

Abstract
Biotin is an essential cofactor for multiple metabolic reactions catalyzed by carboxylases. Biotin is covalently linked to apoproteins by holocarboxylase synthetase (HCS). Accordingly, some mutations in HCS cause holocarboxylase deficiency, a rare metabolic disorder that can be life-threatening if left untreated. However, the long-term effects of HCS deficiency are poorly understood. Here, we report our investigations of bpl-1, which encodes the Caenorhabditis elegans ortholog of HCS. We found that mutations in the biotin-binding region of bpl-1 are maternal-effect lethal and cause defects in embryonic polarity establishment, meiosis, and the integrity of the eggshell permeability barrier. We confirmed that BPL-1 biotinylates four carboxylase enzymes, and we demonstrate that BPL-1 is required for efficient de novo fatty acid biosynthesis. We also show that the lack of larval growth defects as well as nearly normal fatty acid composition in young adult worms is due to sufficient fatty acid precursors provided by dietary bacteria. However, BPL-1 disruption strongly decreased levels of polyunsaturated fatty acids in embryos produced by bpl-1 mutant hermaphrodites, revealing a critical role for BPL-1 in lipid biosynthesis during embryogenesis and demonstrating that dietary fatty acids and lipid precursors are not adequate to support early embryogenesis in the absence of BPL-1. Our findings highlight that studying BPL-1 function in C. elegans could help dissect the roles of this important metabolic enzyme under different environmental and dietary conditions.
Keywords: biotin, Caenorhabditis elegans (C. elegans), cell polarity, development, embryo, polyunsaturated fatty acid (PUFA)
Introduction
Biotin is an essential B vitamin that acts as a critical cofactor in metabolic reactions. Animals cannot synthesize biotin but rather must obtain biotin from plant or bacterial sources. In humans, biotin deficiency is rare because it is available from numerous dietary sources and synthesized by gut microbes (1). However, biotin deficiency does occur rarely in individuals who consume raw egg whites or who are fed parenteral diets without biotin, leading to hair loss, dermatitis, skin rash, ataxia, and neurologic dysfunction. Additionally, subclinical biotin deficiency was shown to be common during pregnancy, a condition that in mice prohibits normal fetal development (2).
Biotin functions primarily as a cofactor in carboxylation reactions. In mammals, five biotin-dependent carboxylases perform diverse metabolic functions in lipid synthesis, gluconeogenesis, amino acid catabolism, and odd-chain fatty acid oxidation. Biotin is covalently attached to carboxylase enzymes by the action of holocarboxylase synthetase (HCS),2 a biotin-ligating protein (1, 3). In humans, mutations in the holocarboxylase synthetase gene result in a disease called holocarboxlase synthetase deficiency, or multiple carboxylase deficiency (MCD) (4, 5). The symptoms of MCD are phenotypically similar to biotin deficiency and present during infancy. If left untreated, MCD can cause coma and death. The underlying mutations causing holocarboxlase synthetase deficiency are well-documented, and the disease can be treated with large doses of biotin in most cases (6). However, the long-term effects of perturbing multiple metabolic pathways in these patients are poorly understood.
The nematode Caenorhabditis elegans offers a powerful model for studying complex metabolic systems. The invertebrate is amenable to forward and reverse genetics, has a short life cycle, and produces large numbers of offspring with well-characterized developmental processes that can be easily studied with light microscopy. Additionally, the C. elegans genome contains many orthologs to mammalian metabolic enzymes. These enzymes include orthologs to carboxylase enzymes and holocarboxlase synthetase, which previously were uncharacterized in C. elegans.
In a forward genetic screen, we obtained two mutations in a biotin ligase orthologous to mammalian holocarboxylase synthetase. These mutations cause maternal-effect embryonic lethality, demonstrating a key role for HCS in embryonic development. We show that BPL-1 biotinylates four key carboxylase enzymes, including acetyl-CoA carboxylase (ACC/POD-2), which is critical for maintaining the levels of embryonic polyunsaturated fatty acids and for the formation of the permeability barrier in the newly formed zygote. In the absence of BPL-1, ACC/POD-2, and fatty acid desaturation, embryos fail to complete meiosis and fail to establish proper polarity during the first zygotic cell divisions. Unlike ACC/POD-2 deficiency, BPL-1 activity is dispensable during larval growth and development, because C. elegans are able to utilize fatty acid precursors from their bacterial diet. Our findings suggest that dietary precursors are partitioned away from embryonic lipids, and thus de novo lipid machinery is critical for the first steps of embryogenesis.
Results
mel-3 mutations correspond to the holocarboxylase synthetase gene
Mutations that defined the mel-3 gene of C. elegans were first identified in a screen for maternal-effect lethal mutants. The mel-3 mutations resulted in 100% lethality in embryos produced by homozygous mutant mothers and mapped to a region of linkage group II uncovered by the mnDf30 deletion (7). Further mapping using deletion strains combined with complementation analysis and transformation rescue allowed us to identify a 7.5-kb BamHI fragment containing a single predicted gene, F13H8.10, that was capable of rescuing the embryo lethality of mel-3(b281) and mel-3(it8).
The F13H8.10 gene encodes a biotin protein ligase, homologous to the enzyme HCS, which is responsible for attaching biotin to carboxylases. For the remainder of this work, the mel-3 gene will be referred to as bpl-1, biotin protein ligase-1. Sequence analysis of b281 and it8 mutants revealed point mutations in the coding sequence of F13H8.10 that result in single amino acid substitutions in the predicted protein product. These mutations affect amino acids within the biotin protein ligase catalytic domain that are highly conserved among all holocarboxylase synthetase genes (8). The b281 mutation causes an A730E amino acid change, and it8 results in G912D substitution in BPL-1 (Fig. 1A). Many human mutations of HCS resulting in multiple carboxylase deficiency disease have also been found in this region (6, 9). The bpl-1 deletion alleles tm5867 and tm6621 remove portions of the conserved biotin protein ligase domain and are predicted to result in frameshift and early termination (Fig. 1A). These deletion mutations also result in 100% maternal-effect lethality (n > 5000 for each allele).
Figure 1.

Phenotypic analysis of bpl-1 mutant embryos. A, mutations in the C. elegans bpl-1 gene alter conserved biotin protein ligase residues. The C. elegans bpl-1 gene encodes three protein isoforms (black lines) differing in length at the N terminus (WormBase WS251). These isoforms all contain the conserved biotin protein ligase/lipoyl protein ligase catalytic domain (BPL_LPL_catalytic domain IPR004143, amino acids 958–1141 in BPL-1a), indicated by a dark blue bar within a larger conserved biotin protein ligase domain (lighter blue bar, PTHR12835, amino acids 526–1215 in BPL-1a). The N-terminal region preceding the BPL domain of C. elegans BPL-1a is not conserved. The positions of the single amino acid changes produced by b281 and it8 mutations are within the conserved biotin-binding pocket of the protein and are indicated with green arrows. tm5867 is a deletion resulting in removal of amino acids 603–711 and subsequent frameshift resulting in premature termination. tm6621 is a deletion that removes codons for amino acids 712–842 and causes a frameshift and premature stop. Thus, neither deletion mutant retains the BPL domain. COBALT alignment between C. elegans BPL-1a and holocarboxylase synthetase proteins of human and zebrafish shows sequence conservation. Amino acid identities are in red. Amino acids changed by C. elegans point mutations are highlighted and indicated with green arrows. b281 changes the codon for alanine 994 (A) to glutamic acid (E); it8 changes the codon for glycine 1056 (G) to aspartic acid (D). Amino acid numbering is with respect to the longest BPL-1 isoform, BPL-1a. B, time-lapse differential interference contrast microscopy shows symmetric first cleavage in bpl-1(tm6621) and synchrony at the second division, compared with control embryos, which have asymmetric first cleavage and asynchronous second division. Both cells of the bpl-1 two-cell embryo are shown divided transverse to the long axis of the embryo; in control embryos, only the posterior P1 cell divides with this orientation. Time-lapse movies from which these images were taken are included as Movie S1 (control) and Movie S2 (bpl-1(tm6621)). C, differential interference contrast (DIC) imaging of bpl-1(b281) two-cell embryo with multinucleation (top) and FM4-64 dye permeability (bottom). D, imaging of PAR-2:: GFP and PAR-6 ::mCherry in live embryos with compromised fatty acid synthesis. The PAR-2 domain is restricted, whereas the PAR-6 domain extends further into the posterior in fasn-1 and pod-2 RNAi and bpl-1 mutant embryos than in controls. Bottom, outline of PAR-6 domain (red) and PAR-2 domain (green). The anterior is to the left and posterior is to the right in all images.
BPL-1 is essential for early embryonic development in C. elegans
Our phenotypic studies demonstrate that bpl-1 plays an essential role in early embryonic development. 100% of embryos produced by homozygous mutant bpl-1 worms fail to hatch. We used time-lapse video microscopy to compare early embryogenesis in bpl-1 mutants with wild type. In wild-type C. elegans, the one-cell embryo divides asymmetrically to produce two daughter cells, AB and P1, which undergo asynchronous second divisions. First, the AB cell divides with a spindle orientation perpendicular to the anterior–posterior axis, followed by division of the P1 cell along the anterior–posterior axis (10). In contrast, bpl-1 embryos show abnormal early division patterns, including synchronous and symmetrical divisions and abnormal spindle orientations (Fig. 1B, Table 1 and Movies S1–S3).
Table 1.
Summary of bpl-1(b281) embryo phenotypes
Biotin was dissolved in water, and 5 μg was added to feeding plates. Oleic acid sodium salts were added to NGM at 0.1 mm with 0.1% Tergitol. Dye permeability was assayed with FM4-64. N, no rescue.
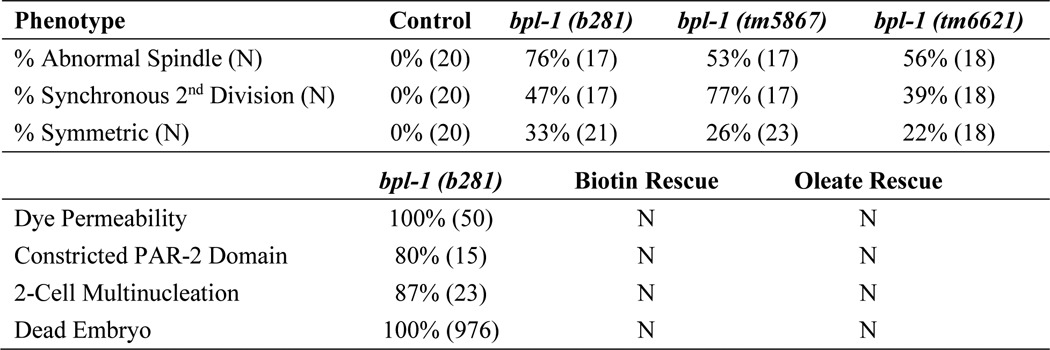
We found that 100% of two-cell stage bpl-1 embryos are permeable to FM4-64 dye, indicating improper formation of the eggshell permeability barrier (Fig. 1C; n = 45). Embryos also display aneuploidy due to earlier meiotic defects (Fig. 1C). We observed extra nuclei in one-cell and two-cell embryos, consistent with failure of meiosis in 90% of bpl-1 embryos (Table 1).
PAR (partitioning-defective) proteins are ancient, conserved proteins that localize asymmetrically and regulate cell polarity (10). We found that bpl-1 embryos fail to segregate PAR proteins properly (Fig. 1D). Specifically, the posterior PAR-2 domain is severely restricted, whereas the anterior PAR-6/PAR-3/PKC-3 domain extends further into the posterior of the embryo. Like b281 and it8, the deletion mutants tm5867 and tm6621 show the same range of embryo phenotypes, with the same penetrance (Table 1). bpl-1 embryos do not undergo morphogenesis but arrest as a mass of many cells.
BPL-1 biotinylates carboxylase enzymes
The predicted amino acid sequence of BPL-1 is orthologous to holocarboxylase synthetase, which in mammals covalently links biotin to multiple carboxylase enzymes, including acetyl-CoA carboxylases 1 and 2, pyruvate carboxylase, 3-methylcrotonyl-CoA carboxylase, and propionyl-CoA carboxylase. These carboxylase enzymes function in a diverse set of metabolic pathways. Both the cytoplasmic acetyl-CoA carboxylase 1 and the mitochondrial acetyl-CoA carboxylase 2 catalyze the rate-limiting step in de novo fatty acid synthesis. Pyruvate carboxylase converts pyruvate to oxaloacetate in gluconeogenesis. Both 3-methylcrotonyl-CoA carboxylase and propionyl-CoA carboxylase are involved in catabolism of branched-chain amino acids, and propionyl-CoA carboxylase also functions in odd-chain fatty acid catabolism.
To determine whether BPL-1 biotinylates carboxylase enzymes in C. elegans, we probed total protein blots with streptavidin-horseradish peroxidase conjugates. Blots of proteins from wild-type control worms display prominent protein bands at ∼240 and 130 kDa and two at ∼70 kDa (Fig. 2A). The intensity of each of these bands is reduced in bpl-1(b281) homozygous adult worms, indicating a reduction in biotinylation of the proteins. To determine whether the biotinylated protein bands correspond to carboxylase enzymes, we knocked down pod-2, pyc-1, pcca-1, and mccc-1 with RNAi.
Figure 2.

BPL-1 biotinylates carboxylase enzymes. A, Western blot probed with streptavidin to reveal biotinylated proteins. Total protein extracts from young adult bpl-1(b281) worms show reduced biotinylation compared with protein extracts from wild-type control. Also shown are biotinylated proteins in worms where each of the four carboxylase enzymes were knocked down with RNAi; in bpl-1(b281), these carboxylase enzymes have reduced biotinylation. The same blot was probed with an antibody to histone 3 to serve as a loading control. B, percentage of hatched and unhatched embryos upon knockdown of genes encoding carboxylase enzymes and genes encoding mitochondrial fatty acid synthase II components by RNAi. Only knockdown of pod-2 produced embryonic lethality similar to bpl-1(b281). C, percentage of permeable and impermeable embryos upon knockdown of genes encoding carboxylase enzymes and genes encoding mitochondrial fatty acid synthase II components. Only knockdown of pod-2 produced embryonic permeability defects similar to bpl-1(b281). D, efficiency of RNAi knockdown. RNA was extracted from two biological replicates, RNAi 1 and RNAi 2, in young adult worms exposed to feeding RNAi constructs. Expression of the gene corresponding to the RNAi knockdown was determined by quantitative RT-PCR, using two reference genes as controls, shown relative to expression in the empty vector control. The RNAi knockdowns resulted in 3–100-fold reduction of transcript abundance. Error bars, S.E.
The pod-2 gene is predicted to encode acetyl-CoA carboxylase isoforms of 91.4, 230.6, and 242.6 kDa. When we knocked down pod-2 with RNAi, the highest molecular mass band was absent from the protein blot (Fig. 2A). The same band is absent in bpl-1 homozygotes, indicating that BPL-1 biotinylates the 242.6-kDa POD-2 isoform. We did not detect the lower molecular mass isoforms in our assay, which implies that the 242.6-kDa isoform is the most abundant biotinylated form under laboratory growth conditions.
The pyc-1 gene encodes pyruvate carboxylase, which catalyzes the carboxylation of pyruvate to oxaloacetate in gluconeogenesis. The predicted molecular mass of PYC-1 is 129.3 and 67.8 kDa. Knockdown of pyc-1 with RNAi resulted in reduction of intensity of the ∼130 kDa protein band, and it is also reduced in bpl-1 homozygotes.
Propionyl-CoA carboxylase converts propionyl-CoA to d-methylmalonyl-CoA in the breakdown of the branched-chain amino acids valine and isoleucine and in the metabolism of odd-chain fatty acids. Subsequent steps of d-malonylmalonyl-CoA metabolism provide succinyl-CoA to the tricarboxylic acid cycle. The presence of this pathway has been confirmed in C. elegans (11). The C. elegans genome contains orthologs for propionyl-CoA carboxylase subunits A and B (pcca-1 and pccb-1). The pcca-1 gene is predicted to encode a 79.7-kDa protein. Only PCCA-1 is predicted to contain a biotin-binding domain. Knockdown of pcca-1 with RNAi caused a decrease in intensity of biotinylated protein bands at ∼75 kDa (Fig. 2A). This protein band could contain a modified PCCA-1 protein, and it is reduced in bpl-1 homozygotes.
The 3-methylcrotonyl-CoA carboxylase is also involved in branched-chain amino acid metabolism, converting 3-methylcrotonyl-CoA (derived from leucine) to 3-methylglutaconyl-CoA, which is further metabolized to acetyl-CoA and acetoacetate (1). Knockdown of mccc-1 reduced intensity of the 73-kDa biotinylated protein band (Fig. 2A), indicating that mccc-1 is the major biotinylated protein at 73 kDa.
To determine whether loss of carboxylase function explained the bpl-1 embryonic phenotypes, we assessed viability and permeability of embryos when each of the carboxylases were knocked down by RNAi (Fig. 2, B and C). Mutations in the pod-2 gene were previously shown to cause phenotypes very similar to those of bpl-1 embryos (12, 13). We found that treating worms with RNAi against pyc-1, pcca-1, and mccc-1 for 2 generations resulted in normal growth, impermeable embryos, and very low levels of embryonic lethality, similar to worms treated with an empty RNAi vector. Conversely, RNAi against pod-2 starting at the L3 larval stage caused 100% permeable embryos and 100% embryonic lethality. Thus, these data provide evidence that loss of POD-2 biotinylation is responsible for embryonic defects caused by perturbed BPL-1 function.
Cytosolic, but not mitochondrial, fatty acid synthesis is required for C. elegans embryogenesis
Acetyl-CoA carboxylase, encoded by the pod-2 gene in C. elegans (12), produces malonyl-CoA, which is a required substrate for several reactions, including cytosolic fatty acid synthesis, elongation of C16 and C18 fatty acids in the endoplasmic reticulum, and mitochondrial fatty acid synthesis. Interestingly, embryo phenotypes very similar to those we observe in bpl-1 mutants have also been reported in a pod-2 mutant and for RNAi knockdown of fasn-1, the major cytosolic fatty acid synthase gene of C. elegans. Early embryos of pod-2(−) or fasn-1(RNAi) exhibit egg shell permeability, multiple nuclei, cell cycle synchrony, and PAR protein mislocalization (Figs. 1D and 2C) (12).
In contrast, RNAi knockdown for genes required for mitochondrial fatty acid synthesis failed to show abnormal embryo phenotypes, although the disruption of mitochondrial synthesis of α-lipoic acid, which requires mitochondrial fatty acid synthesis, causes embryonic lethality in mice (14). Because malonyl-CoA is required for both cytosolic and mitochondrial fatty acid synthesis, we examined the embryo phenotypes of RNAi knockdowns for mecr-1, the mitochondrial trans-2-enoyl-CoA reductase (15). This RNAi knockdown did not result in embryo lethality or permeability, indicating that the fatty acid synthesis defects in bpl-1 and pod-2 mutants are probably mediated by the limitations of malonyl-CoA substrates required for cytosolic fatty acid synthesis and fatty acid elongation in the endoplasmic reticulum.
BPL-1 functions in fatty acid biosynthesis
To directly assess BPL-1's role in fatty acid synthesis, we used a stable isotope-labeling strategy that allows the identification of newly synthesized fatty acids as opposed to those obtained from the diet (16). We measured the relative proportion of newly synthesized fatty acids that animals accumulated over a 12-h period during the L4 larval stage and young adulthood. Knockdown of bpl-1 led to reduced synthesis of several fatty acids, including the long-chain fatty acids 18:0, 18:1(n-7), and 18:2 by 25–50% (Fig. 3A), confirming that BPL-1 plays an important role in de novo fat synthesis. RNAi knockdown of pod-2 also causes a reduction in de novo fat synthesis (17).
Figure 3.
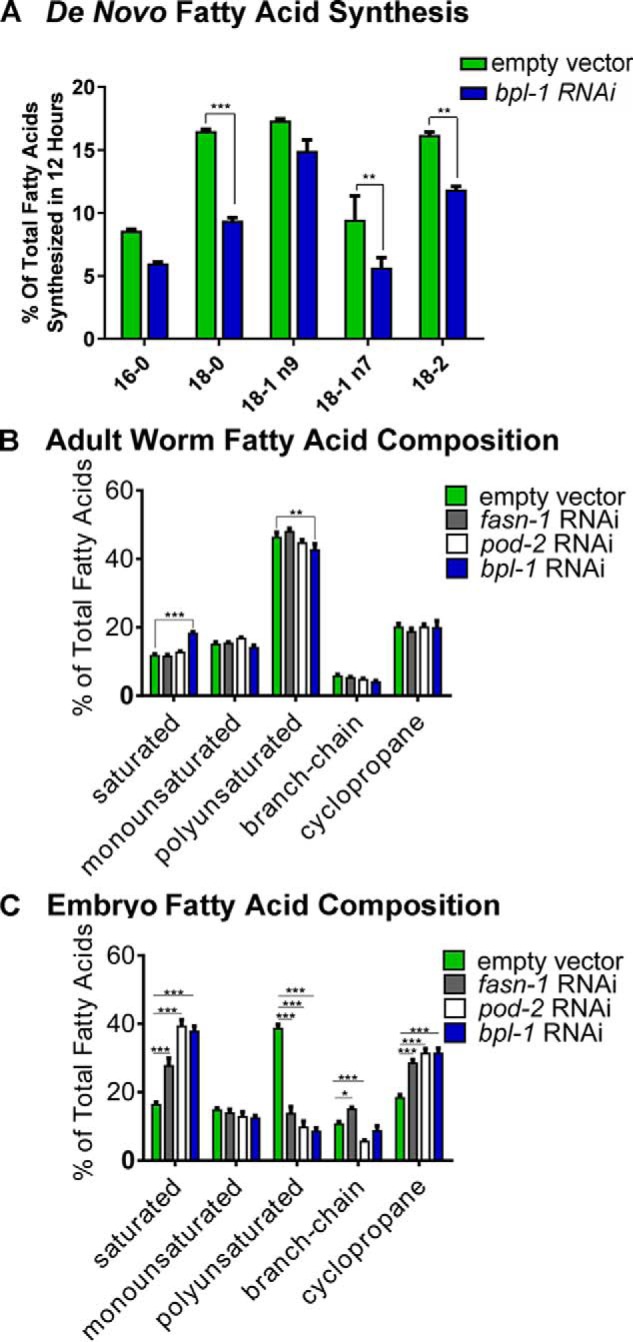
Loss of BPL-1 reduces de novo synthesis of fatty acids and alters embryonic fatty acid composition. A, the proportion of individual fatty acids produced by de novo synthesis was determined by labeling of the RNAi-hypersensitive rrf-3(pk1426) strain treated with RNAi feeding vectors or the empty vector control plasmid starting at the L4 larval stage. Labeling was achieved by feeding the worms a 1:1 (v/v) mixture of bacterial cultures that were grown in medium containing 13C or in unlabeled medium containing 12C. The percentage of de novo synthesized fatty acids was calculated after GC/MS analysis of isolated fatty acid methyl esters, where de novo synthesized isotopologs have masses corresponding to carbon chains consisting of a mixture of 12C and 13C carbons, whereas dietary fatty acids that are incorporated intact contain 100% 12C or 13C fatty acids. Bars, mean and S.E. (error bars). Knockdown of bpl-1 reduced de novo synthesis of 16- and 18-carbon fatty acids. B, relative fatty acid composition of young adult worms grown on fasn-1, pod-2, or bpl-1 RNAi bacteria compared with L4440 (empty vector) controls. C, fatty acid composition of embryos isolated from adult worms by hypochlorite treatment. Relative values of individual fatty acids are reported in Table 2. Saturated fatty acids include 14:0, 16:0, and 18:0; polyunsaturated fatty acids include 18:2, 18:3, 20:3, 20:4, 20:4(n-3), and 20:5; branched-chain fatty acids include 15:iso and 17:iso; and cyclopropane fatty acids include 17Δ and 19Δ. Bars, means with S.E. (error bars). *, p < 0.05; **, p < 0.005; ***, p < 0.0005.
Knockdown of BPL-1 and other fat synthesis genes reduces embryonic polyunsaturated fats
We asked whether the reduction in fatty acid synthesis when bpl-1, fasn-1, or pod-2 is knocked down causes changes to the overall proportions of fatty acids in the worm. Wild-type C. elegans contain a mix of saturated fatty acids, cyclopropane fatty acids, branched-chain fatty acids, and long-chain polyunsaturated fatty acids (PUFAs) (18). These fats are obtained both from the bacterial diet and by de novo synthesis (16). We used GC/MS to measure the fatty acid composition of late L4/young adult worms. Knockdown of bpl-1 by RNAi slightly, but significantly, reduced the relative amounts of PUFAs and increased saturated fatty acids in worms (Fig. 3B and Table 2). Many of the fatty acids in C. elegans, including the 14- and 16-carbon saturated fatty acids, which are the end product of de novo synthesis, are obtained directly from the bacterial diet (19, 20), and these precursors can be elongated and desaturated to form a range of 18- and 20-carbon PUFAs (18). Our data demonstrate that nearly normal fatty acid composition of adult worms can be attained under conditions in which de novo fat synthesis is reduced.
Table 2.
Fatty acid composition of adult worms and embryos
Fatty acid nomenclature is as follows: x:y(n-z), where x represents the number of carbons, y is the number of double bonds, and n-z is the location of terminal double bonds, z carbons from the methyl end. 15:iso, 13-methyltetradecanoic acid; 17:iso, 15-methylhexanoic acid; 17Δ, cis-9,10-methylenehexadecanoic acid; 19Δ, cis-11,12-methylene octadecanoic acid. *, p < 0.05; **, p < 0.005; ***, p < 0.0005.
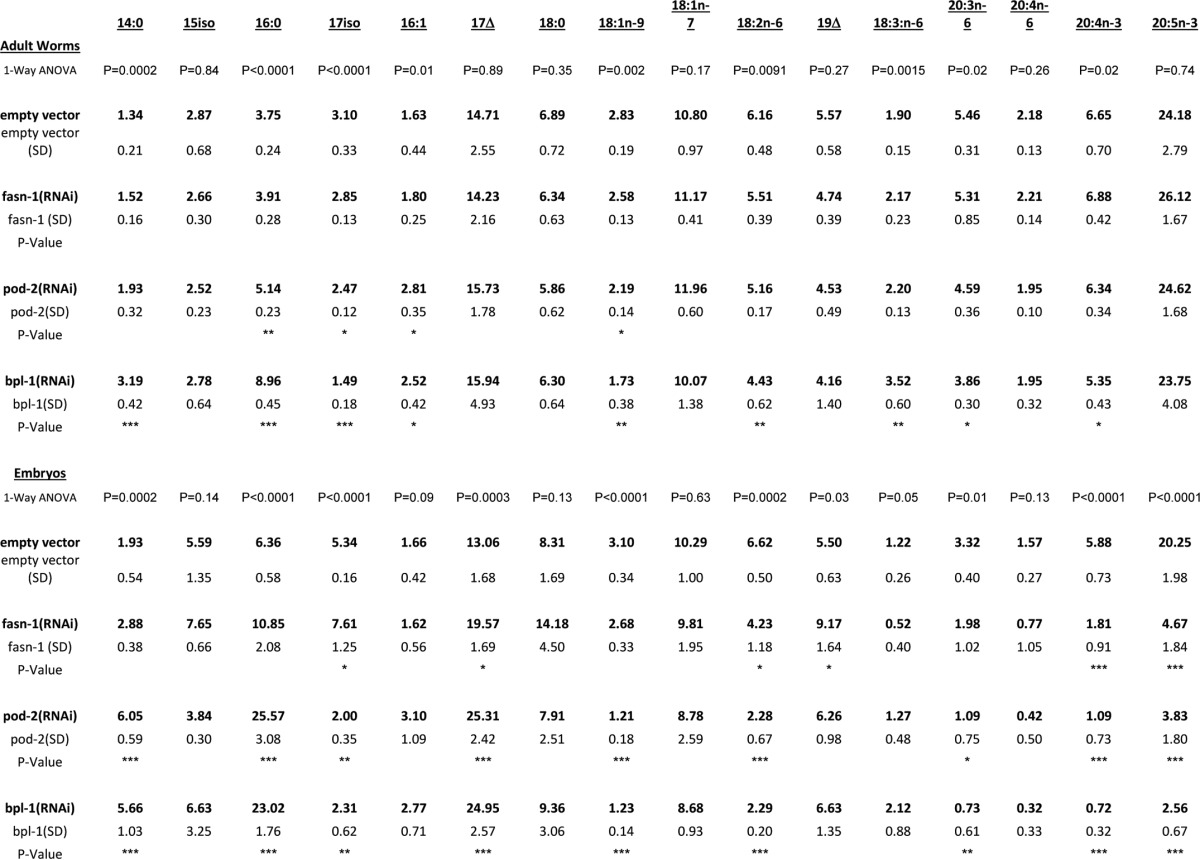
Because bpl-1 and pod-2 depletion lead to maternal-effect lethality, we asked whether reduction of de novo fatty acid biosynthesis enzymes cause changes in embryonic lipids. We carried out parallel GC/MS analysis of embryos obtained by hypochlorite treatment of semi-synchronous, RNAi-treated C. elegans. Contrary to the subtle fatty acid changes in the whole worm lipid profile, knockdown of bpl-1, pod-2, or fasn-1 resulted in drastic changes to the embryonic fatty acid composition: a >2-fold decrease in the total amount of PUFAs and an increased proportion of saturated fatty acids and cyclopropane fatty acids (Fig. 3C and Table 2). The dramatic decrease in embryonic PUFAs, which primarily are components of membrane phospholipids (21), suggests that membrane biophysical properties such as fluidity may be compromised.
Polyunsaturated fatty acids are required for early embryonic development
To test whether any of the embryonic defects observed in bpl-1 were caused by loss of embryonic PUFAs, we examined mutants in the PUFA biosynthesis pathway. Synthesis of PUFAs requires the sequential addition of two double bonds to a saturated fatty acid by Δ9 and Δ12 desaturase enzymes (18). The C. elegans genome encodes three redundant Δ9 desaturases, FAT-5, FAT-6, and FAT-7 (22, 23), but only a single Δ12 desaturase, FAT-2. Mutations in the fat-2 gene result in the accumulation of monounsaturated fatty acids and a nearly total loss of PUFAs (18), whereas the double mutant strain fat-6;fat-7 has greatly increased saturated fatty acids and reduced PUFA composition, consisting of unusual PUFAs that are not normally synthesized (24).
We found that similar to bpl-1 mutants, mutations in fat-2 and, to a lesser extent, fat-6;fat-7 resulted in embryonic lethality, disruption of the permeability barrier, and aneuploidy (Fig. 4, A and B). The penetrance of these embryonic abnormalities was incomplete, suggesting that disruption of PUFA synthesis cannot entirely explain the embryonic defects caused by mutations in the genes encoding earlier components of the fat biosynthesis pathway. However, these genetic data provide strong evidence that PUFAS are important for the formation of the embryonic permeability barrier and that the embryonic defects resulting from loss of BPL-1, POD-2, and FASN-1 are at least partially due to reduction of PUFAs.
Figure 4.

Polyunsaturated fatty acids are required for meiosis and synthesis of the embryonic permeability barrier. A, representative image of fat-2(wa17) loss of function mutant, compared with wild type (control), showing multiple nuclei in the anterior blastomere at the two-cell stage. B, quantification of FM4-64 dye permeability in fatty acid desaturase mutants fat-6;fat-7 and fat-2. C and D, treatment of wild-type (control), bpl-1(it8), or fat-2(wa17) mutant worms with 0.1 mm oleic acid (18:1), linoleic acid (18:2), or carrier 0.1% Tergitol. Treatment with 18:2 rescues embryonic lethality (C) and FM4-64 dye permeability (D) in fat-2 but not bpl-1. Error bars, S.E.
To further investigate the role of PUFAs in the early embryo, we supplemented the C. elegans diet with either the monounsaturated fatty acid, oleic acid, or with the PUFA, linoleic acid, and assessed lethality and permeability in embryos of fat-supplemented animals compared with controls. Treatment with the 18-carbon PUFA linoleic acid, but not the monounsaturated fatty acid oleic acid, rescued embryonic lethality and permeability barrier defects in fat-2, confirming that PUFAs, but not MUFAs, are critical for the early embryo (Fig. 4, C and D). Interestingly, neither supplemental MUFAs nor PUFAs rescued embryonic lethality or permeability barrier defects inbpl-1(it8). That bpl-1 is not rescued by supplemental PUFAs probably means that simpler fatty acid precursors, such as malonyl-CoA, are needed for de novo synthesis of complex permeability barrier components. Given the requirement for cytochrome P450s, cytochrome P450 reductase, and the putative sugar-modifying enzyme PERM-1 for the formation of the permeability barrier (25, 26), malonyl-CoA synthesis could be required for de novo synthesis and/or elongation of PUFAs as well as for synthesis of a complex lipid, such as a hydroxylated glycosphingolipid. Because these types of complex lipids are digested by lipases upon ingestion by C. elegans, it is not possible to test these molecules for rescue by dietary supplementation.
Bacterial sources of malonyl-CoA or other fatty acid precursors compensate for BPL-1 activity during larval growth and development but not during embryogenesis
Previous studies showed that both ACC/POD-2 and FASN-1 are needed for synthesis of specific sphingolipids during larval development. Knockdown of either gene caused defects in the intestinal lumen and early embryonic arrest due to a defect in the synthesis of glucosylceramides (27, 28). Thus, the ability of homozygous bpl-1 deletion mutant strains to develop normally throughout their larval growth stages was puzzling, because malonyl-CoA is required for fatty acid synthesis reactions catalyzed by ACC/POD-2 and FASN-1.
The finding that bpl-1 depletion only modestly reduced de novo lipid synthesis in young adult nematodes (Fig. 3A) led us to hypothesize that C. elegans obtains precursors for de novo fatty acid synthesis, such as malonyl-CoA, malonate, or short-chain fatty acids, directly from their bacterial diet to support lipid biosynthesis. Malonyl-CoA levels are highly regulated in animal cells; however, malonyl-CoA has been shown to be present in micromolar concentrations in the intracellular pool of Escherichia coli (29). To test whether bacterial fatty acid precursors were providing sufficient metabolites for lipid synthesis during larval development, we provided a malonyl-CoA–restricted food source by feeding K12-derived E. coli L8, containing a temperature-sensitive mutation in the accB gene, which encodes the β-subunit of the E. coli acetyl-CoA carboxylase. Growth of the bacteria at restrictive temperature depletes malonyl-CoA stores and impedes new bacterial lipid synthesis compared with L8 grown at non-restrictive temperature.
Growth and lipid composition of both wild-type and bpl-1 mutant C. elegans larvae fed L8 bacteria grown at non-restrictive temperature were comparable with those of C. elegans grown on the E. coli strain OP50. In contrast, we found that bpl-1(b281) mutants were much more severely affected than wild type when fed acetyl-CoA carboxylase–restricted L8 bacteria that were grown at restrictive temperature; bpl-1(b281) worms arrested growth at the L3 larval stage, whereas wild-type worms reached adulthood but had a smaller size. A small proportion of wild type arrested similarly to bpl-1 (Fig. 5, A–C). Growth of both bpl-1 and wild type was completely rescued by supplementation with biotin (Fig. 5, A and B), but not oleate, malonate, or malonyl-CoA (not shown). This is in contrast to the embryonic phenotypes described above, where neither biotin nor supplemented fatty acid precursors or fatty acids are able to rescue the permeability barrier and polarity defects of embryos produced by bpl-1 mutant mothers.
Figure 5.

Dietary fatty acid precursors support larval growth in the absence of BPL-1. A, images show WT or bpl-1(b281) adults grown on either non- restricted E. coli (L8 grown at permissive temperature) or ACC-restricted bacteria (L8 grown at restrictive temperature) for 66 h. Larval development is arrested in bpl-1(b281) when bacterial fatty acid precursors are restricted. However, growth is rescued by the addition of 5 μg of biotin to feeding plates. B, a diet of L8 bacteria grown at restrictive temperature causes arrested development in bpl-1(b281) and, to a lesser extent, in wild type. C, a diet of L8 bacteria grown at restrictive temperature reduces body size of wild-type and bpl-1(b281) worms at 42 h of growth. D, de novo synthesis of individual fatty acids is increased in wild type when fatty acid precursors are restricted from the diet, but not in bpl-1(b281). The RNAi-hypersensitive rrf-3(pk1426) strain was treated with RNAi or the empty vector control plasmid starting at the L4 larval stage. Their progeny were fed at L4 larval stage for 12 h on HT115 bacteria grown in medium containing a mix of 12C and 13C media (1:1, v/v). E and F, model depicting dietary versus synthetic sources of malonyl-CoA during somatic development and during the oocyte–embryo transition.
Finally, we tested the ability of worms to synthesize fatty acids when dietary malonyl-CoA and other fatty acid precursors were restricted. Interestingly, wild-type worms significantly increased de novo fat synthesis in response to the L8 diet (Fig. 5D), suggesting that the worms can detect dietary malonate and other fatty acid precursors and increase de novo synthesis to compensate when these dietary substrates are limited. Together, these data indicate that C. elegans maintains a balance of malonate or other precursors obtained from the diet and malonyl-CoA synthesized de novo from acetyl-CoA to support lipid synthesis for larval growth and development but that dietary lipids and precursors are not available or are not utilized during oogenesis and embryogenesis, and thus de novo synthesized lipids are required at this stage (Fig. 5, E and F).
Discussion
We have demonstrated that the C. elegans gene bpl-1 encodes a biotin-ligating enzyme homologous to mammalian holocarboxylase synthetase. BPL-1 is required for normal biotinylation of at least four conserved carboxylase enzymes. Mutations in the conserved biotin-binding domain of BPL-1 are detrimental to embryonic and larval development. Further, by knocking down each of four carboxylase genes with RNAi, we showed that reduction of the POD-2 acetyl-CoA carboxylase produces embryonic defects similar to bpl-1 mutants, whereas reduction of other carboxylase gene products showed no detrimental developmental effects. Thus, under laboratory conditions when nutrients are abundantly available, de novo fatty acid synthesis and fatty acid elongation, reactions that required malonyl-CoA produced by acetyl-CoA carboxylase, are the primary biotin-dependent pathways affected by loss of BPL-1.
BPL-1 plays a central role in lipid de novo fatty acid synthesis and elongation and is required for the synthesis of the embryonic permeability barrier and polarity in the embryo. Embryo production is a major consumer of nutrients, and young adult hermaphrodites convert their body mass to embryos every 24 h (30). Bacterial nutrients provide intact fatty acids as well as precursors, such as malonyl-CoA, for growth and development. Our finding that loss of de novo synthesis genes causes a large reduction of PUFAs in embryos, but not in the adult worm, supports a model in which dietary fatty acid precursors and de novo synthesized malonyl-CoA and fatty acids contribute to the lipid requirements for rapid growth and development during larval stages. However, in the sequestered environment of the early embryo, dietary support is unavailable and lipid incorporation is dependent on de novo synthesis and elongation, particularly for PUFA production. Stable isotope labeling studies indicate that PUFAs are preferentially synthesized from de novo derived, as opposed to dietary, precursors (16, 17). The inaccessibility of dietary fats during the oocyte–embryo transition perhaps ensures that fresher, less oxidized fatty acids are present at the start of embryonic life. In addition to providing precursors for membrane and storage lipid synthesis, recent studies revealed that fatty acid levels signal environmental conditions that influence sex determination in nematodes (31), and microbial metabolites, including lipid precursors, affect host metabolism, influencing reproduction and lifespan (32–34).
The embryonic permeability barrier performs the critical function of maintaining osmotic integrity of the embryo. Depletions of the lipid biosynthetic genes pod-2 and fasn-1 both cause osmotic integrity defects, as does loss of EMB-8, the NADPH reductase, and two lipid-modifying cytochrome P450 enzymes, as well as the putative sugar-modifying enzyme PERM-1 (12, 25, 26), which suggests that the permeability barrier may contain glycosylated and hydroxylated lipids. We have demonstrated that permeability barrier defects caused by depletion of lipid biosynthesis genes result, at least partially, from loss of embryonic PUFAs, which could be incorporated into the barrier as a component of complex hydroxylated glycolipids. In mammals, the PUFAs linoleic and linolenic acid are required for the synthesis of the impermeable corneocyte lipid envelope of the skin, in which PUFAs are esterified to the ω-hydroxyl of the amide-linked very-long-chain fatty acid of a unique class of esterified ceramides (35). A cytochrome P450 enzyme is required for the ω-hydroxylation, and lipoxygenase enzymes are proposed to further oxidize the esterified ceramides to allow binding of the lipid to proteins of the cell envelope, forming the permeability barrier (35, 36).
Similar to other permeability barrier genes, bpl-1 is important for embryonic polarity. We found that loss of bpl-1 caused symmetric first divisions, restricted PAR-2 domains, synchronous second divisions, and transverse spindle orientations during the second division. The relationship between the permeability barrier and polarity is not understood. The permeability barrier could affect polarity via direct interactions between the barrier and the cell membrane or cell cortex, or polarity defects could result from loss of osmotic regulation. Alternatively, the polarity defects associated with fat synthesis could be independent of permeability barrier function. Previous work suggested that pod-2– and emb-8–depleted embryos displayed weakened interaction between the paternal pronucleus and the cell cortex, which correlated with symmetric first divisions (12). This weakened interaction with the cortex could be explained by either a defective interaction with the permeability barrier or altered embryonic membrane composition. Future work is needed to determine the structure and function of the specific lipids that compose the permeability barrier and how they influence polarity.
The rate-limiting step in de novo fatty acid synthesis is carboxylation of acetyl-CoA by acetyl-CoA carboxylase to generate malonyl-CoA. In humans, holocarboxylase synthetase deficiency is an autosomal recessive disorder that usually manifests early in life, with patients showing defects in fatty acid biosynthesis, gluconeogenesis, and amino acid metabolism that lead to severe conditions of the skin, damage to the nervous system, coma, and early death (37). Fortunately, continual supplementation with biotin is sufficient to treat the syndrome in most cases, suggesting that excess biotin can drive carboxylase reactions in the absence of fully functioning holocarboxylase synthetase (5). Due to rapid turnover, it is not generally believed that appreciable malonyl-CoA pools accumulate during fatty acid synthesis (38, 39). Despite this, our work demonstrates that C. elegans uses both dietary fatty acid precursors and de novo synthesized malonyl-CoA for lipid synthesis. Restricting dietary fatty acid precursors by feeding E. coli deficient in acetyl-CoA carboxylase activity caused severe arrest during larval development in worms without functioning BPL-1 and even showed a measurable effect on growth in wild-type worms. Dietary biotin rescues these growth deficiencies in both wild type and the bpl-1 mutants.
Our studies indicate that C. elegans obtain malonyl-CoA from both dietary precursors and from acetyl-CoA carboxylase reactions. Our observation that supplemental biotin rescued larval development is consistent with the success of treatments for holocarboxylase synthetase deficiency in humans, although it is not known whether holocarboxylase synthetase–deficient humans treated with dietary biotin experience fertility loss or birth defects in their offspring. Furthermore, malonyl-CoA is an important regulator of lipid metabolism. In hepatocytes, malonyl-CoA levels regulate the balance between fat synthesis and fatty acid oxidation (40, 41). We found that in C. elegans, loss of dietary fatty acid precursors caused an increase in de novo fatty acid synthesis, demonstrating that de novo fat synthesis is regulated depending on the availability of dietary precursors.
This study is the first to characterize the C. elegans holocarboxylase synthetase, BPL-1. Holocarboxylase synthetase is a critical regulator of biotin utilization at the epicenter of multiple metabolic pathways. Here, we demonstrated that BPL-1 function is critical for maintaining fatty acid composition during embryonic development, establishment of embryonic polarity, integrity of the embryo permeability barrier, and support of post-embryonic development. In addition to the role of HCS in metabolism, the enzyme also appears to play a role in gene regulation through biotinylation and direct interaction with histones (42–44). Study of HCS function in C. elegans will provide a powerful tool for dissecting the diverse roles of HCS in a multicellular organism under different environmental and dietary conditions.
Experimental procedures
C. elegans strains and RNAi
Nematodes were maintained on nematode growth medium (NGM) at 20 °C seeded with E. coli (OP50) unless otherwise stated (45). The wild-type strain was N2, and the mel-3(b281) and mel-3(it8) mutants were isolated in screens for maternal-effect lethal mutations (7) and maintained as heterozygotes over the mnC1 balancer as mel-3(b281 or it8) unc-4(e120)/mnC1 II. Strains carrying deletions for deletion mapping with mel-3 mutants were provided by the Caenorhabditis Genetics Center. Two deletion alleles, tm5867 and tm6621, were obtained from the Mitani laboratory, Tokyo Women's Medical University School of Medicine. These were outcrossed, marked with unc-4(e120), and balanced with mnC1. Deletion end points of tm5867 and tm6621 were confirmed by DNA sequence analysis.
Transformation rescue of b281 and it8 mutants with cosmid or plasmid DNA was performed as described (46). Approximately 6.5 kb of the bpl-1 gene, including 600 bp upstream of the first predicted transcription start point, were sequenced from b281 and it8 homozygous worms. Only a single point mutation was found for each mutant.
Feeding RNAi experiments used NGM supplemented with 100 μg/ml ampicillin, 2 mm isopropyl β-d-1-thiogalactopyranoside and seeded with the appropriate HT115 RNAi bacteria. Feeding RNAi was performed by plating embryos of the RNAi-sensitive strain rrf-3(pk1426) onto seeded RNAi plates, with the exception of RNAi of pod-2 and fasn-1, in which feeding RNAi was initiated at the L3 larval stage. The pod-2, fasn-1, mecr-1, and pyc-1 feeding constructs were obtained from the Ahringer RNAi library and verified by sequencing (47) (Source BioSource). For RNAi of bpl-1, pcca-1, and mccc-1, a 250–550-bp portion of each gene was amplified from N2 DNA using primer sequences TTCTTCGATTTGCATTTGGGAGTC and GTTTCCGGGCCATTATCCTCAT for bpl-1, GGTAGTGTGGGGCCAGGAT and TTTTGAAGGGCAGCCAAAGC for mccc-1, and CTTGAGAAGGCTGGAGCCAA and CGAGGAATTCGACGGTT for pcca-1. Amplified DNA was cloned into L4440, and RNAi constructs were sequence-verified. Empty vector L4440 was used as the control.
RNA extraction and quantitative RT-PCR
RNA was extracted from ∼2000 nematodes using 1 ml of TRIzol reagent and multiple freeze–thaw cycles to break up nematodes. After chloroform addition and phase separation, the aqueous phase was precipitated with isopropyl alcohol, and RNA was further purified using a Qiagen RNeasy Plus minikit with genomic DNA eliminator column as per the manufacturer's instructions. cDNA was synthesized from 1 μg of RNA using oligo(dT) primer and superscript IV. Real-time quantitative PCR was performed in triplicate on each of two biological replicates using PowerUp SYBR Green master mix (Applied Biosystems) using an Applied Biosystems 7300 real-time PCR system. The ΔΔCt method was used to determine the relative expression of the target gene in the RNAi knockdowns compared with empty vector controls. The ΔCt values were calculated using the geometric mean of the Ct values for two reference genes, tbb-2 and Y45F10D.4 (Fig. 2D).
Microscopy
To image live embryos, adult worms were dissected with a syringe needle in 0.8× egg buffer on a glass coverslip and compressed against a pad of 1% agarose dissolved in 0.8× egg buffer and cushioned with petroleum jelly (48). To visualize eggshell defects, 2–6 μm FM4-64 (Thermo Fisher catalog no. T-3166) dye was added to the dissecting buffer (26). Imaging was performed on a Leica DM RA2 microscope with Hamamatsu Orca-ER using OpenLab (Improvision) or BioVision software. Additional imaging was performed on an Olympus BX53 microscope with a QImaging Retiga 2000R CCD camera and analyzed with the QCapture software and ImageJ. Embryo permeability was confirmed for all bpl-1 alleles using Nile Blue A uptake (49).
PAR-2 and PAR-6 localization in living early embryos was visualized with the transgenes itIs272[Ppar-6:: par-6::mCherry] (50), and itIs297[Ppie-1:: par-2::gfp + unc-119(+)] (provided by Aaron Schetter). The unc-4–marked bpl-1 mutants b281, tm5867, and tm6621 were each crossed into KK1213 itIs272[Ppar-6:: par-6::mCherry + unc-119(+)]; itIs297[Ppie-1:: par-2::gfp + unc-119(+)] to create the strains KK1205, KK1212, and KK1230, respectively. To confirm PAR protein mislocalization phenotypes in b281, it8, tm5867, and tm6621 without transgenes, we fixed unc-4 bpl-1 and control unc-4 worms and embryos on slides in methanol at −20 °C and stained with primary antibodies to PAR-2 (51) and PAR-3 (52). AF488– or Cy-3–labeled secondary antibodies were from Invitrogen and Jackson ImmunoResearch.
Meiosis was examined in bpl-1 oocytes and fertilized zygotes in utero using strain KK1193 bpl-1(b281) unc-4(e120)/mnC1 II; unc-119(ed3) ruIs32[Ppie-1:: gfp::H2B + unc-119(+)] III and their heterozygous siblings from the same strain, which have fully viable embryos. ruIs32 was derived from strain AZ212 (53).
Protein blot detection of biotinylated proteins
To extract worm protein, 300 young adult worms were collected in M9 buffer and allowed to settle on ice before washing one time with M9, resettling on ice, and then removing the supernatant until ∼20 μl of worm pellet remained. We then added 20 μl of 2× Laemmli buffer and boiled the samples for 5 min. After boiling, each sample was Dounce-homogenized 100× with the melted end of a pipette tip and frozen at −20 °C. Before use, samples were reheated to 90 °C.
Proteins were separated by SDS-PAGE with Bio-Rad 7% Mini-Protean precast gels and transferred with Towbin buffer with no SDS for 20 min using semidry transfer onto PVDF membranes. Membranes were blocked with 2% BSA in PBST (0.05% Tween 20). We probed for biotinylated proteins by incubating the membranes with 2 μl of streptavidin-horseradish peroxidase conjugate (Abcam ab7403; 1 mg/ml) in 10 ml of PBST (0.1% Tween 20) for 30 min, followed by washing with PBST (six 7-min washes). Bands were visualized using the Pierce ECL Western blotting substrate with a 2-min incubation time. Biotinylated proteins were visualized on X-ray film, and protein standards (Bio-Rad dual color standards) were visualized using a Bio-Rad imager.
Lipid extraction for fatty acid composition
Lipids were extracted as described previously (21). Briefly, worms were washed from feeding plates with water, washed once to remove residual bacteria, and allowed to settle on ice. After residual water was removed, the worm pellet was incubated for 1 h at 70 °C in 2.5% sulfuric acid in methanol. After stopping the reaction with water, the fatty acid methyl esters were extracted with hexane.
Stable isotope labeling assay
As described elsewhere (16, 17), to quantify the amount of de novo fatty acid synthesis, worms were fed a mixed culture of E. coli, where bacteria was grown in either LB broth or 13C-labeled Isogro medium (Sigma-Aldrich). Overnight cultures of each were pelleted and mixed (1:1, v/v) after resuspension to 0.075 mg/ml in M9. The RNAi-hypersensitive rrf-3(pk1426) strain was treated with RNAi or the empty vector control plasmid starting at the L4 larval stage. Their progeny were fed for 12 h starting at the L4 larval stage on HT115 bacteria grown in medium containing a mix of 12C and 13C media (1:1, v/v).
Lipid analysis
Fatty acid methyl esters were analyzed by GC/MS using an Agilent 7890 GC/5975C MS. For de novo synthesis experiments, we operated the MS in scanning mode to monitor ions m/z 260–300 for 16 carbon fatty acids and m/z 290–320 for 18 carbon fatty acids. The percentages of individual fatty acids that derived completely via de novo synthesis from acetyl-CoA were determined using the ratio of ions m/z 273–283 to total isotopologs for 16 carbon fatty acids and the ratio of ions m/z 290–314 for 18 carbon fatty acids.
Isolation of embryos for lipid analysis
To isolate early embryos for lipid analysis, worms were synchronized with hypochlorite treatment, and isolated eggs were allowed to hatch and develop on bpl-1 RNAi plates until adult stage. Embryos were collected from the uteri of adult worms by hypochlorite treatment and washed twice with water. The embryo pellet was then flash-frozen in liquid nitrogen and stored at −80 °C before lipid analysis. For pod-2 and fasn-1 RNAi treatment, the semisynchronized offspring were allowed to develop for 48 h on empty vector plates to avoid larval arrest and were then washed with M9 buffer and moved to pod-2 or fasn-1 RNAi plates for 24 h before embryo collection.
Malonyl-CoA restriction plates
To restrict dietary malonyl-CoA or malonic acid, we used the E. coli K12-derived strain L8. The L8 strain contains a temperature-sensitive mutation in the acetyl-CoA carboxylase β-subunit gene, accB22 (54). The L8 strain normally grows at 30 °C but does not grow above 37 °C due to inhibited malonyl-CoA synthesis. For our feeding experiments, we grew overnight cultures of L8 at 30 °C until the A600 of the culture was 2.0. We then collected half of the overnight culture, centrifuged, and concentrated 10× in M9 buffer; plated 300 μl of the bacterial suspension on nutrient-free plates (control); and exposed it to 15 min of UV light in a cell culture hood to prevent further growth. The remaining culture was incubated for an additional 6 h at 40 °C before plating (malonyl-CoA–restricted). Nutrient-free plates contained 1.5% agarose, 50 mm NaCl, 1 mm CaCl2, 1 mm MgSO4, and 25 mm KPO4.
Biotin, malonyl-CoA, malonic acid, and fatty acid supplementation
Biotin (Sigma-Aldrich, B4501) was dissolved in water at 22 mg/100 ml and filter-sterilized before top-dressing directly onto bacterial feeding plates at 0.05–50 μg/plate. We found that 0.5 μg/plate was sufficient to rescue growth of worms grown on an ACC-restricted diet. Malonic acid (Sigma-Aldrich, M1296) and malonyl-CoA lithium salt (Sigma-Aldrich, M4263) were dissolved in water at 100 mg/ml and then added to the top of bacterial lawns in concentrations ranging from 1 to 200 μg/plate for malonyl-CoA and up to 1000 μg/plate for malonic acid. The 1000-μg/plate replicates caused toxicity and growth arrest of nematodes, which was alleviated by adding NaOH solution to correct the pH; however, no rescue of embryo lethality of bpl-1(b281) was observed. Individual fatty acid sodium salts (Nu-Check Prep, Elysian, MN) dissolved in water were added to NGM at 0.1 mm with 0.1% Tergitol before plating as described previously (55).
Author contributions
J. S. W. conducted most of the experiments and wrote most of the paper. D. G. M. and K. J. K. isolated the bpl-1 mutant strains. D. G. M. performed the mapping and complementation tests and carried out the embryonic phenotype characterization shown in Fig. 1, including filming of the movies. J. S. W., D. G. M., K. J. K., and J. L. W. conceived ideas for the experiments. D. G. M., K. J. K., and J. L. W. edited the paper.
Supplementary Material
Acknowledgments
Bacterial strains were provided by the Coli Genetic Stock Center, supported by National Science Foundation/Biological Infrastructure/Living Collections program Grant DBI-0742708. Nematode strains were provided by the Caenorhabditis Genetics Center, funded by National Institutes of Health Office of Research Infrastructure Programs Grant P40 OD010440. We thank Liam Coyne for assistance with early bpl-1 RNAi experiments.
This work was supported by National Institutes of Health Grants T32GM083864 (to J. S. W.), R01DK074114 (to J. L. W.), and R01GM079112 and R01HD27689 (to K. J. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Movies S1–S3.
- HCS
- holocarboxylase synthetase
- MCD
- multiple carboxylase deficiency
- PUFA
- polyunsaturated fatty acid
- ACC
- acetyl-CoA carboxylase
- NGM
- nematode growth medium.
References
- 1. Zempleni J., Liu D., Camara D. T., and Cordonier E. L. (2014) Novel roles of holocarboxylase synthetase in gene regulation and intermediary metabolism. Nutr. Rev. 72, 369–376 10.1111/nure.12103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mock D. M. (2009) Marginal biotin deficiency is common in normal human pregnancy and is highly teratogenic in mice. J. Nutr. 139, 154–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sternicki L. M., Wegener K. L., Bruning J. B., Booker G. W., and Polyak S. W. (2017) Mechanisms governing precise protein biotinylation. Trends Biochem. Sci. 42, 383–394 10.1016/j.tibs.2017.02.001 [DOI] [PubMed] [Google Scholar]
- 4. Rios-Avila L., Prince S. A., Wijeratne S. S., and Zempleni J. (2011) A 96-well plate assay for high-throughput analysis of holocarboxylase synthetase activity. Clin. Chim. Acta 412, 735–739 10.1016/j.cca.2010.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wolf B., Hsia Y. E., Sweetman L., Feldman G., Boychuk R. B., Bart R. D., Crowell D. H., Di Mauro R. M., and Nyhan W. L. (1981) Multiple carboxylase deficiency: clinical and biochemical improvement following neonatal biotin treatment. Pediatrics 68, 113–118 [PubMed] [Google Scholar]
- 6. Suzuki Y., Yang X., Aoki Y., Kure S., and Matsubara Y. (2005) Mutations in the holocarboxylase synthetase gene HLCS. Hum. Mutat. 26, 285–290 10.1002/humu.20204 [DOI] [PubMed] [Google Scholar]
- 7. Kemphues K. J., Kusch M., and Wolf N. (1988) Maternal-effect lethal mutations on linkage group II of Caenorhabditis elegans. Genetics 120, 977–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reche P. A. (2000) Lipoylating and biotinylating enzymes contain a homologous catalytic module. Protein Sci. 9, 1922–1929 10.1110/ps.9.10.1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang X., Aoki Y., Li X., Sakamoto O., Hiratsuka M., Kure S., Taheri S., Christensen E., Inui K., Kubota M., Ohira M., Ohki M., Kudoh J., Kawasaki K., Shibuya K., Shintani A., Asakawa S., Minoshima S., Shimizu N., Narisawa K., Matsubara Y., and Suzuki Y. (2001) Structure of human holocarboxylase synthetase gene and mutation spectrum of holocarboxylase synthetase deficiency. Hum. Genet. 109, 526–534 10.1007/s004390100603 [DOI] [PubMed] [Google Scholar]
- 10. Rose L., and Gonczy P. (December 30, 2014) Polarity establishment, asymmetric division and segregation of fate determinants in early C. elegans embryos. WormBook 10.1895/wormbook.1.30.2 [DOI] [PubMed] [Google Scholar]
- 11. Chandler R. J., Aswani V., Tsai M. S., Falk M., Wehrli N., Stabler S., Allen R., Sedensky M., Kazazian H. H., and Venditti C. P. (2006) Propionyl-CoA and adenosylcobalamin metabolism in Caenorhabditis elegans: evidence for a role of methylmalonyl-CoA epimerase in intermediary metabolism. Mol. Genet. Metab. 89, 64–73 10.1016/j.ymgme.2006.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rappleye C. A., Tagawa A., Le Bot N., Ahringer J., and Aroian R. V. (2003) Involvement of fatty acid pathways and cortical interaction of the pronuclear complex in Caenorhabditis elegans embryonic polarity. BMC Dev. Biol. 3, 8 10.1186/1471-213X-3-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tagawa A., Rappleye C. A., and Aroian R. V. (2001) Pod-2, along with pod-1, defines a new class of genes required for polarity in the early Caenorhabditis elegans embryo. Dev. Biol. 233, 412–424 10.1006/dbio.2001.0234 [DOI] [PubMed] [Google Scholar]
- 14. Yi X., and Maeda N. (2005) Endogenous production of lipoic acid is essential for mouse development. Mol. Cell. Biol. 25, 8387–8392 10.1128/MCB.25.18.8387-8392.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gurvitz A. (2009) A C. elegans model for mitochondrial fatty acid synthase II: the longevity-associated gene W09H1.5/mecr-1 encodes a 2-trans-enoyl-thioester reductase. PLoS One 4, e7791 10.1371/journal.pone.0007791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Perez C. L., and Van Gilst M. R. (2008) A 13C isotope labeling strategy reveals the influence of insulin signaling on lipogenesis in C. elegans. Cell Metab. 8, 266–274 10.1016/j.cmet.2008.08.007 [DOI] [PubMed] [Google Scholar]
- 17. Dancy B. C., Chen S. W., Drechsler R., Gafken P. R., and Olsen C. P. (2015) 13C- and 15N-labeling strategies combined with mass spectrometry comprehensively quantify phospholipid dynamics in C. elegans. PLoS One 10, e0141850 10.1371/journal.pone.0141850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Watts J. L., and Browse J. (2002) Genetic dissection of polyunsaturated fatty acid synthesis in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 99, 5854–5859 10.1073/pnas.092064799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brooks K. K., Liang B., and Watts J. L. (2009) The influence of bacterial diet on fat storage in C. elegans. PLoS One 4, e7545 10.1371/journal.pone.0007545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tanaka T., Ikita K., Ashida T., Motoyama Y., Yamaguchi Y., and Satouchi K. (1996) Effects of growth temperature on the fatty acid composition of the free-living nematode Caenorhabditis elegans. Lipids 31, 1173–1178 10.1007/BF02524292 [DOI] [PubMed] [Google Scholar]
- 21. Shi X., Li J., Zou X., Greggain J., Rødkaer S. V., Faergeman N. J., Liang B., and Watts J. L. (2013) Regulation of lipid droplet size and phospholipid composition by stearoyl-CoA desaturase. J. Lipid Res. 54, 2504–2514 10.1194/jlr.M039669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brock T. J., Browse J., and Watts J. L. (2006) Genetic regulation of unsaturated fatty acid composition in C. elegans. PLoS Genet. 2, e108 10.1371/journal.pgen.0020108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Watts J. L., and Browse J. (2000) A palmitoyl-CoA-specific Δ9 fatty acid desaturase from Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 272, 263–269 10.1006/bbrc.2000.2772 [DOI] [PubMed] [Google Scholar]
- 24. Brock T. J., Browse J., and Watts J. L. (2007) Fatty acid desaturation and the regulation of adiposity in Caenorhabditis elegans. Genetics 176, 865–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benenati G., Penkov S., Müller-Reichert T., Entchev E. V., and Kurzchalia T. V. (2009) Two cytochrome P450s in Caenorhabditis elegans are essential for the organization of eggshell, correct execution of meiosis and the polarization of embryo. Mech. Dev. 126, 382–393 10.1016/j.mod.2009.02.001 [DOI] [PubMed] [Google Scholar]
- 26. Olson S. K., Greenan G., Desai A., Muller-Reichert T., and Oegema K. (2012) Hierarchical assembly of the eggshell and permeability barrier in C. elegans. J. Cell Biol. 198, 731–748 10.1083/jcb.201206008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu H., Shen H., Sewell A. K., Kniazeva M., and Han M. (2013) A novel sphingolipid-TORC1 pathway critically promotes postembryonic development in Caenorhabditis elegans. Elife 2, e00429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang H., Abraham N., Khan L. A., Hall D. H., Fleming J. T., and Göbel V. (2011) Apicobasal domain identities of expanding tubular membranes depend on glycosphingolipid biosynthesis. Nat. Cell Biol. 13, 1189–1201 10.1038/ncb2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takamura Y., and Nomura G. (1988) Changes in the intracellular concentration of acetyl-CoA and malonyl-CoA in relation to the carbon and energy metabolism of Escherichia coli K12. J. Gen. Microbiol. 134, 2249–2253 [DOI] [PubMed] [Google Scholar]
- 30. Hirsh D., Oppenheim D., and Klass M. (1976) Development of the reproductive system of Caenorhabditis elegans. Dev. Biol. 49, 200–219 10.1016/0012-1606(76)90267-0 [DOI] [PubMed] [Google Scholar]
- 31. Tang H., and Han M. (2017) Fatty acids regulate germline sex determination through ACS-4-dependent myristoylation. Cell 169, 457–469.e13 10.1016/j.cell.2017.03.049 [DOI] [PubMed] [Google Scholar]
- 32. Watson E., MacNeil L. T., Ritter A. D., Yilmaz L. S., Rosebrock A. P., Caudy A. A., and Walhout A. J. (2014) Interspecies systems biology uncovers metabolites affecting C. elegans gene expression and life history traits. Cell 156, 759–770 10.1016/j.cell.2014.01.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Watson E., Olin-Sandoval V., Hoy M. J., Li C. H., Louisse T., Yao V., Mori A., Holdorf A. D., Troyanskaya O. G., Ralser M., and Walhout A. J. (2016) Metabolic network rewiring of propionate flux compensates vitamin B12 deficiency in C. elegans. Elife 5, e17670 10.7554/eLife.17670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lin C. J., and Wang M. C. (2017) Microbial metabolites regulate host lipid metabolism through NR5A-Hedgehog signalling. Nat. Cell Biol. 19, 550–557 10.1038/ncb3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Uchida Y., and Holleran W. M. (2008) ω-O-acylceramide, a lipid essential for mammalian survival. J. Dermatol. Sci. 51, 77–87 10.1016/j.jdermsci.2008.01.002 [DOI] [PubMed] [Google Scholar]
- 36. Zheng Y., Yin H., Boeglin W. E., Elias P. M., Crumrine D., Beier D. R., and Brash A. R. (2011) Lipoxygenases mediate the effect of essential fatty acid in skin barrier formation: a proposed role in releasing ω-hydroxyceramide for construction of the corneocyte lipid envelope. J. Biol. Chem. 286, 24046–24056 10.1074/jbc.M111.251496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Burri B. J., Sweetman L., and Nyhan W. L. (1981) Mutant holocarboxylase synthetase: evidence for the enzyme defect in early infantile biotin-responsive multiple carboxylase deficiency. J. Clin. Invest. 68, 1491–1495 10.1172/JCI110402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moore J. H., and Christie W. W. (1979) Lipid metabolism in the mammary gland of ruminant animals. Prog. Lipid Res. 17, 347–395 10.1016/0079-6832(79)90012-0 [DOI] [PubMed] [Google Scholar]
- 39. Post-Beittenmiller D., Roughan G., and Ohlrogge J. B. (1992) Regulation of plant fatty acid biosynthesis: analysis of acyl-coenzyme a and acyl-acyl carrier protein substrate pools in spinach and pea chloroplasts. Plant Physiol. 100, 923–930 10.1104/pp.100.2.923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McGarry J. D., Leatherman G. F., and Foster D. W. (1978) Carnitine palmitoyltransferase I: the site of inhibition of hepatic fatty acid oxidation by malonyl-CoA. J. Biol. Chem. 253, 4128–4136 [PubMed] [Google Scholar]
- 41. McGarry J. D., Mannaerts G. P., and Foster D. W. (1977) A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J. Clin. Invest. 60, 265–270 10.1172/JCI108764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bao B., Wijeratne S. S. K., Rodriguez-Melendez R., and Zempleni J. (2011) Human holocarboxylase synthetase with a start site at methionine-58 is the predominant nuclear variant of this protein and has catalytic activity. Biochem. Biophys. Res. Commun. 412, 115–120 10.1016/j.bbrc.2011.07.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Narang M. A., Dumas R., Ayer L. M., and Gravel R. A. (2004) Reduced histone biotinylation in multiple carboxylase deficiency patients: a nuclear role for holocarboxylase synthetase. Hum. Mol. Genet. 13, 15–23 [DOI] [PubMed] [Google Scholar]
- 44. Trujillo-Gonzalez I., Cervantes-Roldan R., Gonzalez-Noriega A., Michalak C., Reyes-Carmona S., Barrios-Garcia T., Meneses-Morales I., and Leon-Del-Rio A. (2014) Holocarboxylase synthetase acts as a biotin-independent transcriptional repressor interacting with HDAC1, HDAC2 and HDAC7. Mol. Genet. Metab. 111, 321–330 10.1016/j.ymgme.2013.10.016 [DOI] [PubMed] [Google Scholar]
- 45. Stiernagle T. (2006) Maintenance of C. elegans (February 11, 2006) WormBook (The C. elegans Research Community, ed) 10.1895/wormbook.1.1010.1 [DOI] [Google Scholar]
- 46. Mello C. C., Kramer J. M., Stinchcomb D., and Ambros V. (1991) Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fraser A. G., Kamath R. S., Zipperlen P., Martinez-Campos M., Sohrmann M., and Ahringer J. (2000) Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature 408, 325–330 10.1038/35042517 [DOI] [PubMed] [Google Scholar]
- 48. Verbrugghe K. J., and Chan R. C. (2011) Imaging C. elegans embryos using an epifluorescent microscope and open source software. J. Vis. Exp. 10.3791/2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rappleye C. A., Tagawa A., Lyczak R., Bowerman B., and Aroian R. V. (2002) The anaphase-promoting complex and separin are required for embryonic anterior-posterior axis formation. Dev. Cell 2, 195–206 10.1016/S1534-5807(02)00114-4 [DOI] [PubMed] [Google Scholar]
- 50. Brennan L. D., Roland T., Morton D. G., Fellman S. M., Chung S., Soltani M., Kevek J. W., McEuen P. M., Kemphues K. J., and Wang M. D. (2013) Small molecule injection into single-cell C. elegans embryos via carbon-reinforced nanopipettes. PLoS One 8, e75712 10.1371/journal.pone.0075712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Boyd L., Guo S., Levitan D., Stinchcomb D. T., and Kemphues K. J. (1996) PAR-2 is asymmetrically distributed and promotes association of P granules and PAR-1 with the cortex in C. elegans embryos. Development 122, 3075–3084 [DOI] [PubMed] [Google Scholar]
- 52. Etemad-Moghadam B., Guo S., and Kemphues K. J. (1995) Asymmetrically distributed PAR-3 protein contributes to cell polarity and spindle alignment in early C. elegans embryos. Cell 83, 743–752 10.1016/0092-8674(95)90187-6 [DOI] [PubMed] [Google Scholar]
- 53. Robert V. J., Sijen T., van Wolfswinkel J., and Plasterk R. H. (2005) Chromatin and RNAi factors protect the C. elegans germline against repetitive sequences. Genes Dev. 19, 782–787 10.1101/gad.332305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Harder M. E., Beacham I. R., Cronan J. E. Jr., Beacham K., Honegger J. L., and Silbert D. F. (1972) Temperature-sensitive mutants of Escherichia coli requiring saturated and unsaturated fatty acids for growth: isolation and properties. Proc. Natl. Acad. Sci. U.S.A. 69, 3105–3109 10.1073/pnas.69.11.3105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Deline M. L., Vrablik T. L., and Watts J. L. (2013) Dietary supplementation of polyunsaturated fatty acids in Caenorhabditis elegans. J. Vis. Exp. 10.3791/50879 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.