

Abstract
Bacterial resistance has become a worldwide concern after the emergence of metallo-β-lactamases (MBLs). They represent one of the major mechanisms of bacterial resistance against beta-lactam antibiotics. Among MBLs, New Delhi metallo-β-lactamase-1 NDM-1, the most prevalent type, is extremely efficient in inactivating nearly all-available antibiotics including last resort carbapenems. No inhibitors for NDM-1 are currently available in therapy, making the spread of NDM-1 producing bacterial strains a serious menace. With this perspective, we performed a structure-based in silico screening of a commercially available library using FLAPdock and identified several, non-β-lactam derivatives as promising candidates active against NDM-1. The binding affinities of the highest scoring hits were measured in vitro revealing, for some of them, low micromolar affinity toward NDM-1. For the best inhibitors, efficacy against resistant bacterial strains overexpressing NDM-1 was validated, confirming their favorable synergistic effect in combination with the carbapenem Meropenem.
Keywords: NDM-1 Metallo-β-Lactamase; carbapenemases; bacterial resistance; structure-based virtual screening; enzyme inhibition; 4H-1,2,4-triazole-3-thiol derivatives
The extensive use of antibiotics has created major resistance problems leading to increased morbidity, mortality, and healthcare costs.1 Among several mechanisms of resistance pathogenic bacteria have developed against antimicrobial therapy, the hydrolysis of β-lactam antibiotics by β-lactamase enzymes is one of the most prevalent in resistant strains.2,3 BLs have emerged in Gram-positive and -negative bacteria and evolved in extended spectrum β-lactamases (ESBLs) first and carbapenemases later, leaving not many alternatives to microbial infections.2−5 BLs belong to two main groups: serine-based BLs (SBLs class A, C, D) and metallo-BLs (class B).6 With respect to SBLs, for which approved inhibitors are available in combination with beta-lactam antibiotics7 and few others are in the development pipeline,8 at present no drug capable of inhibiting any of the class B zinc-dependent metallo-β-lactamases is available in therapy.
MBLs use a zinc-bound hydroxyl group as nucleophile to catalyze the hydrolysis of a very broad range of β-lactam antibiotics including penicillins, cephalosporins, carbapenems, and BL inhibitors, with the only exception of monobactams.5 Therefore, the emergence of these enzymes is particularly concerning for the future treatment of bacterial infections.
Among MBLs, the clinically relevant metallo-carbapenemase NDM-1 (New Delhi metallo-β-lactamase) is the most worrisome in light of its substrate promiscuity, its resistance to nearly all available drugs, and its easiness of variants appearance and transferability. NDM-1 belongs to the B1 family of metallo-β-lactamases, and since its discovery in bacterial infections harboring plasmid-encoded NDM-1, this carbapenemase has emerged as a global health threat.9 It has caused multiple epidemics and it is capable of easy propagation to other species.10 Moreover, most plasmids harboring the NDM-1 gene are often associated with other resistance markers, making NDM-1 positive strains resistant to multiple drugs.10−12
As a consequence, there is a growing need for novel inhibitors against NDM-1, targeted in this study.
To identify novel MBLs inhibitors, starting from the available NDM-1 crystal structures [PDB code 3q6x and 3spu], a structure-based virtual screening (SBVS) of a large database of available chemicals was conducted, searching for potential inhibitors able to highly complement NDM-1 binding site. For the best inhibitors biological assays against clinical strains overexpressing NDM-1 were conducted, confirming their ability to synergize last generation antibiotics and to restore susceptibility in resistant strains.
The virtual screening was performed with FLAPdock implemented within FLAP (Fingerprints for Ligands and Proteins).13 A specific version of FLAPdock was developed ad hoc to deal with MBL’s active site.14
Starting from an initial library of ∼300,000 Specs commercially available compounds (www.specs.net) opportunely filtered for LogP lower than 2.5, a library of 100 compounds was selected according to the score assigned by the native scoring function, the number of hydrogen bonds formed within the binding site, their chemical diversity, and their ability to complement the NDM-1 active site. The compounds were first rapidly screened in vitro for inhibitory activity against NDM-1 through a spectrophotometric assay. Based on the determined percentage of inhibition against NDM-1 (cut off % inhibition <50 at 200 μM) a more focused library of 31 derivatives was selected (Table S1). Each compound was further validated for its capability to inhibit NDM-1: binding affinities and mechanism of inhibition were determined. In inhibition assays, the activity of NDM-1 was monitored following the hydrolysis rate of the substrate nitrocefin at λ = 480 nm, after 10 min incubation of each compound with the enzymes.
We noted that the 31 active candidates are characterized by the presence of electron donor chemotype able to coordinate the zinc ions, that is, triazol-thiole, amino-triazole, tetrazole, carboxylates, sulfonyl, and sulfonamide groups.16−18 Most active inhibitors in the library for which the binding mode is herein described are reported in Table 1 (for the complete list of focused library, see Table S1).
Table 1. Ki Values for the Most Representative NDM-1 Inhibitors.

Estimated Ki as per competitive inhibitor.15 Assays were conducted vs NDM-1 (0.5 nM), 10 min incubation, at 25 °C, inhibitor concentration ranging from 0 to 500 μM in HEPES 20 mM (pH 7.0) + 20 μM ZnCl2, using Nitrocefin 30 μM as reporter substrate. All the experiments were performed in triplicate.
Summarizing, among the total 31 derivatives (Table S1), we identified two compounds showing Ki of 0.72 and 1 μM (Table 1, compounds 1 and 2). Other 22 compounds, exploiting diverse chemical functionalities, showed Kis between 52 and 99 μM. All compounds behave as competitive inhibitors with respect to reporter substrate nitrocefin. The most active compounds 1 and 2 share the common 4H-1,2,4-triazole-3-thiol core able to advantageously interact with both Zn ions present in the binding site. The binding poses of compounds 1 and 2 are reported in Figure 1a,b.
Figure 1.
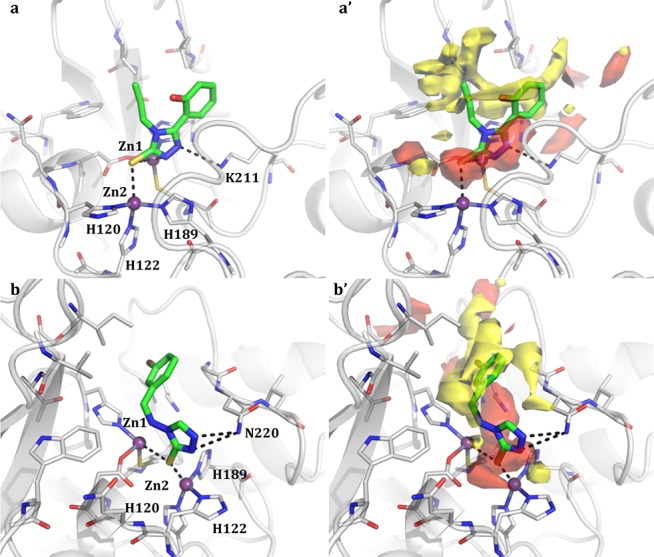
Binding poses of compounds 1 and 2 in NDM-1 binding site (a and b, respectively). Compounds are shown as capped sticks, zinc ions as purple spheres, and H-bonds as dashed lines. The binding site Molecular Interaction Fields are shown in panels a′ and b′. For clarity, only the hydrophobic (yellow) and hydrogen-bond acceptor (red) contours are shown. Pictures were prepared using Pymol (http://www.pymol.org).
The thiol group is a known zinc chelator and a chemical feature often shared by other reported MBL inhibitors.18 At the moment of completing this Letter, Sevaille et al. discussed the activity of ligands sharing a similar triazole-3-thiole core toward a panel of dizinc MBL, including NDM-1.19
The interactions of our compounds 1 and 2 with the zinc ions are mediated by S3 and N2, as reported for the similar 1,2,4-triazole-3-thiole compound cocrystallized with L1MBL reported by Sevaille et al.20 However, in compounds 1 and 2, the substituent on C5 assumes a different orientation given to the diverse architecture and composition of the binding site, and in particular, of the hydrophobic hot spots. Further interactions are made by N1 with Lys211, a residue normally involved in substrate recognition, orientation, and hydrolysis. This strongly supports the value of our results and the potential of the procedure, able to identify 1,2,4-triazole-3-thiole derivatives among the best binders in a filtered library of more than 20,000 compounds.
Compound 2 contacts both zinc ions with S3 and forms another interaction with Asn220, a key residue involved, along with Lys211, in substrate binding and in the intermediate product stabilization via formation of an oxyanion hole in conjunction with Zn1 (Figure 1b). Both molecules properly fit the H-bond acceptor region identified by GRID (Figure 1a′,b′) lined by the two ions and Lys211, and the large hydrophobic contour lined by Ile35, Val73, Trp93, and Gly219 and usually involved in hydrophobic interactions with β-lactam R groups.
According to the literature21 and crystallographic evidence,20,22 thiols groups were modeled as thiolates. In the MBL binding site, sulphydryl groups exist as anions, mimicking the catalytic hydroxide, because of the zinc effect, which lowers the pKa of bound thiol groups by about two orders of magnitude.22,23
In light of their Ki (0.72 and 1 μM, respectively), their reversible mechanism of inhibition, their ligand efficiencies (0.5 kcal mol–1), and their lead-like physical properties, they represent promising candidates for chemical derivatization.24
Compounds 5 and 6 present a triazol-thiole moiety as well, thus able to coordinate the zinc ions (Table S1). Compound 6 bears a carboxylate also involved in the coordination of one of the two ions. In the complexes formed by the other compounds, the metals are coordinated by different electron donor groups. Compounds 3 and 4 interact with the ions by means of the sulfonyl or sulfonamide moiety (see Table 1, Table S1). In particular, compound 3 contacts both of them through the carbonyl and the sulfonyl moiety, also interacting with Lys211 and Asn220 (Figure 2a).
Figure 2.
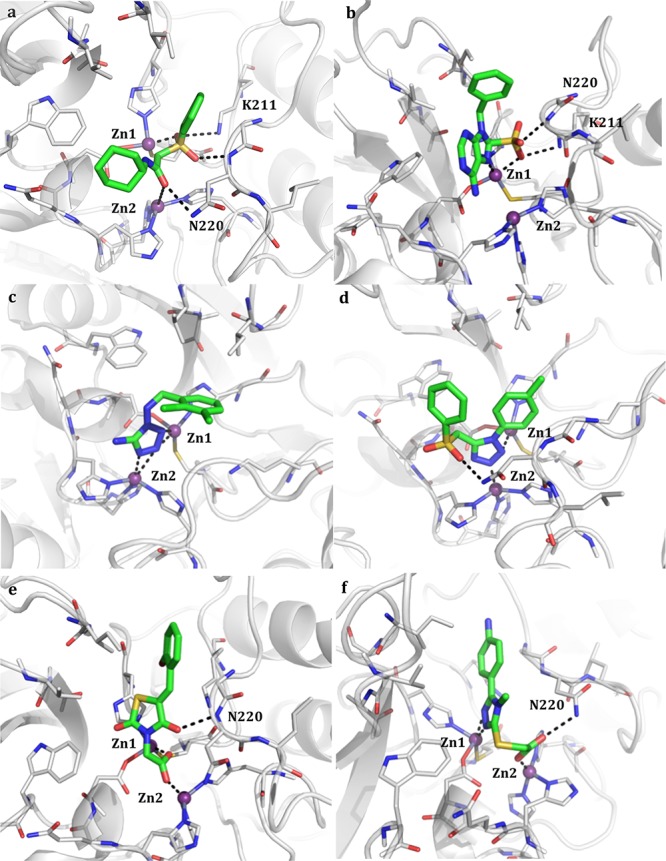
Predicted binding pose of the most representative functionalities in the 31-candidate focused library (Table S1): compounds 3 (a), 7 (b), 8 (c), 11 (d), 12 (e), and 15 (f) in the NDM-1 binding site (Table 1). The compounds are shown as capped sticks, zinc ions as purple spheres, and H-bonds as dashed lines. Some residues have been removed for clarity. Pictures were prepared using Pymol (http://www.pymol.org).
In the case of compound 7 (Table 1, Figure 2b), the coordination contacts are made by both the purine ring and the sulfonic acid, still forming hydrogen bonds with Lys211 and Asn220, while for compound 10 only the sulfonylic amid moiety is responsible for the metal coordination (Table S1). Compounds 8 presents a tetrazol-amine moiety, with N2, N3, and N4 coordinating both Zn1 and Zn2 (Table 1, Figure 2c).
Similarly, compounds 11 (Table 1, Figure 2d) and 14 (Table S1) exploit the presence of a tetrazol ring and, also, of the sulfonyl moiety to bind the protein active site. In the case of compounds 9 (Table S1), 12 (Table 1, Figure 2e), and 13 (Table S1), the coordination is mediated by carboxylic functions. In particular, compound 12 (Figure 2e) is able to contact Zn1 and Zn2 with the single carboxylic acid and to further stabilize its orientation in the pocket through the formation of an additional hydrogen bond with Asn220. Compound 15 combines a carboxylic function and a triazol-thiole group, both involved in the metals coordination. The carboxylic moiety also contacts Asn220 (Table 1, Figure 2f). Compounds 16 and 17 are characterized by the presence of a triazole carboxamide and a triazole amine, respectively (Table S1).
Apparently, the presence of a mercapto-triazole moiety one/two atom spaced from an aromatic ring represents a suitable scaffold to stably bind the enzyme active site and inhibit its activity. The identified compounds could be further optimized to better fit the target binding site shape and MIFs.
To verify the ability of the compounds to synergically interact with therapeutically available antibiotics, a microdilution drug–drug interaction assay was conducted both on E. coli clinical isolate expressing NDM-1 enzyme and E. coli BL21 (DE3) recombinant strain harboring the vector pET-24blaNDM-1 overproducing NDM-1 enzyme. The checkerboard microdilution assay makes possible to test in a 96-well microplate 77 possible combinations of two drugs simultaneously, allowing the estimation of the fractional inhibitory concentration index (FICI). Since it was not possible to determine the MIC values of the tested NDM-1 inhibitors in the used range (maximum concentration 256 μg/mL), the FICI index was estimated as the maximum value that can be obtained assigning to the inhibitors a MIC value equal to 512 (μg/mL). As reported in Tables 2 and 3, compounds 1 and 2 synergically interact with Meropenem, the preferential substrate of NDM-1 enzyme. At a concentration of 128 μg/mL, 1 reduces the MIC value of Meropenem in the clinical isolate and the recombinant strain 8- and 16-fold, respectively.
Table 2. In Vitro Interaction between Antibiotics and Novel NDM-1 Inhibitors Determined by FICI in E. coli BL21 (DE3) pET-24a-blaNDM-1.
| FIC
indexa |
||||||
|---|---|---|---|---|---|---|
| inhibitor | antibiotic | MICb antibiotic (μg/mL) | MICb combination (μg/mL) | inhibitorb concentration (μg/mL) | ΣFICmin | INT |
| 1 | AMP | 1024 | 1024 | >256 | >1.0 | IND |
| 0.0625 | 256 | <0.563 | IND | |||
| MEM | 1 | 0.25 | 16 | <0.281 | SYN | |
| 0.5 | 0.25 | <0.501 | IND | |||
| CFZ | 32 | 32 | >256 | >1.0 | IND | |
| 1 | 256 | <0.625 | IND | |||
| CTX | 8 | 2 | 4 | <0.258 | SYN | |
| 4 | 0.25 | <0.505 | IND | |||
| 2 | AMP | 2048 | 1024 | 0.25 | <0.501 | IND |
| MEM | 1 | 0.25 | 64 | <0.375 | SYN | |
| 0.5 | 0.5 | <0.501 | IND | |||
| CFZ | >32 | 32 | 0.25 | <0.501 | IND | |
| CTX | 8 | 4 | 0.25 | <0.501 | IND | |
INT, interpretation; IND, indifference; SYN, synergy; ANT, antagonism. Synergy is defined when the FICI is ≤0.5, antagonism when the FICI is >4, and indifference when the FICI is >0.5 and ≤4.
MIC values were determined as the median of three independent experiments. N.D.: not determined.
Table 3. In Vitro Interaction between Antibiotics and NDM-1 Inhibitors Determined by FICI in E. coli Clinical Strain Overexpressing NDM-1.
| FIC
indexa |
||||||
|---|---|---|---|---|---|---|
| inhibitor | antibiotic | MICb antibiotic (μg/mL) | MICb combination (μg/mL) | inhibitorb concentration (μg/mL) | ΣFICmin | INT |
| 1 | AMP | >32768 | >32768 | >256 | N.D. | |
| 16 | 128 | <0.375 | SYN | |||
| MEM | 128 | 32 | 32 | <0.313 | SYN | |
| 64 | 2 | <0.504 | IND | |||
| CFZ | 2048 | 2048 | >256 | >1.0 | IND | |
| CTX | 1024 | 1024 | >256 | >1.0 | IND | |
| 2 | AMP | >32768 | >32768 | >256 | N.D. | |
| MEM | 128 | 32 | 32 | <0.313 | SYN | |
| 64 | 0.5 | <0.501 | IND | |||
| CFZ | 2048 | 2048 | >256 | >1.0 | IND | |
| CTX | 1024 | 1024 | >256 | >1.0 | IND | |
INT, interpretation; IND, indifference; SYN, synergy; ANT, antagonism. Synergy is defined when the FICI is ≤0.5, antagonism when the FICI is >4, and indifference when the FICI is >0.5 and ≤4.
MIC values were determined as the median of three independent experiments. N.D.: not determined.
A 4-fold reduction is observed in both strains when Meropenem is combined with 2. With ampicillin, cefazolin, and cefotaxime, no synergic activity was observed with the exception of compound 1 in combination with cefotaxime in the recombinant strain, where a 4-fold reduction of the antibiotic MIC value is reported. The obtained results show that compounds 1 and 2 display a significant synergistic effect when in combination with Meropenem, against clinical strain overexpressing NDM-1.
We report the successful application of structure-based virtual screening to the discovery of novel ligands active against NDM-1. Several recurrent chemical classes of MBL inhibitors have been reported over the years, here efficaciously recognized by the virtual strategy applied: thiols, N-arylsulphonyl hydrazones, succinic acids, and mercaptocarboxylate acids were included among our candidates.16−18
With respect to other disclosed inhibitors active against NDM-1, undoubtedly, our results stand out for the variety of chemical functionality, correct modeling prediction of the interaction with the crucial Zn atoms, high complementarity with the target together with drug likeness and ligand efficiency LE (Table S2).
Among the compounds selected by means of SBVS approaches, several demonstrated low micromolar activity toward the target (Ki for the best selected inhibitor equal to 0.72 μM). The identified inhibitors all share a common competitive inhibition mechanism vs NDM-1 (Figure S1). In microbiological tests, the synergic activity in association with Meropenem was demonstrated also against clinical resistant strains.
The ability of the best performing compounds to synergize antibiotics against pathogenic resistant bacteria reverting resistance in clinical isolates is encouraging. For the best two compounds, studies for improving their affinity and confirming their binding mode via X-ray crystallography are ongoing.
In the emergence of bacteria resistance new strategies, targets and compounds are urgently needed. Despite the source of structural information on NDM-1 at the atomic level has raised, the challenge to design a valid NDM-1 inhibitor is wide open.
Our results demonstrate the feasibility of applying virtual screening methodologies in identifying novel inhibitors for NDM-1, despite is malleable active site, providing a mechanism base for rational design of NDM-1 inhibitors.
Experimental Procedures
Structure-Based Virtual Screening
The crystallographic structures of NDM-1 from Klebsiella pneumoniae in the apo form and complexed with ampicillin (PDB codes 3spu and 3q6x, respectively) were used as templates for SBVS experiments. The binding site was identified and defined by flapsite, implemented in FLAP.13 The Specs database (www.specs.net) was chosen as the starting library. This database is part of the ZINC archive (www.zinc.docking.org) and, according to previous experiences,14 contains molecules with significant chemical and geometric diversity and good purity and availability. A set of about 300,000 compounds was downloaded and filtered according to LogP values calculated by Moka.25 In order to ensure sufficient solubility, only molecules with LogP < 2.5 were retained, amounting, in this experiment, to 22,634. Tautomers and protomers were added. The virtual screening was performed with FLAPdock implemented within FLAP software, developed and licensed by Molecular Discovery Ltd. (www.moldiscovery.com). FLAP is based on the Molecular Interaction Fields (MIFs) calculated by GRID.26 A specific version of FLAPdock was developed to deal with the MBL active site. The native docking scoring function was modified to add a classical electrostatic term and also a soft Lennard-Jones term. These terms were scaled for comparison with the FLAPdock MIF similarity score. Additional MIFs for the DRY, O, and N1 fields were also added, but cut at the most favorable 10% to emphasize the strongest hotspots in the site. The modified docking algorithm was validated on the DUD data set.27 Molecules were ranked according to the FLAP-S Score function, containing four terms to evaluate the similarity of the H (shape), O (H-bond acceptor), N1 (H-bond donor), and DRY (Hydrophobic) MIFs between the template pocket on NDM-1 and the candidates, a Lennard-Jones potential, an electrostatic term, and a penalization term to penalize H-bond donor groups in H-bond acceptor MIFs, and vice versa. After the first VS, the highest score compounds (∼1% of the initial library) were redocked in the binding site with a more accurate modality, taking more computational time but providing more reliable results. All compounds in the pocket were visually inspected, and on the basis of (i) the FLAP S-score, the superposition with the pocket’s MIFs, (ii) the interactions with the surrounding residues, and (iii) the chemical diversity, 100 compounds were selected for testing.
Protein Sovra Expression and Purification
E. coli JM109(DE3) cells containing the recombinant plasmids pFM-NDM-1 were grown in 1 L of tryptic soy broth (TSB) medium with kanamycin (50 μg/mL) at 37 °C in an orbital shaker (180 rpm). Each culture was grown to achieve an A600 of approximately 0.5, and 0.4 mM IPTG (isopropyl-β-d-thiogalac-topyranoside) was added. After the addition of IPTG, the cultures were incubated for 16 h at 22 °C, under aerobic conditions. Cells were harvested by centrifugation at 8000 rpm for 10 min at 4 °C and washed twice with 20 mM Tris-HCl buffer (pH 7.0) (buffer A). Crude enzymes were obtained by treatment with lysozyme at a concentration of 2 mg/mL for 30 min at 30 °C followed by sonication on ice (five cycles at 60 W for 1 min and 2 min of break). The lysate was centrifuged at 30 000 rpm for 30 min, and the cleared supernatant was recovered and loaded onto a Sepharose-Q fast-flow column equilibrated with buffer A. The column was extensively washed to remove unbound proteins, and the β-lactamase was eluted with a linear gradient of 20 mM Tris-HCl (pH 7.0)–NaCl (0.5 M) (buffer B). The fractions containing β-lactamase activity were pooled, concentrated 20-fold using an Amicon concentrator (YM 10 membrane; Millipore, Bedford, MA, USA), and loaded onto Sephacryl S-100 equilibrated with buffer A. The pure fractions were pooled and dialyzed in 20 mM HEPES buffer (pH 7.0) containing 0.01 mg/mL bovine serum albumin (BSA) (buffer C) for further experiments.28
Inhibition Assays
All molecules (from Specs; www.specs.net, tested without further purification; compounds purity was always above 90% as declared by Specs) were tested toward NDM-1 enzyme purified at homogeneity as previously described.28 For kinetic experiments the molecules were dissolved in 100% DMSO to have a final concentration in cuvettes of 1%. The inhibition assays were carried out by preincubating, for 10 min at 25 °C, 0.5 nM pure enzyme with various concentrations of inhibitors (from 0 to 500 μM) in 20 mM HEPES buffer (pH 7.0) containing 20 μM ZnCl2. The rate of inhibition was measured by the reporter substrate method15 by using 30 μM nitrocefin at the following conditions:
| 1 |
where vi and v0 represent the initial rates of hydrolysis of nitrocefin with or without inhibitor, respectively; I is the concentration of inhibitor, Ki is the inhibition constant, Km is the Henri–Michaelis constant, and S is the concentration of the reporter substrate. The plot v0/vi versus [I] yielded a straight line of slope Km/(Km + S)Ki.15 The data were collected with a Lambda 25 spectrophotometer (PerkinElmer Italia).
Checkerboard Microdilution Assay
The in vitro interactions between ampicillin, Meropenem (MEM), cefazolin (CFZ), and cefotaxime (CTX) and best inhibitors 1 and 2 were investigated by a two-dimensional checkerboard microdilution assay, using 96-well microtitration plates as previously described.29 The microtiter plates were incubated at 37 °C for 18 h. The growth in each well was quantified spectrophotometrically at 595 nm by a microplate reader iMark, BioRad (Milan, Italy). The percentage of growth in each well was calculated as
where the background was obtained from the microorganism-free plates, processed as the inoculated plates. The MIC for each combination of drugs was defined as the concentration of drug that reduced bacterial growth by 80% compared to that of organisms grown in the absence of drug. All experiments were performed in triplicate.
Drug Interaction Models
In order to assess the nature of the in vitro interactions between the compounds and antibiotics against the recombinant E. coli BL21 (DE3) pET-24a-blaNDM-1 strain and against the E. coli clinical strain overexpressing NDM-1, the data obtained from the checkerboard assay were analyzed to calculate the fractional inhibitory concentration index.30 The fractional inhibitory concentration index (FICI) is the mathematical expression of the effect of the combination of antibacterial agents expressed as
where MICA and MICB are the MICs of drugs A and B when acting alone and MICAB and MICBA are the MICs of drugs A and B when acting in combination. Among all ΣFICs calculated for each microplate, the FICI was determined as the lowest ΣFIC (ΣFICmin) when synergy is supposed, or the highest ΣFIC (ΣFICmax) when antagonism is evident. Since in MIC determination the variation in a single result places a MIC value in a three-dilution range (±1 dilution), therefore, the reproducibility errors in the MIC checkerboard assays are considerable. For that reason, the interpretation of FICI data should be done taking into consideration values well below or above the theoretical cutoff (1.0) defined by Berenbaum. Synergy was, therefore, defined when FICI ≤ 0.5, while antagonism was defined when FICI > 4. A FIC index between 0.5 and 4 (0.5< FICI ≤ 4) was considered indifferent.31
Glossary
ABBREVIATIONS
- NDM-1
New Delhi Metallo-β-lactamase-1
- ESBLs
extended spectrum β-lactamases
- SBLs
serine based BLs
- MBLs
metallo-β-lactamases
- FLAP
Fingerprints for Ligands and Proteins
- FICI
fractional inhibitory concentration index
- MIC
minimum inhibitory concentration
- LE
ligand efficiency
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00428.
Focused library: 31 candidates selected for further validation vs NDM-1 in Table S1. In silico properties and ligand efficiencies for candidates in Table S2. Kinetic characterization of NDM-1 inhibition by compounds 1 and 2 in Figure S1. SDS-PAGE gel of purified NDM-1 in Figure S2. (PDF)
Author Present Address
⊥ Dipartimento di Scienza e Tecnologia del Farmaco, Università degli Studi di Torino, Via Pietro Giuria 9, 10125 Torino, Italy.
Author Contributions
The manuscript was written through contributions of all authors and all have given approval to the final version of the manuscript.
This work was supported by Finanziamento per la Ricerca di Ateneo (FAR 2014), Università degli Studi di Modena e Reggio Emilia to D.T.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO. Antimicrobial Resistance: Global Report on Surveillance, 2014. http://apps.who.int/iris/bitstream/10665/112642/1/9789241564748_eng.pdf.
- Frere J. M.; Sauvage E.; Kerff F. From ″An Enzyme Able to Destroy Penicillin″ to Carbapenemases: 70 Years of Beta-lactamase Misbehaviour. Curr. Drug Targets 2016, 17 (9), 974–82. 10.2174/1389450116666151001112859. [DOI] [PubMed] [Google Scholar]
- Tondi D.; Cross S.; Venturelli A.; Costi M. P.; Cruciani G.; Spyrakis F. Decoding the Structural Basis For Carbapenem Hydrolysis By Class A beta-lactamases: Fishing For A Pharmacophore. Curr. Drug Targets 2016, 17 (9), 983–1005. 10.2174/1389450116666151001104448. [DOI] [PubMed] [Google Scholar]
- Tondi D.; Venturelli A.; Bonnet R.; Pozzi C.; Shoichet B. K.; Costi M. P. Targeting class A and C serine beta-lactamases with a broad-spectrum boronic acid derivative. J. Med. Chem. 2014, 57 (12), 5449–58. 10.1021/jm5006572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica M. F.; Bonomo R. A.; Fast W. B1-Metallo-beta-Lactamases: Where Do We Stand?. Curr. Drug Targets 2016, 17 (9), 1029–50. 10.2174/1389450116666151001105622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambler R. P. The structure of beta-lactamases. Philos. Trans. R. Soc., B 1980, 289 (1036), 321–31. 10.1098/rstb.1980.0049. [DOI] [PubMed] [Google Scholar]
- Farina D.; Spyrakis F.; Venturelli A.; Cross S.; Tondi D.; Costi M. P. The inhibition of extended spectrum beta-lactamases: hits and leads. Curr. Med. Chem. 2014, 21 (12), 1405–34. 10.2174/09298673113206660323. [DOI] [PubMed] [Google Scholar]
- Bush K.; Page M. G. P. What we may expect from novel antibacterial agents in the pipeline with respect to resistance and pharmacodynamic principles. J. Pharmacokinet. Pharmacodyn. 2017, 44, 113–132. 10.1007/s10928-017-9506-4. [DOI] [PubMed] [Google Scholar]
- Walsh T. R.; Toleman M. A.; Poirel L.; Nordmann P. Metallo-beta-lactamases: the quiet before the storm?. Clin Microbiol Rev. 2005, 18 (2), 306–25. 10.1128/CMR.18.2.306-325.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fair R. J.; Tor Y. Antibiotics and bacterial resistance in the 21st century. Perspect. Med. Chem. 2014, 6, 25–64. 10.4137/PMC.S14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirel L.; Lagrutta E.; Taylor P.; Pham J.; Nordmann P. Emergence of metallo-beta-lactamase NDM-1-producing multidrug-resistant Escherichia coli in Australia. Antimicrob. Agents Chemother. 2010, 54 (11), 4914–6. 10.1128/AAC.00878-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordmann P.; Poirel L.; Toleman M. A.; Walsh T. R. Does broad-spectrum beta-lactam resistance due to NDM-1 herald the end of the antibiotic era for treatment of infections caused by Gram-negative bacteria?. J. Antimicrob. Chemother. 2011, 66 (4), 689–92. 10.1093/jac/dkq520. [DOI] [PubMed] [Google Scholar]
- Baroni M.; Cruciani G.; Sciabola S.; Perruccio F.; Mason J. S. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for Ligands and Proteins (FLAP): theory and application. J. Chem. Inf. Model. 2007, 47 (2), 279–94. 10.1021/ci600253e. [DOI] [PubMed] [Google Scholar]
- Spyrakis F.; Cellini B.; Bruno S.; Benedetti P.; Carosati E.; Cruciani G.; Micheli F.; Felici A.; Cozzini P.; Kellogg G. E.; Voltattorni C. B.; Mozzarelli A. Targeting cystalysin, a virulence factor of treponema denticola-supported periodontitis. ChemMedChem 2014, 9 (7), 1501–11. 10.1002/cmdc.201300527. [DOI] [PubMed] [Google Scholar]
- De Meester F.; Joris B.; Reckinger G.; Bellefroid-Bourguignon C.; Frere J. M.; Waley S. G. Automated analysis of enzyme inactivation phenomena. Application to β-lactamases and DD-peptidases. Biochem. Pharmacol. 1987, 36, 2393–2403. 10.1016/0006-2952(87)90609-5. [DOI] [PubMed] [Google Scholar]
- Chen A. Y.; Thomas P. W.; Stewart A. C.; Bergstrom A.; Cheng Z.; Miller C.; Bethel C. R.; Marshall S. H.; Credille C. V.; Riley C. L.; Page R. C.; Bonomo R. A.; Crowder M. W.; Tierney D. L.; Fast W.; Cohen S. M. Dipicolinic Acid Derivatives as Inhibitors of New Delhi Metallo-beta-lactamase-1. J. Med. Chem. 2017, 60 (17), 7267–7283. 10.1021/acs.jmedchem.7b00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingler F. M.; Wichelhaus T. A.; Frank D.; Cuesta-Bernal J.; El-Delik J.; Muller H. F.; Sjuts H.; Gottig S.; Koenigs A.; Pos K. M.; Pogoryelov D.; Proschak E. Approved Drugs Containing Thiols as Inhibitors of Metallo-beta-lactamases: Strategy To Combat Multidrug-Resistant Bacteria. J. Med. Chem. 2015, 58 (8), 3626–30. 10.1021/jm501844d. [DOI] [PubMed] [Google Scholar]
- Groundwater P. W.; Xu S.; Lai F.; Varadi L.; Tan J.; Perry J. D.; Hibbs D. E. New Delhi metallo-beta-lactamase-1: structure, inhibitors and detection of producers. Future Med. Chem. 2016, 8 (9), 993–1012. 10.4155/fmc-2016-0015. [DOI] [PubMed] [Google Scholar]
- Sevaille L.; Gavara L.; Bebrone C.; De Luca F.; Nauton L.; Achard M.; Mercuri P.; Tanfoni S.; Borgianni L.; Guyon C.; Lonjon P.; Turan-Zitouni G.; Dzieciolowski J.; Becker K.; Benard L.; Condon C.; Maillard L.; Martinez J.; Frere J. M.; Dideberg O.; Galleni M.; Docquier J. D.; Hernandez J. F. 1,2,4-Triazole-3-thione Compounds as Inhibitors of Dizinc Metallo-beta-lactamases. ChemMedChem 2017, 12 (12), 972–985. 10.1002/cmdc.201700186. [DOI] [PubMed] [Google Scholar]
- Nauton L.; Kahn R.; Garau G.; Hernandez J. F.; Dideberg O. Structural insights into the design of inhibitors for the L1 metallo-beta-lactamase from Stenotrophomonas maltophilia. J. Mol. Biol. 2008, 375 (1), 257–69. 10.1016/j.jmb.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Jin W.; Arakawa Y.; Yasuzawa H.; Taki T.; Hashiguchi R.; Mitsutani K.; Shoga A.; Yamaguchi Y.; Kurosaki H.; Shibata N.; Ohta M.; Goto M. Comparative study of the inhibition of metallo-beta-lactamases (IMP-1 and VIM-2) by thiol compounds that contain a hydrophobic group. Biol. Pharm. Bull. 2004, 27 (6), 851–6. 10.1248/bpb.27.851. [DOI] [PubMed] [Google Scholar]
- King D. T.; Worrall L. J.; Gruninger R.; Strynadka N. C. New Delhi metallo-beta-lactamase: structural insights into beta-lactam recognition and inhibition. J. Am. Chem. Soc. 2012, 134 (28), 11362–5. 10.1021/ja303579d. [DOI] [PubMed] [Google Scholar]
- Antony J.; Gresh N.; Olsen L.; Hemmingsen L.; Schofield C. J.; Bauer R. Binding of D- and L-captopril inhibitors to metallo-beta-lactamase studied by polarizable molecular mechanics and quantum mechanics. J. Comput. Chem. 2002, 23 (13), 1281–96. 10.1002/jcc.10111. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Keseru G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13 (2), 105–21. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Milletti F.; Storchi L.; Goracci L.; Bendels S.; Wagner B.; Kansy M.; Cruciani G. Extending pKa prediction accuracy: high-throughput pKa measurements to understand pKa modulation of new chemical series. Eur. J. Med. Chem. 2010, 45 (9), 4270–9. 10.1016/j.ejmech.2010.06.026. [DOI] [PubMed] [Google Scholar]
- Wade R. C.; Goodford P. J. The role of hydrogen-bonds in drug binding. Prog. Clin Biol. Res. 1989, 289, 433–44. [PubMed] [Google Scholar]
- Huang N.; Shoichet B. K.; Irwin J. J. Benchmarking sets for molecular docking. J. Med. Chem. 2006, 49 (23), 6789–801. 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcoccia F.; Bottoni C.; Sabatini A.; Colapietro M.; Mercuri P. S.; Galleni M.; Kerff F.; Matagne A.; Celenza G.; Amicosante G.; Perilli M. Kinetic Study of Laboratory Mutants of NDM-1 Metallo-beta-Lactamase and the Importance of an Isoleucine at Position 35. Antimicrob. Agents Chemother. 2016, 60 (4), 2366–72. 10.1128/AAC.00531-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellio P.; Segatore B.; Mancini A.; Di Pietro L.; Bottoni C.; Sabatini A.; Brisdelli F.; Piovano M.; Nicoletti M.; Amicosante G.; Perilli M.; Celenza G. Interaction between lichen secondary metabolites and antibiotics against clinical isolates methicillin-resistant Staphylococcus aureus strains. Phytomedicine 2015, 22 (2), 223–30. 10.1016/j.phymed.2014.12.005. [DOI] [PubMed] [Google Scholar]
- Greco W. R.; Bravo G.; Parsons J. C. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev. 1995, 47 (2), 331–85. [PubMed] [Google Scholar]
- Odds F. C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003, 52 (1), 1. 10.1093/jac/dkg301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.