

Abstract

5′-Adenosine monophosphate-activated protein kinase (AMPK) is a key regulator of mammalian energy homeostasis and has been implicated in mediating many of the beneficial effects of exercise and weight loss including lipid and glucose trafficking. As such, the enzyme has long been of interest as a target for the treatment of Type 2 Diabetes Mellitus. We describe the optimization of β1-selective, liver-targeted AMPK activators and their evolution into systemic pan-activators capable of acutely lowering glucose in mouse models. Identifying surrogates for the key acid moiety in early generation compounds proved essential in improving β2-activation and in balancing improvements in plasma unbound fraction while avoiding liver sequestration.
Keywords: AMPK, activator, T2DM, ACC, liver-selectivity
Type 2 Diabetes Mellitus (T2DM) is a chronic, progressive disease characterized by insulin resistance leading to reduced peripheral glucose uptake and increased hepatic glucose production. The incidence of T2DM has reached epidemic proportions with 642 million people projected to suffer from the disease by 2040.1
Several classes of antihyperglycemic agents are prescribed for the treatment of T2DM, either singly or in combination (e.g., sulfonylureas, metformin, PPARγ-selective agonists, GLP-1 analogs, DPP-4 inhibitors, SGLT2 inhibitors, insulin, and insulin analogs). Despite the available options, most diabetic patients fail to achieve their therapeutic goal. In addition, many current therapies have significant limitations and liabilities such as hypoglycemia, weight gain, edema, fractures, lactic acidosis, and gastrointestinal intolerance.2
Exercise, combined with weight loss, is a powerful means to lower blood glucose levels in T2DM patients and forms the foundation for effective treatment and disease management.3 Numerous beneficial effects of exercise are thought to be mediated through 5′-adenosine monophosphate-activated protein kinase (AMPK).4 These include alterations in cellular fuel utilization as well as changes in gene transcription.
AMPK is a Ser/Thr protein kinase that serves as the primary mechanism for maintenance of energy homeostasis in eukaryotic cells.5 The enzyme is a heterotrimer comprising a catalytic α-subunit (two isoforms), a β-subunit (two isoforms), and a regulatory γ-subunit (three isoforms); it is activated by changes in cellular adenine nucleotide or calcium levels.6
However, there are potential safety concerns associated with AMPK activation. Mutations in the AMPK γ2-subunit are found in a percentage of Wolff–Parkinson–White syndrome patients, leading to PRKAG2 cardiomyopathy. These mutations appear to result in intrinsic activation of AMPK.7,8 It is unclear which clinical manifestations of PRKAG2 cardiomyopathy have developmental origins, but cardiac safety is a major concern for the therapeutic application of systemic AMPK activators.
Despite a growing understanding of the link between AMPK and exercise and the myriad potential applications of an AMPK activator,7 to date, there have been few reports of small molecule direct activators.9,10 This likely reflects the complexity of the enzyme as well as, in our case (and presumably others), the failure of traditional high throughput screening campaigns to identify activator hits. Many of the existing small molecule tools are indirect activators that work by increasing the AMP/ATP ratio in cells. By far the most widely used direct activator is the prodrug AICAR (1a), which is 5′-phosphorylated intracellularly to the AMP-mimetic ZMP. Unfortunately, the compound has poor selectivity and is rapidly cleared in vivo. A-769662 (1b, Figure 1) was developed by Abbott and has stood as the most widely investigated drug-like small molecule, non-nucleoside activator. A-769662 selectively activates the β1-containing AMPK complexes and does not activate the 6 β2-containing complexes.11 The molecule also suffers from some off-target activities, and while clearance is sufficiently low for mechanistic work, permeability is poor and the compound has low oral bioavailability, limiting its utility for in vivo studies.
Figure 1.

Structure of A-769662.
With the potential cardiac adverse effects in mind, we initially focused on identifying liver-selective molecules. Liver AMPK activation is anticipated to provide some benefits, in particular in addressing dyslipidemia and improving insulin sensitization.12
After failing to identify any progressive leads from multiple high throughput screens of the Merck compound collection, we turned to a fragment screening approach. From a library of ca. 25,000 compounds, compound 2 (Figure 2) was identified as a weak (EC50 ≈ 30 μM) activator of AMPK subtype 1 (α1β1γ1). A rapid hit-to-lead effort identified compound 3 with significantly improved potency at AMPK1 (α1β1γ1), and the other five β1 containing AMPK complexes. However, activation of the β2-containing complexes remained low. Compound 3a is an analog of compound 3 with very similar properties that became a standard in our in vitro enzyme assays. To address any potential variability in the assay, enzyme activation data (both EC50 and %Max) (Table 1) are reported relative to data for this compound (Supporting Information). Since activity for β1 containing complexes was usually comparable (and similarly activity for β2-containing complexes was comparable), screening was typically run with one representative β1-containing enzyme (AMPK1) and one representative β2-containing enzyme [AMPK10 (α2β2γ1)]; data provided a gauge as to the relative β1- vs β2-complex selectivity for each analog. X-ray crystallographic data for compound 3a bound to AMPK has been reported.13
Figure 2.

Hit-to-lead progression of the series and evolution from hepatic to systemic β1-selective activators.
Table 1. Profile of Compounds 3–6: Fold Change in Phosphorylated AMPK1 (pAMPK1) and pAMPK10 Activation vs. Compound 3a, Effects on DNL in 3T3-L1 Adipocytes, Cell Permeability, Unbound Fraction in Human Plasma, Fold Change in Muscle pACC, and Liver/Muscle Exposure Ratio in db/+ Mice.
| cmpd | AMPK1 fold change vs 3a EC50a (%Max)b | AMPK10 fold change vs 3a EC50a (%Max)b | DNL IC50 (μM)a | Papp (× 10–6 cm/s)d | human Fu: 10% serum (100% serum)d | liver pacc/acc fold change vs vehiclee | muscle pACC/ACC fold change vs vehiclee | Cliver/Cmusclee |
|---|---|---|---|---|---|---|---|---|
| 3 | 1.8 (1.0) | 0.52 (0.8) | 11 | 9.4 | 0.2 | 1.7 | 1.1 | 51 |
| 4 | 7.5 (1.0) | 18 (0.8) | 1.4 | 12.9 | 0.3 | 1.4 | 2.0 | 4 |
| 4a | 6.3 (0.9) | 100 (0.7) | 5.3 | NDc | 1.0 | 1.8 | 2.3 | 7 |
| 4b | 4.3 (1.0) | 6.0 (1.1) | 5.4 | 21.5 | 5.4 (0.6) | 2.0 | 1.5 | 650 |
| 5 | 3.3 (1.0) | 3.3 (1.5) | 3.9 | NDc | 0.2 | 1.5 | 1.2 | 190 |
| 6 | 1.0 (0.5) | 0.3 (1.9) | NDc | 23.6 | 4.6 (0.1) | 1.9 | 2.9 | 3 |
Average of at least two experiments, each using a 10-point titration.
Highest activation achieved at titrations to 50 μM compared to the maximal activation achieved for AMP.
Not determined.
Average of at least three experiments.
N = 7 animals in a single experiment.
We developed robust in vitro assays and in vivo pharmacodynamic screens utilizing the site-specific phosphorylation of acetyl CoA carboxylase (ACC) isoforms 1 and 2 by AMPK. The assays either directly measured the ratio of pACC to total ACC as a robust proximate target engagement readout for AMPK activation, or measured suppression of de novo lipogenesis (DNL) or activation of fatty acid oxidation (FAO) as functional consequences of increased ACC phosphorylation/inhibition.14 Following incubation with 3T3-L1 adipocytes, compound 3 suppressed DNL with an IC50 of 1.1 μM (Table 1).
Our primary in vivo pharmacodynamic screen assessed target engagement (TE) (pACC/ACC elevations) in liver and muscle in db/+ mice 2 h after a single oral dose. Tissue concentrations of compound were also collected, and the combination of pACC elevation and exposure ratios in the two tissues were used to infer the degree of liver selectivity.
Compound 3 (30 mpk) caused robust increases in liver TE but did not elevate pACC/ACC in muscle in db/+ mice (Table 1). The 51-fold higher exposure in liver vs muscle further suggested that the compound preferentially distributes to the liver.
Despite a relatively short plasma half-life and a free fraction in plasma below the level of quantification, compound 3 served as a useful tool to interrogate the effects of β1-selective AMPK activation in the liver. While reductions in plasma insulin were evident following chronic administration in an insulin resistant mouse model (diet-induced obese mouse), compound 3 failed to cause reproducible glucose lowering in rodent models of T2DM.15
As discussed, several studies have implicated activation of AMPK as a principle mediator of many of the beneficial effects of exercise on glucose and lipid utilization.4,16 Our studies demonstrated that beneficial effects on lipids can be realized with hepatic β1-selective activation in rodents; however, glucose homeostasis remains unaffected. Reports have suggested that β2-containing AMPK complexes predominate in skeletal muscle.17 This suggested that enhancing distribution to muscle and increasing the degree of β2-containing AMPK activation had the potential to provide greater efficacy in resolving hyperglycemia.
Nothing in the SAR we had generated thus far suggested a means to improve β2 activation. With no relevant structural information available to rationally modulate activity, the team focused on addressing the ADME/PK challenges while looking for SAR that might be used to drive improved pan-activation.
We initially focused on two key goals: dialing-out liver selectivity and enhancing the free fraction in plasma, something that we anticipated would be needed to avoid difficulties in clinical development and translation.
In looking at the properties that might contribute to liver distribution, we focused on two areas: susceptibility to liver-uptake transporters and membrane permeability. Cell-based studies confirmed that compound 3 is a substrate of OATP1B1 and 1B2. In addition, assessment in LLC-PK1 cells determined that permeability of the compound was relatively low (Papp ≈ 9 × 10–6 cm/s). We therefore hypothesized that active uptake into hepatocytes was more rapid than passive diffusion into/out of the cells resulting in compound sequestration in liver.
Liver-selectivity was reduced in a series of aliphatic acids represented by compound 4. Compound 4 was more β1-selective than compound 3 with similar activity at AMPK1 but reduced potency at AMPK10. However, cell activity was similar (IC50 = 1.4 μM for suppression of DNL). In vivo, the compound caused significant elevation of liver and muscle TE with comparable exposure in the two tissues (liver/muscle concentration ratio ≈ 4) (Table 1).
Similar to compound 3, compound 4 remained a substrate of the OATP transporters. Permeability for the compound was similar to compound 3 (Papp ≈ 13 × 10–6 cm/s) suggesting that compound 4, while still a substrate, may have reduced susceptibility for active uptake. Plasma protein binding for the compound was little improved relative to compound 3 (Table 1) so we turned our attention toward altering physicochemical properties to address this.
Efforts to increase polarity focused on changes to the benzimidazole core and the lipophilic biphenyl tail (Figure 3). Replacement of the terminal phenyl ring with a pyrrolidine led to the identification of compound 4a. cLogP was improved relative to compound 4 (5.8 vs 7.3), and we observed a commensurate improvement in plasma protein binding with ∼1% free in 10% human serum. Introduction of a heteroatom in the core led to compound 4b with further improvements in cLogP (4.3). Again, these enhancements to polarity translated to improved plasma protein binding with ∼5.4% free in 10% human serum. When we looked at 100% human serum, we were gratified to find that the unbound fraction was indeed measurable (∼0.6%) (Table 1). Compounds 4a and 4b had comparable activity to compound 3 at AMPK1 but degraded AMPK10 potency. Both compounds had lower cell potency (as measured by reduction of DNL in adipocytes) vs compound 4 with IC50s of 5.3 and 5.4 μM, respectively. In vivo, the behavior of the two compounds diverged. Compound 4a maintained a similar profile to compound 4 with significant TE in liver and muscle and a low liver/muscle exposure ratio (∼7). Exposure of compound 4b was almost exclusively limited to liver (liver/muscle concentration ratio ∼649) with no increase in muscle pACC despite robust TE in the liver (Table 1).
Figure 3.

Evolution of compounds with improved unbound fraction in plasma and identification of systemic pan-activator MK-8722.
Another approach to enhancing polarity is represented by compound 5 where we attempted to add heteroatoms in the carbocyclic acid moiety. This had a more limited impact on PPB relative to the changes to the biphenyl and core regions, but resulted in a similar in vivo distribution pattern to compound 4b with liver-specific TE and a liver/muscle exposure ratio of ∼192. One serendipitous finding focused on the in vitro selectivity profile of the compound. While activity at AMPK1 was still comparable to the other analogs, compound 5 was the first in the series where we saw modest increases in the maximal activation of β2 containing complex AMPK10 (Table 1).
Permeability for the analog with measurable free fraction in plasma (compound 4b) was enhanced relative to earlier analogs (Papp ≈ 22 × 10–6 cm/s) suggesting that the degree of liver selectivity again likely rested on rates of active transport into the liver. The SAR raised the prospect that the physicochemical properties that we anticipated would drive to improved PPB (increased polarity) were running counter to those that would enhance systemic compound distribution. This suggested that we needed to overhaul our strategy if we wanted to accomplish both goals. Since polar acid moieties serve as key recognition elements for the OATP transporters, this became a major focus of optimization efforts. Replacement of the acid was also anticipated to reduce binding to albumin and increase the unbound fraction of drug in plasma. In bile-duct cannulated rat studies, the primary routes of elimination of compound 3 were biliary excretion of the parent molecule and parent glucuronidation followed by biliary excretion of the glucuronide (data not shown). This provided additional rationale for the strategy of replacing the acid moiety.
We capitalized on the slight improvement in β2 activation observed for compound 5 by focusing on furan and pyran based moieties and replaced the acid moiety with alcohols and ethers. This led to a series of alcohol- and sugar-based analogs that eventually identified MK-8722 (Figure 3).
The medicinal chemistry route for MK-8722 is outlined in Scheme 1. The aza-benzimidazole ring was constructed from commercially available 5-chloro-3-nitropyridin-2-amine via halogenation of the 6-position, reduction of the nitro group, and cyclization with thiophosgene. Subsequent methylation and oxidation to the sulfoxide afforded the fully functionalized core. Palladium catalyzed coupling with biphenylboronic acid and SEM protection prepared the way for substitution with the protected sugar moiety. A final deprotection afforded MK-8722.
Scheme 1. Synthesis of MK-8722.

Reagents and conditions: (a) NCS, AcOH, 80 °C, 18 h; (b) NaI, AcOH, 90 °C, 2 h; (c) SnCl2·2H2O, EtOH, 70 °C, 0.5 h; (d) thiophosgene, DMAP, THF, 22 °C, 1 h; (e) KOH, MeI, EtOH, 22 °C, 0.5 h; (f) oxone, MeCN/H2O, 22 °C, 18 h; (g) 4-biphenylboronic acid, K3PO4, Pd(OAc)2, n-butyldiadamantylphosphine, THF/H2O, 45 °C, 18 h; (h) Et3N, SEMCl, THF, 0–22 °C, 15 min; (i) 1,4:3,6-dianhydro-2-O-[tert-butyl(dimethyl)silyl]-d-mannitol, DBU, DMA, 22 °C, 18 h; (j) KHSO4, HCO2H, 22 °C, 18 h then NaOH, THF, 0–22 °C, 18 h.
The acid-replacement strategy proved successful in allowing moderation of both liver sequestration and plasma protein binding while increasing activation of the β2-containing AMPK subtypes. MK-8722 is a potent, systemic activator of all 12 AMPK complexes (supplemental). The compound has high permeability (Papp ≈ 24 *10–6 cm/s), is not a substrate of OATP1B1 or OATP1B2, and has measurable free fraction in plasma (Table 1).
In order to assess the impact of increased β2 activity on glycemic effects, we compared the effects of 10 and 30 mpk of MK-8722 and the systemic β1-selective compound 4 in eDIO mice (Figure 4). These mice are hyperinsulinemic with slightly elevated blood glucose levels, serving as a useful model of insulin resistance/prediabetes. The assay examined acute compound effects on fasting blood glucose levels over the course of 2 h. Compounds were administered in 0.25% MC, 5% Tween-80, and 0.02% SDS. Plasma exposure and both liver and muscle pACC were assessed at the end of the study (approximately 2–3 h postdose). Both compounds achieved high exposure in plasma (MK-8722, 18 and 59 μM; compound 4, 43 and 124 μM). Given the high plasma protein binding of compound 4, it was not possible to compare the free fraction for the two compounds so we relied on target engagement assays to confirm we had attained similar degrees of AMPK activation. Rodent liver is almost exclusively made up of β1-containing AMPK complexes.18 Given the similar AMPK1 potency and maximal activation for both compounds as well as their comparable, balanced tissue distribution, we anticipated that liver pACC would provide a means to compare the systemic target engagement attained by each compound. Both compounds caused a similar elevation of liver pACC/ACC (Figure 4A). However, the compounds diverged in their effects on muscle pACC (Figure 4B). Muscle contains significant amounts of β2-derived AMPK complexes.17,18 Compound 4 was able to elevate pACC/ACC in muscle at the higher 30 mpk dose, presumably due to robust activation of β1 containing AMPK complexes and a modest degree of β2-complex AMPK activation. However, MK-8722 increased pACC/ACC to a much greater extent, reflecting the ability of the compound to robustly activate both β1 and β2-containing complexes. Importantly, the compounds also caused profoundly different effects on glucose (Figure 4C). Relative to vehicle treated controls, compound 4 administration had no significant effect on glucose at either dose (albeit with some variability in the high dose animals). By contrast, MK-8722 induced a significant reduction in glucose at both doses.
Figure 4.
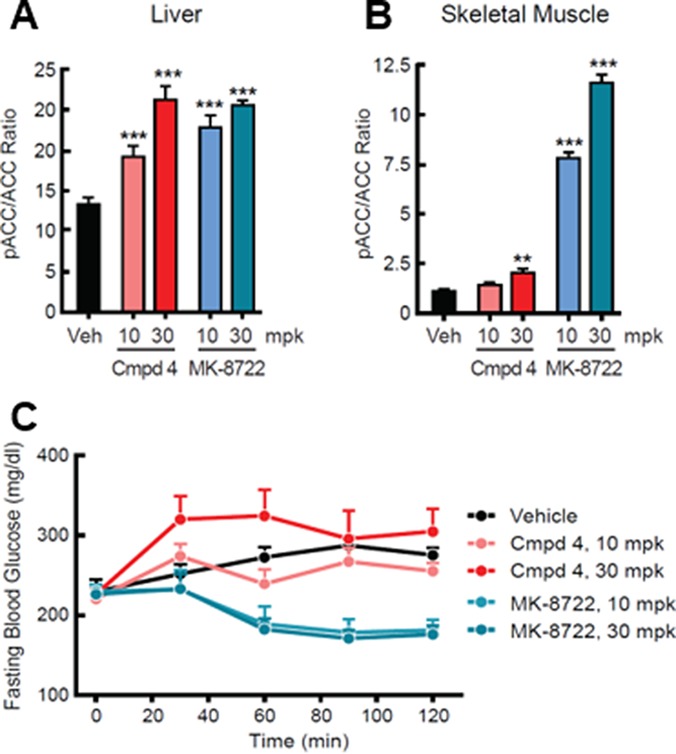
Effects of compound 4 and MK-8722 in eDIO mice. (A−B) Effects on tissue pACC/ACC. (C) Effects on fasting blood glucose. N = 8 in each group.
We subsequently assessed the effects of MK-8722 administration in an oral glucose tolerance test (oGTT) in eDIO mice (Figure 5A,B). The compound dose-dependently lowered fasting glucose as well as the glycemic excursion following the glucose challenge. We confirmed that these effects were not due to reduced glucose absorption by running a similar study where xylose was coadministered with the glucose challenge. While the effects on glucose were recapitulated (Figure 5C), there was no change in xylose absorption relative to vehicle treated controls (Figure 5D). Similar effects on fasting blood glucose and glucose excursion during a GTT have been observed for the compound across a range of normal, insulin resistant, and frankly diabetic rodent models.19 The minimum efficacious exposure in eDIO mice was ∼1 μM (∼1 nM unbound), although exposures of ∼3 μM (3 nM unbound) are required for significant glucose lowering in other rodent models.
Figure 5.
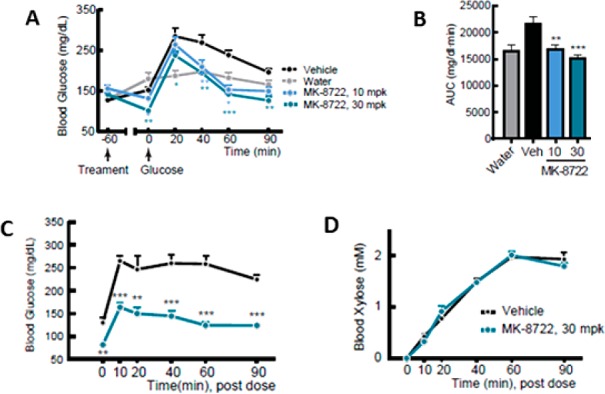
Effects of MK-8722 on glucose and xylose excursion during an oGTT in lean C57BL/6 mice. (A) Dose dependent effects on the blood glucose/time curve. (B) AUC of the glucose/time curve in panel A. (C) Effects on the blood glucose/time curve. (D) Effects on the blood xylose/time curve. N = 8 in each group.
In a screen of 115 enzymes, transporters, and receptors, MK-8722 had 3–10 μM activity at four common off-targets [serotonin 5-hydroxytryptamine (5-HT2A), dopamine transporter (DAT), norepinephrine transporter (NET), monoamineoxidase-A (MOA-A), and monoamine oxidase-B (MOA-B)]. None of these activities are anticipated to result in any significant pharmacology at the (unbound) exposures relevant for AMPK-mediated efficacy. Pharmacokinetics in preclinical species were supportive of QD dosing in the clinic.19
In summary, MK-8722 is a selective, small molecule, direct, pan-AMPK activator. Our efforts to arrive at the molecule also identified several tool compounds that have been useful in assessing the in vitro and in vivo effects of liver-selective and/or β1-selective AMPK activation. Based on our studies, both β1- and liver-selective activators have robust effects on lipid metabolism in rodents. While these may prove to be useful for various disease conditions, they were not sufficient to improve glucose homeostasis in rodents. Data obtained using MK-8722 suggest that β2 activation is required to modulate glycemic control, reinforcing the link between glucose uptake following exercise and activation of β2 AMPK complexes.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00417.
Syntheses of compounds 3–6; in vitro enzyme activation data for compounds 3a and 6 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Federation I. D.IDF Diabetes Atlas, 7th ed.; International Diabetes Federation: Belgium, 2015. [Google Scholar]
- Sterrett J. J.; Bragg S.; Weart C. W. Type 2 Diabetes Medication Review. Am. J. Med. Sci. 2016, 351, 342–55. 10.1016/j.amjms.2016.01.019. [DOI] [PubMed] [Google Scholar]
- Sigal R. J.; Kenny G. P.; Boule N. G.; Wells G. A.; Prud’homme D.; Fortier M.; Reid R. D.; Tulloch H.; Coyle D.; Phillips P.; Jennings A.; Jaffey J. Effects of aerobic training, resistance training, or both on glycemic control in type 2 diabetes: a randomized trial. Ann. Intern. Med. 2007, 147, 357–69. 10.7326/0003-4819-147-6-200709180-00005. [DOI] [PubMed] [Google Scholar]
- Richter E. A.; Hargreaves M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 2013, 93, 993–1017. 10.1152/physrev.00038.2012. [DOI] [PubMed] [Google Scholar]
- Hardie D. G. AMP-activated protein kinase: maintaining energy homeostasis at the cellular and whole-body levels. Annu. Rev. Nutr. 2014, 34, 31–55. 10.1146/annurev-nutr-071812-161148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg G. R.; Kemp B. E. AMPK in health and disease. Physiol. Rev. 2009, 89, 1025–1078. 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- Grahame Hardie D. AMP-activated protein kinase: a key regulator of energy balance with many roles in human disease. J. Intern. Med. 2014, 276, 543–59. 10.1111/joim.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arad M.; Seidman C. E.; Seidman J. G. AMP-activated protein kinase in the heart: role during health and disease. Circ. Res. 2007, 100, 474–88. 10.1161/01.RES.0000258446.23525.37. [DOI] [PubMed] [Google Scholar]
- Rana S.; Blowers E. C.; Natarajan A. Small molecule adenosine 5′-monophosphate activated protein kinase (AMPK) modulators and human diseases. J. Med. Chem. 2015, 58, 2–29. 10.1021/jm401994c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron K. O.; Kurumbail R. G. Recent progress in the identification of adenosine monophosphate activated protein kinase (AMPK) activators. Bioorg. Med. Chem. Lett. 2016, 26, 5139–5148. 10.1016/j.bmcl.2016.09.065. [DOI] [PubMed] [Google Scholar]
- Treebak J. T.; Birk J. B.; Hansen B. F.; Olsen G. S.; Wojtaszewski J. F. A-769662 activates AMPK beta1-containing complexes but induces glucose uptake through a PI3-kinase-dependent pathway in mouse skeletal muscle. Am. J. Physiol. Cell Physiol. 2009, 297, C1041–52. 10.1152/ajpcell.00051.2009. [DOI] [PubMed] [Google Scholar]
- Viollet B.; Guigas B.; Leclerc J.; Hebrard S.; Lantier L.; Mounier R.; Andreelli F.; Foretz M. AMP-activated protein kinase in the regulation of hepatic energy metabolism: from physiology to therapeutic perspectives. Acta Physiol. 2009, 196, 81–98. 10.1111/j.1748-1716.2009.01970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B.; Sanders M. J.; Carmena D.; Bright N. J.; Haire L. F.; Underwood E.; Patel B. R.; Heath R. B.; Walker P. A.; Hallen S.; Giordanetto F.; Martin S. R.; Carling D.; Gamblin S. J. Structural basis of AMPK regulation by small molecule activators. Nat. Commun. 2013, 4, 3017. 10.1038/ncomms4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton M. D.; Galic S.; Marcinko K.; Sikkema S.; Pulinilkunnil T.; Chen Z. P.; O’Neill H. M.; Ford R. J.; Palanivel R.; O’Brien M.; Hardie D. G.; Macaulay S. L.; Schertzer J. D.; Dyck J. R.; van Denderen B. J.; Kemp B. E.; Steinberg G. R. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–54. 10.1038/nm.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan P.; Romero F. A.; Wodka D.; Kassick A. J.; Zhou G.; Chen Y.; Zhang X.; Zhang A.; Ying L.; Trujillo M. E.; Shao Q.; Wu M.; Xu S.; He H.; Chapman K. T.; Weber A.; Sebhat I. K.; Makara G. M. Hit-to-lead optimization and discovery of 5-((5-([1,1′-biphenyl]-4-yl)-6-chloro-1H-benzo[d]imidazol-2-yl)oxy)-2-methylbenzoic acid (MK-3903): a novel class of benzimidazole-based activators of AMP-activated protein kinase. J. Med. Chem. 2017, 60, 9040–9052. 10.1021/acs.jmedchem.7b01344. [DOI] [PubMed] [Google Scholar]
- O’Neill H. M. AMPK and Exercise: Glucose Uptake and Insulin Sensitivity. Diabetes Metab. J. 2013, 37, 1–21. 10.4093/dmj.2013.37.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszewski J. F.; Birk J. B.; Frosig C.; Holten M.; Pilegaard H.; Dela F. 5′AMP activated protein kinase expression in human skeletal muscle: effects of strength training and type 2 diabetes. J. Physiol. 2005, 564, 563–73. 10.1113/jphysiol.2005.082669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Heierhorst J.; Mann R. J.; Mitchelhill K. I.; Michell B. J.; Witters L. A.; Lynch G. S.; Kemp B. E.; Stapleton D. Expression of the AMP-activated protein kinase beta1 and beta2 subunits in skeletal muscle. FEBS Lett. 1999, 460, 343–348. 10.1016/S0014-5793(99)01371-X. [DOI] [PubMed] [Google Scholar]
- Myers R. W.; Guan H.-P.; et al. Systemic Pan-AMPK Activator MK-8722 Improves Glucose Homeostasis But Induces Cardiac Hypertrophy. Science 2017, 357, 507–511. 10.1126/science.aah5582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.