

Abstract
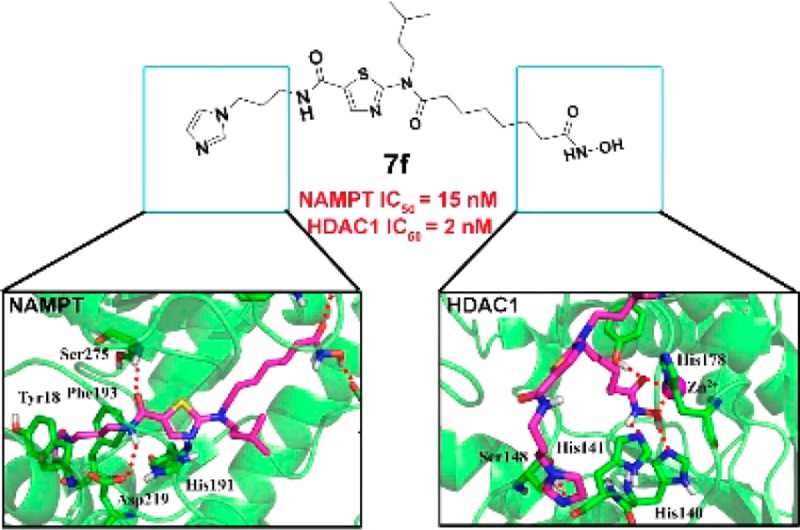
Novel dual nicotinamide phosphoribosyltransferase (NAMPT) and histone deacetylase (HDAC) inhibitors were designed by a pharmacophore fusion approach. The thiazolocarboxamide inhibitors were highly active for both targets. In particular, compound 7f (NAMPT IC50 = 15 nM, HDAC1 IC50 = 2 nM) showed potent in vivo antitumor efficacy in the HCT116 xenograft model. The study offers a new strategy for multitarget antitumor drug discovery by simultaneously acting on cancer metabolism and epigenetics.
Keywords: Nicotinamide phosphoribosyltransferase, histone deacetylase, dual inhibitors, cancer metabolism, cancer epigenetics
Single-target cancer chemotherapy often suffers from limited efficacy, drug resistance, significant adverse effects, and toxicities.1 Currently, specific drug combinations are widely used to achieve more durable disease control. However, the employment of drug cocktails also lead to complicated doses/schedule, unpredictable pharmacokinetic profile, drug–drug interactions, and reduced patient compliance.1,2 To overcome these limitations, multitargeting antitumor agents have been considered as an effective strategy and gained major attention in drug discovery.3,4 Ideally, multitargeting agents can simultaneously modulate a network of disease-relevant targets and result in a synergistic effect. They can also offer the potential for higher efficacy and less adverse effects.
Epigenetic post-translational modification, especially lysine deacetylation, plays an important role in the onset and progression of cancer.5 Histone deacetylases (HDACs) are a family of epigenetic enzymes that remove acetyl groups from lysine on histones and other proteins.6 HDACs have key effects on numerous cellular functions such as regulation of gene, transcription and cell proliferation, differentiation, and death.6,7 HDACs are found to be overexpressed in different human cancers, which are regarded as promising drug targets for cancer therapy.5 So far, five HDAC inhibitors (HDACis), namely, vorinostat (SAHA), romidepsin (FK228), belinostat (PXD-101), panobinostat (LBH-589), and chidamide have been a launched on the market for the treatment of hematologic cancer.8−11 However, most of the current HDACis have poor efficacy against solid tumors, and the discovery of HDACi-based multitarget antitumor agents is emerging as an effective strategy.12,13
Nicotinamide adenine dinucleotide (NAD+) is an important cofactor that plays a central role in cellular redox reactions and serves as a second messenger essential for the survival of a number of cellular process.14 There are multiple biological pathways for NAD+ biosynthesis and the primary salvage pathway using nicotinamide (NAM) as the precursor accounts for the main source of cellular NAD+ in human cells.15,16 Nicotinamide phosphoribosyltransferase (NAMPT) is the rate-limiting enzyme in NAD+ biosynthesis. Because of the high demand for NAD+ in cancer cells, inhibition of NAMPT can dramatically repress cancer proliferation. Thus, NAMPT has been considered as an attractive target for the development of anticancer agents. A number of NAMPT inhibitors has been reported, and two of them, FK866 and CHS828, have entered human clinical trials (Figure 1A).17−20 However, their clinical development has been hampered by dose-limiting thrombocytopenia.19,20
Figure 1.

Pharmacophoric model and chemical structures of the NAMPT (A) and HDAC (B) inhibitors.
NAMPT inhibitor FK866 was proven to synergistically enhance the inhibitory effect of HDACi,21 suggesting that dual NAMPT/HDAC inhibitors are likely to possess promising antitumor profiles. Analysis of the pharmacophore of NAMPT and HDAC inhibitors indicated that they share similar structural features, providing a basis for designing dual inhibitors. Recently, we designed the first class of dual NAMPT/HDAC inhibitors, which showed promising features as novel antitumor lead compounds.21 Thus, discovering diverse dual-targeting inhibitors with novel chemotypes and exploring systemic structure–activity relationships (SARs) are highly desirable.
NAMPT inhibitors are generally linear in shape (Figure 1A) and consist of a cap group, a linker, and a tail group. Similarly, the pharmacophores of HDACis contain a hydrophobic cap group, a linker, and a zinc-binding group (ZBG) (Figure 1B). Inspired by novel NAMPT (1, Figure 1A) and HDAC (2, Figure 1B) inhibitors,22−24 herein novel thiazolocarboxamide dual NAMPT/HDAC inhibitors were rationally designed following a pharmacophore fusion strategy (Figure 2) in which propyl imidazole and hydroxamic acid side chain were used as the NAM mimic and ZBG, respectively.
Figure 2.

Design strategy of dual NAMPT/HDAC inhibitors.
The synthetic route of the designed compounds is shown in Scheme 1. Condensation of commercially available compound 3 (2-bromothiazole-5-carboxylic acid) with 3-(1H-imidazol-1-yl)propan-1-amine or 3-(aminomethyl)pyridine gave intermediates 4a–b, which was treated with (4-fluorophenyl)methanamine or 3-methylbutan-1-amine to afford intermediates 5a–d. Alkylation of 5a–d with the bromo esters gave compounds 6a–i, which was reacted with freshly prepared hydroxylamine methanol solution to yield target compounds 7a–i. Initially, the inhibitory activity of the target compounds against human recombinant NAMPT and HDAC1 was tested using the assay as previously described.25,26 As shown in Table 1, all the target compounds were proven to be dual NAMPT/HDAC1 inhibitors and exhibited excellent inhibitory activity. Compound 7a with a six-carbon alkyl linker was found to be optimal for the inhibition of NAMPT (IC50 = 7 nM), which was slightly better than FK866 (IC50 = 9 nM). However, it was less potent against HDAC1 with the IC50 value of 0.1 μM. When the alkyl carbon number was increased to seven, a balanced NAMPT/HDAC1 inhibitory activity was achieved for compound 7b, whose IC50 value against NAMPT and HDAC1 was 19 and 36 nM, respectively. When 4-fluorobenzyl group of compound 7b was replaced by the isopentyl group (7f), the best anti-HDAC1 activity was observed. Compound 7f (HDAC1 IC50 = 2 nM) was 10-fold more active than SAHA (HDAC1 IC50 = 26 nM). Also, it was a potent NAMPT inhibitor (IC50 = 15 nM). For the cap group, classical meta-substituted pyridine cap (compounds 7h and 7i) led to obvious decrease of the NAMPT/HDAC1 inhibitory activity, indicating that this kind of design strategy did not work for the present NAMPT/HDAC1 dual inhibitors.
Scheme 1. Chemical Synthesis of Compound 7a–i.

Reagents and conditions: (a) 3-(1H-imidazol-1-yl)propan-1-amine or 3-(aminomethyl)pyridine, HBTU, Et3N, DMF, rt, 3 h, yield 64–69%; (b) C2H5OH, 150 °C, microwave, 2 h, yield 57–68%; (c) Br-R3, NaH, DMF, rt, 0.5 h, yield 47–60%; (d) NH2OH/NH2OK, MeOH, 45 °C, 0.5 h, yield 74–83%.
Table 1. Enzyme Inhibition and in Vitro Antitumor Activity of Target Compounds.
| IC50 (μM) |
|||||
|---|---|---|---|---|---|
| compds | NAMPT | HDAC1 | HCT116 | A549 | HepG2 |
| 7a | 0.007 ± 0.001 | 0.10 ± 0.012 | 19 ± 1.2 | 54 ± 5.3 | 30 ± 2.9 |
| 7b | 0.019 ± 0.005 | 0.036 ± 0.003 | 9.7 ± 0.87 | 9.7 ± 8.1 | 9.4 ± 0.88 |
| 7c | 0.018 ± 0.003 | 0.044 ± 0.005 | 63 ± 4.2 | 6.6 ± 0.56 | 26 ± 3.2 |
| 7d | 0.014 ± 0.004 | 0.046 ± 0.005 | 99 ± 9.1 | 7.3 ± 0.77 | 79 ± 8.4 |
| 7e | 0.014 ± 0.002 | 0.091 ± 0.007 | 83 ± 7.9 | 2.7 ± 0.21 | 13 ± 1.5 |
| 7f | 0.015 ± 0.004 | 0.002 ± 0.001 | 30 ± 2.2 | 5.2 ± 0.48 | 24 ± 2.7 |
| 7g | 0.020 ± 0.009 | 0.060 ± 0.004 | 85 ± 7.7 | 13 ± 1.2 | 106 ± 19 |
| 7h | 0.47 ± 0.023 | 0.095 ± 0.009 | 17 ± 1.3 | 22 ± 3.0 | 14 ± 1.0 |
| 7i | 0.22 ± 0.015 | 0.036 ± 0.002 | 12 ± 1.0 | 19 ± 2.5 | 7.0 ± 0.92 |
| SAHA | NTa | 0.026 ± 0.002 | 9.6 ± 0.89 | 4.4 ± 2.1 | 9.1 ± 0.83 |
| FK866 | 0.009 ± 0.001 | NTa | 1.6 ± 0.17 | 3.7 ± 3.3 | 0.89 ± 0.076 |
NT = not tested.
In order to explore the HDAC isoform selectivity profile, the inhibitory activity of compounds 7b and 7f were examined against recombinant HDAC1, HDAC2, HDAC3, HDAC4, HDAC6, and HDAC8. As depicted in Table 2, the HDAC profiling revealed that compounds 7b and 7f were potent inhibitors against HDACs. They showed nanomolar activity against all the tested HDAC isoforms except HDAC4 and were more active than SAHA. Consistent with SAHA’s selectivity, compounds 7b and 7f were almost inactive against HDAC4. They also exhibited low potency against HDAC8. In particular, compound 7f exhibited excellent inhibitory activity against HDAC3 and HDAC6, with an IC50 value of 0.67 and 0.64 nM, respectively.
Table 2. HDAC Profiling of Compounds 7b, 7f, and SAHA.
| IC50 (nM) |
|||
|---|---|---|---|
| SAHA | 7b | 7f | |
| HDAC1 | 26 ± 0.21 | 36 ± 3.0 | 2.0 ± 0.10 |
| HDAC2 | 40 ± 0.2 | 33 ± 0.85 | 6.8 ± 0.35 |
| HDAC3 | 9.8 ± 0.5 | 4.1 ± 0.15 | 0.67 ± 0.03 |
| HDAC4 | >100 μM | 21 ± 0.55 μM | 12 ± 0.15 μM |
| HDAC6 | 21 ± 1.4 | 6.2 ± 0.45 | 0.64 ± 0.05 |
| HDAC8 | 7300 ± 300 | 351 ± 16 | 204 ± 11 |
The effect of the compounds 7b and 7f on the acetylation level of histone 3 and histone 4 is shown in Figure 3A. Both compounds caused a dramatic increase in acetyl-histone H3 and acetyl-histone H4 in a dose-dependent manner. As depicted in Figure 3B, compounds 7b and 7f decreased the cellular NAD+ level effectively after incubation with human HCT116 cells for 24 h.
Figure 3.

Inhibitory effects of compounds 7b and 7f against NAMPT and HDAC. (A) Western blot analysis of acetylated histone H3 and acetylated histone H4 after 24 h treatment with 7b and 7f (at 1 μM and 5 μM) in HCT116 cells. (B,C) Concentration response curves of 7b and 7f on NAD+ level of HCT116 cells after 24 h treatment.
Cellular thermal shift assay (CETSA)25 was performed to investigate whether NAMPT is the direct binding target of the target compounds in intact cells. The results indicated that NAMPT protein levels from cells incubated with 7b and 7f were more stable as compared with the control, indicating a good binding affinity between the tested compounds and NAMPT (Figure 4A). As shown in Figure 4B, CESTA melt curves in intact cells for NAMPT target with compounds 7b and 7f moved to the right obviously, with the ΔTm value of 7.36 and 3.01 °C, respectively. Thus, NAMPT was confirmed as the direct binding target of compounds 7b and 7f in living cells.
Figure 4.
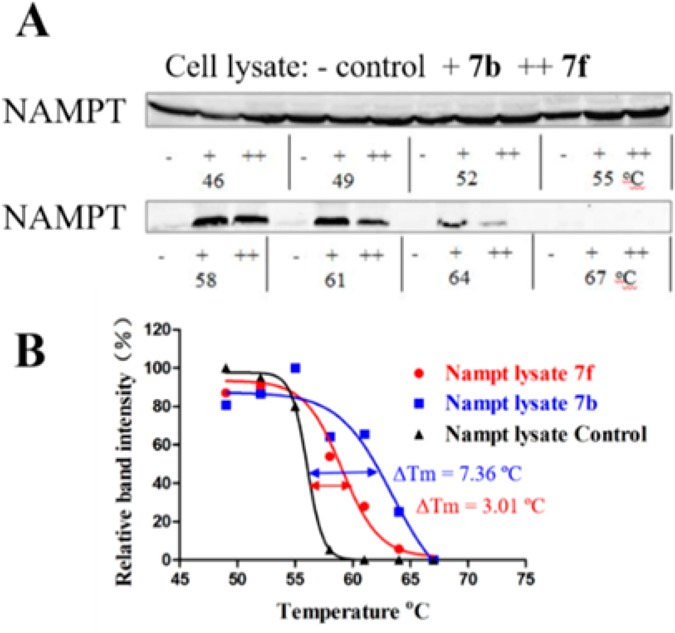
(A) Western blot of cellular thermal shift assay in intact cells for NAMPT with compounds 7b and 7f (at 10 μM). (B) CESTA melt curves in intact cells for NAMPT with 7b and 7f (at 10 μM).
Molecular docking studies were performed to predict the binding mode of the NAMPT/HDAC inhibitors. The result revealed that compounds 7b and 7f bound the same pocket to that of FK866 in the active site of NAMPT (Figure S1). As shown in Figure 5A, compound 7b interacted with NAMPT in an extended conformation. Imidazole group formed face-to-face π–π interactions with Phe193 and Tyr18, respectively. Amide group formed hydrogen bonding interactions with Asp219 and Ser275. The terminal hydroxamic acid moiety was located at the outside of the active site and faced to the solvent, forming hydrogen-bonding interactions with Arg349. The alkyl linker was found to interact with Ile309 and Ala379 mainly through hydrophobic and van der Waals interactions. The binding mode of compound 7f with NAMPT was very similar to that of compound 7b (Figure 5B). For the binding mode of compounds 7b and 7f with HDAC1, the hydroxamic acid moiety was observed deep into the active site and established a hydrogen bonding network with Tyr303, His140, and Hisl41, chelating the Zn2+ in a bidentate way (Figure 5C,D). The remaining part of the molecule was found to complement the surface of the cap region of HDAC1 very closely, leading to hydrophobic interactions with Pro29 and Phe205 (Figure S2 in the Supporting Information). In addition, the imidazole nitrogen atom of compound 7f formed a hydrogen bond with Ser148 (Figure 5D). This observation may explain why compound 7f was significantly more potent against HDAC1.
Figure 5.
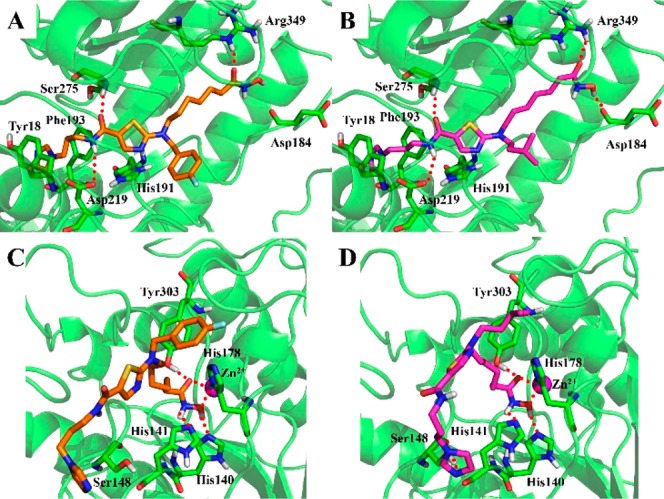
Predicted binding pose of compounds 7b (orange) and 7f (purple) in the active site of NAMPT (A,B) and HDAC1 (C,D). Zinc (C,D) is shown as a sphere (purple). The figure was generated using PyMol (http://www.pymol.org/).
Finally, in vitro and in vivo antitumor activity of the target compounds was evaluated. As shown in Table 1, these compounds generally showed moderate cytotoxicity against HCT116 (colon cancer), A549 (lung cancer), and HepG2 (liver cancer) human cancer cell lines. Among them, compound 7f was selected for evaluating the in vivo antitumor efficacy in HCT116 tumor xenografts in nude mice by measuring tumor growth inhibition (TGI) and relative increment ratio (T/C).27 Compound 7f was administered intraperitoneally (ip) at 25 mg/kg twice a day for 21 consecutive days. The positive control included SAHA (25 mg/kg, ip, bid) and FK866 (15 mg/kg, ip, bid). The experiment was performed in the same animal model reported by our group.21 As shown in Figure 6A, treatment with compound 7f caused a dramatic reduction in tumor growth. Moreover, compound 7f demonstrated superior or comparable antitumor activity (TGI = 42%, T/C = 58%) to that of the reference drugs SAHA (TGI = 33%, T/C = 67%) and FK866 (TGI = 39%, T/C = 61%), respectively. The final tumor tissue size visualized in Figure 6C also explicitly showed a good antineoplastic activity of compound 7f. Importantly, mice treated with a high dose of 7f (25 mg/kg, ip, bid) showed no significant body weight loss, the same as those treated with SAHA or FK866 (Figure 6B). Also, it should be noted that compound 7f remained to be further optimized to improve the in vivo antitumor efficacy.
Figure 6.

Growth curve of implanted HCT116 xenograft in nude mice. (A) Compound 7fin vivo efficacy results in HCT116 tumor xenograft model. Data are expressed as the mean ± standard deviation. **P < 0.01, versus the control group, determined with Student’s t test. (B) Body weight loss as a result of compound 7f administration in HCT116 xenograft experiment. (C) Picture of dissected HCT116 tumor tissues.
In summary, the present investigation discovered novel NAMP/HDAC dual inhibitors. Compound 7f was highly active against both NAMPT and HDAC and showed significant in vivo antitumor efficacy in the HCT116 xenograft model without significant toxicity. Further optimization is currently in progress.
Glossary
ABBREVIATIONS
- NAMPT
nicotinamide phosphoribosyltransferase;
- HDAC
histone deacetylase
- HDACis
histone deacetylase inhibitors
- SAR
structure–activity relationship
- NAD+
nicotinamide adenine dinucleotide
- NAM
nicotinamide
- ZBG
zinc-binding group
- CETSA
cellular thermal shift assay
- TGI
tumor growth inhibition.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00414.
Chemical synthesis and structural characterization of the target compounds; protocols of biological assays (PDF)
Author Contributions
† W.C., G.D., and Y.W. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the National Natural Science Foundation of China (Grant 81402796 to G.D. and 81725020 to C.S.) and Shanghai Education Development Foundation and Shanghai Municipal Education Commission (Chenguang Program, Grant 15CG42).
The authors declare no competing financial interest.
Supplementary Material
References
- Anighoro A.; Bajorath J.; Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- Peters J. U. Polypharmacology: foe or friend?. J. Med. Chem. 2013, 56, 8955–8971. 10.1021/jm400856t. [DOI] [PubMed] [Google Scholar]
- Haber D. A.; Gray N. S.; Baselga J. The evolving war on cancer. Cell 2011, 145, 19–24. 10.1016/j.cell.2011.03.026. [DOI] [PubMed] [Google Scholar]
- Trusolino L.; Bertotti A. Compensatory pathways in oncogenic kinase signaling and resistance to targeted therapies: six degrees of separation. Cancer Discovery 2012, 2, 876–880. 10.1158/2159-8290.CD-12-0400. [DOI] [PubMed] [Google Scholar]
- Conway S. J.; Woster P. M.; Shen J. K.; Georg G.; Wang S. Epigenetics: novel therapeutics targeting epigenetics. J. Med. Chem. 2015, 58, 523–524. 10.1021/jm501941q. [DOI] [PubMed] [Google Scholar]
- Falkenberg K. J.; Johnstone R. W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discovery 2014, 13, 673–691. 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Bountra C.; Fish P. V.; Lee K.; Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discovery 2012, 11, 384–400. 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- Ververis K.; Hiong A.; Karagiannis T. C.; Licciardi P. V. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Biologics 2013, 7, 47–60. 10.2147/BTT.S29965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann B. S.; Johnson J. R.; Cohen M. H.; Justice R.; Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- Bailey H.; Stenehjem D. D.; Sharma S. Panobinostat for the treatment of multiple myeloma: the evidence to date. J. Blood Med. 2015, 6, 269–276. 10.2147/JBM.S69140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manal M.; Chandrasekar M. J.; Gomathi Priya J.; Nanjan M. J. Inhibitors of histone deacetylase as antitumor agents: A critical review. Bioorg. Chem. 2016, 67, 18–42. 10.1016/j.bioorg.2016.05.005. [DOI] [PubMed] [Google Scholar]
- Lai C. J.; Bao R.; Tao X.; Wang J.; Atoyan R.; Qu H.; Wang D. G.; Yin L.; Samson M.; Forrester J.; Zifcak B.; Xu G. X.; DellaRocca S.; Zhai H. X.; Cai X.; Munger W. E.; Keegan M.; Pepicelli C. V.; Qian C. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010, 70, 3647–3656. 10.1158/0008-5472.CAN-09-3360. [DOI] [PubMed] [Google Scholar]
- Papavassiliou K. A.; Papavassiliou A. G. Histone deacetylases inhibitors: conjugation to other anti-tumour pharmacophores provides novel tools for cancer treatment. Expert Opin. Invest. Drugs 2014, 23, 291–294. 10.1517/13543784.2014.857401. [DOI] [PubMed] [Google Scholar]
- Galluzzi L.; Kepp O.; Vander Heiden M. G.; Kroemer G. Metabolic targets for cancer therapy. Nat. Rev. Drug Discovery 2013, 12, 829–846. 10.1038/nrd4145. [DOI] [PubMed] [Google Scholar]
- Sampath D.; Zabka T. S.; Misner D. L.; O’Brien T.; Dragovich P. S. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol. Ther. 2015, 151, 16–31. 10.1016/j.pharmthera.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Galli U.; Travelli C.; Massarotti A.; Fakhfouri G.; Rahimian R.; Tron G. C.; Genazzani A. A. Medicinal chemistry of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors. J. Med. Chem. 2013, 56, 6279–6296. 10.1021/jm4001049. [DOI] [PubMed] [Google Scholar]
- Watson M.; Roulston A.; Belec L.; Billot X.; Marcellus R.; Bedard D.; Bernier C.; Branchaud S.; Chan H.; Dairi K.; Gilbert K.; Goulet D.; Gratton M. O.; Isakau H.; Jang A.; Khadir A.; Koch E.; Lavoie M.; Lawless M.; Nguyen M.; Paquette D.; Turcotte E.; Berger A.; Mitchell M.; Shore G. C.; Beauparlant P. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol. Cell. Biol. 2009, 29, 5872–5888. 10.1128/MCB.00112-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasmann M.; Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003, 63, 74367442. [PubMed] [Google Scholar]
- Holen K.; Saltz L. B.; Hollywood E.; Burk K.; Hanauske A. R. The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Invest. New Drugs 2008, 26, 45–51. 10.1007/s10637-007-9083-2. [DOI] [PubMed] [Google Scholar]
- von Heideman A.; Berglund A.; Larsson R.; Nygren P. Safety and efficacy of NAD depleting cancer drugs: results of a phase I clinical trial of CHS 828 and overview of published data. Cancer Chemother. Pharmacol. 2010, 65, 1165–1172. 10.1007/s00280-009-1125-3. [DOI] [PubMed] [Google Scholar]
- Dong G.; Chen W.; Wang X.; Yang X.; Xu T.; Wang P.; Zhang W.; Rao Y.; Miao C.; Sheng C. Small molecule inhibitors simultaneously targeting cancer metabolism and epigenetics: discovery of novel nicotinamide phosphoribosyltransferase (NAMPT) and histone deacetylase (HDAC) dual inhibitors. J. Med. Chem. 2017, 60, 7965–7983. 10.1021/acs.jmedchem.7b00467. [DOI] [PubMed] [Google Scholar]
- Brazeau J. F.; Rosse G. Thiazolocarboxamide Analogues as NAMPT Inhibitors. ACS Med. Chem. Lett. 2014, 5, 277. 10.1021/ml500046t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terracciano S.; Chini M. G.; Riccio R.; Bruno I.; Bifulco G. Design, synthesis, and biological activity of hydroxamic tertiary amines as histone deacetylase inhibitors. ChemMedChem 2012, 7, 694–702. 10.1002/cmdc.201100531. [DOI] [PubMed] [Google Scholar]
- Terracciano S.; Chini M. G.; Bifulco G.; D’Amico E.; Marzocco S.; Riccio R.; Bruno I. Synthesis of new mono and bisamides projected as potential histone deacetylase (HDAC) inhibitors. Tetrahedron 2010, 66, 2520–2528. 10.1016/j.tet.2010.01.061. [DOI] [Google Scholar]
- Xu T. Y.; Zhang S. L.; Dong G. Q.; Liu X. Z.; Wang X.; Lv X. Q.; Qian Q. J.; Zhang R. Y.; Sheng C. Q.; Miao C. Y. Discovery and characterization of novel small-molecule inhibitors targeting nicotinamide phosphoribosyltransferase. Sci. Rep. 2015, 5, 10043. 10.1038/srep10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S.; Dong G.; Wang Z.; Chen W.; Huang Y.; Li Z.; Jiang Y.; Liu N.; Yao J.; Miao Z.; Zhang W.; Sheng C. Discovery of Novel Multiacting Topoisomerase I/II and Histone Deacetylase Inhibitors. ACS Med. Chem. Lett. 2015, 6, 239–243. 10.1021/ml500327q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong G.; Wang S.; Miao Z.; Yao J.; Zhang Y.; Guo Z.; Zhang W.; Sheng C. New tricks for an old natural product: discovery of highly potent evodiamine derivatives as novel antitumor agents by systemic structure-activity relationship analysis and biological evaluations. J. Med. Chem. 2012, 55, 7593–7613. 10.1021/jm300605m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.