

Abstract
The extracellular pH (pHe) is a key determinant of the cellular (micro)environment and needs to be maintained within strict boundaries to allow normal cell function. Here we used HEK293 cells to study the effects of pHe acidification (24 h), induced by mitochondrial inhibitors (rotenone, antimycin A) and/or extracellular HCl addition. Lowering pHe from 7.2 to 5.8 reduced cell viability by 70% and was paralleled by a decrease in cytosolic pH (pHc), hyperpolarization of the mitochondrial membrane potential (Δψ), increased levels of hydroethidine-oxidizing ROS and stimulation of protein carbonylation. Co-treatment with the antioxidant α-tocopherol, the mitochondrial permeability transition pore (mPTP) desensitizer cyclosporin A and Necrostatin-1, a combined inhibitor of Receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and Indoleamine 2,3-dioxygenase (IDO), prevented acidification-induced cell death. In contrast, the caspase inhibitor zVAD.fmk and the ferroptosis inhibitor Ferrostatin-1 were ineffective. We conclude that extracellular acidification induces necroptotic cell death in HEK293 cells and that the latter involves intracellular acidification, mitochondrial functional impairment, increased ROS levels, mPTP opening and protein carbonylation. These findings suggest that acidosis of the extracellular environment (as observed in mitochondrial disorders, ischemia, acute inflammation and cancer) can induce cell death via a ROS- and mPTP opening-mediated pathogenic mechanism.
Abbreviations: Δψ, mitochondrial membrane potential; AA, antimycin A; CI, complex I or NADH:Ubiquinone oxidoreductase; CIII, complex III or Ubiquinol-cytochrome c oxidoreductase; CsA, cyclosporin A; ETC, electron transport chain; Fer-1, ferrostatin-1; HEt, hydroethidine; mPTP, mitochondrial permeability transition pore; Nec-1, necrostatin-1; OXPHOS, oxidative phosphorylation; pHc, cytosolic pH; pHe, extracellular pH; ROS, reactive oxygen species; ROT, rotenone; TCA, tricarboxylic acid; TMRM, tetramethyl rhodamine methyl ester; TOC, α-tocopherol
Keywords: Acidosis, Mitochondria, Membrane potential, Permeability transition pore
Graphical abstract
Highlights
-
•
Extracellular acidification induces mitochondrial dysfunction.
-
•
Extracellular acidification increases intracellular ROS levels.
-
•
Extracellular acidification stimulates protein carbonylation.
-
•
Extracellular acidification induces mPTP opening- and ROS-dependent cell death.
-
•
Acidosis-induced oxidative stress likely contributes to various pathologies.
1. Introduction
Changes in the cellular (micro)environment profoundly affect cell physiology and are associated with induction of pathology [64], [9]. A key property of the extracellular environment is its pH (pHe), which has to be maintained within strict boundaries to allow proper cellular function and prevent cell death [43], [51]. Alterations in cellular energy metabolism often induce extracellular acidification, the rate and mechanism of which depend on the cell type and used energy substrate [37]. In most mammalian cells, cellular energy in the form of ATP is generated by the integrated action of the glycolysis pathway in the cytosol, and the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) system in the mitochondrion [30], [31], [62]. These systems not only produce ATP by catabolizing energy substrates (e.g. glucose, fatty acids and glutamine), but also generate protons (H+) and lactate during pyruvate metabolism. Moreover, CO2 is produced inside the mitochondrion during the conversion of pyruvate into acetyl Coenzyme A (acetyl CoA) and by the TCA cycle. Once formed, the CO2 enters the extracellular environment via the cytosol, where its reaction with water (H2O) generates carbonic acid (H2CO3), which then dissociates into hydrogen carbonate (HCO3-) and H+ [37].
Extracellular acidification was demonstrated in various cell models of inherited and inhibitor-induced OXPHOS dysfunction [16], [47], [50], [60]. In this respect, using C2C12 myoblasts, we recently demonstrated that acute OXPHOS inhibition stimulates steady-state cellular glucose uptake, which compensates for the reduction in mitochondrial ATP production [36]. The latter study further revealed that increased glucose uptake was associated with increased cellular lactate release and extracellular acidification due to a higher glycolytic flux. Similarly, acidification of the extracellular environment (pHe 6.2–6.8) is also a characteristic feature of cancer cells [44], [59], linked to their predominantly glycolytic mode of ATP generation [23], [62]. Other pathologies associated with extracellular acidification are severe ischemia (pHe < 6.3; [54]), heart arrhythmia [7] and inflammation (pHe 5.4; [56]). Interestingly, mitochondrial dysfunction and extracellular acidification have been associated with an increase in the cellular level of reactive oxygen species (ROS; [45], [18], [26], [70], [5]). ROS can serve as signalling molecules (for instance in the activation of antioxidant defence systems), but when their level exceeds a certain threshold value, oxidative stress is induced [1], [52], [65], [66]. Cancer cells generally display a reduced pHe and increased ROS levels that are likely involved in maintaining the cancer phenotype and providing these cells with a survival advantage relative to non-cancer cells [42], [8]. For instance, in breast cancer cells extracellular acidosis stimulates the pentose phosphate pathway to increase NADPH production and enhance the cell's resistance to oxidative stress [35]. ROS can induce various modifications in proteins including metal-catalysed carbonylation, oxidation of aromatic and sulphur-containing amino acid residues, oxidation of the protein backbone, or even protein fragmentation due to backbone breakage [11], [41], [55]. Protein carbonylation appears to be irreversible and has been observed under conditions of increased ROS production and/or inefficient antioxidant systems, associated with a reduced removal capacity for oxidized proteins [11], [22], [67]. Potentially due to their protein-modifying ability, ROS can also trigger opening of the mitochondrial permeability transition pore (mPTP; [6]), which is associated with induction of various modes of cell death [1], [49]. We previously used HEK293 cells [19] to demonstrate that chronic (24 h) inhibition of OXPHOS complex I (CI) and complex III (CIII) by 100 nM rotenone (ROT) or 100 nM antimycin A (AA), respectively, stimulates oxidation of the ROS sensor hydroethidine (HEt). Using the genetic pH-sensor SypHer, we observed that this increased HEt oxidation was paralleled by a lowering of the cytosolic and intra-mitochondrial pH and a minor reduction in cell viability [21]. The latter study also revealed that CI and CIII inhibition were not accompanied by a detectable increase in protein carbonylation or changes in mitochondrial and cytosolic superoxide dismutase (SOD) protein levels. However, evidence in the literature suggests that acidic stress is linked to cell death induction (e.g. [51]) and pHe acidification triggers intracellular damage and reduces the replicative lifespan of chronologically aging budding yeast [38]. Triggered by the above findings, we here investigated whether HEK293 cell viability is affected by extracellular acidification. Our results support a mechanism in which pHe acidification induces necroptotic cell death by decreasing intracellular pH (pHc), increasing cellular ROS levels and stimulating mitochondrial permeability transition pore (mPTP) opening.
2. Materials and methods
2.1. Cell culture and treatment regime
HEK293 cells were cultured in DMEM medium (Invitrogen, Breda, The Netherlands) containing 25 mM glucose, 2 mM L-glutamine, no pyruvate, 10% (v/v) fetal calf serum, phenol red and 1% penicillin/streptomycin in a humidified atmosphere (5% CO2, 37 °C). HEK293 cells were seeded (25,000 cells/cm2) and allowed to proliferate for 24 h during which they reached 40–60% confluence. Next, cells were incubated for another 24 h in the presence of vehicle (0.1% ethanol; CT), the complex I (CI) inhibitor rotenone (ROT; 100 nM) or the complex III (CIII) inhibitor antimycin A (AA; 100 nM) in “normal” DMEM or “acidified” DMEM culture medium (“+H+”; by adding 25 µmol HCl/ml at the start of the 24 h incubation).
2.2. Extracellular pH (pHe) measurements
The culture medium was centrifuged to remove cells (1000 g, 5 min,) and the extracellular pH (pHe) was measured in the clear supernatant using a Sension-PH31 pH meter (Sanhai, Shila Instrument, China).
2.3. Cytosolic pH (pHc) measurements
Cells were cultured in 48-well plates (25,000 cells/cm2) (VWR, Vila Nova de Gaia, Portugal) as described above and subjected to the various treatments. Thereafter, cells were loaded with the pH-sensitive reporter molecule BCECF-AM (2′,7′-Bis-(2-Carboxyethyl)-5-(and-6)-Carboxyfluorescein acetoxymethyl ester) by incubating them in HBSS medium (containing 5 μM BCECF-AM, 137 mM NaCl, 5.4 mM KCl, 4.2 mM NaHCO3, 0.3 mM Na2HPO4, 0.4 mM KH2PO4, 1.3 mM CaCl2, 0.5 mM MgCl2, 0.6 mM MgSO4, and 5.6 mM D-glucose, pH 7.4) for 15 min at 37 °C and 5% CO2 in the dark. Then, cells were washed 3 times with HBSS without BCECF and fluorescence signals were quantified using a microplate reader (Cytation 3; BioTek US, Winooski, VT, USA). The protonated and deprotonated forms of BCECF were excited at 440 nm and 490 nm, respectively, and BCECF fluorescence was quantified at 530 nm. The ratio between the emission signal obtained following 490 and 440 excitation was used as a measure of cytosolic pH (pHc).
2.4. Imaging of mitochondrial TMRM fluorescence
Cells were seeded in 6-well plates (VWR) at a density of 237,000 cells/well and subjected to the various treatments. Thirty minutes prior the end of the incubation time, HEK293 cells were incubated in HBSS containing 30 nM tetramethyl rhodamine methyl ester (TMRM; Invitrogen) for 25 min at 37 °C and 5% CO2 in the dark. Next, cells were washed with HBSS and images were acquired using a Nikon Eclipse TE2000U microscope (Nikon Instruments, Amsterdam, The Netherlands) equipped with a x40 Plan Fluor 0.6 NA objective (Nikon). TMRM was excited during 600 ms at 535 nm using a monochromator (pE-2; CoolLED Ltd., Andover, UK). The excitation light intensity was minimized using two neutral density filters (ND8, ND4). TMRM fluorescence emission was directed onto a DS-Qi1Mc monochrome camera (Nikon), using a TRITC HYQ fluorescence filter box (excitation filter: 530–550 nm; dichroic mirror: 565 nm; emission filter: 590–650 nm; Nikon). For each individual cell, the mitochondrial TMRM fluorescence signal was quantified by placing a region of interest (ROI) on a mitochondria-dense part as described previously [21]. For each well, ten random fields of view were routinely analysed.
2.5. Imaging of live-cell hydroethidine (HEt) oxidation
Cells were seeded (100,000 cells per coverslip) on 24-mm coverslips (Thermo Scientific, Etten-Leur, The Netherlands), placed in 35-mm CellStar tissue culture dishes (Sigma-Aldrich, Zwijndrecht, The Netherlands), two days prior to experiments to achieve a 70% confluence at the day of imaging. Cells were incubated with 10 µM HEt (Invitrogen) in harvested culture medium for 10 min at 37 °C and 5% CO2 in the dark. The HEt oxidation reaction was terminated by thoroughly washing the cells with PBS to remove excess HEt. Subsequently, the cells were covered by a colourless HEPES-Tris (HT) buffer (containing 132 mM NaCl, 10 mM HEPES, 4.2 mM KCl, 1 mM MgCl2, 1 mM CaCl2 and 25 mM D-glucose, adjusted to pH 7.4 with Tris salt). To quantify fluorescent HEt oxidation products, coverslips were mounted in an incubation chamber that was placed on the temperature-controlled (37 °C) stage of an inverted microscope (Axiovert 200 M, Carl Zeiss, Jena, Germany) equipped with a Zeiss x40 1.3 NA Fluar objective. The cells were excited for 100 ms at 490 nm using a monochromator (Polychrome IV; TILL Photonics, Gräfelfing, Germany). Fluorescence light from the cells was directed onto a CoolSNAP HQ monochrome CCD-camera (Roper Scientific, Vianen, The Netherlands) using a 525DRLP dichroic mirror (Omega Optical Inc., Brattleboro, VT, USA) and a 565ALP emission filter (Omega). The hardware was controlled using Metafluor 6.0 software (Universal Imaging Corporation, Downingtown, PA, USA). For each individual cell HEt signals were quantified by drawing a region of interest (ROI) around the entire nucleus and by an ROI placed on a mitochondria-dense part as described previously [19], [20]. For each coverslip, ten random fields of view were routinely analysed.
2.6. Oxyblot analysis
HEK293 cells were seeded (25,000 cells/cm2) and allowed to proliferate for 24 h after which they had reached 40–60% confluence. Then, cells were treated for 24 h with vehicle (CT), ROT, AA or 3 mM hydrogen peroxide (H2O2) in “normal” DMEM or “acidified” DMEM culture medium. Next, cells were harvested by centrifugation (1000 g, 5 min) and the cell pellet was washed once with 1 ml PBS (containing 1 mM DTT) and re-suspended in 10 mM Tris-EDTA buffer containing 10 µg/ml DNase. After three freeze/thaw cycles in liquid nitrogen the suspension was stored at −20 °C. Protein carbonyl levels were determined by immunoblotting using the Oxyblot assay kit (Merck-Millipore, Temecula, CA, USA) with slight modifications. In brief, 10 µl samples (containing ~50 µg protein) were denatured with 10 µl 12% SDS and then derivatized with 20 µl 2,4-dinitrophenylhydrazine (DNPH) to the corresponding 2,4-dinitrophenylhydrazone (DNP). After 15 min at room temperature the reaction was stopped with 15 µl Neutralization buffer and the samples were 2-fold diluted with 2× SDS-sample buffer, further neutralized with TRIS, and 20–40 µl was run on a 10% SDS-PAGE gel. Next, protein was transferred electrophoretically to a PVDF membrane using the iBlot system (Life Technologies, Carlsbad, CA, USA). After blotting, membranes were blocked with Odyssey Blocking Buffer (Li-Cor Biosciences, Lincoln, NE, USA) mixed 1:1 with PBS-Tween20 (0.1%, w/v) for 1 h at room temperature. Next, blots were incubated overnight at room temperature with the polyclonal antibody directed against DNP (Merck-Millipore) and the monoclonal antibody against β-actin (Sigma-Aldrich) in the Odyssey-PBS-Tween buffer. Subsequently, blots were washed (3–5 times) with PBS-Tween and incubated with the secondary goat antibodies against rabbit and mouse IRdye 680 and IRdye 800 (Li-Cor) in the above-mentioned buffer for 45 min at room temperature. After washing with PBS-Tween, PBS and water, the PVDF membranes were air dried in the dark. Blots were scanned using an Odyssey Imaging system (Li-Cor).
2.7. Crystal violet assay
Cells were cultured in 48-well plates (25,000 cells/cm2) (Corning B.V., Amsterdam, The Netherlands) as described above and subjected to the various treatments. Following treatment the cells were washed with PBS and incubated for 10 min at RT in a staining solution consisting of 0.5% (v/v) crystal violet solution, 30% (v/v) ethanol and 3% (v/v) formaldehyde. After staining, plates were rinsed with tap water followed by addition of a 1% (w/v) SDS solution for 30 min on an orbital shaker. The amount of re-solubilized crystal violet was used as a readout of relative cell number [53] by absorbance measurements (550 nm) using a microplate spectrophotometer (Infinite M100; Tecan, Crailsheim, Germany).
2.8. Chemicals
(±)-α-tocopherol (#T3251; TOC), antimycin A (#A8674; AA), Crystal Violet solution (#HT90132), Cyclosporin A (#30024; CsA), Ferrostatin-1 (#SML0583; Fer-1) and rotenone (#R8875; ROT) were obtained from Sigma-Aldrich. N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (#ALX260020; zVAD.fmk) was obtained from Enzo Life Sciences (Raamsdonkveer, The Netherlands) and Necrostatin-1 (#SC200142; Nec-1) from Santa Cruz Biotech (Dallas, TX, USA).
2.9. Data analysis
Microscopy images and Western blots were processed and quantified using MetaMorph 6.1 (Universal Imaging Corporation), Image Pro Plus 6.3 (Media Cybernetics, Silver Spring, MD, USA) and ImageJ software (version 1.49 v; https://imagej.nih.gov/ij/). Graphing, curve fitting and statistical analysis were performed using Origin Pro 6.1 software (OriginLab Corp., Northampton, MA, USA). Values from multiple experiments are expressed as mean±SEM unless stated otherwise. Statistical significance was assessed using an unpaired Student's t-test (Bonferroni-corrected).
3. Results
3.1. Extracellular acidification decreases cytosolic pH and cell viability
To induce various degrees of extracellular acidification, HEK293 cells were treated for 24 h with vehicle (CT), rotenone (ROT; 100 nM) or antimycin A (AA; 100 nM) in the absence and presence of 25 µmol added HCl/ml (H+). Regarding the ROT- and AA-treatment conditions, these were identical to those used in our previous studies [19], [21]. In this sense, the used ROT and AA concentrations were the minimal ones that maximally inhibited mitochondrial oxygen consumption [21]. The above treatment regimens yielded a variable drop in extracellular pH (pHe) ranging between 7.2 (CT) and 5.8 (H++AA; Fig. 1A and Table 1). In particular, extracellular H+ addition decreased pHe (Fig. 1A; black bars), and the latter was further decreased by the presence of ROT and AA (Fig. 1A; white bars). Next, the dual-excitation ratiometric pH indicator BCECF [3] was used to analyse whether the drop in pHe affected cytosolic pH (pHc). Control experiments (Fig. 1B) revealed that BCECF correctly reported pHc changes upon extracellular addition and removal of NH4+, associated with alkalinisation and acidification respectively [36]. Under steady-state conditions, BCECF fluorescence analysis revealed that the drop in pHe (Fig. 1A) was paralleled by a drop in pHc (Fig. 1C). As presented previously (data taken from [19]), the viability of ROT- or AA-treated HEK293 cells was reduced by only 10–19% (Fig. 1D; three columns on the left). In contrast, extracellular acidification by itself reduced cell viability by 46% (Fig. 1D; black columns), a value that dropped further upon co-incubation with ROT (by 60%) or AA (by 71%). Taken together, these results demonstrate that the extent of pHe acidification is linked to the degree of reduction in cell viability.
Fig. 1.

Effect of mitochondrial inhibition and extracellular H+addition on cytosolic pH (pHc) and cell viability. (A) Extracellular pH (pHe) of HEK293 cells after 24 h treatment with vehicle (CT), rotenone (ROT; 100 nM) or antimycin A (AA; 100 nM) in the absence and presence of 25 µmol added HCl/ml (H+). The data presented is the average of two independent experiments (N=2). (B) Dynamic behaviour of the fluorescence ratio signal of the cytosolic pH sensor BCECF in HEK293 cells. Subsequent extracellular addition and removal of NH4+ induced cytosolic alkalinisation and acidification, respectively. (C) BCECF fluorescence ratio signal for HEK293 cells cultured under the conditions in panel A. (D) Cell viability of HEK293 cells cultured in the conditions in panel A. Statistics: Data was expressed as percentage of average control value measured on the same day (panel C) or as control value measured on the same day (panel D). Numerical data is provided in Table 1. Statistically significant differences with the indicated columns are marked by * (P < 0.05), ** (P < 0.01) and *** (P < 0.001). n.s. indicates “not significant”.
Table 1.
Measured cell parameters.
| +H+ | +H+ | +H+ | ||||
|---|---|---|---|---|---|---|
| PARAMETER | CT | CT+ROT | CT+AA | CT | CT+ROT | CT+AA |
| pHe | 7.20 | 6.84 | 6.90 | 6.70 | 6.00 | 5.76 |
| (N = 2) | (N = 2) | (N = 2) | (N = 2) | (N = 2) | (N = 2) | |
| apHc | 100 ± 2.98 | 69.0 ± 5.21 | 62.7 ± 6.17 | 48.1 ± 1.20 | 50.3 ± 1.83 | 50.3 ± 1.21 |
| (N = 1, n = 6) | (N = 1, n = 5) | (N = 1, n = 4) | (N = 1, n = 6) | (N = 1, n = 6) | (N = 1, n = 6) | |
| bTMRMmito | 100 ± 1.96 | 101 ± 2.02 | 91.8 ± 1.61 | 111 ± 1.93 | 94.5 ± 1.78 | 97.1 ± 1.70 |
| (N = 2, n = 196) | (N = 2, n = 183) | (N = 2, n = 227) | (N = 2, n = 221) | (N = 2, n = 210) | (N = 2, n = 228) | |
| bHEtmito | 100 ± 4.13 | 181 ± 5.37 | 137 ± 4.36 | 164 ± 5.99 | 274 ± 14.5 | 227 ± 9.45 |
| (N = 3, n = 238) | (N = 3, n = 118) | (N = 101, n = 3) | (N = 3, n = 120) | (N = 3, n = 60) | (N = 93, n = 3) | |
| bHEtnuc | 100 ± 2.21 | 273 ± 7.99 | 228 ± 6.74 | 243 ± 10.9 | 512 ± 30.0 | 405 ± 16.4 |
| (N = 3, n = 238) | (N = 3, n = 118) | (N = 101, n = 3) | (N = 3, n = 120) | (N = 3, n = 60) | (N = 93, n = 3) | |
| cOxyblot signal | 50.5 | 52.6 | 48.9 | 60.8 | 68.0 | 69.2 |
| (N = 1) | (N = 1) | (N = 1) | (N = 1) | (N = 1) | (N = 1) | |
| Cell Viability | 100 | 95 ± 7.6 | 82 ± 6.3 | 54 ± 3.6 | 40 ± 6.5 | 29 ± 4.1 |
| (N = 3, n = 12) | (N = 2, n = 8) | (N = 2, n = 8) | (N = 3, n = 8) | (N = 2, n = 8) | (N = 2, n = 8) |
HEK293 cells were cultured for 24 h in the presence of vehicle (CT), the complex I inhibitor ROT (100 nM) or the complex III inhibitor AA (100 nM) in “normal” or “acidified” culture medium (“+H+”).
Statistics: Statistically significant differences between the conditions are indicated in the figures. N, number of independent experiments, n, number of individual cells or assays.
BCECF ratio signal expressed as percentage of average CT value.
Expressed as percentage of average CT value.
Expressed as percentage of the H2O2-induced (maximal) value and normalized to β-actin.
3.2. Mitochondrial inhibition and extracellular acidification alter mitochondrial membrane potential
To determine whether pHe/pHc acidification affected mitochondrial function, we estimated mitochondrial membrane potential (Δψ) by quantifying the mitochondrial fluorescence intensity of tetramethylrhodamine methyl ester (TMRM; [32]). The average TMRM fluorescence signal was quantified at the subcellular single cell level using a mitochondria-dense (“m”) region of interest (ROI; Fig. 2A). These signals were background-corrected using an extracellular ROI (“b”). We observed a small increase (ROT) and significant drop (AA) in mitochondrial TMRM fluorescence (Fig. 2B; three columns on the left). This suggests that ROT and AA induce Δψ hyperpolarization and depolarization, respectively, which confirms our previous results [21]. By itself, extracellular H+ addition induced a higher mitochondrial TMRM fluorescence, suggesting that Δψ becomes hyperpolarized (Fig. 2B; black columns). This phenomenon was prevented by ROT and AA treatment (Fig. 2B; two columns on the right), suggesting that pHe acidification induces Δψ hyperpolarization by a mechanism that requires active CI and/or CIII.
Fig. 2.

Effect of mitochondrial inhibition and extracellular H+addition on mitochondrial TMRM fluorescence. (A) Typical fluorescence images of HEK293 cells stained with TMRM after 24 h treatment with vehicle (CT), rotenone (ROT; 100 nM) and antimycin A (AA; 100 nM) in the absence (upper panels) and presence (lower panels) of 25 µmol added HCl/ml (H+). Circles (top left panel) indicate a typical mitochondria-dense (“m”) region of interest (ROI) used for quantification of fluorescence intensity. A typical ROI marked “b” was used for background correction. (B) Average intensity of the mitochondrial TMRM signal for the conditions in panel A. Data was expressed as percentage of the average value measured in CT cells on the same day. Statistics: The images in panel A were contrast-optimized using identical settings for visualization purposes. Fluorescence intensity was quantified using the original images. Numerical data is provided in Table 1. Statistically significant differences with the indicated columns (panel B) are marked by * (P < 0.05), ** (P < 0.01) and *** (P < 0.001).
3.3. Mitochondrial inhibition and extracellular acidification increase the levels of hydroethidine-oxidizing ROS
OXPHOS dysfunction and changes in Δψ are often paralleled by alterations in mitochondrial ROS production and increased cellular ROS levels (e.g. [33], [65]). To assess the potential role of ROS in pHe acidification-induced cell death, cells were incubated with the ROS-sensor hydroethidine (HEt). Fluorescent HEt-oxidation products are positively charged [33], [46]. Therefore they accumulate in the mitochondrial matrix (due to the inside-negative Δψ) and bind to negatively-charged DNA in nucleoli, which enhances their fluorescence signal (e.g. [4], [20]). As a consequence, the distribution of HEt-oxidation products between the mitochondrial and nuclear compartment is Δψ-dependent. This means that a (sufficiently large) Δψ depolarization will lead to translocation of HEt oxidation products from the mitochondrial to the nuclear compartment, as induced by the mitochondrial uncoupler carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) in HEK293 cells [20]. Importantly, our TMRM results strongly suggest that Δψ is differentially affected by the various treatments (Fig. 2B). This heterogeneity could also differentially affect the subcellular distribution of the HEt-oxidation products. To minimize the effect of this potential artefact we quantified the fluorescence signal of HEt-oxidation products for each individual cell using a mitochondria-dense (“m”) and nuclear (“n”) region of interest (ROI; Fig. 3A). These signals were background-corrected using an extracellular ROI (“b”). Confirming our previous results in HEK293 cells [19], ROT and AA treatment stimulated HEt oxidation (Fig. 3B). By itself, extracellular H+ addition also stimulated HEt oxidation (Fig. 3B; black columns), which was further increased by ROT and AA (Fig. 3B). Of note, in vitro experiments revealed that ethidium cation (Et+) fluorescence decreased at lower pH (Supplementary Fig. S1). This rules out that the increased fluorescence signals in our HEt experiments are due to the observed decrease in pHc (Fig. 1C). Linear regression analysis revealed that the average mitochondrial and nuclear fluorescence signals increased proportionally for the various conditions, as demonstrated by a highly linear correlation (Fig. 3C). This strongly suggests that the HEt-oxidation products detected in the nucleoplasm are of mitochondrial origin [33]. The fact that the mitochondrial and nuclear fluorescence signals are on the same straight line, demonstrates that the level of HEt-oxidizing ROS increases in the order: CT < AA < CT + H+ < ROT < AA + H+ < ROT + H+. Taken together, these results demonstrate that extracellular acidification by itself suffices to increase the levels of HEt-oxidizing ROS and that these levels are further increased by ROT and AA treatment.
Fig. 3.

Effect of mitochondrial inhibition and extracellular H+addition on hydroethidine oxidation. (A) Typical fluorescence images of HEK293 cells stained with hydroethidine (HEt) after treatment for 24 h with vehicle (CT), rotenone (ROT; 100 nM) and antimycin A (AA; 100 nM) in the absence (upper panels) and presence (lower panels) of 25 µmol added HCl/ml (H+). Circles (top left panel) indicate a typical mitochondria-dense (“m”) and nuclear (“n”) region of interest (ROI) used for quantification of fluorescence intensity. A typical ROI marked “b” was used for background correction. (B) Average fluorescence intensity of HEt oxidation products in mitochondrial (“Mito”) and nuclear (“Nuc”) ROIs for the conditions in panel A. Data was expressed as percentage of the average value in the mitochondrial and nuclear ROIs measured on the same day in CT cells. (C) Average fluorescence intensity of HEt oxidation products in the mitochondrial vs. nuclear ROIs for the conditions in panel A and B (data taken from panel B). A linear fit of the data (thick line) and its 95% confidence limits (thin lines) are indicated. Individual data points were weighed according to their SEM value. Statistics: The images in panel A were contrast-optimized using identical settings for visualization purposes. Fluorescence intensity was quantified using the original images. Numerical data is provided in Table 1. Statistically significant differences with the indicated columns (panel B) are marked by ** (P < 0.01) and *** (P < 0.001).
3.4. Extracellular acidification increases protein carbonylation
Increased intracellular ROS levels have been linked to non-enzymatic post translational modifications (PTMs) of proteins including carbonylation [67]. The latter occurs when specific amino acids are oxidized, leading to formation of stable carbonyl (-CO) groups (i.e. aldehydes and ketones; [12], [13]). When carbonylated, proteins generally display a loss of function and/or are degraded by the proteasome pathway [67]. To determine whether pHe/pHc acidification and increased ROS levels were associated with increased protein carbonylation, we performed Oxyblot analysis. In this approach, protein carbonyl groups react with 2,4-dinitrophenylhydrazine (DNPH) to form 2,4-dinitrophenylhydrazone (DNP), which can be quantified using a DNP-specific antibody and Western blotting [63]. As argued previously [57], proper analysis of Oxyblot signals requires inclusion of a positive control to exogenously stimulate protein carbonylation (i.e. 3 mM H2O2). To further facilitate Oxyblot quantification and interpretation equal amounts of protein were loaded in each lane (i.e. exactly 8 µg). Next, the integrated optical density (IOD) of the Oxyblot signal was determined for each condition (lane) and normalized on the β-actin signal. Then, Oxyblot signals were expressed as percentage of the maximal signal obtained for the H2O2-treated condition. Addition of H2O2 did not induce pHe acidification (i.e. in this case pHe equalled 7.5). ROT and AA treatment did not detectably increase the Oxyblot signal (Fig. 4A and Table 1), as previously observed in HEK293 cells [19]. In contrast, extracellular H+ addition increased the Oxyblot signal from 51% to 61% of the H2O2-induced condition (Fig. 4B and Table 1). In the presence of extracellular H+, AA and ROT treatment further increased the Oxyblot signal to 68% and 69%, respectively (Fig. 4B and Table 1). These findings demonstrate that extracellular acidification stimulates cellular protein carbonylation.
Fig. 4.

Effect of mitochondrial inhibition and extracellular H+addition on cellular protein carbonylation (A) Typical Oxyblot signals (“DNP-proteins”) of whole HEK293 cell homogenates. The latter were obtained for cells treated for 24 h with vehicle (CT), rotenone (ROT; 100 nM), antimycin A (AA; 100 nM) or hydrogen peroxide (H2O2; 3 mM). β-actin was used as a loading control. (B) Same as panel A but now in the presence of 25 µmol added HCl/ml (H+). The images in panel A and B were linearly contrast optimized using identical settings for visualization purposes. Whole-lane signals were quantified using the original images, expressed as % of the H2O2-induced signal and normalized to β-Actin levels. Numerical data is provided in Table 1.
3.5. Linear regression analysis of the measured cell parameters
To highlight potential patterns in our cell data (Table 1) we performed an explorative linear regression analysis. To this end we generated a scatter matrix and calculated two established estimators of linear correlation for each pair-wise comparison of the parameters (Fig. 5): the adjusted R2-value (“R^2”) and Pearson's r-value (“r”). R^2 is a modified version of R2, adjusted for the number of predictors in the fitted line, and quantifies how well the linear model fits the data. Pearson's r measures the strength of linear relationship between paired data. This r-value is always between −1 and 1 and the closer its value is to these extremes, the stronger linear correlation is. To facilitate visual interpretation, the degree of correlation between parameters (Fig. 5) was categorized as positive (green ovals), negative (red ovals) and absent (black ovals). A positive correlation was found between pHe and pHc, between pHc and cell viability, between pHe and cell viability and between the mitochondrial (HEtmito) and nuclear (HEtnuc) fluorescence signal of HEt oxidation products. This suggests that a decrease in pHe stimulates cell death via decreasing pHc and increasing the levels of HEt-oxidizing ROS. Similarly, the Oxyblot signal positively correlated with the levels of HEt-oxidizing ROS, suggesting that these ROS play a role in stimulating protein carbonylation. A negative correlation was observed between pHe and HEt oxidation, between pHc and HEt oxidation, between viability and HEt oxidation, and between the Oxyblot signal and cell viability. No apparent correlation was observed between the mitochondrial TMRM signal (Δψ) and the other parameters, meaning that the former is not predictive for the latter.
Fig. 5.

Correlation analysis of measured cell parameters. HEK293 cells were treated for 24 h with vehicle (CT), rotenone (ROT; 100 nM) and antimycin A (AA; 100 nM) in the absence and presence of 25 µmol added HCl/ml (H+). This figure depicts the linear correlation between the various parameters analysed in this study being pH (pHe, pHc), mitochondrial TMRM fluorescence (TMRMm), HEt fluorescence in the mitochondrial and nuclear compartment (HEtmito, HEtnuc), protein carbonylation (Oxyblot) and cell viability. The types of linear correlation are indicated by ellipses in green (positive correlation), red (negative correlation) and black (no correlation). Statistics: Ellipses indicate the 95% confidence limits. The degree of linear correlation was determined by calculating the adjusted R2-value (“R^2) and Pearson's r-value (“r”). All numerical data was taken from Table 1.
3.6. Extracellular acidification induces cell death in HEK293 cells via ROS and mPTP opening
Regression analysis suggested a central role of ROS in the acidification-induced cell death mechanism. Supporting this hypothesis, cell death induced by extracellular H+ addition was fully prevented by the antioxidant α-tocopherol (TOC; Fig. 6). We recently demonstrated that CI inhibition triggers cell death in melanoma cancer cell lines by a mechanism involving ROS and opening of the mPTP [1]. Here we observed that 2 h pre-treatment with 0.5 µM cyclosporin A (CsA), which reduces the mPTP open probability [2], inhibited acidification-induced cell death (Fig. 6). Finally, we investigated the mode of cell death triggered by pHe acidification. Application of a relatively high concentration of the broad-spectrum caspase inhibitor (zVAD.fmk) was without effect, arguing against involvement of apoptosis [1]. The inhibitor Necrostatin-1 (Nec-1), a dual inhibitor of receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and indoleamine 2,3-dioxygenase (IDO; [61], [10], [15]), fully prevented acidification-induced cell death, whereas the ferroptosis inhibitor Ferrostatin-1 (Fer-1; [17], [29]) was ineffective. Taken together, these results suggest a mechanism in which extracellular acidification increases ROS levels and stimulates mPTP opening to induce necroptotic cell death.
Fig. 6.

Mode-of-action by which extracellular H+addition reduces cell viability. The viability of HEK293 cells was determined after 24 h treatment in the absence (-H+) or presence (+H+) of 25 µmol added HCl/ml (CT condition). H+-induced cell death was prevented by the antioxidant α-tocopherol (TOC), the mitochondrial permeability transition pore (mPTP) desensitizer cyclosporin A (CsA) and the RIPK1/IDO inhibitor Necrostatin-1 (Nec-1). The caspase inhibitor zVAD.fmk and the ferroptosis inhibitor Ferrostatin-1 (Fer-1) were ineffective. Statistics: Data was expressed as percentage of average control value (CT) measured on the same day. Statistically significant differences with the indicated columns are marked by ** (P < 0.01) and *** (P < 0.001).
4. Discussion
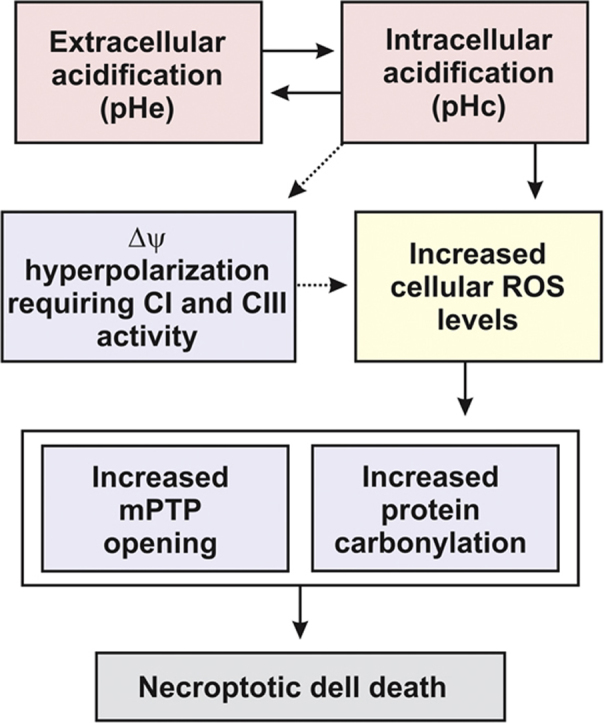
Using exogenous H+ addition in the absence or presence of mitochondrial inhibitors, we here provide evidence that a drop in extracellular pH (pHe) decreases cytosolic pH (pHc), leading to hyperpolarization of the mitochondrial membrane potential (Δψ), increased levels of hydroethidine-oxidizing ROS, stimulation of protein carbonylation and triggering of cell death (Fig. 7). Mechanistically, our results suggest that the latter is ROS-dependent, requires mPTP opening and involves necroptosis.
Fig. 7.

Proposed mechanism linking extracellular acidification to induction of intracellular oxidative stress and cell death. Extracellular acidification (a drop in pHe) leads to intracellular acidification (a drop in pHc) and vice versa. The latter induces hyperpolarization of the mitochondrial membrane potential (Δψ) and increases cellular ROS levels. As a consequence, opening of the mitochondrial permeability transition pore (mPTP) and protein carbonylation are triggered, ultimately inducing necroptotic cell death.
4.1. Mitochondrial inhibition by ROT and AA moderately decreases pHe and pHc, increases ROS levels but neither increases protein carbonylation nor greatly reduces HEK293 cell viability
Chronic (24 h) inhibition of mitochondrial CI (using ROT) or CIII (using AA) lowered pHe from 7.2 to 6.8–6.9 (Fig. 1A). This result is compatible with our previous studies revealing that OXPHOS inhibition and inherited CI deficiency stimulate glycolytic ATP production, associated with increased lactate release [16], [21], [36], [60]. In case of ROT treatment, the drop in pHe was paralleled by an increase in the levels of HEt-oxidizing ROS to 181% (mitochondrial signal) and 273% (nuclear signal) of control (Fig. 3B). For AA treatment, HEt-oxidation increased to 137% (mitochondrial signal) and 228% (nuclear signal) of control (Fig. 3B). Under these conditions cell viability was only marginally affected (Fig. 1D), demonstrating that HEK293 cells can tolerate this drop in pHe and increase in HEt oxidizing ROS levels. The latter is compatible with our observation that ROT and AA treatment do not greatly affect the level of cellular and mitochondrial lipid peroxidation [19], do not induce protein carbonylation (Fig. 4A and [19]), and do not trigger upregulation of superoxide dismutases at the protein level [19]. ROT and AA similarly decreased pHc as measured by the cytosolic pH sensor BCEFC (Fig. 1C). This result agrees with our previous study using the protein-based pH-reporting molecule SypHer and confirms that both ROT and AA treatment induce cytosolic acidification [19].
4.2. Exogenous H+ addition greatly decreases pHe and pHc, increases ROS levels, increases protein carbonylation and greatly reduces HEK293 cell viability
Addition of exogenous H+ to the culture medium lowered pHe to a value of 6.7 (Fig. 1A). Similarly low pHe values were also observed in other pathological models (see Introduction), including cancer cells (pHe = 6.2–6.8), severe ischemia (pHe < 6.3) and acute inflammation (pHe 5.4). Exogenous H+ addition reduced pHc to a value below that induced by ROT and AA alone (Fig. 1C), increased the level of HEt-oxidizing ROS (Fig. 3B), stimulated protein carbonylation (Fig. 4B), and greatly reduced cell viability to 54% of control (Fig. 1D). When exogenous H+ addition was combined with ROT or AA, pHe (Fig. 1A) and cell viability (Fig. 1D) were further reduced but pHc was not (Fig. 1C). However, BCECF displays a pKa value of 6.97, making it a less quantitative readout of pHc at low pH values. Combined analysis of mitochondrial and nuclear signals (Fig. 3C) suggested that the levels of HEt-oxidizing ROS increased in the order: CT < AA < CT + H+ < ROT < AA + H+ < ROT + H+. This confirms our previous finding that ROT and AA treatment stimulate the level of HEt-oxidizing ROS in HEK293 cells [21] and demonstrates that these levels are further increased by an additional drop in pHe.
4.3. Role of
In the absence of exogenously added H+, mitochondrial TMRM accumulation was slightly increased and decreased for ROT and AA treatment, respectively (Fig. 2B; left panel). This agrees with our previous study [21], which revealed that the pH gradient (ΔpH) across the mitochondrial inner membrane increased (ROT-treated cells) and decreased (AA-treated cells). By itself, addition of exogenous H+ induced mitochondrial hyperpolarization, reflected by increased mitochondrial TMRM accumulation (Fig. 2B; right panel). The latter was inhibited by ROT and AA, suggesting that CI and CIII activity is required. This result makes it highly unlikely that the observed increased in TMRM accumulation upon exogenous H+ addition is an experimental artefact. In contrast to our results, extracellular acidification (pHe = 6.5) induced a diffuse cytosolic TMRM staining pattern in pulmonary neuroendocrine (PNEC) cells [28]. The latter study also revealed that TMRM accumulated 1.7- and 2.5-fold less in mitochondria at acidic pH (pHe = 6.5), relative to normal (pHe = 7.4) and alkaline pH (pHe = 8.5), respectively. Similarly, evidence in rat brain synaptosomes suggests that a drop in pHe induced Δψ depolarization [40]. We speculate that the degree of pHe acidification, as well as its effect on pHc and other downstream effectors, might depend on the type of the cell, its metabolic state, and/or mode of ATP generation.
4.4. Role of protein carbonylation
Increased protein carbonylation is considered a marker of oxidative stress (e.g. [11], [22]) and was observed in a large variety of human disorders including cardiac tissue following ischemia/reperfusion [48] and in cholangiocarcinoma during chronic inflammation [58]. Interestingly, extracellular acidification also was observed under these pathological conditions [54], [56]. Here, we used exogenous H2O2 as a positive control to induce protein carbonylation (Fig. 4). Inclusion of such a control is essential since cells have the ability to eliminate H2O2 from the medium, depending on the cell number and incubation time [25]. Since exogenous H2O2 application did not lower pHe, it is unlikely that exogenous H2O2 addition stimulates protein carbonylation via inducing pHc acidification. Applying the genetic H2O2-sensor HyPer [19], we previously demonstrated that H2O2 levels were increased upon 24 h treatment with ROT (10% increase in HyPer signal) and AA (50% increase). However, these treatments were unable to stimulate protein carbonylation (Fig. 4A), to trigger cellular/mitochondrial lipid peroxidation or to increase superoxide dismutase levels [19], [21]. The fact that exogenous H2O2 application was able to stimulate protein carbonylation in the absence of exogenously added H+ (Fig. 4A), suggests that the HyPer-detected increase in cytosolic H2O2 levels is below the oxidative stress inducing-level in ROT- and AA-treated cells. In contrast, protein carbonylation was stimulated upon addition of exogenous H+ (Fig. 4B), thereby lowering pHe from 7.2 to 6.7 (Fig. 1A). In the presence of added H+, ROT and AA further lowered pHe to 6.0 (ROT) and 5.8 (AA), respectively, which was paralleled by a further increase in protein carbonylation. These results indicate that full inhibition of CI or CIII, when not accompanied by a sufficiently large pHe acidification, does not induce oxidative stress in HEK293 cells. It was proposed that reversible protein carbonylation potentially plays a role in cellular signalling [67]. However, here we observed that increased carbonylation was not limited to a specific protein (i.e. the overall Oxyblot signal increased), suggesting this carbonylation reflects oxidative damage, rather than a specific redox signalling event.
4.5. Proposed mechanism
Cell viability linearly decreased with decreasing pHe and pHc, increasing HEt oxidation and increasing protein carbonylation, whereas no obvious linear correlation was observed with Δψ (Fig. 5). This is compatible with a mechanism in which pHe acidification triggers pHc acidification and increases cellular ROS levels, followed by induction of oxidative stress (protein carbonylation) and cell death induction. Mechanistically, pHe acidification can increase cellular ROS levels by stimulating ROS generation, inhibiting ROS removal, or a combination of the two (e.g. [65]). Our current and previous results obtained with ROT and AA in the absence of added exogenous H+ indicate that pHc acidification leads to pHe acidification. Conversely, pHe acidification by exogenous H+ addition induces pHc acidification, compatible with a study in rat brain synaptosomes that reported the same phenomenon [40]. In agreement with our findings, the latter study also demonstrated that extracellular acidification (pHe = 6.0 and pHe = 7.0) stimulated intracellular HEt oxidation. In our HEt experiments, the strong linear correlation between the mitochondrial and nuclear fluorescence signals provides evidence that the increased HEt-oxidation occurs in the mitochondrion [33]. This suggests that pHe acidification induces pHc acidification and that the latter affects mitochondrial function, thereby stimulating generation of mitochondrial HEt-oxidizing ROS (Fig. 7). Alternatively, evidence in breast cancer cells suggests that acidosis can inhibit the synthesis of glutathione (GSH), a key cellular antioxidant [35], which might be responsible for or contribute to the observed increase in HEt oxidation. In the presence of the antioxidant α-tocopherol (TOC), exogenous H+ addition was unable to reduce cell viability (Fig. 6), strongly suggesting that increased ROS, lipid peroxidation and/or oxidative stress induction are responsible for cell death induction. Among other factors, increased ROS levels have been identified as driving factors of ferroptosis, a distinct non-apoptotic and iron-dependent form of cell death [17], [29], [68], [69]. It was previously demonstrated that a TOC-variant (Trolox) inhibited ferroptotic cell death by reducing ROS levels whereas CsA, zVAD.fmk and Nec-1 were unable to prevent ferroptosis induction [17]. However, in our experiments the ferroptosis inhibitor Fer-1 did not prevent acidification-induced cell death (Fig. 6), suggesting that the ferroptosis pathway is not involved. Similarly, the (continued) opening the mPTP has been linked to (apoptotic) cell death induction and the pathogenesis of many human diseases [27]. In the presence of CsA, extracellular acidification did not reduce cell viability (Fig. 6), suggesting that mPTP opening is required for cell death induction. In contrast, application of a relatively high concentration of the broad-spectrum caspase inhibitor (zVAD.fmk) was without effect (Fig. 6), arguing against involvement of apoptosis in pHe acidification-induced cell death. Conversely, this death was fully prevented by Nec-1, a combined RIPK1/IDO inhibitor, suggesting a necroptotic mode of cell death.
4.6. Conclusions
Using a HEK293 cell model we here provide evidence that pHe acidification induces pHc acidification followed by ROS- and mPTP opening-dependent necroptosis (Fig. 7). To the best of our knowledge, there are no mechanistic studies linking increased protein carbonylation to necroptosis induction. In this sense, our results may suggest that stimulation of mPTP opening rather than increased protein carbonylation is most relevant for necroptosis induction. Although the exact molecular mechanism by which pHe/pHc acidification increases cellular ROS levels remains to be determined, we propose that this link could represent a way for inside-out signalling between neighbouring cells or tissue-domains. Within tissues, this signalling likely occurs locally thereby mostly affecting cells within the acidified region [34], [39], [44]. Inside-out pH signalling would provide a new conceptual framework that allows a better understanding of organ- or pathway-specific diseases. For instance, cancer cells display an intrinsic resistance against low pHe and elevated ROS levels [14], [24]. This suggests that cancer cells could trigger ROS-induced cell death in neighbouring non-cancer cells by acidifying the extracellular environment, whereas maintaining their own replication and survival potential. Acidosis-induced oxidative stress might also contribute to pathology in patients suffering OXPHOS disorders and other diseases/conditions in which pHe is sufficiently reduced and/or ROS levels are sufficiently increased.
Acknowledgements
This research was supported by a grant from the Netherlands Organization for Scientific Research (NWO, No: 911-02-008, instrumentation) and a grant from the Institute for Genetic and Metabolic Disease (IGMD) of the Radboudumc (to WJHK). Grants to JT (PTDC/DTP-FTO/2433/2014 and NORTE-01-0145-FEDER-000028) are supported by the European Regional Development Fund (ERDF) through the COMPETE 2020 - Operational Programme for Competitiveness and Internationalisation and Portuguese national funds via FCT–Fundação para a Ciência e a Tecnologia. Further support was provided by European Regional Development Fund (ERDF) through the COMPETE 2020 - Operational Programme for Competitiveness and Internationalisation and Portuguese national funds via FCT–Fundação para a Ciência e a Tecnologia (projects: POCI-01-0145-FEDER-007440, PTDC/DTP-FTO/2433/2014 and POCI-01-0145-FEDER-016659).
Acknowledgments
Conflict of interest
The authors declare that there is no conflict of interest.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2017.12.018.
Appendix A. Supplementary material
Supplementary material
References
- 1.Basit F., van Oppen L.M.P.E., Schoeckel L., Bossenbroek H.M., van Emst-deVries S.E., Hermeling J.C.W., Grefte S., Kopitz C., Heroult M., Willems P.H.G.M., Koopman W.J.H. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017;8:e2716. doi: 10.1038/cddis.2017.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basso E., Fante L., Fowlkes J., Petronilli V., Forte M.A., Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J. Biol. Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 3.Boyer M.J., Hedley D.W. Measurement of intracellular pH. Methods Cell Biol. 1994;41:135–148. doi: 10.1016/s0091-679x(08)61714-8. [DOI] [PubMed] [Google Scholar]
- 4.Budd S.L., Castilho R.F., Nicholls D.G. Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett. 1997;415:21–24. doi: 10.1016/s0014-5793(97)01088-0. [DOI] [PubMed] [Google Scholar]
- 5.Caito S.W., Aschner M. Mitochondrial redox dysfunction and environmental exposures. Antioxid. Redox Signal. 2015;23:578–595. doi: 10.1089/ars.2015.6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carraro M., Bernardi P. Calcium and reactive oxygen species in regulation of the mitochondrial permeability transition and of programmed cell death in yeast. Cell Calcium. 2016;60:102–107. doi: 10.1016/j.ceca.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cascio W.E., Yang H., Muller-Borer B.J., Johnson T.A. Ischemia-induced arrhythmia: the role of connexins, gap junctions, and attendant changes in impulse propagation. J. Electrocardiol. 2005;38:55–59. doi: 10.1016/j.jelectrocard.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 8.Cerutti P.A. Prooxidant states and tumor promotion. Science. 1985;227:375–381. doi: 10.1126/science.2981433. [DOI] [PubMed] [Google Scholar]
- 9.Charras G., Sahai E. Physical influences of the extracellular environment on cell migration. Nat. Rev. Mol. Cell Biol. 2014;15:813–824. doi: 10.1038/nrm3897. [DOI] [PubMed] [Google Scholar]
- 10.Conrad M., Angeli J.P., Vandenabeele P., Stockwell B.R. Regulated necrosis: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2016;15:348–366. doi: 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dalle-Donne I., Aldini G., Carini M., Colombo R., Rossi R., Milzani A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell Mol. Med. 2006;10:389–406. doi: 10.1111/j.1582-4934.2006.tb00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dalle-Donne I., Giustarini D., Colombo R., Rossi R., Milzani A. Protein carbonylation in human diseases. Trends Mol. Med. 2003;9:169–176. doi: 10.1016/s1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 13.Dalle-Donne I., Rossi R., Giustarini D., Milzani A., Colombo R. Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta. 2003;329:23–38. doi: 10.1016/s0009-8981(03)00003-2. [DOI] [PubMed] [Google Scholar]
- 14.Damaghi M., Wojtkowiak J.W., Gillies R.J. pH sensing and regulation in cancer. Front Physiol. 2013;4:370. doi: 10.3389/fphys.2013.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Degterev A., Huang Z., Boyce M., Li Y., Jagtap P., Mizushima N., Cuny G.D., Mitchison T.J., Moskowitz M.A., Yuan J. Chemical inhibitor of nonaptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 16.Distelmaier F., Valsecchi F., Liemburg-Apers D.C., Lebiedzinska M., Rodenburg R.J., Heil S., Keijer J., Fransen J., Imamura H., Danhauser K., Seibt A., Viollet B., Gellerich F.N., Smeitink J.A., Wieckowski M.R., Willems P.H.G.M., Koopman W.J.H. Mitochondrial dysfunction in primary human fibroblasts triggers an adaptive cell survival program that requires AMPK-alpha. Biochim. Biophys. Acta. 2015;1852:529–540. doi: 10.1016/j.bbadis.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 17.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S., Morrison B., Stockwell B.R. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Figueira T.R., Barros M.H., Camargo A.A., Castilho R.F., Ferreira J.C., Kowaltowski A.J., Sluse F.E., Souza-Pinto N.C., Vercesi A.E. Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid. Redox Signal. 2013;18:2029–2074. doi: 10.1089/ars.2012.4729. [DOI] [PubMed] [Google Scholar]
- 19.Forkink M., Basit F., Teixeira J., Swarts H.G., Koopman W.J.H., Willems P.H.G.M. Complex I and complex III inhibition specifically increase cytosolic hydrogen peroxide levels without inducing oxidative stress in HEK293 cells. Redox Biol. 2015;6:607–616. doi: 10.1016/j.redox.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forkink M., Willems P.H.G.M., Koopman W.J.H., Grefte S. Live-cell assessment of mitochondrial reactive oxygen species using dihydroethidine. Methods Mol. Biol. 2015;1264:161–169. doi: 10.1007/978-1-4939-2257-4_15. [DOI] [PubMed] [Google Scholar]
- 21.Forkink M., Manjeri G.R., Liemburg-Apers D.C., Nibbeling E., Blanchard M., Wojtala A., Smeitink J.A., Wieckowski M.R., Willems P.H.G.M., Koopman W.J.H. Mitochondrial hyperpolarization during chronic complex I inhibition is sustained by low activity of complex II, III, IV and V. Biochim. Biophys. Acta. 2014;1837:1247–1256. doi: 10.1016/j.bbabio.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 22.Fritz K.S., Petersen D.R. Exploring the biology of lipid peroxidation-derived protein carbonylation. Chem. Res. Toxicol. 2011;24:1411–1419. doi: 10.1021/tx200169n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gatenby R.A., Gillies R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 24.Griffiths J.R., Stevens A.N., Iles R.A., Gordon R.E., Shaw D. 31P NMR investigation of solid tumours in the living rat. Biosci. Rep. 1981;1:319–325. doi: 10.1007/BF01114871. [DOI] [PubMed] [Google Scholar]
- 25.Gülden M., Jess A., Kammann J., Maser E., Seibert H. Cytotoxic potency of H2O2 in cell cultures: impact of cell concentration and exposure time. Free Radic. Biol. Med. 2010;49:1298–1305. doi: 10.1016/j.freeradbiomed.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 26.Gupta S.C., Singh R., Pochampally R., Watabe K., Mo Y.Y. Acidosis promotes invasiveness of breast cancer cells through ROS-AKT-NF-kappaB pathway. Oncotarget. 2014;5:12070–12082. doi: 10.18632/oncotarget.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hurst S., Hoek J., Sheu S.S. Mitochondrial Ca2+ and regulation of the permeability transition pore. J. Bioenerg. Biomembr. 2017;49:27–47. doi: 10.1007/s10863-016-9672-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ippolito J.E., Brandenburg M.W., Ge X., Crowley J.R., Kirmess K.M., Som A., D. A. D′Avignon, Arbeit J.M., Achilefu S., Yarasheski K.E., Milbrandt J. Extracellular pH modulates neuroendocrine prostate cancer cell metabolism and susceptibility to the mitochondrial inhibitor niclosamide. PLoS One. 2016;11:e0159675. doi: 10.1371/journal.pone.0159675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang L., Kon N., Li T., Wang S.J., Su T., Hibshoosh H., Baer R., Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koopman W.J.H., Distelmaier F., Smeitink J.A., Willems P.H.G.M. OXPHOS mutations and neurodegeneration. EMBO J. 2013;32:9–29. doi: 10.1038/emboj.2012.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koopman W.J.H., Willems P.H.G.M., Smeitink J.A. Monogenic mitochondrial disorders. N. Engl. J. Med. 2012;366:1132–1141. doi: 10.1056/NEJMra1012478. [DOI] [PubMed] [Google Scholar]
- 32.Koopman W.J.H., Distelmaier F., Esseling J.J., Smeitink J.A., Willems P.H.G.M. Computer-assisted live cell analysis of mitochondrial membrane potential, morphology and calcium handling. Methods. 2008;46:304–311. doi: 10.1016/j.ymeth.2008.09.018. [DOI] [PubMed] [Google Scholar]
- 33.Koopman W.J.H., Verkaart S., Visch H.J., van der Westhuizen F.H., Murphy M.P., van den Heuvel L.W., Smeitink J.A., Willems P.H.G.M. Inhibition of complex I of the electron transport chain causes O2-. -mediated mitochondrial outgrowth. Am. J. Physiol. Cell Physiol. 2005;288:1440–1450. doi: 10.1152/ajpcell.00607.2004. [DOI] [PubMed] [Google Scholar]
- 34.Kweon H.J., Suh B.C. Acid-sensing ion channels (ASICs): therapeutic targets for neurological diseases and their regulation. BMB Rep. 2013;46:295–304. doi: 10.5483/BMBRep.2013.46.6.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.LaMonte G., Tang X., Chen J.L., Wu J., Ding C.K., Keenan M.M., Sangokoya C., Kung H.N., Ilkayeva O., Boros L.G., Newgard C.B., Chi J.T. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab. 2013;1:23. doi: 10.1186/2049-3002-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liemburg-Apers D.C., Schirris T.J., Russel F.G., Willems P.H.G.M., Koopman W.J.H. Mitoenergetic dysfunction triggers a rapid compensatory increase in steady-state glucose flux. Biophys. J. 2015;109:1372–1386. doi: 10.1016/j.bpj.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mookerjee S.A., Goncalves R.L., Gerencser A.A., Nicholls D.G., Brand M.D. The contributions of respiration and glycolysis to extracellular acid production. Biochim. Biophys. Acta. 2015;1847:171–181. doi: 10.1016/j.bbabio.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 38.Murakami C., Delaney J.R., Chou A., Carr D., Schleit J., Sutphin G.L., An E.H., Castanza A.S., Fletcher M., Goswami S., Higgins S., Holmberg M., Hui J., Jelic M., Jeong K.S., Kim J.R., Klum S., Liao E., Lin M.S., Lo W., Miller H., Moller R., Peng Z.J., Pollard T., Pradeep P., Pruett D., Rai D., Ros V., Schuster A., Singh M., Spector B.L., Vander Wende H., Wang A.M., Wasko B.M., Olsen B., Kaeberlein M. pH neutralization protects against reduction in replicative lifespan following chronological aging in yeast. Cell Cycle. 2012;11:3087–3096. doi: 10.4161/cc.21465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Öörni K., Rajamäki K., Nguyen S.D., Lähdesmäki K., Plihtari R., Lee-Rueckert M., Kovanen P.T. Acidification of the intimal fluid: the perfect storm for atherogenesis. J. Lipid Res. 2015;56:203–214. doi: 10.1194/jlr.R050252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pekun T.G., Lemeshchenko V.V., Lyskova T.I., Waseem T.V., Fedorovich S.V. Influence of intra- and extracellular acidification on free radical formation and mitochondria membrane potential in rat brain synaptosomes. J. Mol. Neurosci. 2013;49:211–222. doi: 10.1007/s12031-012-9913-3. [DOI] [PubMed] [Google Scholar]
- 41.Petrov D., Zagrovic B. Microscopic analysis of protein oxidative damage: effect of carbonylation on structure, dynamics, and aggregability of villin headpiece. J. Am. Chem. Soc. 2011;133:7016–7024. doi: 10.1021/ja110577e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pisoshi A.M., Pop A. The role of antioxidants in the chemistry of oxidative stress: a review. Eur. J. Med. Chem. 2015;97:55–74. doi: 10.1016/j.ejmech.2015.04.040. [DOI] [PubMed] [Google Scholar]
- 43.Rajamaki K., Nordstrom T., Nurmi K., Akerman K.E., Kovanen P.T., Oorni K., Eklund K.K. Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome. J. Biol. Chem. 2013;288:13410–13419. doi: 10.1074/jbc.M112.426254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reshkin S.J., Greco M.R., Cardone R.A. Role of pHi, and proton transporters in oncogene-driven neoplastic transformation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014;369:20130100. doi: 10.1098/rstb.2013.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Riemann A., Schneider B., Ihling A., Nowak M., Sauvant C., Thews O., Gekle M. Acidic environment leads to ROS-induced MAPK signaling in cancer cells. PLoS One. 2011;6:e22445. doi: 10.1371/journal.pone.0022445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robinson K.M., Janes M.S., Pehar M., Monette J.S., Ross M.F., Hagen T.M., Murphy M.P., Beckman J.S. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc. Natl. Acad. Sci. USA. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson B.H. Lactic acidemia and mitochondrial disease. Mol. Genet. Metab. 2006;89:3–13. doi: 10.1016/j.ymgme.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 48.Rodrigo R., Libuy M., Feliú F., Hasson D. Oxidative stress-related biomarkers in essential hypertension and ischemia-reperfusion myocardial damage. Dis. Markers. 2013;35:773–790. doi: 10.1155/2013/974358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rottenberg H., Hoek J.B. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell. 2017;16:943–955. doi: 10.1111/acel.12650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schoeckel L., Glasauer A., Basit F., Bitschar K., Truong H., Erdmann G., Algire C., Hagebarth A., Willems P.H.G.M., Kopitz C., Koopman W.J.H., Heroult M. Targeting mitochondrial complex I using BAY 87-2243 reduces melanoma tumor growth. Cancer Metab. 2015;3:11. doi: 10.1186/s40170-015-0138-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharma V., Kaur R., Bhatnagar A., Kaur J. Low-pH-induced apoptosis: role of endoplasmic reticulum stress-induced calcium permeability and mitochondria-dependent signaling. Cell Stress Chaperones. 2015;20:431–440. doi: 10.1007/s12192-014-0568-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sieprath T., Corne T.D., Willems P.H.G.M., Koopman W.J.H., De Vos W.H. Integrated high-content quantification of intracellular ROS levels and mitochondrial morphofunction. Adv. Anat. Embryol. Cell Biol. 2016;219:149–177. doi: 10.1007/978-3-319-28549-8_6. [DOI] [PubMed] [Google Scholar]
- 53.Sliwka L., Wiktorska K., Suchocki P., Milczarek M., Mielczarek S., Lubelska K., Cierpial T., Lyzwa P., Kielbasinski P., Jaromin A., Flis A., Chilmonczyk Z. The comparison of MTT and CVS assays for the assessment of anticancer agent interactions. PLoS One. 2016;11:e0155772. doi: 10.1371/journal.pone.0155772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith M.L., von Hanwehr R., Siesjo B.K. Changes in extra- and intracellular pH in the brain during and following ischemia in hyperglycemic and in moderately hypoglycemic rats. J. Cereb. Blood Flow Metab. 1986;6:574–583. doi: 10.1038/jcbfm.1986.104. [DOI] [PubMed] [Google Scholar]
- 55.Stadtman E.R. Protein oxidation and aging. Science. 1992;257:1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- 56.Steen D.L., O’Donoghue M.L. Lp-PLA2 inhibitors for the reduction of cardiovascular events. Cardiol. Ther. 2013;2:125–134. doi: 10.1007/s40119-013-0022-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stiburek L., Cesnekova J., Kostkova O., Fornuskova D., Vinsova K., Wenchich L., Houstek J., Zeman J. YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol. Biol. Cell. 2012;23:1010–1023. doi: 10.1091/mbc.E11-08-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thanan R., Oikawa S., Yongvanit P., Hiraku Y., Ma N., Pinlaor S., Pairojkul C., Wongkham C., Sripa B., Khuntikeo N., Kawanishi S., Murata M. Inflammation-induced protein carbonylation contributes to poor prognosis for cholangiocarcinoma. Free Radic. Biol. Med. 2012;52:1465–1472. doi: 10.1016/j.freeradbiomed.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 59.Tian L., Bae Y.H. Cancer nanomedicines targeting tumor extracellular pH. Colloids Surf. B Biointerfaces. 2012;99:116–126. doi: 10.1016/j.colsurfb.2011.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valsecchi F., Monge C., Forkink M., de Groof A.J., Benard G., Rossignol R., Swarts H.G., van Emst-deVries S.E., Rodenburg R.J., Calvaruso M.A., Nijtmans L.G., Heeman B., Roestenberg P., Wieringa B., Smeitink J.A., Koopman W.J.H., Willems P.H.G.M. Metabolic consequences of NDUFS4 gene deletion in immortalized mouse embryonic fibroblasts. Biochim. Biophys. Acta. 2012;1817:1925–1936. doi: 10.1016/j.bbabio.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 61.Vandenabeele P., Grootjans S., Takahashi N. Necrostatin-1 blocks both RIPK1 and IDO: consequences for the study of cell death in experimental disease models. Cell Death Differ. 2013;20:185–187. doi: 10.1038/cdd.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vander Heiden M.G., Cantley L.C., Thompson C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wehr N.B., Levine R.L. Quantification of protein carbonylation. Methods Mol. Biol. 2013;965:265–281. doi: 10.1007/978-1-62703-239-1_18. [DOI] [PubMed] [Google Scholar]
- 64.Wei W.C., Jacobs B., Becker E.B., Glitsch M.D. Reciprocal regulation of two G protein-coupled receptors sensing extracellular concentrations of Ca2+ and H. Proc. Natl. Acad. Sci. USA. 2015;112:10738–10743. doi: 10.1073/pnas.1506085112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Willems P.H.G.M., Rossignol R., Dieteren C.E., Murphy M.P., Koopman W.J.H. Redox homeostasis and mitochondrial dynamics. Cell Metab. 2015;22:207–218. doi: 10.1016/j.cmet.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 66.Wong C.M., Cheema A.K., Zhang L., Suzuki Y.J. Protein carbonylation as a novel mechanism in redox signaling. Circ. Res. 2008;102:310–318. doi: 10.1161/CIRCRESAHA.107.159814. [DOI] [PubMed] [Google Scholar]
- 67.Wong C.M., Marcocci L., Liu L., Suzuki Y.J. Cell signaling by protein carbonylation and decarbonylation. Antioxid. Redox Signal. 2010;12:393–404. doi: 10.1089/ars.2009.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang W.S., Stockwell B.R. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang W.S., SriRamaratnam R., Welsch M.E., Shimada K., Skouta R., Viswanathan V.S., Cheah J.H., Clemons P.A., Shamji A.F., Clish C.B., Brown L.M., Girotti A.W., Cornish V.W., Schreiber S.L., Stockwell B.R. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zorov D.B., Juhaszova M., Sollott S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014;94:909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material