

Abstract
CXCR4 is the most common chemokine receptor expressed on the surface of many cancer cell types. In comparison to normal cells, cancer cells overexpress CXCR4, which correlates with cancer cell metastasis, angiogenesis, and tumor growth. CXCR4 antagonists can potentially diminish the viability of cancer cells by interfering with CXCL12-mediated pro-survival signaling and by inhibiting chemotaxis. Herein, we describe a series of CXCR4 antagonists that are derived from (S)-5,6,7,8-tetrahydroquinolin-8-amine that has prevailed in the literature. This series removes the rigidity and chirality of the tetrahydroquinoline providing 2-(aminomethyl)pyridine analogs, which are more readily accessible and exhibit improved liver microsomal stability. The medicinal chemistry strategy and biological properties are described.
Keywords: CXCR4, GPCR, antagonist, chemokine, anticancer, tetrahydroisoquinoline, muscarinic, biotransformation
Chemokines and chemokine receptors are essential to immune cell trafficking.1 The human chemokine system contains four types of ligands differentiated by their cysteine residue pattern at the amino termini.2 Of particular interest is the C–X–C motif containing chemokine receptor type 4 (CXCR4). CXCR4 is a seven transmembrane G protein-coupled receptor (GPCR).3 In normal embryonic and adult tissue, CXCR4 expression is primarily associated with hematopoietic and immune systems; however, it can be found in several other tissues including the thymus, brain, and small intestine. When CXCR4 is bound to its specific natural ligand CXCL12 (also known as stromal cell derived factor 1, SDF-1), downstream signals mediated by heterotrimeric G proteins trigger various cellular responses including increased intracellular calcium flux, gene transcription, chemotaxis, cell survival, and proliferation.1 CXCR4 stands as the most widely overexpressed chemokine receptor in human tumor cells.4−7 In several types of cancer, CXCR4 activation stimulates proliferation, angiogenesis, migration, adhesion, invasion, metastasis, and survival.8 Because cancer cells utilize the CXCR4/CXCL12 mechanism for survival, metastasis, and immune evasion, identification of new and effective small molecule CXCR4 inhibitors would be beneficial.
Several small molecule CXCR4 antagonists exemplified by AMD3100 (Plerixafor, Mozobil),9 AMD070 (X4P-001),10,11 and GSK 81239712,13 have been reported in the literature (Figure 1). The bicyclam AMD3100 was the first generation CXCR4 antagonist developed by AnorMED but exhibited poor oral bioavailability and dose-limiting adverse events. Subsequently, AMD070 improved oral bioavailability and eliminated the adverse interactions associated with cyclams. However, this structure suffered from cytochrome P450 2D6 inhibition,14 likely due to the 2-substituted benzimidazole.15 The 2D6 isoform is primarily expressed in the liver and is responsible for the metabolism of nearly 25% of all drugs.16 Strong inhibition of 2D6 leads to drug–drug interactions and detracts from future therapeutic value. Exchanging the benzimidazole moiety with other heterocycles was expected to reduce such off-target effects.
Figure 1.

Literature examples of small molecule CXCR4 antagonists.
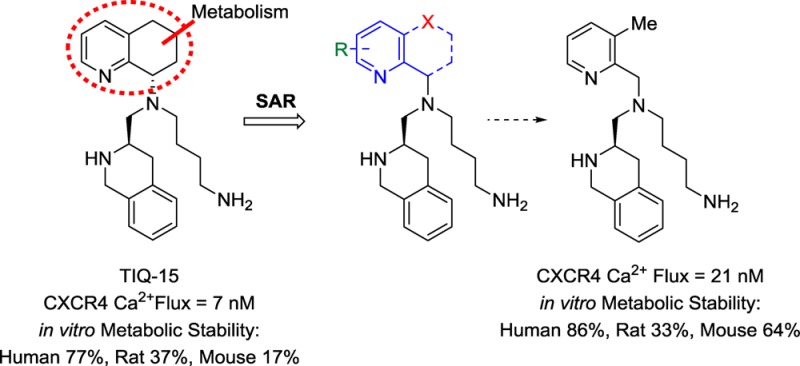
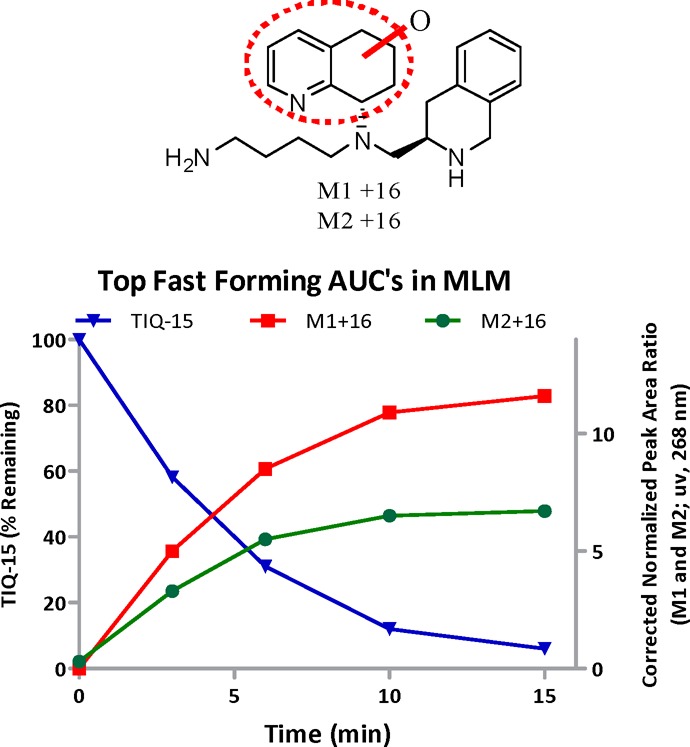
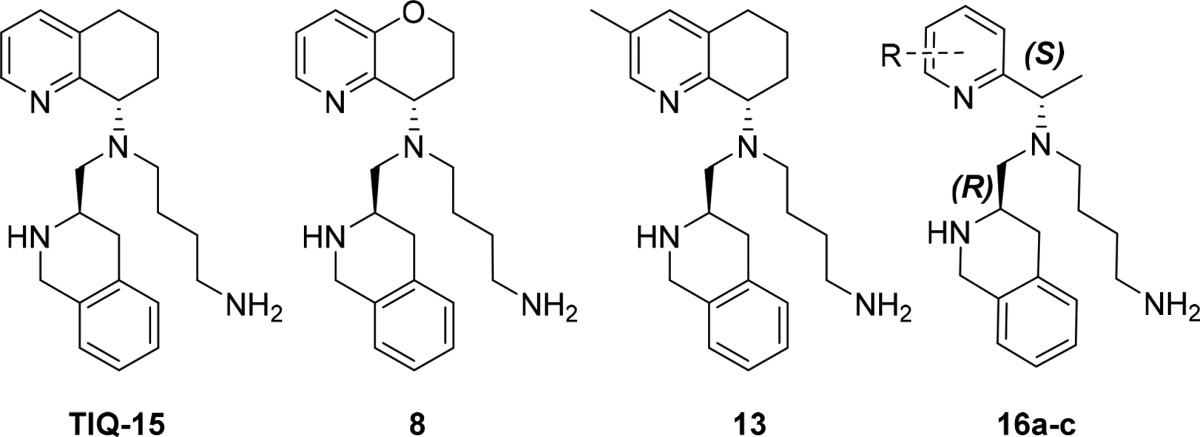
Recently, our group disclosed the discovery of TIQ-15, a potent small molecule 1,2,3,4-tetrahydroisoquinoline (THIQ)-based CXCR4 antagonist (Figure 2).17 Subsequent synthetic efforts focused on increasing lipophilicity by attenuating the basicity of the 1° and 2° amine moieties of TIQ-15. Various analogs bearing alkyl, amido, benzyl, and heteroaryl substitutions on the 2° nitrogen or the 1° butyl amine of TIQ-15 were synthesized; nevertheless, the parent compound remained the most attractive analog. A critical aspect of this investigation was to improve upon TIQ-15 as an orally administrable compound. To address this, we conducted biotransformation studies of TIQ-15 in human liver microsomes (HLM). Interestingly, these studies revealed no metabolites over 120 min when monitored by LC–MS/MS. Upon incubation with mouse liver microsomes (MLM), rapid metabolism of the tetrahydroquinoline (THQ) ring was observed. In order for an accurate evaluation of our compounds in a mouse model, it was essential to determine the first pass metabolites in MLM for TIQ-15 and tune out that metabolic liability. The biotransformation study with MLM exposed oxidation metabolites of the THQ moiety (M1 and M2, Chart 1) and was monitored by LC–MS/MS.
Figure 2.
Derivatization of TIQ-15 and new approach to tetrahydroquinoline replacement.
Chart 1. Metabolism of TIQ-15 in MLM.
We sought to inhibit MLM metabolism of the THQ by incorporating an oxygen into the ring. Our goal was to improve upon the metabolic stability in both human and mouse liver microsomes while maintaining CXCR4 antagonist potency. Commercially available ketone 1 was transformed to pyran 8 in five synthetic steps (Scheme 1). Reductive amination of ketone 1 and (R)-1-(4-methoxyphenyl)ethan-1-amine 2 followed by trifluoroacetic acid (TFA) mediated removal of the para-methoxybenzyl (PMB) moiety delivered chiral amine 3.18,19 Aldehyde 5 was produced from commercially available carboxylic acid 4 via borane reduction to the alcohol and Swern conditions20 for oxidation. Amine 3 and aldehyde 5 were treated with sodium triacetoxyborohydride in 1,2-dichloroethane to yield secondary amine 6. A final reductive amination of butyraldehyde 7 and amine 6 with subsequent removal of the carbamate protecting groups revealed pyran analog 8.
Scheme 1. Synthesis of Pyran Analog 8.

Reagents: (a) Ti(OiPr)4, NaBH(OAc)3, THF, 35%; (b) TFA, DCM 85%, 39%; (c) BH3·Me2S, THF, 90%; (d) (COCl)2, DMSO, Et3N, CH2Cl2, 63%; (e) NaBH(OAc)3, 1,2-DCE, 3, 77%; (f) NaBH(OAc)3, 1,2-DCE, 6, 51%; (g) TFA, DCM, 39%.
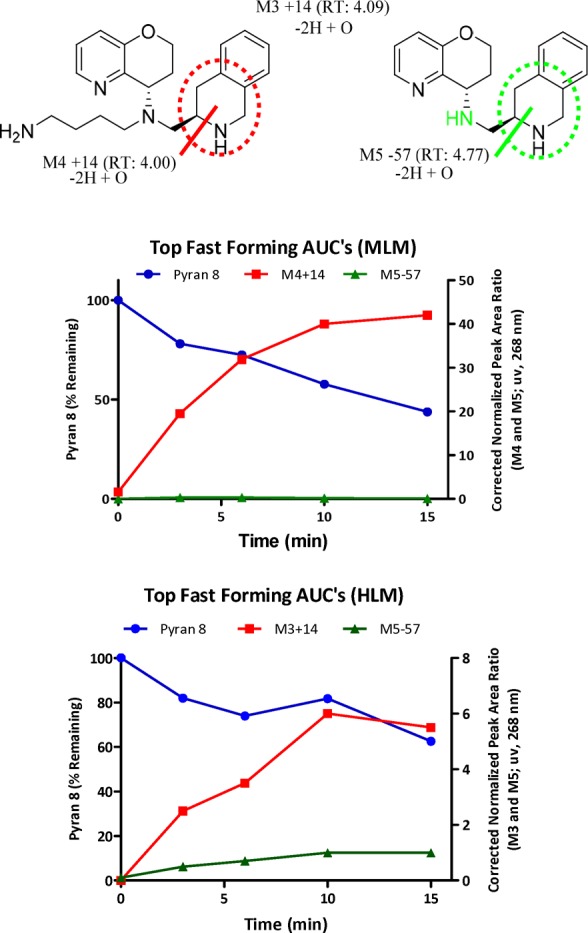
We discovered that our strategy to block metabolism in MLM with pyran 8 shifted the metabolic liability to the THIQ moiety (Chart 2a, MLM). However, comparing TIQ-15 with pyran 8, more parent compound remained at 15 min in Chart 2a vs Chart 1. Metabolism of pyran 8 with MLM generated a soft spot that was subjected to oxidation and dehydrogenation as identified by LC–MS fragmentation (M4). Incubation of pyran 8 in HLM revealed two distinct metabolites: M3 and M5. M3 formed due to oxidation and dehydrogenation of the THIQ ring. The formation of M5 was due to oxidation and dehydrogenation of the THIQ, in combination with a dealkylation of the tertiary amine (M5, green circle). The observed metabolism of pyran 8 in MLM and additional HLM metabolites warranted a more extensive SAR to improve microsomal stability.
Chart 2. Metabolism of Pyran 8 in MLM and HLM.
Incorporation of substituents onto the THQ required long synthetic sequences and limited our ability to conduct efficient SAR studies. For instance, in order to produce a methyl substitution at the 3-position we used the following strategy (Scheme 2). N-Oxidation of 3-methyl tetrahydroquinoline 9 followed by a Boekelheide rearrangement21 and hydrolysis of the acetyl ester provided alcohol 10. Swern oxidation followed by the Boggs12 protocol delivered chiral amine 11. Two consecutive reductive aminations, first with aldehyde 5 and then butyl aldehyde 7, delivered the carbamate intermediate that was treated with TFA to afford the desired compound 13. The final compound 13 was synthesized in nine total steps only to discover that, although CXCR4 potency was retained, mouse liver microsomal stability was significantly worse than TIQ-15 and pyran 8 (Table 1, entries 1–3).
Scheme 2. Synthesis of 3-Methyl THQ Compound 13.

Reagents: (a) AcOH, H2O2; (b) Ac2O; (c) K2CO3, MeOH, 35% over 3 steps; (d) (COCl)2, DMSO, Et3N, CH2Cl2, 75%; (e) NaBH(OAc)3, 2, 1,2-DCE, 61%; (f) TFA, 93%; (g) NaBH(OAc)3, 5, 1,2-DCE, 70%; (h) NaBH(OAc)3, 7, 1,2-DCE; (i) TFA, CH2Cl2, 74% over 2 steps.
Table 1. SAR for Benzylic Methyl (S,R) Analogs.
| entry | R | CXCR4 | mAChR | LM %a | ||
|---|---|---|---|---|---|---|
| Ca2+ flux (IC50) (nM) | Ca2+ flux (IC50) (nM) | H | R | M | ||
| 1 | TIQ-15 | 7 | >30000 | 77 | 37 | 17 |
| 2 | 8 | 6 | >16700 | 91 | 3 | 39 |
| 3 | 13 | 11 | >16700 | 84 | 7 | 2 |
| 4 | H (16a) | 62 | 28500 | 97 | 77 | 79 |
| 5 | 3-Me (16b) | 1710 | 33000 | 100 | 22 | 11 |
| 6 | 5-Me (16c) | 24 | 6100 | 79 | 1 | 5 |
Liver microsomal stability measured as percent remaining by LC–MS/MS after 10 min in human (H), rat (R), and mouse (M).
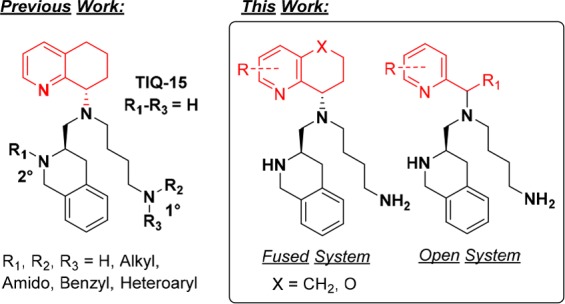
Given the lengthy synthesis required to incorporate substitutions onto the conformationally restricted THQ moiety (8 and 13), we chose to access new molecules more rapidly by opening the THQ into substituted 2-(aminomethyl)pyridines, quinolines, and isoquinolines, which were more readily available. Similar studies with pyridine analogs have been conducted using AMD070 as the parent structure, and we anticipated the CXCR4 activity of this new series of compounds to follow a similar trend.22 However, there have been no reports on microsomal stability for these molecules. The properties that we sought to improve upon were (1) inhibition of SDF-1 induced Ca2+ flux and (2) liver microsomal stability while avoiding off target interactions, such as mAChR23 inhibition. Intracellular calcium release is a basic functional assay that measures GPCR activity and inhibition thereof. Calcium flux assays were chosen over ligand binding assays as the primary on-target screen for several reasons. First, calcium flux assays are functional assays that give information regarding receptor activation, potentiation, inhibition, etc., as opposed to competition ligand binding assays, which convey no functional information. Since inhibition of CXCR4 signaling was desired, calcium flux assays were more appropriate for initial measurements of on-target activity. Second, allosteric binders can evade detection by competition ligand binding assays that employ labeled orthosteric binders, commonly leading to false negatives. The compounds described herein block the functional response of SDF-1/CXCR4 ligand/receptor interactions as either orthosteric or allosteric antagonists.
Analogs with benzylic methyl substitution maintaining the same chiral configuration as TIQ-15 (S,R) were analyzed first (Scheme 3). These compounds were prepared using the procedure adapted from Boggs et al.12 Reductive aminations with 2 and 2-acetylpyridines 14a–c provided a mixture of diastereomers with an approximate ratio of 4:1 favoring the desired (S,S) conformation. The (S,S) diastereomer was separated via column chromatography followed by recrystallization in hexanes to provide X-ray quality crystals, which were used to assign the absolute stereochemistry (see Supporting Information). TFA mediated removal of the PMB moiety revealed chiral amino pyridines with the desired (S) configuration 15a–c. The chiral amines 15a–c were reacted as described previously in successive reductive aminations with aldehyde 5, then 7, followed by carbamate deprotection protocol to furnish compounds 16a–c.
Scheme 3. Synthesis of Three (S,R) Analogs.

Reagents: (a) NaBH(OAc)3, CH2Cl2, 2 (27–45%); (b) TFA (49–98%); (c) NaBH(OAc)3, 5, 1,2-DCE (67–80%); (d) NaBH(OAc)3, 7, 1,2-DCE; (e) TFA, CH2Cl2 (36–61% over 2 steps).
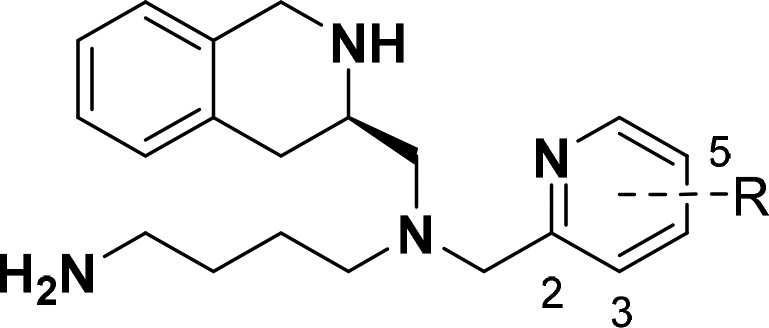
The assay results from compounds 16a–c (Table 1, entries 4–6) provided unexpected data. The potency (IC50) for these compounds was defined as the concentration of the compound required to inhibit 50% of the SDF-1 induced Ca2+ signaling. Unsubstituted pyridine 16a revealed a 10-fold decrease in CXCR4 potency in comparison to TIQ-15, while significantly increasing microsomal stability (Table 1, entry 4). The 3-methylpyridine 16b exhibited nearly a 100-fold loss in CXCR4 potency and a substantial reduction in RLM and MLM stability. The loss in CXCR4 potency could be due to steric clash between the benzylic methyl and the 3-methyl substituent, which might hinder the molecule from attaining the appropriate conformation for receptor binding. Conversely, the 5-methylpyridine 16c showed a 3-fold improvement in potency compared to 16a, but liver microsomal stability suffered. Initially, the pyridine moiety 16a validated that an open framework could improve metabolic stability and maintain decent potency; however, introducing additional methyl groups onto this framework (16b and 16c) proved to be deleterious to RLM and MLM stability. Compound 13 corroborates this result by showing a marked decrease in mouse liver microsomal stability compared to pyran 8. Continuing with our analysis of pyridine substituents, we chose to remove the (S) chiral center and utilized substituted pyridine-2-carboxaldehyde moieties as starting materials. These building blocks provided access to monosubstituted 2-(aminomethyl)pyridines, eliminating two synthetic steps and providing a more rapid production and analysis pipeline.
In our open scaffold series, compounds 19a–s could be obtained after a simple reductive amination with commercially available pyridine carboxaldehydes 17a–s and butyl amine 18 using STAB-H in 1,2-dichloroethane (Scheme 4). A subsequent reductive amination with the secondary amines 19a–s and aldehyde 5 followed by carbamate removal furnished final compounds 20a–s. The 3,5-disubstituted pyridine series 20t–v was obtained via the 2-cyano-pyridine derivatives 21t–v. The cyano group was reduced using nickel boride prepared in situ in the presence of di-tert-butyl dicarbonate to yield the Boc-protected primary amines.24 TFA mediated deprotection followed by consecutive reductive aminations with 5, then 7, and a final deprotection provided the finished compounds 20t–v.
Scheme 4. Synthetic Sequence To Access Open Scaffold.

Reagents: (a) NaBH(OAc)3, H2N(CH2)4NHBoc (18), 1,2-DCE (29–83%); (b) 5, NaBH(OAc)3, 1,2-DCE; (c) TFA, CH2Cl2 (56–86% over 2 steps); (d) NiCl2·6H2O, NaBH4, Boc2O, MeOH, 0 °C to rt; (e) TFA, CH2Cl2 (55–78% over 2 steps); (f) 5, NaBH(OAc)3, 1,2-DCE (57–82%); (g) NaBH(OAc)3, 7, 1,2-DCE; (h) TFA, CH2Cl2 (31–58% over 2 steps).
Overall, 22 molecules with various functional group substitutions on the pyridine moiety were synthesized and screened for (1) improved potency in a CXCR4 Ca2+ flux assay, (2) limited off-target interaction with mAChR receptor, and (3) increased liver microsomal stability (Table 2). In comparison to the constrained moieties (Table 1, entries 1–3), the open scaffold markedly improved upon metabolic stability albeit with a slight drop in CXCR4 antagonist activity, exemplified by 20h.
Table 2. Open Scaffold SAR.
| compound | R | CXCR4 | mAChR | LM %b | ||
|---|---|---|---|---|---|---|
| Ca2+ flux (IC50) (nM)a | Ca2+ flux (IC50) (nM) | H | R | M | ||
| 20a | H | 230 | 11500 | 100 | 33 | 77 |
| 20b | 2-quin | 5751 | >16700 | 88 | 8 | 35 |
| 20c | 1-isoquin | 8721 | 4350 | 88 | 5 | 62 |
| 20d | 3-isoquin | 3307 | >16700 | 100 | 31 | 45 |
| 20e | 6-Me | 2902 | 10700 | 100 | 34 | ND |
| 20f | 5-Me | 71 | 23200 | 100 | 3 | 67 |
| 20g | 4-Me | 698 | 3000 | 87 | 30 | 74 |
| 20h | 3-Me | 21 | 5900 | 86 | 33 | 64 |
| 20i | 5-Cl | 631 | >16700 | 89 | 5 | 56 |
| 20j | 4-Cl | >33000 | >33000 | 89 | 31 | ND |
| 20k | 3-Cl | 1855 | >16700 | 100 | 14 | 43 |
| 20l | 5-F | 260 | >33000 | 88 | 10 | 76 |
| 20m | 3-F | 2116 | >16700 | 83 | 9 | 64 |
| 20n | 5-OMe | >23000 | >27000 | 92 | 13 | 69 |
| 20o | 3-OMe | >33000 | >33000 | 100 | 38 | 99 |
| 20p | 5-CF3 | >33000 | >21000 | 98 | 8 | 85 |
| 20q | 3-CF3 | 406 | >33000 | 100 | 66 | 82 |
| 20r | 3-cPr | 163 | >16700 | 90 | 29 | 56 |
| 20s | 3-vinyl | 484 | >16700 | 86 | 4 | 44 |
| 20t | 3,5-diMe | 69 | >33000 | 100 | 39 | 79 |
| 20u | 3-Me-5-F | 127 | >33000 | 100 | 36 | 91 |
| 20v | 5-Me-3-F | 323 | 11,630 | 86 | 18 | 51 |
See Supporting Information for standard deviation.
Liver microsomal stability measured as percent remaining by LC–MS/MS after 10 min in human (H), rat (R), and mouse (M). ND = not determined.
When comparing the unsubstituted pyridine analog 20a with the unsubstituted quinoline analogs 20b–20d, the pyridine was determined to be the superior antagonist. Establishing the pyridine as higher priority, we began by substituting a methyl group at each position of the pyridine 20e–20h. We observed that the 5-methyl (20f, 71 nM) and 3-methyl (20h, 21 nM) substituents demonstrated the greatest potency and improved MLM stability. At this point, our focus was to test different functional groups (electron donating and electron withdrawing) at the 3- and 5-positions to evaluate the effect on CXCR4 potency. Halogen (Cl, F) substitution was best suited at the 5-position (20i, 20l), whereas 3- or 5-methoxy (20n, 20o) and 5-trifluoromethyl 20p substituted analogs were inactive. Interestingly, a 3-trifluoromethyl 20q substituent retained moderate activity as well as 3-vinyl 20s and 3-cyclopropyl 20r, which were alkyl modifiers, in an attempt to improve upon the properties associated with the 3-methyl substituted compound 20h. The 3,5-dimethyl analog 20t was equipotent to 20f but elicited weaker inhibition of muscarinic acetylcholine receptor compared to 20h. Compound 20t also had an enhanced stability in HLM and MLM. It was posited that a synergistic effect would be observed between analogs 20h and 20l if the 5-methyl group was exchanged with a fluorine 20u. We observed a 5-fold reduction in CXCR4 activity for 20u, compared to 20h, but we benefitted from a loss in muscarinic activity and increased microsomal stability. Substituted thiazoles were also tested, but compounds containing these moieties were inactive in the CXCR4 Ca2+ flux assay (see Supporting Information).
Additional parameters including parallel artificial membrane permeability assay (PAMPA) and cytochrome P450 (CYP450) inhibition were analyzed. Unfortunately, we observed little to no passive membrane permeability. In the CYP450 assays, inhibition resulting from this chemotype was solely observed on isoform 2D6 (see Supporting Information). Overall, the open methyl-substituted pyridine analogs showed a modest decrease in CXCR4 activity; however, they showed promise due to the increased microsomal stability compared to TIQ-15. This suggests that a more robust compound could be derived from an open pyridine analog.
In summary, we have developed a series of CXCR4 antagonists that possess increased liver microsomal stability in human and mouse models compared to the parent molecule TIQ-15. Specifically, compounds 20c, 20e, and 20t bearing methyl substituents around a pyridylmethyl moiety seem to be the most promising. Ultimately, we have significantly progressed this scaffold and intend to continue to improve upon the membrane permeability (PAMPA, Caco-2) and reduce CYP450 activity of this series. This and other studies will be presented in due course.
Acknowledgments
The authors would like to acknowledge Dr. Shaoxiang Wu of Emory NMR facility, Dr. John Bacsa for determination of crystal structure, and Dr. Fred Strobel for mass spectrometry.
Glossary
ABBREVIATIONS
- CXCR4
CXC chemokine receptor type 4
- GPCR
G protein-coupled receptor
- CXCL12
CXC chemokine ligand type 12
- THQ
5,6,7,8-tetrahydroquinoline
- THIQ
1,2,3,4-tetrahydroisoquinoline
- HLM
human liver microsomes
- RLM
rat liver microsomes
- MLM
mouse liver microsomes
- PMB
para-methoxybenzyl
- TFA
trifluoroacetic acid
- DCE
1,2-dichloroethane
- mAChR
muscarinic acetylcholine receptor
- STAB-H
sodium triacetoxyborohydride
- PAMPA
parallel artificial membrane permeability assay
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00381.
Experimental and characterization data for all new compounds and all biological data (PDF)
Author Contributions
The manuscript was written by R.J.W. and through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors acknowledge the use of shared instrumentation provided by grants from NSF (CHE1531620).
The authors declare the following competing financial interest(s): D.C.L. is the principle investigator on a research grant from Bristol-Myers Squibb Research and Development to Emory University. D.C.L., L.J.W., E.J.M., E.J., H.H.N., Y.A.T., R.J.W., V.M.T., and M.B.K. are co-inventors on Emory-owned Intellectual Property that includes CXCR4 antagonists.
Supplementary Material
References
- Busillo J. M.; Benovic J. L. Regulation of CXCR4 signaling. Biochim. Biophys. Acta, Biomembr. 2007, 1768 (4), 952–63. 10.1016/j.bbamem.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wald O.; Shapira O. M.; Izhar U. CXCR4/CXCL12 axis in non small cell lung cancer (NSCLC) pathologic roles and therapeutic potential. Theranostics 2013, 3 (1), 26–33. 10.7150/thno.4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath B.; Xu S.; Grande F.; Garofalo A.; Neamati N. Small molecule inhibitors of CXCR4. Theranostics 2013, 3 (1), 47–75. 10.7150/thno.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishan M. A.; Ahmadiankia N.; Bahrami A. R. CXCR4 and CCR7: Two eligible targets in targeted cancer therapy. Cell Biol. Int. 2016, 40 (9), 955–967. 10.1002/cbin.10631. [DOI] [PubMed] [Google Scholar]
- Zlotnik A.; Burkhardt A. M.; Homey B. Homeostatic chemokine receptors and organ-specific metastasis. Nat. Rev. Immunol. 2011, 11 (9), 597–606. 10.1038/nri3049. [DOI] [PubMed] [Google Scholar]
- Rubin J. B. Chemokine signaling in cancer: One hump or two?. Semin. Cancer Biol. 2009, 19 (2), 116–122. 10.1016/j.semcancer.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow M. T.; Luster A. D. Chemokines in Cancer. Cancer Immunol. Res. 2014, 2 (12), 1125–1131. 10.1158/2326-6066.CIR-14-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teicher B. A.; Fricker S. P. CXCL12 (SDF-1)/CXCR4 Pathway in Cancer. Clin. Cancer Res. 2010, 16 (11), 2927–2931. 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- De Clercq E. The bicyclam AMD3100 story. Nat. Rev. Drug Discovery 2003, 2 (7), 581–587. 10.1038/nrd1134. [DOI] [PubMed] [Google Scholar]
- Skerlj R. T.; Bridger G. J.; Kaller A.; McEachern E. J.; Crawford J. B.; Zhou Y.; Atsma B.; Langille J.; Nan S.; Veale D.; Wilson T.; Harwig C.; Hatse S.; Princen K.; De Clercq E.; Schols D. Discovery of Novel Small Molecule Orally Bioavailable C–X–C Chemokine Receptor 4 Antagonists That Are Potent Inhibitors of T-Tropic (X4) HIV-1 Replication. J. Med. Chem. 2010, 53 (8), 3376–3388. 10.1021/jm100073m. [DOI] [PubMed] [Google Scholar]
- Mosi R. M.; Anastassova V.; Cox J.; Darkes M. C.; Idzan S. R.; Labrecque J.; Lau G.; Nelson K. L.; Patel K.; Santucci Z.; Wong R. S. Y.; Skerlj R. T.; Bridger G. J.; Huskens D.; Schols D.; Fricker S. P. The molecular pharmacology of AMD11070: An orally bioavailable CXCR4 HIV entry inhibitor. Biochem. Pharmacol. 2012, 83 (4), 472–479. 10.1016/j.bcp.2011.11.020. [DOI] [PubMed] [Google Scholar]
- Boggs S.; Elitzin V. I.; Gudmundsson K.; Martin M. T.; Sharp M. J. Kilogram-Scale Synthesis of the CXCR4 Antagonist GSK812397. Org. Process Res. Dev. 2009, 13 (4), 781–785. 10.1021/op9000675. [DOI] [Google Scholar]
- Jenkinson S.; Thomson M.; McCoy D.; Edelstein M.; Danehower S.; Lawrence W.; Wheelan P.; Spaltenstein A.; Gudmundsson K. Blockade of X4-tropic HIV-1 cellular entry by GSK812397, a potent noncompetitive CXCR4 receptor antagonist. Antimicrob. Agents Chemother. 2010, 54 (2), 817–24. 10.1128/AAC.01293-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyunt M. M.; Becker S.; MacFarland R. T.; Chee P.; Scarborough R.; Everts S.; Calandra G. B.; Hendrix C. W. Pharmacokinetic effect of AMD070, an Oral CXCR4 antagonist, on CYP3A4 and CYP2D6 substrates midazolam and dextromethorphan in healthy volunteers. JAIDS, J. Acquired Immune Defic. Syndr. 2008, 47 (5), 559–65. 10.1097/QAI.0b013e3181627566. [DOI] [PubMed] [Google Scholar]
- Cao Y. J.; Flexner C. W.; Dunaway S.; Park J. G.; Klingman K.; Wiggins I.; Conley J.; Radebaugh C.; Kashuba A. D.; MacFarland R.; Becker S.; Hendrix C. W. Effect of low-dose ritonavir on the pharmacokinetics of the CXCR4 antagonist AMD070 in healthy volunteers. Antimicrob. Agents Chemother. 2008, 52 (5), 1630–4. 10.1128/AAC.01460-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh L. K.; Bertilsson L. Pharmacogenomics of CYP2D6: Molecular Genetics, Interethnic Differences and Clinical Importance. Drug Metab. Pharmacokinet. 2012, 27 (1), 55–67. 10.2133/dmpk.DMPK-11-RV-121. [DOI] [PubMed] [Google Scholar]
- Truax V. M.; Zhao H.; Katzman B. M.; Prosser A. R.; Alcaraz A. A.; Saindane M. T.; Howard R. B.; Culver D.; Arrendale R. F.; Gruddanti P. R.; Evers T. J.; Natchus M. G.; Snyder J. P.; Liotta D. C.; Wilson L. J. Discovery of Tetrahydroisoquinoline-Based CXCR4 Antagonists. ACS Med. Chem. Lett. 2013, 4 (11), 1025–1030. 10.1021/ml400183q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudmundsson K.Preparation of N-[(imidazo[1,2-a]pyridin-2-yl)methyl]-3,4-dihydro-2H-pyrano[3,2-b]pyridin-4-amines that bind to chemokine receptors for use against HIV and other disorders. WO2006076131A2, 2006.
- Bridger G.; Kaller A.; Harwig C.; Skerlj R.; Bogucki D.; Wilson T. R.; Crawford J.; McEachern E. J.; Atsma B.; Nan S.; Zhou Y.; Schols D.; Smith C. D.; Di Fluri M. R.. Preparation of chemokine receptor binding (benzimidazol-2-ylmethyl)(5,6,7,8-tetrahydroquinolin-8-yl)amines and related heterocyclic compounds with enhanced efficacy against AIDS and other disorders. US20040019058A1, 2004.
- Omura K.; Swern D. Oxidation of alcohols by “activated” dimethyl sulfoxide. a preparative, steric and mechanistic study. Tetrahedron 1978, 34 (11), 1651–1660. 10.1016/0040-4020(78)80197-5. [DOI] [Google Scholar]
- Boekelheide V.; Linn W. J. Rearrangements of N-Oxides. A Novel Synthesis of Pyridyl Carbinols and Aldehydes. J. Am. Chem. Soc. 1954, 76 (5), 1286–1291. 10.1021/ja01634a026. [DOI] [Google Scholar]
- Skerlj R.; Bridger G.; McEachern E.; Harwig C.; Smith C.; Kaller A.; Veale D.; Yee H.; Skupinska K.; Wauthy R.; Wang L.; Baird I.; Zhu Y.; Burrage K.; Yang W.; Sartori M.; Huskens D.; Clercq E. D.; Schols D. Design of novel CXCR4 antagonists that are potent inhibitors of T-tropic (X4) HIV-1 replication. Bioorg. Med. Chem. Lett. 2011, 21 (5), 1414–1418. 10.1016/j.bmcl.2011.01.021. [DOI] [PubMed] [Google Scholar]
- mAChR inhibition was measured routinely due to previously observed off-target antagonism of these receptors amongst similar classes of compounds (internal observations).
- Caddick S.; Judd D. B.; Lewis A. K. d. K.; Reich M. T.; Williams M. R. V. A generic approach for the catalytic reduction of nitriles. Tetrahedron 2003, 59 (29), 5417–5423. 10.1016/S0040-4020(03)00858-5. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.