

Abstract

p300 and its paralog CBP can acetylate histones and other proteins and have been implicated in a number of diseases characterized by aberrant gene activation, such as cancer. A novel, highly selective, orally bioavailable histone acetyltransferase (HAT) domain inhibitor has been identified through virtual ligand screening and subsequent optimization of a unique hydantoin screening hit. Conformational restraint in the form of a spirocyclization followed by substitution with a urea led to a significant improvement in potency. Replacement of the hydantoin moiety with an oxazolidinedione followed by fluoro substitution led to A-485, which exhibits potent cell activity, low clearance, and high oral bioavailability.
Keywords: p300, CBP, histone acetyl transferase
Reversible protein acetylation has emerged as a key signaling mechanism for regulating cellular function and, in particular, transcription regulation.1 Acetylation of protein lysine residues is mediated by a family of histone acetyltransferases (HATs), whereas removal of acetyl groups is catalyzed by histone deacetylases (HDACs).2,3 Small molecule HDAC inhibitors such as panobinostat and vorinostat have been successfully developed as novel therapeutic agents for the treatment of certain cancers; however, progress on HAT inhibitors has been limited due to the lack of selective, drug-like inhibitors.4,5 Two of the best described HATs are p300 and the closely related paralog CBP.6 p300/CBP acetylates histones on lysines 18 and 27 of histone H3 (H3K18, H3K27), as well as numerous transcription factors to facilitate gene activation programs important for cell growth and differentiation.7,8 Inhibition of p300/CBP has been proposed as a therapeutic strategy in diseases driven by gene activation such as cancer, Alzheimer’s disease, diabetes, and cardiovascular diseases.9,10
In addition to the enzymatic HAT domain, p300/CBP has multiple other domains including three cysteine-histidine rich domains (CH1, CH2, and CH3), a KIX domain, a bromodomain, and a steroid receptor coactivator interaction domain (SRC-1 interaction domain). Efforts at modulating the activity of p300/CBP have focused on inhibition of the N-terminal region that binds beta-catenin,11 the HIF1α binding KIX domain,12 the acetyl-Lys binding bromodomain,13 and the histone acetyltransferase (HAT) domain.14 Recent reports describe several examples of potent and selective p300/CBP bromodomain inhibitors, such as I-CBP112 and CPI-637.15,16 These inhibitors exhibited potentially therapeutically relevant cellular activities, such as inhibition of AML-ETO driven transcription in leukemia and IRF4 driven transcription in multiple myeloma.17
Due to the multiple biologically relevant domains of p300 described above as well as its scaffolding functions, genetic knockdown approaches cannot be used to study the consequences of HAT domain inhibition alone. Thus, selective small molecule inhibitor compounds are needed to understand the therapeutic potential of p300 HAT inhibition. Several early literature reports of p300/CBP HAT inhibitors describe either natural products (e.g., garcinol, Figure 1) or compounds based on bisubstrate analogs.14 These compounds display only modest potency and selectivity and have poor cell permeability.14 Compound C646, identified by virtual screening18 (Figure 1), has been widely used as a tool compound, but its thiol reactive functionality may limit its pharmacologic specificity.19,20 There is thus a pressing need for a highly selective, optimized HAT inhibitor that can be used to unequivocally interrogate the biology of p300/CBP.14,21 We have recently reported on the biological characterization of A-485, a potent, highly selective p300/CBP inhibitor suitable for in vitro and in vivo target validation studies.22 We now wish to disclose the medicinal chemistry optimization efforts that led to the discovery of A-485 (24).
Figure 1.

p300 HAT inhibitors.
A previously described virtual ligand screening approach, based on compound docking to the Lys-CoA binding pocket present in the X-ray structure of p300 HAT was used in the discovery of C646.23 In order to identify novel chemical matter, we pursued an alternative virtual screening approach based on the hypothesis that the published X-ray structure represents a closed form of the enzyme and that this enzyme can undergo conformational changes upon binding of Ac-CoA and the histone substrate, akin to other Ac-CoA binding proteins, such as citrate synthase24,25 and serotonin N-acetyl transferase.26 The initial challenge then was to predict the open form of p300 HAT. We decided to focus on the loop 1438–1458, which lines one side of the tunnel, based on the critical catalytic role of Tyr1446 and the known conformational changes of the Ac-CoA binding proteins mentioned above. Normal mode analyses and Monte Carlo searches of loop conformations allowed multiple hypotheses to be formed. One conformation was distinctive, in terms of its consistency with most known data on p300 HAT, which we hypothesized to be the open conformation.
We applied a variety of computational approaches to this hypothesized open conformation, to determine likely subpockets where ligands may bind. Various constellations of subpockets were used to form five distinct pharmacophore hypotheses. Each approach was used to search an 800,000 compound set of commercially available compounds, and approximately 1300 compounds were purchased and tested in a radioactive enzymatic assay measuring inhibition of p300 HAT mediated acetylation of a peptide substrate. This led to two confirmed hits, one of which, compound 1, was successfully optimized as described below.
Compound 1 (Figure 2) is a micromolar inhibitor of p300 HAT with reasonable ligand and lipid efficiency (IC50 = 5.1 μM, LE = 0.23, LipE = 3), devoid of reactive functionalities and thus was an attractive starting point for hit-to-lead chemistry.27 Initial SAR development focused on the indole and amide terminal portions of 1. Extensive SAR looking at both substitution and replacement of the indole and benzyl (2) either led to a loss in activity or did not provide a boost in potency. Attempts at significantly reducing molecular weight and increasing binding efficiency were not fruitful, although the phenyl substituted hydantoin 3a (IC50 = 7.6 μM) was found to be equipotent. ortho-Methyl substitution (3b) provided a modest increase in potency (IC50 = 2.7 μM, LE = 0.27) suggesting that a nonplanar orientation between the phenyl and hydantoin group is preferred. We hypothesized that restricting the conformational mobility of the pendant phenyl might provide an improvement in potency and more rigid vectors for substitution. Thus, the ring-constrained spiro indane hydantoin compound 4 was prepared. The compound was equipotent to 3b (IC50 = 1.6 μM) but ca. 5-fold more potent than the parent phenyl hydantoin 3a, and provided the starting point for a successful lead optimization effort as described below.
Figure 2.

Discovery of spiroindane hydantoin lead.
Compound 4 is a diastereomeric mixture; so to fully understand the SAR and the effect of the spiro indane rigidification, we synthesized all four diastereomers (Figure 3). As might be expected given the similar steric size of the methyl and cyclopropyl groups, the stereochemistry of the side chain had a modest (2–3-fold) effect on potency (cf. 5 vs 7 and 6 vs 8), while a more significant (10–13 fold) difference was observed between the diastereomers at the spiro carbon (cf. 5 vs 6 and 7 vs 8). The observed differences in diastereomer activity, suggesting specific enzyme interactions and the favorable ligand efficiency of the (S,S) diastereomer 5 (LE = 0.29, LipE = 2.8), prompted us to pursue an extensive optimization of this spiroindane series.
Figure 3.

Spiroindane hydantoin diastereomers.
In the absence of any structural information about the binding mode, we undertook a systematic investigation of all portions of the molecule including indane and amide substitutions or replacements. Ultimately, the most fruitful avenue of investigation was substitution on the phenyl portion of the indane core. A variety of substituents including lipophilic, hydrogen bond acceptors and donors were incorporated and select results are shown in Table 1. In order to quickly obtain the SAR, compounds were initially prepared and tested as mixtures of either all four possible diastereomers or mixtures of the two diastereomers at the spiro indane center. In general, substitution at C5′ was better tolerated than C4′ (cf. 9 vs 10 and 11 vs 12). Substitution with ethers (13), small alkyl (e.g., cyclopropyl 15), or carboxamide (11) was tolerated but did not provide a boost in potency. A modest boost in potency was observed when a hydrogen bond donor was directly attached to the indane core such as an amide (16). Following up on this observation we prepared the corresponding carbamate (17), sulfonamide (18), and urea (19) in the more active (S)-cyclo-propyl ethyl motif. The urea 19 was found to provide a further boost in activity and significantly improved lipid efficiency (IC50 = 245 nM, LE = 0.26, LipE = 5.24).
Table 1. SAR of Indane Substitution.
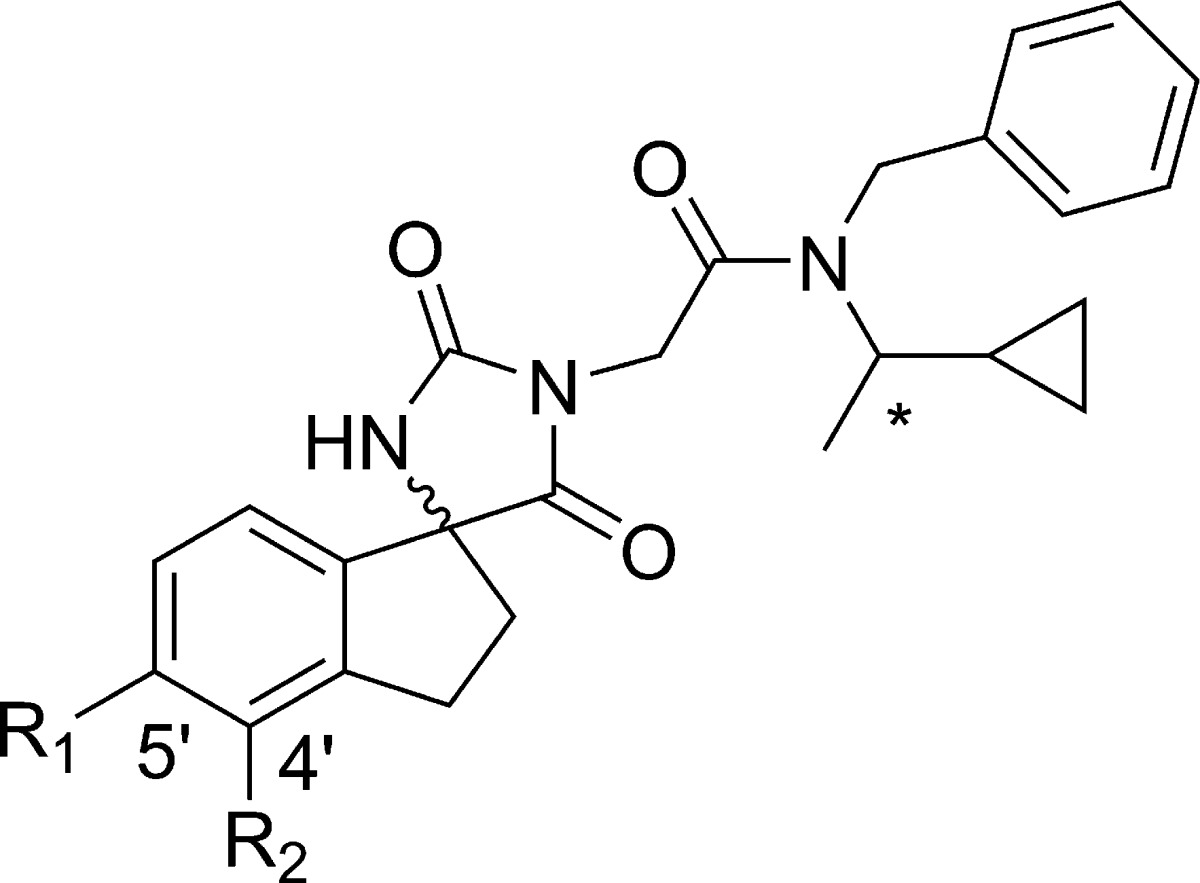
| compd | *stereochemistry | R1 | R2 | p300 HAT IC50 (μM) |
|---|---|---|---|---|
| 4 | R,S | H | H | 1.6 |
| 9 | R,S | CN | H | 2.5 |
| 10 | R,S | H | CN | 27.8 |
| 11 | R,S | CONH2 | H | 4.86 |
| 12 | R,S | H | CONH2 | 17.9 |
| 13 | R,S | OMe | H | 1.6 |
| 14 | R,S | CH2 OH | H | 14.0 |
| 15 | R,S | cyclopropyl | H | 1.71 |
| 16 | R,S | NHCOMe | H | 0.78 |
| 17 | S | NHCO2-Me | H | 1.0 |
| 18 | S | NHSO2-Me | H | 16.0 |
| 19 | S | NHCONH-Me | H | 0.24 |
The improved potency of the urea analog 19 prompted us to prepare the corresponding single diastereomer with the desired (S)-stereochemistry at the spiro center (20). As expected, the compound potently inhibited p300 HAT domain acetyltransferase activity (IC50 = 170 nM). Gratifyingly, the compound also inhibited acetylation of histone substrates in cells, as measured by a decrease in H3K27Ac levels in PC-3 cells (IC50 = 470 nM). Given the promising in vitro profile of 20, we evaluated its pharmacokinetic properties in mice. The compound exhibited very high clearance (CLp = 6.6 L/h/kg) in excess of liver blood flow and no oral exposure. We hypothesized that reducing the number of H-bond donors could be beneficial in improving permeability, which in conjunction with improved microsomal stability could lead to better oral exposure. To this end, we prepared the oxazolidinedione analog 21, which maintained potency in both the enzymatic and cellular assays (Figure 4). We also prepared the diastereomeric (R)-spiro center analog 22 and surprisingly found that the oxazolidinedione and hydantoin series had the opposite preferred stereochemistry at the spiro center. Thus, compound 22 was ca. 5-fold more potent in inhibiting p300 HAT than the (S)-spiro center analog 21. This improved enzyme inhibitory activity also translated into improved cellular activity (H3K27Ac IC50 = 47 nM). The pharmacokinetic profile in mice was significantly improved relative to the hydantoin 20 with improvements in both clearance (CLp 1.62 vs 6.6 L/h/kg) and oral exposure (AUC = 2.5 μg·h/mL vs no exposure; 10 mg/kg oral dose). The improved oral bioavailability of the oxazolidinedione 22 relative to the hydantoin analog 20 is consistent with improved permeability as measured by PAMPA (2.35 × 10–6 cm/s for 22 vs 0.47 × 10–6 cm/s for 20).
Figure 4.

From spirohydantoins to spirooxazolidinediones.
In light of the potent in vitro profile and promising pharmacokinetic properties of 22 we focused our lead optimization efforts on the urea substituted oxazolidinediones. The primary objective was to improve microsomal stability, which we hoped would translate into reduced plasma clearance. We hypothesized that the low microsomal stability of 22 was due to oxidative metabolism of the side chain phenyl and/or amide N-alkyl substituents. Thus, we prepared a number of fluorinated derivatives. Substitution with one or two fluorine atoms on the terminal phenyl was tolerated (data not shown), and the site of substitution was not critical for potency; we focused our optimization on the para-substituted mono-fluorophenyl analogs, and representative compounds are shown in Table 2. Replacement of the methyl group with trifluoromethyl (23) maintained potency, but microsomal stability was not improved when corrected for nonspecific microsomal binding.28 Total plasma clearance for 23 was improved (Clp = 0.4 L/h/kg); however, unbound clearance was still high and comparable to the parent 22 in accordance with the in vitro data.29 However, replacement of cyclopropyl with trifluoromethyl (24; A-485) led to significantly improved microsomal stability, which importantly translated into low unbound plasma clearance and very high oral exposure. Its pharmacokinetic properties were also evaluated in rats and dogs, species that could be potentially used for tolerability studies. The compound had modest clearance in rat (1.23 L/h/kg) and very low clearance in dogs (0.09 L/h/kg) with good oral bioavailability in both species (Table 3).
Table 2. Oxazolidinedione Ureas.
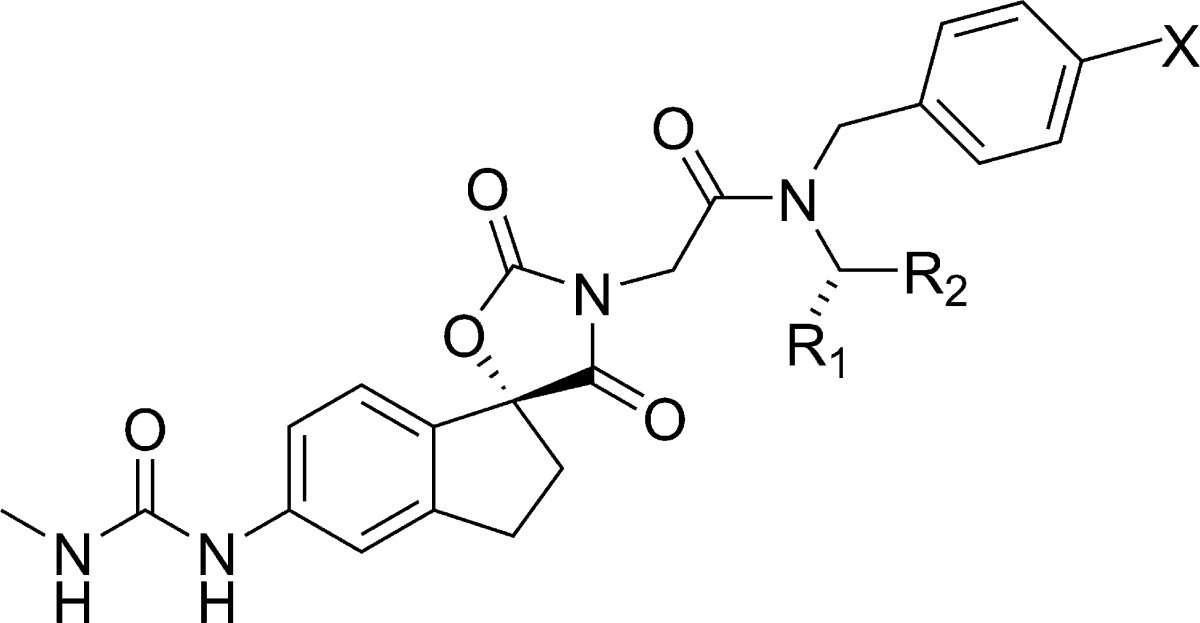
| mouse
microsomal stability |
mouse
pharmacokinetics |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | X | p300 HAT IC50 (μM) | H3K27 Ac IC50 (μM) | CL scaled (L/h/kg) | fu, mica | Clint,u (L/h/kg) | CLp, ivb,f (L/h/kg) | PPB % | CLp,u (L/h/kg) | F % oral | oral AUC (μg·h/mL) |
| 22 | Me | cyclopropyl | H | 0.025 | 0.047 | 107 | 0.48 | 188 | 1.62 | 99.1 | 186 | 40 | 2.5c |
| 23 | CF3 | cyclopropyl | F | 0.032 | 0.045 | 41 | 0.22 | 181 | 0.4 | 99.8 | 252 | 45 | 1.1d |
| 24 | Me | CF3 | F | 0.060 | 0.101 | 21.1 | 0.56 | 37.6 | 0.56 | 99.2 | 75 | >100 | 1.94,d11.6e |
Fraction unbound in human microsomes.
1 mg/kg dose.
10 mg/kg dose.
1 mg/kg dose.
12.5 mg/kg dose.
1L/h/kg = 16.6 mL/min/kg.
Table 3. Pharmacokinetic Properties of 24 (A-485).
| mouse | rat | dog | |
|---|---|---|---|
| CLp (L/h/kg) | 0.56 | 1.23 | 0.09 |
| CLp,u (L/h/kg) | 75 | 47 | 4.4 |
| t1/2 (h) | 0.7 | 1.2 | 7 |
| Vss (L/kg) | 0.5 | 1.5 | 0.8 |
| F (%) | 110 | 63 | 74 |
| Fa × Fg30 | 1 | 0.93 | 0.78 |
The synthesis of 24 (A-485) is typical of the general route used to prepare analogs (Scheme 1). The chirality at the spiro center was established via an enantioselective TMSCN addition to indanone 25. Nitrile hydrolysis followed by triphosgene-mediated cyclization led to the oxazolidinedione 27 in 99.8% ee after recrystallization (74% overall yield from 25). Stereochemistry of the spiro center was unequivocally established via an X-ray crystal structure. The versatile intermediate 28 was alkylated with the bromide 30, and the urea was introduced via a Buchwald amination, followed by urea formation with triphosgene and methyl amine.
Scheme 1. Synthesis of 24 (A-485).

Reagents and conditions. (a) Et3Al, TMSCN, 2% 2,2′-((1E,1′E)-(((1S,2S)-1,2-diphenylethane-1,2-diyl)bis(azanylylidene))bis(methanylylidene))bis(4-bromophenol), N,N-dimethylaniline oxide; (b) HCl, EtOH; (c) triphosgene, Et3N, 0 °C, then 2 N HCl; (d) K2CO3, DMF; (e) 1. Pd(OAc)2, BINAP, diphenylmethanimine; 2. 2 N HCl; 3. triphosgene, MeNH2.
A-485 has been extensively characterized for its selectivity within the HAT family and against nonepigenetic off-targets, and the binding mode has been definitively established through an X-ray crystal structure in complex with the fully active p300 HAT domain, as previously described.22 Notably, the compound was evaluated for its CYP inhibitory activity in anticipation of possible preclinical in vivo combination studies. The compound exhibited modest inhibition of CYP2C8 (IC50 = 0.99 μM) and CYP2C9 (IC50 = 1.6 μM) but significantly weaker inhibition of CYP3A4 (IC50 = 24 μM).

In summary, we have described the optimization of a micromolar hit from a VLS screen into a potent (mid-nanomolar), selective, drug-like, orally bioavailable small molecule inhibitor of p300/CBP HAT. Compound 24 (A-485) has cellular and pharmacokinetic properties that make it suitable for definitive biological interrogation of the effects of p300/CBP HAT inhibition in both the in vitro and in vivo settings.
Acknowledgments
We are grateful to Saul Rosenberg for helpful discussions and project guidance. We thank Amanda Olson for the pharmacokinetic analyses. We thank Stella Doktor, Sue Swanson, Nicole Richwine, and Xiaomei Zhang for the in vitro ADME data. We thank Vivek Abraham and Morey Smith for high content microscopy assistance. P.A.C. was supported by the NIH and FAMRI foundation.
Glossary
ABBREVIATIONS
- CYP
cytochromes P450
- PPB
plasma protein binding
- TMS
trimethylsilyl
- BINAP
2,2′-bis(diphenylphospino)-1,1′- binaphthyl
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00395.
Experimental details are provided that pertain to the biological assays and the synthesis and characterization of compounds 1–24 (PDF)
Author Present Address
§ I-to-D, Inc., P.O. Box 6177, Lincoln, Massachusetts 01773, United States.
Author Present Address
∥ Mitobridge, Inc., 1030 Massachusetts Avenue, Second Floor, Cambridge, Massachusetts 02139, United States.
Author Present Address
⊥ China Novartis Institutes for BioMedical Research, No. 4218 Jinke Road, Zhangjiang Hi-Tech Park, Pudong District, Shanghai 201203, China.
Author Present Address
# Petra Pharma Corporation, 430 East 29th Street, Suite 506, New York, New York 10016, United States.
Author Present Address
∇ BASF Corporation, 26 Davis Drive, Research Triangle Park, North Carolina 27709, United States.
Author Present Address
○ Accelerator Corporation, 430 East 29th Street, New York, New York 10016, United States.
Author Present Address
◆ Van Drie Research, 109 Millipond, Andover, Massachusetts 01845, United States.
Author Present Address
¶ Perelman School of Medicine, University of Pennsylvania, 421 Curie Blvd., Philadelphia, Pennsylvania 19104, United States.
Author Present Address
△ Johns Hopkins University, 725 North Wolfe Street, Baltimore, Maryland 21205, United States.
Author Present Address
● Cascadian Therapeutics, Inc., 2601 Fourth Avenue, Suite 500, Seattle, Washington 98121, United States.
Author Present Address
□ Faraday Pharmaceuticals, 1616 Eastlake Avenue E, Suite 560, Seattle, Washington 98102, United States.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): This study was sponsored by AbbVie. AbbVie contributed to the study design, research, and interpretation of data, writing, reviewing, and approving the publication. M.R.M., T.M.H., R.M.R., R.Man., H.L., K.D.B., L.L., and A.L. are employees of AbbVie. D.M., E.A.K., K.K., P.d.V., and J.W.L were employees of Acylin, which provided assets to Abbvie at the time of the study. R.Mar. and P.A.C. are co-founders of Acylin and consultants for AbbVie.
Supplementary Material
References
- Tessarz P.; Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. 10.1038/nrm3890. [DOI] [PubMed] [Google Scholar]
- Haberland M.; Montgomery R. L.; Olson E. N. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H.; Marmorstein R. Histone acetyltransferases: Rising an- cient counterparts to protein kinases. Biopolymers 2013, 99, 98–111. 10.1002/bip.22128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R. P.; Robaa D.; Alhalabi Z.; Sippl W.; Jung M. KATch- ing-up on small molecule modulators of lysine acetyltransferases. J. Med. Chem. 2016, 59, 1249–1270. 10.1021/acs.jmedchem.5b01502. [DOI] [PubMed] [Google Scholar]
- Richters A.; Koehler A. N. Epigenetic modulation using small molecules-targeting histone acetyl transferases in Disease. Curr. Med. Chem. 2017, 24, 4121–4150. 10.2174/0929867324666170223153115. [DOI] [PubMed] [Google Scholar]
- Attar N.; Kurdistani S. K. Exploitation of EP300 and CREBB lysine acetyltransferase by cancer. Cold Spring Harbor Perspect. Med. 2017, 7, a026534. 10.1101/cshperspect.a026534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Tang Y.; Cole P. A.; Marmorstein R. Structure and chemistry of the p300/CBP and Rtt109 histone acetyl transferases: implications for histone acetyltransferase evolution and function. Curr. Opin. Struct. Biol. 2008, 18, 741–747. 10.1016/j.sbi.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Q.; Yu L.-R.; Wang L.; Zhang Z.; Kasper L. H.; Lee J.-E.; Wang C.; Brindle P. K.; Dent S. Y.R.; Ge K. Distinct roles of GCN5/PCAF PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011, 30, 249–262. 10.1038/emboj.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farria A.; Li W.; Dent S. Y. R. KATs in Cancer: functions and therapies. Oncogene 2015, 34, 4901–4913. 10.1038/onc.2014.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valor L. M.; Viosca J.; Lopez-Atalaya J. P.; Barco A. Lysine acetyltransferases CBP and p300 as therapeutic targets in cognitive and neurodegenerative disorders. Curr. Pharm. Des. 2013, 19, 5051–64. 10.2174/13816128113199990382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gang E. J.; Hsieh Y. T.; Pham J.; Zhao Y.; Nguyen C.; Huantes S.; Park E.; Naing K.; Klemm L.; Swaminathan S.; Con- way E. M.; Pelus L. M.; Crispino J.; Mullighan C. G.; McMillan M.; Müschen M.; Kahn M.; Kim Y. M. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene 2014, 33, 2169–2178. 10.1038/onc.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao B. B.; Grishagin I.; Mesallati H.; Brewer T. F.; Olenyuk B. Z.; Arora P. S. In vivo modulation of hypoxia-inducible signaling by topographical helix mimetics. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 7531–6. 10.1073/pnas.1402393111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay D. A.; Fedorov O.; Martin S.; Singleton D. C.; Tallant C.; Wells C.; Picaud S.; Philpott M.; Monteiro O. P.; Rogers C. M.; Conway S. J.; Rooney T. P. C.; Tumber A.; Yapp C.; Filippakopou- los P.; Bunnage M. E.; Muller S.; Knapp S.; Schofield C. J.; Brennan P. E. Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J. Am. Chem. Soc. 2014, 136, 9308–9319. 10.1021/ja412434f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancy B. M.; Cole P. A. Protein lysine acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2452. 10.1021/cr500452k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picaud S.; Fedorov O.; Thanasopoulou A.; Leonards K.; Jones K.; Meier J.; Olzscha H.; Monteiro O.; Martin S.; Philpott M.; Tumber A.; Filippakopoulos P.; Yapp C.; Wells C.; Che K. H.; Bannister A.; Robson S.; Kumar U.; Parr N.; Lee K.; Lugo D.; Jeffrey P.; Taylor S.; Vecellio M. L.; Bountra C.; Brennan P. E.; O’Mahony A.; Velichko S.; Muller S.; Hay D.; Daniels D. L.; Urh M.; La Thangue N. B.; Kouzarides T.; Prinjha R.; Schwaller J.; Knapp S. Generation of a selective small molecule inhibitor of the CBP/p300 bromodomain for leukemia therapy. Cancer Res. 2015, 75, 5106–5119. 10.1158/0008-5472.CAN-15-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor M. A.; Cote A.; Hewitt M. C.; Pastor R.; Leblanc Y.; Nasveschuk C. G.; Romero F. A.; Crawford T. D.; Cantone N.; Jayaram H.; Setser J.; Murray J.; Beresini M. H.; de Leon Boenig G. L.; Chen Z.; Conery A. R.; Cummings R. T.; Dakin L. A.; Flynn E. M.; Huang O. W.; Kaufman S.; Keller P. J.; Kiefer J. R.; Lai T.; Li Y.; Liao J.; Liu W.; Lu H.; Pardo E.; Tsui V.; Wang J.; Wang Y.; Xu Z.; Yan F.; Yu D.; Zawadzke L.; Zhu X.; Zhu X.; Sims R. J.; Cochran A. G.; Bellon S.; Audia J. E.; Magnuson S.; Albrecht B. K. Fragment-based discovery of a selective and cell-active benzodiazepinone CBP/EP300 bromodomain inhibitor (CPI-637). ACS Med. Chem. Lett. 2016, 7, 531–536. 10.1021/acsmedchemlett.6b00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conery A. R.; Centore R. C.; Neiss A.; Keller P. J.; Joshi S.; Spillane K. L.; Sandy P.; Hatton C.; Pardo E.; Zawadzke L.; Bommi- Reddy A.; Gascoigne K. E.; Bryant B. M.; Mertz J. A.; Sims R. J. III Bromodomain inhibition of the transcriptional coactivators CBP/EP300 as a therapeutic strategy to target the IRF4 network in multiple myeloma. eLife 2016, 10483. 10.7554/eLife.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers E. M.; Yan G.; Mukherjee C.; Orry A.; Wang L.; Hol- bert M. A.; Crump N. T.; Hazzalin C. A.; Liszczak G.; Yuan H.; Larocca C.; Saldanha S. A.; Abagyan R.; Sun Y.; Meyers D. J.; Marmorstein R.; Mahadevan L. C.; Alani R. M.; Cole P. A. Virtual ligand screening of the p300/CBP histone acetyltransferase: identifi- cation of a selective small molecule inhibitor. Chem. Biol. 2010, 17, 471–482. 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrimp J. H.; Sorum A. W.; Garlick J. M.; Guasch L.; Nicklaus M. C.; Meier J. L. Characterizing the covalent targets of a small molecule inhibitor of the lysine acetyltransferase P300. ACS Med. Chem. Lett. 2016, 7, 151–155. 10.1021/acsmedchemlett.5b00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bosch T.; Boichenko A.; Leus N. G. J.; Ourailidou M. E.; Wapenaar H.; Rotili D.; Mai A.; Imhof A.; Bischoff R.; Haisma H. J.; Dekker F. J. The histone acetyltransferase p300 inhibi- tor C646 reduces pro-inflammatory gene expression and inhibits his- tone deacetylases. Biochem. Pharmacol. 2016, 102, 130–140. 10.1016/j.bcp.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagg J.; Workman P. Choose and use your chemical probe wisely to explore cancer biology. Cancer Cell 2017, 32, 9–25. 10.1016/j.ccell.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasko L. M.; Jakob C. S.; Edalji R. P.; Qiu W.; Montgomery D.; Digiammarino E. L.; Hansen T. M.; Risi R. M.; Frey R.; Manaves V.; Shaw B.; Algire M.; Hessler P.; Lam L. T.; Uziel T.; Faivre E.; Ferguson D.; Buchanan F. G.; Martin R. L.; Torrent M.; Chiang G. G.; Karukurichi K.; Langston J. W.; Weinert B. T.; Choudhary C.; DeVries P.; Van Drie J. H.; McElligott D.; Kesicki E.; Marmorstein R.; Sun C.; Cole P. A.; Rosenberg S.; Michaelides M. R.; Lai A.; Bromberg K. D. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. 10.1038/nature24028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Wang L.; Zhao K.; Thompson P. R.; Hwang Y.; Mar- morstein R.; Cole P. A. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 2008, 451, 846–850. 10.1038/nature06546. [DOI] [PubMed] [Google Scholar]
- Marques O.; Sanejouand Y. H. Hinge-bending motion in citrate synthase arising from normal mode calculations. Proteins: Struct., Funct., Bioinf 1995, 23, 557–560. 10.1002/prot.340230410. [DOI] [PubMed] [Google Scholar]
- Wiegand G.; Remington S. J. Citrate Synthase: structure, control, and mechanism. Annu. Rev. Biophys. Biophys. Chem. 1986, 15, 97–117. 10.1146/annurev.bb.15.060186.000525. [DOI] [PubMed] [Google Scholar]
- Hickman A. B.; Namboodiri M. A.; Klein D. C.; Dyda F. The structural basis of ordered substrate binding by serotonin N- acetyltransferase enzyme complex at 1.8 Å resolution with a bisub- strate analog. Cell 1999, 97, 361–369. 10.1016/S0092-8674(00)80745-X. [DOI] [PubMed] [Google Scholar]
- LE = −1.4 log Ki/# heavy atoms; LipE = −log Ki – clogP.
- Zhang Y.; Yao L.; Lin J.; Gao H.; Wilson T. C.; Giragossian C. Lack of appreciable species differences in nonspecific microsomal binding. J. Pharm. Sci. 2010, 99, 3620–3627. 10.1002/jps.22124. [DOI] [PubMed] [Google Scholar]
- Liu X.; Wright M.; Hop C. E. Rational use of plasma binding and tissue binding data in drug design. J. Med. Chem. 2014, 57, 8238–48. 10.1021/jm5007935. [DOI] [PubMed] [Google Scholar]
- Fa × Fg = fraction absorbed × fraction escaping gut metabolism = F/(1 – CLp/liver blood flow).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.