements were continued. Water molecules and ions (chloride and acetate) were added and refined. Selected refinement parameters are summarized in Table 2. All structural illustrations were prepared with the program UCSF Chimera (51).
Molecular docking
Models of complexes between the zebrafish P450 17A1-ETCMM mutant protein and 17α-OH progesterone, 17α-OH pregnenolone, 16,17α-(OH)2 progesterone, 16,17α-(OH)2 pregnenolone, and 6β,16,17α-(OH)3 progesterone (Fig. 9) were built with Coot (67) and taking into account the structures of the complex described in the present contribution, the complex between human P450 17A1 and pregnenolone (PDB code 4NKW) (50), and the complex between human P450 21A2 and 17α-OH progesterone (PDB code 5VBU) (69). Missing hydroxyl groups were added, and the substrate/product molecules were oriented in the active site such that hydroxyl oxygen atoms were at a distance of between ∼4 Å (16α-/17α-OH) and ∼5 Å (6β-OH) from the Fe(III) atom. Hydrogen atoms were added automatically, and the initial models were then refined using Amber ff14SB in UCSF Chimera (51), by taking into account steric and hydrogen bonding interactions and applying a minimization protocol of 100 steepest descent steps with a step size of 0.02 Å, followed by 10 conjugate gradient steps with a step size of 0.02 Å.
Author contributions
E.G., F.K.Y., M.E., and F.P.G. conceptualization; E.G., K.M.J., P.S.P., Z.W., and F.P.G. data curation; E.G., K.M.J., P.S.P., and M.E. formal analysis; E.G., K.M.J., P.S.P., T.T.N.P., W.Z., L.L., Z.W., and M.E. investigation; E.G., L.L., and F.P.G. methodology; E.G., K.M.J., F.K.Y., M.E., and F.P.G. writing-review and editing; P.S.P. visualization; M.E. supervision; M.E. and F.P.G. writing-original draft; M.E. and F.P.G. project administration; F.P.G. funding acquisition.
Supplementary Material
Acknowledgment
We thank K. Trisler for assistance in preparation of the manuscript.
This work was supported by National Institutes of Health Grants R01 GM118122 (to F. P. G.), R01 GM103937 (to M. E. and F. P. G.), and T32 ES007028 (to E. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2 and Figs. S1–S17.
The atomic coordinates and structure factors (code 6B82) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- P450
- cytochrome P450 (also CYP)
- b5
- cytochrome b5
- Andro
- androstenedione
- DHEA
- dehydroepiandrosterone
- ESI
- electrospray ionization
- HRMS
- high-resolution mass spectrometry
- OH
- hydroxy
- -OOH
- hydroperoxy
- P450 17A1-ETCMM
- zebrafish P450 17A1 containing the mutations D309E, V321T, R369C, L372M, and L407M (Fig. S1)
- P450 17A2-DVRLL
- zebrafish P450 17A2 containing the mutations E298D, T310V, C358R, M361L, and M396L (Fig. S1)
- r.m.s.d.
- root mean square deviation
- PDB
- Protein Data Bank.
References
- 1. Rendic S., and Guengerich F. P. (2015) Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals. Chem. Res. Toxicol. 28, 38–42 10.1021/tx500444e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guengerich F. P. (2015) Human cytochrome P450 enzymes. In Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th Ed (Ortiz de Montellano P. R., ed) pp. 523–785, Springer, New York [Google Scholar]
- 3. Auchus R. J., and Miller W. L. (2015) P450 enzymes in steroid processing. In Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th Ed (Ortiz de Montellano P. R., ed) pp. 851–879, Springer, New York [Google Scholar]
- 4. Auchus R. J. (2017) Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J. Steroid Biochem. Mol. Biol. 165, 71–78 10.1016/j.jsbmb.2016.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DeVore N. M., and Scott E. E. (2012) Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 482, 116–119 10.1038/nature10743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Njar V. C., and Brodie A. M. (1999) Inhibitors of 17α-hydroxylase/17,20-lyase (CYP17): potential agents for the treatment of prostate cancer. Curr. Pharm. Des. 5, 163–180 [PubMed] [Google Scholar]
- 7. Njar V. C. O., and Brodie A. M. H. (2015) Discovery and development of galeterone (TOK-001 or VN/124–1) for the treatment of all stages of prostate cancer. J. Med. Chem. 58, 2077–2087 10.1021/jm501239f [DOI] [PubMed] [Google Scholar]
- 8. Brock B. J., and Waterman M. R. (1999) Biochemical differences between rat and human cytochrome P450c17 support the different steroidogenic needs of these two species. Biochemistry 38, 1598–1606 10.1021/bi9821059 [DOI] [PubMed] [Google Scholar]
- 9. Gilep A. A., Sushko T. A., and Usanov S. A. (2011) At the crossroads of steroid hormone biosynthesis: the role, substrate specificity and evolutionary development of CYP17. Biochim. Biophys. Acta 1814, 200–209 10.1016/j.bbapap.2010.06.021 [DOI] [PubMed] [Google Scholar]
- 10. Mapes S., Corbin C. J., Tarantal A., and Conley A. (1999) The primate adrenal zona reticularis is defined by expression of cytochrome b5, 17α-hydroxylase/17,20-lyase cytochrome P450 (P450c17) and NADPH-cytochrome P450 reductase (reductase) but not 3β-hydroxysteroid dehydrogenase/Δ5–4 isomerase (3β-HSD). J. Clin. Endocrinol. Metab. 84, 3382–3385 10.1210/jcem.84.9.6105 [DOI] [PubMed] [Google Scholar]
- 11. Suzuki T., Sasano H., Takeyama J., Kaneko C., Freije W. A., Carr B. R., and Rainey W. E. (2000) Developmental changes in steroidogenic enzymes in human postnatal adrenal cortex: immunohistochemical studies. Clin. Endocrinol. 53, 739–747 10.1046/j.1365-2265.2000.01144.x [DOI] [PubMed] [Google Scholar]
- 12. Miller S. L., Wright J. N., Corina D. L., and Akhtar M. (1991) Mechanistic studies on pregnene side-chain cleavage enzyme (17α-hydroxylase-17,20-lyase) using 18O. J. Chem. Soc. Chem. Commun. 157–159 10.1039/C39910000157 [DOI] [Google Scholar]
- 13. Gregory M., Mak P. J., Sligar S. G., and Kincaid J. R. (2013) Differential hydrogen bonding in human CYP17 dictates hydroxylation versus lyase chemistry. Angew. Chem. Int. Ed. Engl. 52, 5342–5345 10.1002/anie.201300760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gregory M. C., Denisov I. G., Grinkova Y. V., Khatri Y., and Sligar S. G. (2013) Kinetic solvent isotope effect in human P450 CYP17A1-mediated androgen formation: evidence for a reactive peroxoanion intermediate. J. Am. Chem. Soc. 135, 16245–16247 10.1021/ja4086403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mak P. J., Gregory M. C., Denisov I. G., Sligar S. G., and Kincaid J. R. (2015) Unveiling the crucial intermediates in androgen production. Proc. Natl. Acad. Sci. U.S.A. 112, 15856–15861 10.1073/pnas.1519376113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yoshimoto F. K., and Auchus R. J. (2015) The diverse chemistry of cytochrome P450 17A1 (P450c17, CYP17A1). J. Steroid Biochem. Mol. Biol. 151, 52–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yoshimoto F. K., Gonzalez E., Auchus R. J., and Guengerich F. P. (2016) Mechanism of 17α,20-lyase and new hydroxylation reactions of human cytochrome P450 17A1: 18O labeling and oxygen surrogate evidence for a role of a perferryl oxygen. J. Biol. Chem. 291, 17143–17164 10.1074/jbc.M116.732966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Geller D. H., Auchus R. J., Mendonça B. B., and Miller W. L. (1997) The genetic and functional basis of isolated 17,20-lyase deficiency. Nat. Genet. 17, 201–205 10.1038/ng1097-201 [DOI] [PubMed] [Google Scholar]
- 19. Geller D. H., Auchus R. J., and Miller W. L. (1999) P450c17 mutations R347H and R358Q selectively disrupt 17,20-lyase activity by disrupting interactions with P450 oxidoreductase and cytochrome b5. Mol. Endocrinol. 13, 167–175 10.1210/mend.13.1.0219 [DOI] [PubMed] [Google Scholar]
- 20. Van Den Akker E. L., Koper J. W., Boehmer A. L., Themmen A. P., Verhoef-Post M., Timmerman M. A., Otten B. J., Drop S. L., and De Jong F. H. (2002) Differential inhibition of 17α-hydroxylase and 17,20-lyase activities by three novel missense CYP17 mutations identified in patients with P450c17 deficiency. J. Clin. Endocrinol. Metab. 87, 5714–5721 10.1210/jc.2001-011880 [DOI] [PubMed] [Google Scholar]
- 21. Yamaoka M., Hara T., Hitaka T., Kaku T., Takeuchi T., Takahashi J., Asahi S., Miki H., Tasaka A., and Kusaka M. (2012) Orteronel (TAK-700), a novel non-steroidal 17,20-lyase inhibitor: effects on steroid synthesis in human and monkey adrenal cells and serum steroid levels in cynomolgus monkeys. J. Steroid Biochem. Mol. Biol. 129, 115–128 10.1016/j.jsbmb.2012.01.001 [DOI] [PubMed] [Google Scholar]
- 22. Petrunak E. M., Rogers S. A., Aubé J., and Scott E. E. (2017) Structural and functional evaluation of clinically relevant inhibitors of steroidogenic cytochrome P450 17A1. Drug Metab. Dispos. 45, 635–645 10.1124/dmd.117.075317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bonomo S., Hansen C. H., Petrunak E. M., Scott E. E., Styrishave B., Jørgensen F. S., and Olsen L. (2016) Promising tools in prostate cancer research: selective non-steroidal cytochrome P450 17A1 inhibitors. Sci. Rep. 6, 29468 10.1038/srep29468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Attard G., Reid A. H., Auchus R. J., Hughes B. A., Cassidy A. M., Thompson E., Oommen N. B., Folkerd E., Dowsett M., Arlt W., and de Bono J. S. (2012) Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. J. Clin. Endocrinol. Metab. 97, 507–516 10.1210/jc.2011-2189 [DOI] [PubMed] [Google Scholar]
- 25. Zhou L. Y., Wang D. S., Kobayashi T., Yano A., Paul-Prasanth B., Suzuki A., Sakai F., and Nagahama Y. (2007) A novel type of P450c17 lacking the lyase activity is responsible for C21-steroid biosynthesis in the fish ovary and head kidney. Endocrinology 148, 4282–4291 10.1210/en.2007-0487 [DOI] [PubMed] [Google Scholar]
- 26. Pallan P. S., Nagy L. D., Lei L., Gonzalez E., Kramlinger V. M., Azumaya C. M., Wawrzak Z., Waterman M. R., Guengerich F. P., and Egli M. (2015) Structural and kinetic basis of steroid 17α,20-lyase activity in teleost fish cytochrome P450 17A1 and its absence in cytochrome P450 17A2. J. Biol. Chem. 290, 3248–3268 10.1074/jbc.M114.627265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peng H. M., Liu J., Forsberg S. E., Tran H. T., Anderson S. M., and Auchus R. J. (2014) Catalytically relevant electrostatic interactions of cytochrome P450c17 (CYP17A1) and cytochrome b5. J. Biol. Chem. 289, 33838–33849 10.1074/jbc.M114.608919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Estrada D. F., Laurence J. S., and Scott E. E. (2013) Substrate-modulated cytochrome P450 17A1 and cytochrome b5 interactions revealed by NMR. J. Biol. Chem. 288, 17008–17018 10.1074/jbc.M113.468926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Estrada D. F., Laurence J. S., and Scott E. E. (2016) Cytochrome P450 17A1 interactions with the FMN domain of its reductase as characterized by NMR. J. Biol. Chem. 291, 3990–4003 10.1074/jbc.M115.677294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tan L., Wang H. M., and Falardeau P. (1972) 17-Hydroperoxypregnenes. 3. Studies on their role as possible precursors in the biosynthesis of adrenosteroid hormones. Biochim. Biophys. Acta 260, 731–740 10.1016/0005-2760(72)90022-7 [DOI] [PubMed] [Google Scholar]
- 31. Tan L., Falardeau P., and Rousseau J. (1975) Evidence of an enzymatic 17α leads to 21 hydroxyl transfer in the biosynthesis of cortexolone from 17α-hydroperoxyprogesterone by adrenal cortex microsomes. Hormone Res. 6, 213–225 10.1159/000178695 [DOI] [PubMed] [Google Scholar]
- 32. Tan L., and Rousseau J. (1975) Anaerobic degradation of the progesterone side chain: a possible new pathway for the biosynthesis of androgen hormones. Biochem. Biophys. Res. Commun. 65, 1320–1326 10.1016/S0006-291X(75)80374-3 [DOI] [PubMed] [Google Scholar]
- 33. Pallan P. S., Wang C., Lei L., Yoshimoto F. K., Auchus R. J., Waterman M. R., Guengerich F. P., and Egli M. (2015) Human cytochrome P450 21A2, the major steroid 21-hydroxylase: structure of the enzyme·progesterone substrate complex and rate-limiting C–H bond cleavage. J. Biol. Chem. 290, 13128–13143 10.1074/jbc.M115.646307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kobayashi Y., Saiki K., and Watanabe F. (1993) Characteristics of mass fragmentation of steroids by atmospheric pressure chemical ionization-mass spectrometry. Biol. Pharm. Bull. 16, 1175–1178 10.1248/bpb.16.1175 [DOI] [PubMed] [Google Scholar]
- 35. Yoshimoto F. K., Zhou Y., Peng H. M., Stidd D., Yoshimoto J. A., Sharma K. K., Matthew S., and Auchus R. J. (2012) Minor activities and transition state properties of the human steroid hydroxylases cytochromes P450c17 and P450c21, from reactions observed with deuterium-labeled substrates. Biochemistry 51, 7064–7077 10.1021/bi300895w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gonzalez E., and Guengerich F. P. (2017) Kinetic processivity of the two-step oxidations of progesterone and pregnenolone to androgens by human cytochrome P450 17A1. J. Biol. Chem. 292, 13168–13185 10.1074/jbc.M117.794917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lei L., and Egli M. (2016) In situ proteolysis for crystallization of membrane bound cytochrome P450 17A1 and 17A2 proteins from zebrafish. Curr. Protoc. Protein Sci. 84, 29.16.21–29.16.19 10.1002/0471140864.ps2916s84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mak A. Y., and Swinney D. C. (1992) 17-O-Acetyltestosterone formation from progesterone in microsomes from pig testes: evidence for the Baeyer-Villiger rearrangement in androgen formation catalyzed by Cyp17. J. Am. Chem. Soc. 114, 8309–8310 10.1021/ja00047a063 [DOI] [Google Scholar]
- 39. Swinney D. C., and Mak A. Y. (1994) Androgen formation by cytochrome P450 CYP17: solvent isotope effect and pL studies suggest a role for protons in the regulation of oxene versus peroxide chemistry. Biochemistry 33, 2185–2190 10.1021/bi00174a027 [DOI] [PubMed] [Google Scholar]
- 40. Fischer R. T., Trzaskos J. M., Magolda R. L., Ko S. S., Brosz C. S., and Larsen B. (1991) Lanosterol 14α-methyl demethylase: isolation and characterization of the third metabolically generated oxidative demethylation intermediate. J. Biol. Chem. 266, 6124–6132 [PubMed] [Google Scholar]
- 41. Bailey E. J., Barton D. H. R., Elks J., and Templeton J. F. (1962) Compounds related to the steroid hormones. Part IX. Oxygenation of steroid ketones in strongly basic medium: a new method of preparation of 17α-hydroxypregnan-20-ones. J. Chem. Soc. 1578–1591 [Google Scholar]
- 42. Gardner J. N., Carlon F. E., and Gnoj O. (1968) One-step procedure for the preparation of tertiary α-ketols from the corresponding ketones. J. Org. Chem. 33, 3294–3297 10.1021/jo01272a055 [DOI] [PubMed] [Google Scholar]
- 43. Betancor C., Francisco C. G., Freire R., and Suarez E. (1988) The reaction of enols with superoxide anion radicals: preparation of tertiary α-ketols. J. Chem. Soc. Chem. Commun. 947–948 10.1039/C39880000947 [DOI] [Google Scholar]
- 44. Ortiz de Montellano P. R. (2015) Substrate oxidation. In Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th Ed (Ortiz de Montellano P. R., ed) pp. 111–176, Springer, New York [Google Scholar]
- 45. Katagiri M., Kagawa N., and Waterman M. R. (1995) The role of cytochrome b5 in the biosynthesis of androgens by human P450c17. Arch. Biochem. Biophys. 317, 343–347 10.1006/abbi.1995.1173 [DOI] [PubMed] [Google Scholar]
- 46. Auchus R. J., Lee T. C., and Miller W. L. (1998) Cytochrome b5 augments the 17,20-lyase activity of human P450c17 without direct electron transfer. J. Biol. Chem. 273, 3158–3165 10.1074/jbc.273.6.3158 [DOI] [PubMed] [Google Scholar]
- 47. Estrada D. F., Skinner A. L., Laurence J. S., and Scott E. E. (2014) Human cytochrome P450 17A1 conformational selection: modulation by ligand and cytochrome b5. J. Biol. Chem. 289, 14310–14320 10.1074/jbc.M114.560144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guengerich F. P., Waterman M. R., and Egli M. (2016) Recent structural insights into cytochrome P450 function. Trends Pharmacol. Sci. 37, 625–640 10.1016/j.tips.2016.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Turcu A. F., Rege J., Chomic R., Liu J., Nishimoto H. K., Else T., Moraitis A. G., Palapattu G. S., Rainey W. E., and Auchus R. J. (2015) Profiles of 21-carbon steroids in 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 100, 2283–2290 10.1210/jc.2015-1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Petrunak E. M., DeVore N. M., Porubsky P. R., and Scott E. E. (2014) Structures of human steroidogenic cytochrome P450 17A1 with substrates. J. Biol. Chem. 289, 32952–32964 10.1074/jbc.M114.610998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 52. Bonomo S., Jørgensen F. S., and Olsen L. (2017) Mechanism of cytochrome P450 17A1-catalyzed hydroxylase and lyase reactions. J. Chem. Inform. Model. 57, 1123–1133 10.1021/acs.jcim.6b00759 [DOI] [PubMed] [Google Scholar]
- 53. Kopecky K. R., and Mumford C. (1969) Luminescence in the thermal decomposition of 3,3,4-trimethyl-12,-dioxetane. Can. J. Chem. 47, 709–711 10.1139/v69-114 [DOI] [Google Scholar]
- 54. Richardson W. H., Hodge V. F., Stiggall D. L., Yelvington M. B., and Montgomery F. C. (1974) 1,2-Dioxetane intermediates in the base catalyzed decomposition of α-hydroperoxy ketones. J. Am. Chem. Soc. 96, 6652–6657 10.1021/ja00828a018 [DOI] [Google Scholar]
- 55. Kadlubar F. F., Morton K. C., and Ziegler D. M. (1973) Microsomal-catalyzed hydroperoxide-dependent C-oxidation of amines. Biochem. Biophys. Res. Commun. 54, 1255–1261 10.1016/0006-291X(73)91122-4 [DOI] [PubMed] [Google Scholar]
- 56. Mansuy D., Bartoli J. F., and Momenteau M. (1982) Alkane hydroxylation catalyzed by metalloporphyrins: evidence for different active oxygen species with alkylhydroperoxides and iodosobenzene as oxidants. Tetrahed. Lett. 23, 2781–2784 10.1016/S0040-4039(00)87457-2 [DOI] [Google Scholar]
- 57. Falardeau P., and Tan L. (1974) Labeled steroids of potential biological interest: synthesis and properties of 18O-labeled 17α-hydroperoxyprogesterone. J. Labelled Compd. 10, 239–248 10.1002/jlcr.2590100208 [DOI] [Google Scholar]
- 58. Korde S. S., Katoch R., Udasi R. A., and Trivedi G. K. (1999) Total assignment of 1H and 13C NMR spectra of pregnenolone and progesterone haptens using 2D NMR spectroscopy. Magnet. Reson. Chem. 37, 594–597 10.1002/(SICI)1097-458X(199908)37:8%3C594::AID-MRC484%3E3.0.CO%3B2-J [DOI] [Google Scholar]
- 59. Szendi Z., Forgó P., and Sweet F. (1995) Complete 1H and 13C NMR spectra of pregnenolone. Steroids 60, 442–446 10.1016/0039-128X(94)00047-G [DOI] [PubMed] [Google Scholar]
- 60. Swern D., Clements A. H., and Luong T. M. (1969) Nuclear magnetic resonance spectra of organic peroxides. Anal. Chem. 41, 412–416 10.1021/ac60272a038 [DOI] [Google Scholar]
- 61. Hanna I. H., Teiber J. F., Kokones K. L., and Hollenberg P. F. (1998) Role of the alanine at position 363 of cytochrome P450 2B2 in influencing the NADPH- and hydroperoxide-supported activities. Arch. Biochem. Biophys. 350, 324–332 10.1006/abbi.1997.0534 [DOI] [PubMed] [Google Scholar]
- 62. Guengerich F. P. (2005) Reduction of cytochrome b5 by NADPH-cytochrome P450 reductase. Arch. Biochem. Biophys. 440, 204–211 10.1016/j.abb.2005.06.019 [DOI] [PubMed] [Google Scholar]
- 63. Guengerich F. P. (2014) Analysis and characterization of enzymes and nucleic acids relevant to toxicology. In Hayes' Principles and Methods of Toxicology (Hayes A. W., and Kruger C. L., eds) pp 1905–1964, 6th Ed., CRC Press-Taylor & Francis, Boca Raton, FL [Google Scholar]
- 64. Winter G. (2010) xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190 10.1107/S0021889809045701 [DOI] [Google Scholar]
- 65. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 10.1107/S0907444905036693 [DOI] [PubMed] [Google Scholar]
- 66. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 68. Murshudov G. N., Skubak P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 10.1107/S0907444911001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang C., Pallan P. S., Zhang W., Lei L., Yoshimoto F. K., Waterman M. R., Egli M., and Guengerich F. P. (2017) Functional analysis of human cytochrome P450 21A2 variants involved in congenital adrenal hyperplasia. J. Biol. Chem. 292, 10767–10778 10.1074/jbc.M117.792465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.