

Abstract
This clinical trial developed a personalized dosing model for reducing prostaglandin E2 (PGE2) in colonic mucosa using ω-3 fatty acid supplementation. The model utilized serum eicosapentaenoic acid (EPA, ω-3):arachidonic acid (AA, ω-6) ratios as biomarkers of colonic mucosal PGE2 concentration. Normal human volunteers were given low and high ω-3 fatty acid test doses for 2 weeks. This established a slope and intercept of the line for dose versus serum EPA:AA ratio in each individual. The slope and intercept was utilized to calculate a personalized target dose that was given for 12 weeks. This target dose was calculated on the basis of a model, initially derived from lean rodents, showing a log-linear relationship between serum EPA:AA ratios and colonic mucosal PGE2 reduction. Bayesian methods allowed addition of human data to the rodent model as the trial progressed. The dosing model aimed to achieve a serum EPA:AA ratio that is associated with a 50% reduction in colonic PGE2. Mean colonic mucosal PGE2 concentrations were 6.55 ng/mg protein (SD, 5.78) before any supplementation and 3.59 ng/mg protein (SD, 3.29) after 12 weeks of target dosing. In secondary analyses, the decreases in PGE2 were significantly attenuated in overweight and obese participants. This occurred despite a higher target dose for the obese versus normal weight participants, as generated by the pharmacodynamic predictive model. Large decreases also were observed in 12-hydroxyicosatetraenoic acids, and PGE3 increased substantially. Future biomarker-driven dosing models for cancer prevention therefore should consider energy balance as well as overall eicosanoid homeostasis in normal tissue.
Introduction
Prostaglandin E2 (PGE2) is a proinflammatory mediator formed by the oxygenation of arachidonic acid (AA, 20:4), an ω-6 fatty acid. PGE2 plays a key role in the etiology of colonic proinflammatory milieu that is involved in all stages of colon carcinogenesis (1). Conversely, in vitro data predict that ω-3 fatty acids will have anti-inflammatory effects. When dietary omega (ω)3:ω6 fatty acid ratios are increased by ω-3 fatty acid supplementation or diet modification, ω-3 fatty acids supplant AA and other fatty acids in phospholipids (2). This reduces the availability of AA for cyclooxygenase (COX)-mediated oxygenation to form eicosanoids. In addition, the binding of eicosapentaenoic acid (EPA, 20:5, ω-3) to COX-1 inhibits AA oxygenation (3). Docosahexaenoate (DHA; 22:6, ω-3) and docosapentaenoate (DPA; 22:5ω-3) are poor COX-1 substrates and very modest COX-1 inhibitors, making them less relevant to modulation of COX-1 activity (3). Unlike with COX-1, EPA may serve as a substrate for COX-2, but it is a poor COX-2 substrate as compared with AA (4). DHA and DPA are also ineffective COX-2 inhibitors (5). For COX-2, a key effect of increased ω-3 fatty acids is a reduced AA supply for generation of PGE2.
Given these key roles of EPA and AA on COX-mediated PGE2 production, we modeled these relationships on production of PGE2 in rodents. We fed F344 rats diets with varying EPA:AA ratios for 12 weeks (6). EPA:AA ratios in serum and colon increased proportionately to dietary feeding. PGE2 in the colonic mucosa rapidly declined and then plateaued at EPA:ω-6 FA dietary ratios ≥0.3 (6). We also found a log-linear relationship between serum EPA:AA ratio and colonic PGE2 in distal colon (6).
Here, we translate our in vitro and in vivo data to humans. We tested the hypothesis that ω-3 fatty acid dose could be personalized to reduce colonic mucosal PGE2 concentrations by 50%. Rodent data show that a 50% reduction in PGE2 in both normal-appearing and tumor tissue by dietary agents and aspirin is associated with reduction in colonic neoplasia (7-11). In normal tissue, which is the target for primary prevention, PGE2 has an important role in tissue repair and homeostasis (12). Hence, a basal concentration of PGE2 is required in the colon and systemically at all organ sites. This basal concentration determines inflammatory tone that over time will affect susceptibility to malignancy. This is regulated by substrate availability and by expression of synthetic and catabolic rate-limiting enzymes. The ideal tissue concentration of PGE2 and other eicosanoids that allow for normal function without increasing cancer risk is yet to be determined. Nevertheless, there are important examples of abnormal regulation of PGE2 in the histologically normal colon: expression of COX-1 in normal colonic mucosa of individuals at high risk for colon cancer was 2-fold higher than that in a normal risk group (13), high-fat diets induce COX-2 in colon of rodents (14), and obesity increases COX-2 expression in human colon (15, 16). Genetic polymorphisms of key enzymes in the pathways for synthesis, transport, and degradation of PGE2 are also linked to risk of developing colorectal cancer (17, 18).
The extent of PGE2 reduction required in normal tissue for primary cancer prevention therefore is likely to be highly individual and is currently unknown. The control of PGE2 pools in normal tissues is likely to be related to concentrations of substrate and genetically driven activity of rate-limiting synthetic and catabolic enzymes. In the absence of sufficient human data, we extrapolated from rodent carcinogenesis models, the only data available, to choose a target of reducing colonic PGE2 by 50% for this trial. The goal of this study was to develop and implement a pharmacodynamic model that would personalize ω-3 fatty acid dosing in humans to reduce colon mucosal PGE2 concentration by 50%.
Materials and Methods
Study participants
This phase Ib clinical trial was approved by the University of Michigan Internal Review Board (HUM00051786) and registered at www.clincialtrials.org (NCT# 01860352, Effects of Fish Oil on the Colonic Mucosa). The procedures were conducted in accordance with the ethical standards of the Helsinki Declaration of 1975, as revised in 1983. The study took place at the University of Michigan (Ann Arbor, MI), and all participants gave signed, informed consent to participate. Participants were recruited through a web-based registry, flyers, and word-of-mouth.
Eligibility criteria included being between 25 and 75 years old, a body mass index (BMI) between 18 and 40 kg/mg2, unremarkable medical history, no inflammatory or other colonic diseases, no history of cancer diagnosed within the past 5 years except for basal or squamous cell skin cancer that was surgically excised, and normal complete blood counts, clotting time, and hepatic chemistry. Participants taking medications or supplements that might affect outcomes, such as aspirin or fish oils, were excluded or given the option of a 3-week wash-out period. Pregnant or lactating women or participants contemplating pregnancy for the duration of the protocol were excluded.
Test agent
The ω-3 fatty acid supplement in triglyceride form was a fish oil preparation highly enriched in EPA (EPA-Xtra from Nordic Naturals). The agent was manufactured compliant with Good Manufacturing Practice. The capsules were kept in a cool, dry, and dark place before dispensing, according to the manufacturer’s instructions. Participants were asked to store the capsules in the same manner. The capsules for the 2-week dosing with either low-dose or high-dose ω-3 fatty acids were all dispensed at visits immediately before dosing. For the 90-day period of target dosing, half the capsule supply was provided at the start and half midway through the target dosing period. The capsules each contained 1 g of total fat: 530 mg EPA, 150 mg DHA, and 101 mg other ω-3 fatty acids. Four different lots of the agent were used, and the measured variation for EPA between lots at the end of the study was less than 3%.
Adherence, diet assessment, and adverse events
Participants were assessed for agent adherence every 2 weeks by review of pill consumption diaries and by pill counts at the end of the study. Participants were classified as adherent if 75% or more of the doses were taken as prescribed. Dietary intake was assessed throughout the study using unannounced 24-hour recalls collected by the multi-pass method, twice at baseline and three times during target dosing. The recalls were analyzed with the Nutrition Data System for Research, version 2014 (Nutrition Coordinating Center, University of Minnesota, Minneapolis, MN). The data from 3 to 5 recalls/subject (mean of 4.8 recalls/subject) were averaged.
Participants were assessed for adverse events in person, by e-mail, or by telephone at seven intervals throughout the study. The National Cancer Institute Common Toxicity Scale V 4.0 was used to quantify toxicity (NCI, Common Terminology Criteria for Adverse Events, Version 4.0, U.S. Department of Health and Human Services, Bethesda, MD, 2009).
Dosing overview
Serum EPA:AA ratios were measured after sequential 2-week periods of low and high test doses of ω-3 fatty acids. This resulted in a dose–serum EPA:AA curve for each participant. A personalized dose was then calculated to achieve a serum EPA:AA ratio that is associated with a 50% reduction in colonic PGE2 based on our model. The model for the relationship between serum EPA:AA and colon PGE2 was initially established using our rodent data (6). The model was continuously updated to include human data as the trial progressed in a Bayesian design. The primary endpoint, colonic mucosal PGE2 was measured at study entry and after 12 weeks of target dosing. In secondary analyses, we explored the effects of obesity on changes in colonic PGE2, and we also evaluated the effects of ω-3 fatty acid supplementation on other colonic eicosanoids and epithelial proliferation.
Low- and high-dose ω-3 fatty acid test doses
Individual serum fatty acid responses to 2-week dosing using low and high ω-3 fatty acid test doses were used to define the slope of the dose versus serum EPA:AA relationship. This relationship was needed to calculate a target dose that would be expected to reduce colonic PGE2 by 50% in each study participant based on our model. After eligibility determination, participants were asked to provide a baseline fasting blood sample and to undergo colonic biopsy by flexible sigmoidoscopy without prior preparation of the bowels. Participants were then provided with a low test dose of ω-3 fatty acids designed to approximate a dietary EPA:ω-6 fatty acid ratio of 0.1. This was calculated on the basis of estimated caloric intake from the Harris–Benedict formula (19) and assuming the average American dietary fat intake that is 6.9% of energy from ω-6 fatty acids (based on What We Eat in America at https://www.ars.usda.gov). After 2 weeks of the low test dose, another fasting blood sample was obtained. This was followed by 2 weeks of a high test dose, targeting a dietary EPA:ω-6 fatty acid ratio of 0.3, and another blood sample.
Target ω-3 fatty acid dosing for 12 weeks
The target fatty acid dose calculation utilized the log-linear, pharmacodynamic relationship between serum EPA:AA and colonic mucosal PGE2 concentration that we established in F344 rats (6). The personalized target dose of fish oil was obtained in a two-step manner. In the first step, the natural log (ln)-linear model:
| (A) |
was utilized to calculate the target serum EPA:AA ratio required to reduce the PGE2 by 50% from its baseline value:
| (B) |
In the first stage of the trial, the estimate of slope in Eqs. A and B was from animal data that related serum EPA:AA with colonic PGE2. The estimate of slope was updated following a Bayesian algorithm after data on every 8 to 10 human participants became available until the model did not change appreciably with additional participants.
The two predosing serum EPA:AA measurements taken about 2 weeks apart (at eligibility and baseline) were similar (Spearman correlation = 0.93). Using a simple linear regression of dose on serum EPA:AA predosing and after test low and high ω-3 doses, a model-estimated optimal serum EPA:AA ratio was converted into a personalized target dose that was then given for 12 weeks. Given the participant-specific equation:
| (C) |
where I, M are the individual intercept and slope, and dose is measured in capsules/day, the personalized target dose was calculated as:
| (D) |
where EPA:AAtarget is as in Eq. B. From this model, participants with a steeper dose–serum EPA:AA slope (M) and higher intercept (I) receive a lower target dose. The protocol allowed for accruing participants and updating the model with human data until a stable dosing model derived consisting of human dose data was achieved, up to a maximum of 60 participants. Hence, the dosing model was updated as the trial progressed after every 8 to 10 participants. The trial was designed to be closed when the dosing model stabilized as defined by lack of appreciable changes in the model with additional data from more participants.
Colon mucosal biopsies and serum
Participants underwent flexible sigmoidoscopy at study entry and 20 to 28 hours after the last ω-3 fatty acid target dose. Mucosal biopsies of distal sigmoid colon were taken 2 cm or more away from any sites with visual appearance of trauma. The participants did not prepare their bowels before the procedure, but they were asked to evacuate their rectum within 12 hours of the procedure if possible.
Participants were placed in a left lateral decubitus position and a flexible sigmoidoscope was passed to 20 to 25 cm from the anal sphincter. Tissue samples were taken using Radial Jaw 4 Jumbo forceps by opening and pressing the forceps perpendicular to the mucosal surface with mild pressure. Twelve biopsies (5–10 mg each) were obtained using seven passes of the sigmoidoscope (two biopsies per pass were captured in the jaw for the first five passes and then single biopsies in the last two passes). Eight of the biopsies were placed into sterile 1.5-mL Eppendorf tubes and frozen in liquid nitrogen within 60 seconds, and four biopsies were placed in tubes containing ice-cold, PBS (pH 7.4) for subsequent fixation.
Each of the biopsies in saline was oriented in tissue cassettes on a piece of filter paper and fixed in 10% formalin-90% PBS pH 7.4 within 30 minutes. Biopsies were in formalin for 22 to 25 hours prior to being transferred to 70% ethanol, and they were stored in 70% ethanol up to one week until embedding could take place.
Fasting blood samples from all time points (at eligibility determination, baseline, after low dose, after high dose, midway through target dosing, and after high dose) were delivered to the laboratory wrapped in foil and kept at room temperature for 30 to 60 minutes. Serum was prepared by centrifuging the blood (1,300 × g for 20 minutes). Serum aliquots were frozen immediately at −80°C.
Eicosanoid and fatty acid analyses
Eicosanoids were measured in homogenates of frozen colon mucosal biopsies using chiral high pressure liquid chromatography-tandem mass spectroscopy. Three frozen biopsies were pulverized in liquid nitrogen, and pooled homogenates were prepared in buffered saline containing indomethacin as described previously (20, 21). Deuterated internal standards were added before extraction and analysis of eicosanoids (20). Protein in a portion of the homogenate was determined by the Bradford assay (Bio-Rad).
Fatty acids in serum were analyzed as fatty acid methyl esters by gas chromatography with mass spectral detection (22). Fatty acids were measured in all blood samples obtained, as a percentage of all fatty acids, and the EPA:AA ratios were calculated. The EPA:AA ratios in the serum from eligibility testing and from baseline differed by 1.3% on average.
Colonic proliferation
Proliferation in colon biopsies was determined as described previously (20) using IHC staining for Ki67 with antibodies from Leica Biosystems. Positive nuclei were visualized with horseradish peroxidase–conjugated streptavidin and 3,3′-diaminobenzidine substrate. Image analysis was performed using MCID Elite 7.0 software to enumerate all nuclei and positively stained nuclei. Three values were derived for each side of full-length crypts: (i) height of crypt in number of cells; (ii) labeling index as the total percentage of Ki67-positive cells; and (iii) extent of the proliferative zone as the percentage of crypt height reached by the highest proliferating cell.
Statistical analyses
The effect of fish oil on the changes from baseline in colonic PGE2 concentrations, the primary variable of interest, and other eicosanoids, was assessed using Wilcoxon signed rank tests owing to the extreme skewness in data. There were 12 eicosanoids being analyzed. Using Bonferroni multiple comparison adjustment for potentially inflated type I error probability with experiment-wise error rate of 0.05, P values less than 0.004 (0.05/12) were considered statistically significant. Change in PGE2 was further compared between subgroups characterized by gender and BMI using Mann–Whitney–Wilcoxon (gender) or Kruskal–Wallis (BMI) tests. BMI categories were normal weight (BMI < 25 kg/m2), overweight (BMI, 25–29.9 kg/m2), and obese (BMI ≥ 30 kg/m2).
A linear regression model was fit with change in PGE2 as the outcome, and age, gender, BMI as predictors, controlling for analytic batch. A logistic regression analysis modeling the likelihood of at least 50% reduction in PGE2 was also carried out with the same predictor variables as in the linear regression model. Postdosing change from baseline for other colonic eicosanoids was assessed through a linear mixed effects model with visit (baseline, end of trial) as fixed effects and both analytic batch and participant as random effects. Clustering within participants was incorporated through a random participant intercept. Similar analyses investigated the role of obesity in changes of fatty acids. Linear mixed models were generated with total EPA:AA ratio as the endpoint, and primary covariates were visit, batch, weight strata (normal, overweight, obese) and visit-by-weight interaction. The average slopes from the dose versus serum EPA:AA relationships were compared across BMI categories using ANOVA.
Results
Study participants
We screened 69 individuals, and 59 eligible study participants were enrolled between May 21, 2013, and September 24, 2015 (Fig. 1). Forty-eight participants (81%) completed the study (21 females and 17 males). One participant was excluded due to use of high-dose prednisone for poison ivy. The mean age of the 47 participants analyzed was 47 years (SD, 14; range, 25–75 years). Two participants were African American and 45 were Caucasian. Mean BMI was 27.0 kg/m2 (SD, 4.8; range, 20.1–40.0).
Figure 1.
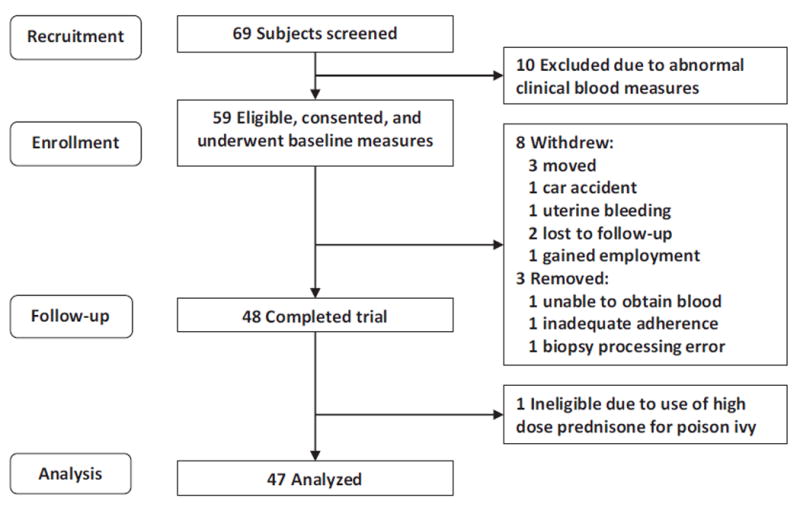
Consolidated Standards of Reporting Trials (CONSORT) diagram of participant flow through the study.
Bayesian modeling of target dose
The study was conducted basing dosing on the relationships between colonic PGE2 and serum EPA:AA ratios in animals. The model was updated by adding human data as it became available such that dosing was calculated using three different models as the study progressed. The initial animal model, Model 1, was updated in a posterior model after data were obtained for the first 10 participants (Model 2), and then again after the next 8 participants had complete data (Model 3). Updating the model further after another 9 participants yielded virtually the same slope parameter as in Model 3 (Fig. 2). The study continued using Model 3 for dosing until all eligible participants who had signed consent completed the study. As study enrollment was continuous as samples were being analyzed and dosing models constructed, the number of participants included under each dosing model varied as shown in Table 1.
Figure 2.
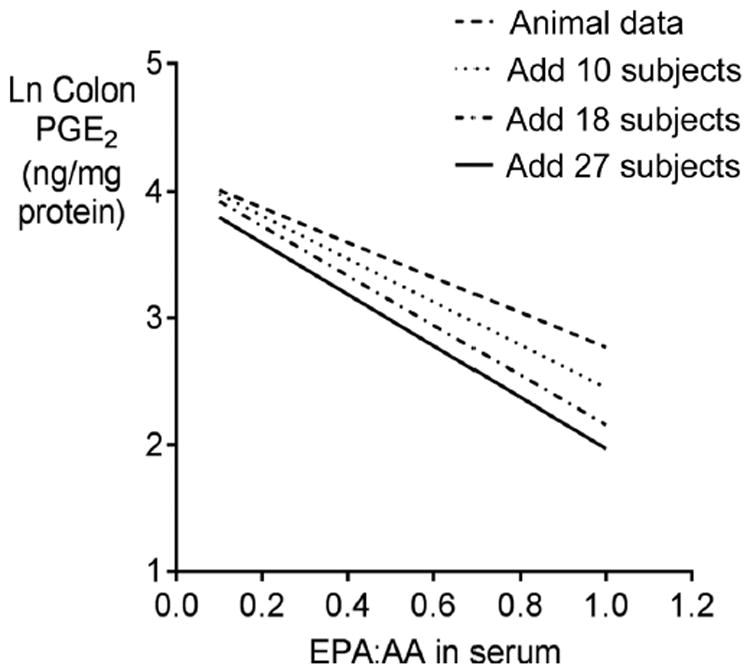
Modeling of colonic PGE2 concentrations as a function of serum EPA:AA ratios. These models allowed subsequent use of the serum EPA:AA ratio as a predictor of colonic PGE2. Initially, the model contained only animal data, and it was updated as human data became available during the course of the trial in an adaptive, Bayesian design. Model 1: animal only data; Model 2: posterior based on adding 10 human participants; Model 3: posterior based on adding 18 participants to the animal data. The fourth model with 27 participants was not utilized for dosing in the present trial as it essentially converged with Model 3. The equations for the lines of natural log of colonic PGE2 (ng/mg protein) as the dependent variable versus serum EPA:AA ratio are: animal data, y = 4.15 – 1.38 × x (Model 1); posterior with 10 participants added, y = 4.15 – 1.70 × x (Model 2); posterior with 18 participants added, y = 4.12 – 1.96 × x (Model 3); posterior with 27 participants added, y = 4.0 – 2.03 × x (Model 4). The models were used to determine the serum EPA:AA ratio needed to achieve the desired colonic PGE2 concentration in each individual.
Table 1.
Target dosing of ω-3 fatty acids
| Model | Number of participants | Number and percent overweight or obese | Target dose (grams ω-3/day) Mean, SD | Pa |
|---|---|---|---|---|
| 1 | 16 | 7 (43.8%) | 6.63 (2.39) | 0.001 |
| 2 | 15 | 4 (26.7%) | 6.00 (1.77) | 0.008 |
| 3 | 16 | 3 (18.8%) | 4.06 (1.53) | Ref. |
NOTE: Shown is the number of participants receiving target dosing and the mean target dose with each model. Each 1 g capsule contained 530 mg EPA, 150 mg DHA, and 101 mg other ω-3 fatty acids.
P values are for the difference in dose with either model 1 or 2 versus model 3.
Doses, adherence, and toxicity
Mean doses of ω-3 fatty acids for the test, 2-week dosing periods were 2.8 g/day (range, 2–4 g/day; SD, 0.7) for the low dose and 8.8 g/day (range, 6–13 g/day; SD, 1.4) for the high dose. The mean target dose overall was 5.5 g/day (range, 2–10; SD, 2) across all three dosing models utilized for this Bayesian design study. The average target dose for dosing Model 3 was significantly lower than that with the other two models (Table 1). The study enrolled a higher percentage of overweight or obese individuals during dosing with the first two models versus the third model (Table 1). One participant had a calculated target dose higher than the high test dose and was dosed with their high dose plus 2 capsules as specified in the protocol.
Adherence by pill counts averaged 97%. There was one adverse event that was likely unrelated to the investigational agent. The grade 1 event was a persistent headache lasting 7 days and nausea. This was relieved by one dose of ibuprofen (600 mg).
Colonic mucosal eicosanoids
The primary study goal was to reduce colonic PGE2 by 50%. The mean concentration of colonic PGE2 in the entire population after supplementation was 45% lower than at baseline (Table 2). When we calculated the reduction in colonic mucosal PGE2 for each participant, the mean reduction was 27% in all 47 participants combined. Twenty of 47 participants had a decrease of 50% or more. One obese individual had a 194% increase in PGE2. Without this outlying observation, the mean of the reductions in colonic mucosal PGE2 for each participant was 31% (n = 46, median, 35%; SD, 51%). We also quantified colonic concentrations of PGE3 (the COX-2 metabolite from EPA), 5, 12, and 15-hydroxyicosatetraenoic acids (HETEs), 13-hydroxyoctadecadienoic acids (HODEs), and 14,15-dihydroxyeicosatrienoic acids (DHET; Table 2). Concentrations of colonic mucosal PGE3 increased in 43 of 47 participants (91%). 12-S- and 12-R-HETEs decreased in 88% and 77% of participants, respectively.
Table 2.
Colonic eicosanoid concentrations (in ng/mg protein) at baseline and after 12 weeks of target ω-3 fatty acid dosing
| Eicosanoid | Major enzymesa | Baseline | After 12 weeks of target dose | Pb |
|---|---|---|---|---|
| PGE2 | Cyclooxygenases 1 and 2, prostaglandin E synthase | 6.55 (5.78) | 3.59 (3.29) | <0.001c |
| Prostaglandin E3 | COX-2, prostaglandin E synthase | 0.25 (0.21) | 0.69 (0.64) | <0.001c |
| 5-S-Hydroxyicosatetraenoic acid | 5-Lipoxygenase | 1.82 (1.30) | 1.90 (1.96) | 0.90 |
| 5-R-Hydroxyicosatetraenoic acid | 5-R-Lipoxygenase | 2.24 (1.24) | 2.90 (1.93) | 0.02 |
| 12-S-Hydroxyicosatetraenoic acid | 12-Lipoxygenase | 3.95 (3.36) | 2.20 (2.43) | <0.001c |
| 12-R-Hydroxyicosatetraenoic acid | 12 (R)-Lipoxygenase or cytochrome P450 | 4.12 (4.23) | 2.70 (2.63) | 0.001c |
| 15-S-Hydroxyicosatetraenoic acid | 15-Lipoxygenase | 892 (616) | 993 (649) | 0.05 |
| 15-R-Hydroxyicosatetraenoic acid | Cytochrome P450 or acetylated COX-2 | 18.0 (12.9) | 15.8 (11.0) | 0.28 |
| 14,15-S-dihydroxyeicosatrienoic acid | Cytochrome P450 and epoxide hydrolase | 1.04 (0.90) | 1.17 (1.05) | 0.008 |
| 14,15-R-dihydroxyeicosatrienoic acid | Cytochrome P450 and epoxide hydrolase | 0.99 (0.79) | 1.13 (0.93) | 0.06 |
| 13-S-Hydroxyoctadecadienoic acid | 15-Lipoxygenase | 12.10 (10.3) | 10.91 (9.0) | 0.18 |
| 13-R-Hydroxyoctadecadienoic acid | Lipoxygenases, or cytochrome P450 | 20.63 (11.0) | 20.96 (15.2) | 0.88 |
Data shown are mean and SD for 47 participants at each time point except for PGE2, where n = 46 was used due to one outlier that was an obese participant with a large increase in PGE2.
All of the eicosanoids shown are formed from arachidonic acid except for PGE3, which is formed from eicosapentaenoic acid, and 13-hydroxyoctadecadienoic acids, which are formed from linoleic acid. Nonenzymatic oxidation of AA is also possible and is not stereospecific.
P values are for comparing baseline and posttarget dose measures and are based on Wilcoxon signed rank test owing to nonnormality of some of the eicosanoids. There were 47 participants eligible for analysis. There was one outlier in the overweight–obese group who had a 3-fold increase in PGE2 and was omitted from the analysis of PGE2 concentrations, resulting in n = 46 (see Results).
Values with P < 0.005 were considered significant to adjust for multiple comparisons.
Colonic mucosal proliferation
Overall Ki67 positivity was 30%, the extent of the proliferative zone was 62%, and crypt height, in the number of cells, was 75. These parameters did not change after target dosing, and the lack of effect was also true in subgroups defined by weight status (not shown). Ki67 positivity decreased by 17% in the 8 participants who consumed a diet containing less than 30% of energy from fat, versus an increase of 14% in the other 39 participants (P < 0.001). Change in Ki67 positivity was negatively associated with change in 13-S-HODE with Spearman r = −0.41 (P = 0.004). There was also a negative correlation of change in Ki67 positivity with 15-S-HETE (r = −0.33, P = 0.024), but this and correlations with changes in other eicosanoids were not significant after correction for multiple comparisons.
Relationship between colonic mucosal PGE2 concentration and BMI
In secondary analyses, we evaluated whether supplementation reduced colonic PGE2 concentration differently based upon BMI category (Fig. 3). In normal weight participants, ω-3 fatty acid supplementation reduced colonic PGE2 concentration by a mean of 46% (median, 60%; SD, 45%; 12/21 decreased by 50% or more). In overweight participants, supplementation reduced colonic PGE2 by a mean of 18% (median, 15%; SD, 51%; 4/12 decreased by 50% or more). In obese participants, the mean reduction also was 18% (median, 24%; SD, 55%; 4/13 decreased by 50% or more) after excluding one outlier with an increase in colonic PGE2 of 194%, as shown in Fig. 3. If the outlying value is included, the mean reduction becomes 3% for the obese group (median, 15%; SD, 77%). This was despite the fact that the mean dose of ω-3 fatty acids was higher in obese versus normal weight participants (P = 0.002 by one-way ANOVA): the means were 4.5 (SD, 1.6), 5.6 (SD, 2.4), and 6.6 (SD, 1.7) g/day in normal, overweight, and obese participant groups, respectively. There was no statistically significant difference in univariate analysis (P = 0.16 based on the Kruskal–Wallis test).
Figure 3.
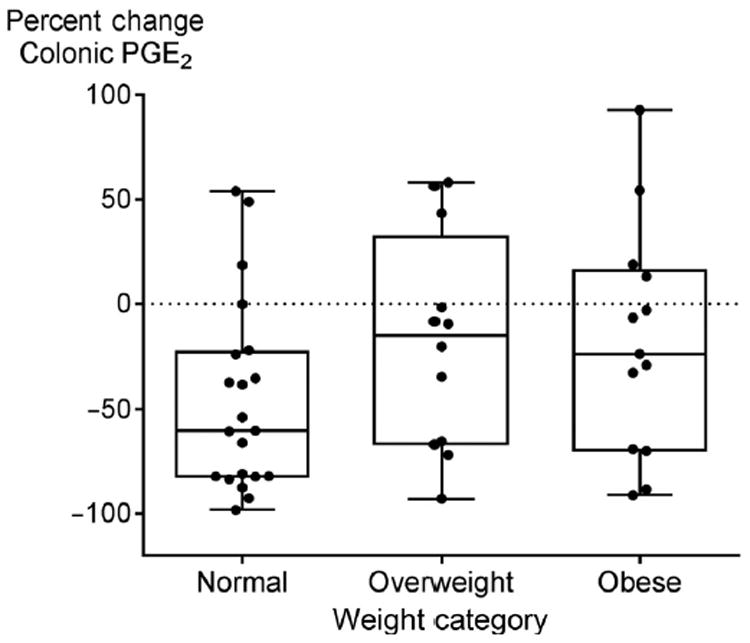
Changes in colonic mucosal PGE2 concentrations by baseline obesity status with a BMI of <25 kg/m2 defined as normal weight, 25–29.9 kg/m2 as overweight, and >30 kg/m2 as obese. Change was calculated for each individual, and data shown are mean change for each body weight group. The box-and-whisker plots show the median indicated by the vertical bar in the middle, box edges indicate 25th to the 75th quartile, and the whiskers extend to the minimum and maximum points. The horizontal dotted line signifies no change (zero percent change) in colonic PGE2 after 12 weeks of target dosing relative to that at study entry. One outlier was excluded in the data shown, and this was an obese subject with a 194% increase in colonic PGE2. Values above the dotted line indicate an increase and values below the dotted line indicate a decrease in colonic PGE2. The mean doses of ω-3 fatty acids were 4.5 (SD, 1.6), 5.6 (SD, 2.4), and 6.6 (SD, 1.7) g/day in normal, overweight, and obese participant groups, respectively (P = 0.002 by one-way ANOVA).
In males, supplementation reduced colonic mucosal PGE2 concentrations minimally (mean, 13%; SD, 58%; median, 3%), whereas in females the reduction was a mean of 41% (SD, 44%; median, 38%). There was a trend toward statistical significance (P = 0.08) in univariate analysis. In a linear regression model with reduction of colonic mucosal PGE2 concentration as the outcome and adjusting for gender, BMI, age, and analytic batch, gender was borderline significant (P = 0.05). In a logistic regression model, the likelihood of at least 50% PGE2 reduction, with age, gender, BMI, and batch as covariates, only BMI was significantly associated with a 50% or more decrease in colonic PGE2 (OR for a decrease with lower BMI was 0.76; 95% CI, 0.61–0.94; P = 0.012).
Effects of obesity on serum fatty acids
We calculated the linear regression slope of individual serum EPA:AA ratio as a function of ω-3 fatty acid dose, in g/day (Fig. 4). The lower the slope, the higher the target dose required to achieve 50% reduction in colonic PGE2 concentrations. In normal weight participants, the mean slope was significantly higher (P = 0.03) than the mean slope in obese participants (0.108, SD 0.027 vs. 0.078, SD 0.028). Overweight participants had an intermediate slope (mean, 0.098; SD, 0.058) that was not significantly different from the other two groups.
Figure 4.
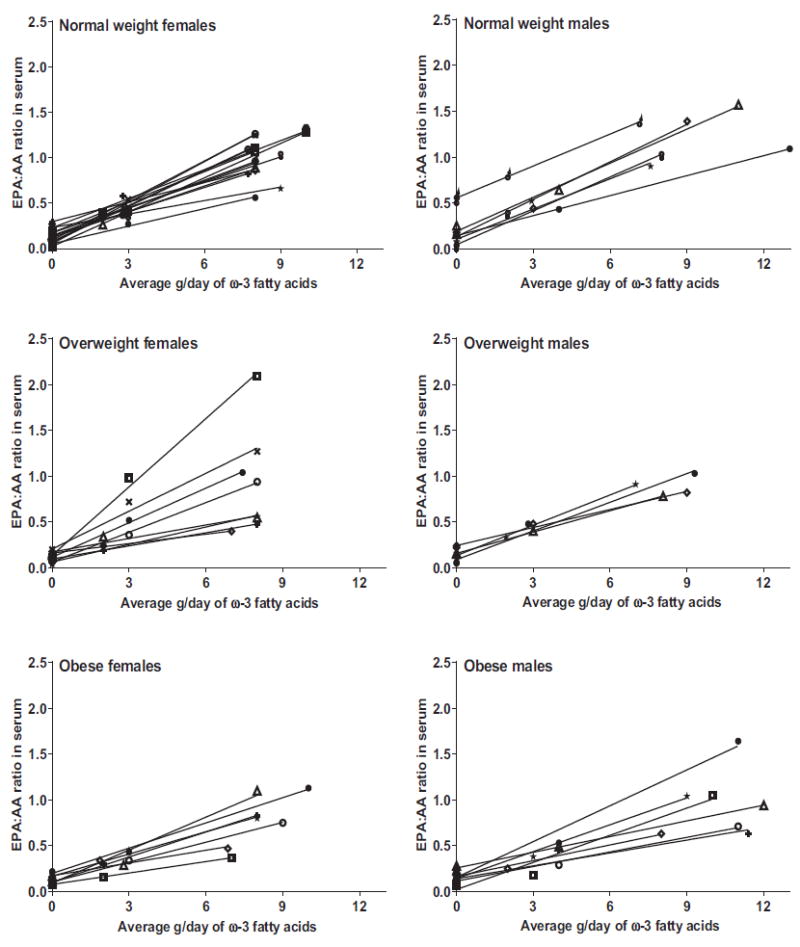
Serum eicosapentaenoic acid to arachidonic acid ratios (EPA:AA) at two predosing time points and after 2 weeks of dosing with low or high test doses of ω-3 fatty acids, shown by gender and weight status in individual participants. The serum EPA:AA responses were linear for each subject, allowing for facile calculation of the target dose needed to achieve the desired serum EPA:AA ratio for each study participant. Each 1 g capsule contained 530 mg EPA, 150 mg DHA, and 101 mg other ω-3 fatty acids. The slope and intercept of each line was used to derive a personalized target dose for each individual based on our model: a higher intercept and a steeper slope resulted in lower target dose for the subsequent 12-week period. The curves show the heterogeneity of responses to ω-3 fatty acid dosing: both the slopes and intercepts vary widely between individuals. This heterogeneity in responses underscores the need for personalized approaches to dosing.
Discussion
We tested the hypothesis that serum EPA:AA fatty acid ratios can be utilized to derive an ω-3 fatty acid dose to humans that reduces colonic PGE2 by 50%. The data validate our preclinical in vitro and in vivo data (3, 4, 6, 21, 23, 24) that support a strong, log-linear relationship between ω-3 fatty acid dosing and colonic mucosal PGE2 concentrations, but only in normal weight human populations.
Relying on serum fatty acid responses as a surrogate for predicting eicosanoid concentrations in colonic tissue takes into account the processes of absorption, distribution, and metabolism. The results showed that serum EPA:AA responses to ω-3 fatty acid dosing varied in each individual (Fig. 4). The serum EPA:AA ratio increased linearly in each participant with no plateau; however, the slope differed among the participants. The lack of a dose–response plateau was surprising because the “high test dose” was higher than doses typically used in supplementation studies: between 6 and 13 g/day. The lack of a high-dose plateau allowed us to utilize the slope of the curve for serum EPA:AA ratio versus dose to calculate a personalized target dose needed to achieve a serum EPA:AA ratio that would reduce colonic mucosal PGE2 concentration by 50%, as predicted by the dosing model. This mathematical dosing model was generated initially with rodent data, and it was enriched with human data as the trial progressed using dynamic dosing adjustments. A stable dosing algorithm was achieved within the first 18 study participants (Fig. 2).
The dosing model used serum EPA:AA ratios as a surrogate biomarker for PGE2 tissue responses based upon biochemical data, demonstrating fatty acid control of COX-1 and COX-2 dimer activities, rate-limiting enzymes in the synthetic pathway of PGE2 (2-5). AA is the only ω-6 fatty acid that serves as a substrate for COX-1, whereas EPA serves to inhibit COX-1 activity allosterically; ω-3 fatty acids other than EPA have little effect on cyclooxygenases (3-5). We cannot exclude the possibility that DHA and other ω-3 fatty acids present in the supplement did not affect formation of PGE2 or other eicosanoids that were not quantified via indirect or unknown effects.
Other eicosanoid metabolites quantified were ω-6 fatty acid metabolites of lipoxygenases and CYP450/epoxide hydrolase (Table 2). Each of these metabolites contributes to eicosanoid homeostasis and ultimately the inflammatory environment of the colonic mucosa (25-27). 14,15-S-DHET is associated with inflammatory stress, induces HIF-1α, activates PPAR-α, and can antagonize COX-2 inhibition (28-31). The DHET metabolites were not significantly changed, but 12-HETEs did decrease. Products from 5- and 12-lipoxygenases have been associated with enhanced carcinogenesis in multiple epithelia (26, 27, 32). Colon 12-S- and 12-R-HETEs were substantially reduced by ω-3 fatty acid supplementation (Table 2). The lack of increases in R isomers indicates that lipid peroxidation was not being augmented by ω-3 fatty acid supplementation. The R-HETE isomers can be non-enzymatically formed from oxidative processes, and excess R isomer formation would represent oxidative stress (33).
There was no effect of supplementation on 15-lipoxygenase metabolites, 15-S-hydroxyeicosatetraenoic acid (HETE) or 13-S-hydroxyoctadecadienoic acid (13-S-HODE). Expression of 15-lipoxygenase-1 increases colonic crypt differentiation, and activity of 15-lipoxygenase is decreased in colonic carcinogenesis (34, 35). The lack of significant decrease in 15-lipoxygenase products can therefore be regarded as beneficial. In addition, a decrease in epithelial proliferation was correlated with an increase in 13-S-HODE (see Results).
The primary goal of the study was to evaluate reduction in colonic PGE2. In secondary analyses, we found that participants with a normal BMI responded with a larger increase in serum EPA: AA ratios and a larger reduction in colonic PGE2 compared with overweight or obese participants. The reduced response in overweight or obese participants occurred despite the relatively higher average dose of ω-3 fatty acids provided to obese participants (Table 1; Fig. 3). This could be due, in part, to the dosing model that retained F344 rat data as the rats were not fed an obesogenic diet. These data support a conclusion that optimal human anticarcinogenic dosing of ω-3 fatty acids differs in obese versus normal weight populations.
Our data confirm an earlier clinical trial that found decreased colonic epithelial proliferation with fish oil supplementation only when combined with a low-fat diet (36). We did not observe an effect on colonic proliferation overall or by BMI; but we found a significant decrease in a subset of individuals consuming less than 30% of calories from fat (Results). In a study using 3 g/day fish oil, obesity substantially altered the effects of ω-3 fatty acids on plasma lysophospholipid profiles, as compared with that in normal weight subjects (37). Future studies might evaluate the dietary and colonic fatty acid milieu in maximizing responses to ω-3 fatty acid dosing.
Data in the literature associating fatty acids and other inflammatory endpoints support our findings that higher BMI reduces the anti-inflammatory and, presumptively, the anticarcinogenic efficacy of ω-3 fatty acid supplementation. In national data, fish intake reduced serum C-reactive protein in normal weight but not obese women (38). In a breast cancer prevention study using ω-3 fatty acid supplementation, high BMI attenuated adipose ω-3 fatty acid concentrations (39). In small human studies, PGE2 in rectal biopsy tissue increased with increasing BMI while weight loss reduced rectal PGE2 (16, 40). Calorie restriction augments the anti-inflammatory effects of ω-3 fatty acids and has strong colonic anticarcinogenic effects in animal models (41). Conversely, obesity is associated with low ω-3:ω-6 fatty acid ratios in blood (42, 43). This is not due to lower dietary ω-3 fatty acid intakes; rather, high dietary ω-6 fatty acid intakes compete with ω-3 and other fatty acids for incorporation into phospholipids (44, 45). Lowering dietary ω-6 FA intakes alone increases ω-3 fatty acids in membranes (46).
Obesity induces COX-2 gene expression and stimulates COX-2 activity, thereby increasing PGE2 concentrations in the human colon (15, 16). Endogenous synthesis of saturated fatty acids and mono-unsaturated fatty acids for storage is also favored under conditions of caloric excess (42). Saturated and monounsaturated fatty acids allosterically increase COX-2 activity, increasing PGE2 production by 1.5- to 4-fold (3, 4). This, together with variations in dietary intakes of other fatty acids, could explain a portion of the variability in the anti-inflammatory responses to ω-3 fatty acid supplements in clinical trials (47). Recent data indicate that metabolic disturbances characteristic of obesity can occur in normal weight individuals, and this metabolic phenotype is associated with increased colorectal cancer risk in women (48).
In summary, we describe a physiologically based approach to personalize the dosing of ω-3 fatty acids based on serum fatty acid responses to short-term dosing with ω-3 fatty acids. This type of pharmacodynamic approach could be used for other preventive agents. The approach was effective for reducing colonic PGE2 by almost 50% in the colon of normal weight participants. The attenuated effects in overweight and obesity are consistent with epidemiologic findings of increased colon cancer risk with increased BMI (49): Preventive strategies may be attenuated by the metabolic effects of obesity. Future models may consider overall eicosanoid balance in the colon, as opposed to PGE2 alone, as a biomarker target to better define the anti-inflammatory effects of ω-3 fatty acids. Another caveat is that our dosing model relied on a lean animal model. The observed results call for increased use of obese animal models in studies of cancer prevention to better represent the varying degree of adiposity that is found in human populations. Weight loss, avoiding a positive energy balance, or use of energy restriction mimetics may be approaches to maximize the preventive effects of ω-3 fatty acids in the setting of overweight and obesity.
Acknowledgments
We thank all the individuals who volunteered for the Fish Oil Study. We also thank Melissa K. Tuck, Chelsea Crofoot, and Brian Kleiner for their roles in assisting with study coordination, participant recruitment, sample collection, and sample processing. We thank Megan Klatt for assistance with fatty acid dosing calculations, Arsh Patel for assisting with proliferation analyses, and Gillian Graifman for assisting with the dietary data. Nordic Naturals provided the EPA-Xtra in bulk for the research study.
Grant Support
This work was supported by grants from the NIH: the Gastro-Intestinal SPORE P50 CA130810, the University of Michigan Comprehensive Cancer Center P30 CA046592, the University of Michigan Clinical Research Center UL1RR024986, the University of Pittsburgh Cancer Institute P30 CA047904, the University of Michigan Clinical Translational Resource Allocation Committee (CTRAC), the Kutsche Family Memorial Endowment (to Dean Brenner), and the Rose and Lawrence C. Page Foundation (to D. Kim Turgeon).
Footnotes
Authors’ Contributions
Conception and design: Z. Djuric, D.K. Turgeon, A. Sen, M.T. Ruffin IV, D.P. Normolle, W.L. Smith, D.E. Brenner
Development of methodology: D.K. Turgeon, A. Sen, M.T. Ruffin IV
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): Z. Djuric, D.K. Turgeon, J. Ren, K. Herman, D. Ramaswamy, M.T. Ruffin IV, D.E. Brenner
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): Z. Djuric, D.K. Turgeon, A. Sen, D. Ramaswamy, L. Zhao, M.T. Ruffin IV, D.E. Brenner
Writing, review, and/or revision of the manuscript: Z. Djuric, D.K. Turgeon, A. Sen, M.T. Ruffin IV, D.P. Normolle, W.L. Smith, D.E. Brenner
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): J. Ren, K. Herman, D.E. Brenner
Study supervision: D.K. Turgeon, A. Sen, K. Herman, D.E. Brenner
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Backlund MG, Mann JR, Dubois RN. Mechanisms for the prevention of gastrointestinal cancer: the role of prostaglandin E2. Oncology. 2005;69(Suppl 1):28–32. doi: 10.1159/000086629. [DOI] [PubMed] [Google Scholar]
- 2.Lands B. Historical perspectives on the impact of n-3 and n-6 nutrients on health. Prog Lipid Res. 2014;55C:17–29. doi: 10.1016/j.plipres.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 3.Dong L, Zou H, Yuan C, Hong YH, Kuklev DV, Smith WL. Different fatty acids compete with arachidonic acid for binding to the allosteric or catalytic subunits of cyclooxygenases to regulate prostanoid synthesis. J Biol Chem. 2016;291:4069–78. doi: 10.1074/jbc.M115.698001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wada M, DeLong CJ, Hong YH, Rieke CJ, Song I, Sidhu RS, et al. Enzymes and receptors of prostaglandin pathways with arachidonic acid-derived versus eicosapentaenoic acid-derived substrates and products. J Biol Chem. 2007;282:22254–66. doi: 10.1074/jbc.M703169200. [DOI] [PubMed] [Google Scholar]
- 5.Yuan C, Sidhu RS, Kuklev DV, Kado Y, Wada M, Song I, et al. Cyclooxygenase allosterism: fatty acid mediated cross-talk between monomers of cyclooxygenase homodimers. J Biol Chem. 2009;284:10042–55. doi: 10.1074/jbc.M808634200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang Y, Djuric Z, Sen A, Ren J, Kuklev D, Waters I, et al. Biomarkers for personalizing omega-3 fatty acid dosing. Cancer Prev Res. 2014;7:1011–22. doi: 10.1158/1940-6207.CAPR-14-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rao CV, Rivenson A, Simi B, Reddy BS. Chemoprevention of colon carcinogenesis by dietary curcumin, a naturally, occuring plant phenolic compound. Cancer Res. 1995;55:259–66. [PubMed] [Google Scholar]
- 8.Cai H, Al-Fayez M, Tunstall RG, Platton S, Greaves P, Steward WP, et al. The rice bran constituent tricin potently inhibits cyclooxygenase enzymes and interferes with intestinal carcinogenesis in ApcMin mice. Mol Cancer Ther. 2005;4:1287–92. doi: 10.1158/1535-7163.MCT-05-0165. [DOI] [PubMed] [Google Scholar]
- 9.Sale S, Tunstall RG, Ruparelia KC, Potter GA, Steward WP, Gescher AJ. Comparison of the effects of the chemopreventive agent resveratrol and its synthetic analog trans 3,4,5,4′-tetramethoxystilbene (DMU-212) on adenoma development in the Apc(Min+) mouse and cyclooxygenase-2 in human-derived colon cancer cells. Int J Cancer. 2005;115:194–201. doi: 10.1002/ijc.20884. [DOI] [PubMed] [Google Scholar]
- 10.Reddy BS, Rao CV, Rivenson A, Kelloff G. Inhibitory effect of aspirin on azoxymethane-induced colon carcinogenesis in F344 rats. Carcinogenesis. 1993;14:1493–7. doi: 10.1093/carcin/14.8.1493. [DOI] [PubMed] [Google Scholar]
- 11.Shen G, Khor TO, Hu R, Yu S, Nair S, Ho CT, et al. Chemoprevention of familial adenomatous polyposis by natural dietary compounds sulforaphane and dibenzoylmethane alone and in combination in ApcMin/+ mouse. Cancer Res. 2007;67:9937–44. doi: 10.1158/0008-5472.CAN-07-1112. [DOI] [PubMed] [Google Scholar]
- 12.Montrose DC, Nakanishi M, Murphy RC, Zarini S, McAleer JP, Vella AT, et al. The role of PGE2 in intestinal inflammation and tumorigenesis. Prostaglandins Other Lipid Mediat. 2015:116–117. 26–36. doi: 10.1016/j.prostaglandins.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang Y, Turgeon DK, Wright BD, Sidahmed E, Ruffin MT, Brenner DE, et al. Effect of ginger root on cyclooxygenase-1 and 15-hydroxyprostaglandin dehydrogenase expression in colonic mucosa of humans at normal and increased risk for colorectal cancer. Eur J Cancer Prev. 2013;22:455–60. doi: 10.1097/CEJ.0b013e32835c829b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim KA, Gu W, Lee IA, Joh EH, Kim DH. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS One. 2012;7:e47713. doi: 10.1371/journal.pone.0047713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delage B, Rullier A, Capdepont M, Rullier E, Cassand P. The effect of body weight on altered expression of nuclear receptors and cyclooxygenase-2 in human colorectal cancers. Nutr J. 2007;6:20. doi: 10.1186/1475-2891-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez ME, Heddens D, Earnest DL, Bogert CL, Roe D, Einspahr J, et al. Physical activity, body mass index, and prostaglandin E2 levels in rectal mucosa. J Natl Cancer Inst. 1999;91:950–3. doi: 10.1093/jnci/91.11.950. [DOI] [PubMed] [Google Scholar]
- 17.Pereira C, Queiros S, Galaghar A, Sousa H, Pimentel-Nunes P, Brandao C, et al. Genetic variability in key genes in prostaglandin E2 pathway (COX-2, HPGD, ABCC4 and SLCO2A1) and their involvement in colorectal cancer development. PLoS One. 2014;9:e92000. doi: 10.1371/journal.pone.0092000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poole EM, Hsu L, Xiao L, Kulmacz RJ, Carlson CS, Rabinovitch PS, et al. Genetic variation in prostaglandin E2 synthesis and signaling, prostaglandin dehydrogenase, and the risk of colorectal adenoma. Cancer Epidemiol Biomarkers Prev. 2010;19:547–57. doi: 10.1158/1055-9965.EPI-09-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roza AM, Shizgal HM. The Harris Benedict equation reevaluated: resting energy requirements and the body cell mass. Am J Clin Nutr. 1984;40:168–82. doi: 10.1093/ajcn/40.1.168. [DOI] [PubMed] [Google Scholar]
- 20.Djuric Z, Turgeon DK, Ren J, Neilson A, Plegue M, Waters IG, et al. Effects of a mediterranean diet intervention on anti- and pro-inflammatory eicosanoids, epithelial proliferation, and nuclear morphology in biopsies of normal colon tissue. Nutr Cancer. 2015;67:721–9. doi: 10.1080/01635581.2015.1029637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neilson AP, Ren J, Hong YH, Sen A, Smith WL, Brenner DE, et al. Effect of fish oil on levels of R- and S-enantiomers of 5-, 12-, and 15-hydroxyeico-satetraenoic acids in mouse colonic mucosa. Nutr Cancer. 2012;64:163–72. doi: 10.1080/01635581.2012.630168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren J, Mozurkewich EL, Sen A, Vahratian AM, Ferreri TG, Morse AN, et al. Total serum fatty acid analysis by GC-MS: assay validation and serum sample stability. Curr Pharm Anal. 2013;9:331–9. doi: 10.2174/1573412911309040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malkowski MG, Thuresson ED, Lakkides KM, Rieke CJ, Micielli R, Smith WL, et al. Structure of eicosapentaenoic and linoleic acids in the cyclooxygenase site of prostaglandin endoperoxide H synthase-1. J Biol Chem. 2001;276:37547–55. doi: 10.1074/jbc.M105982200. [DOI] [PubMed] [Google Scholar]
- 24.Neilson AP, Djuric Z, Ren J, Hong YH, Sen A, Lager C, et al. Effect of cyclooxygenase genotype and dietary fish oil on colonic eicosanoids in mice. J Nutr Biochem. 2012;23:966–76. doi: 10.1016/j.jnutbio.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zuo X, Shureiqi I. Eicosanoid profiling in colon cancer: emergence of a pattern. Prostaglandins Other Lipid Mediat. 2013:104–105. 139–43. doi: 10.1016/j.prostaglandins.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wasilewicz MP, Kolodziej B, Bojulko T, Kaczmarczyk M, Sulzyc-Bielicka V, Bielicki D, et al. Overexpression of 5-lipoxygenase in sporadic colonic adenomas and a possible new aspect of colon carcinogenesis. Int J Colorectal Dis. 2010;25:1079–85. doi: 10.1007/s00384-010-0980-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Honn K, Tang D, Gao X, Butovich I, Liu B, Timar J, et al. 12-lipoxygenases and 12(S)-HETE: role in cancer metastasis. Cancer Metastases Rev. 1994;13:365–96. doi: 10.1007/BF00666105. [DOI] [PubMed] [Google Scholar]
- 28.Pirman DA, Efuet E, Ding XP, Pan Y, Tan L, Fischer SM, et al. Changes in cancer cell metabolism revealed by direct sample analysis with MALDI mass spectrometry. PLoS One. 2013;8:e61379. doi: 10.1371/journal.pone.0061379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang X, Hu S, Xu B, Snyder GD, Harmon S, Yao J, et al. 14,15-Dihydroxyeicosatrienoic acid activates peroxisome proliferator-activated receptor-alpha. Am J Physiol Heart Circ Physiol. 2006;290:H55–63. doi: 10.1152/ajpheart.00427.2005. [DOI] [PubMed] [Google Scholar]
- 30.Park SW, Heo DS, Sung MW. The shunting of arachidonic acid metabolism to 5-lipoxygenase and cytochrome p450 epoxygenase antagonizes the anti-cancer effect of cyclooxygenase-2 inhibition in head and neck cancer cells. Cell Oncol (Dordr) 2012;35:1–8. doi: 10.1007/s13402-011-0051-7. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki S, Oguro A, Osada-Oka M, Funae Y, Imaoka S. Epoxyeicosatrienoic acids and/or their metabolites promote hypoxic response of cells. J Pharmacol Sci. 2008;108:79–88. doi: 10.1254/jphs.08122fp. [DOI] [PubMed] [Google Scholar]
- 32.Tang D, Porter A, Honn K. Critical role of arachindonate lipoxygenases in regulating apoptosis. Adv Exp Med Biol. 1997;407:405–11. doi: 10.1007/978-1-4899-1813-0_61. [DOI] [PubMed] [Google Scholar]
- 33.Gabbs M, Leng S, Devassy JG, Monirujjaman M, Aukema HM. Advances in our understanding of oxylipins derived from dietary PUFAs. Adv Nutr. 2015;6:513–40. doi: 10.3945/an.114.007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shureiqi I, Chen D, Lee JJ, Yang P, Newman RA, Brenner DE, et al. 15-LOX-1: a novel molecular target of nonsteroidal anti-inflammatory drug-induced apoptosis in colorectal cancer cells. J Natl Cancer Inst. 2000;92:1136–42. doi: 10.1093/jnci/92.14.1136. [DOI] [PubMed] [Google Scholar]
- 35.Shureiqi I, Wu Y, Chen D, Yang XL, Guan B, Morris JS, et al. The critical role of 15-lipoxygenase-1 in colorectal epithelial cell terminal differentiation and tumorigenesis. Cancer Res. 2005;65:11486–92. doi: 10.1158/0008-5472.CAN-05-2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bartram HP, Gostner A, Reddy BS, Rao CV, Scheppach W, Dusel G, et al. Missing anti-proliferative effect of fish oil on rectal epithelium in healthy volunteers consuming a high-fat diet: potential role of the n-3:n-6 fatty acid ratio. Eur J Cancer Prev. 1995;4:231–7. doi: 10.1097/00008469-199506000-00004. [DOI] [PubMed] [Google Scholar]
- 37.Del Bas JM, Caimari A, Rodriguez-Naranjo MI, Childs CE, Paras Chavez C, West AL, et al. Impairment of lysophospholipid metabolism in obesity: altered plasma profile and desensitization to the modulatory properties of n-3 polyunsaturated fatty acids in a randomized controlled trial. Am J Clin Nutr. 2016;104:266–79. doi: 10.3945/ajcn.116.130872. [DOI] [PubMed] [Google Scholar]
- 38.Kantor ED, Lampe JW, Kratz M, White E. Lifestyle factors and inflammation: associations by body mass index. PLoS One. 2013;8:e67833. doi: 10.1371/journal.pone.0067833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yee LD, Lester JL, Cole RM, Richardson JR, Hsu JC, Li Y, et al. Omega-3 fatty acid supplements in women at high risk of breast cancer have dose-dependent effects on breast adipose tissue fatty acid composition. Am J Clin Nutr. 2010;91:1185–94. doi: 10.3945/ajcn.2009.29036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pendyala S, Neff LM, Suarez-Farinas M, Holt PR. Diet-induced weight loss reduces colorectal inflammation: implications for colorectal carcinogenesis. Am J Clin Nutr. 2011;93:234–42. doi: 10.3945/ajcn.110.002683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhattacharya A, Chandrasekar B, Rahman MM, Banu J, Kang JX, Fernandes G. Inhibition of inflammatory response in transgenic fat-1 mice on a calorie-restricted diet. Biochem Biophys Res Commun. 2006;349:925–30. doi: 10.1016/j.bbrc.2006.08.093. [DOI] [PubMed] [Google Scholar]
- 42.Micallef M, Munro I, Phang M, Garg M. Plasma n-3 polyunsaturated fatty acids are negatively associated with obesity. Br J Nutr. 2009;102:1370–4. doi: 10.1017/S0007114509382173. [DOI] [PubMed] [Google Scholar]
- 43.Kim JY, Park JY, Kim OY, Ham BM, Kim HJ, Kwon DY, et al. Metabolic profiling of plasma in overweight/obese and lean men using ultra performance liquid chromatography and Q-TOF mass spectrometry (UPLC-Q-TOF MS) J Proteome Res. 2010;9:4368–75. doi: 10.1021/pr100101p. [DOI] [PubMed] [Google Scholar]
- 44.Gronn M, Gorbitz C, Christensen E, Levorsen A, Ose L, Hagve TA, et al. Dietary n-6 fatty acids inhibit the incorporation of dietary n-3 fatty acids in thrombocyte and serum phospholipids in humans: a controlled dietetic study. Scand J Clin Lab Invest. 1991;51:255–63. doi: 10.3109/00365519109091612. [DOI] [PubMed] [Google Scholar]
- 45.Liou YA, King DJ, Zibrik D, Innis SM. Decreasing linoleic acid with constant alpha-linolenic acid in dietary fats increases (n-3) eicosapentaenoic acid in plasma phospholipids in healthy men. J Nutr. 2007;137:945–52. doi: 10.1093/jn/137.4.945. [DOI] [PubMed] [Google Scholar]
- 46.Taha AY, Cheon Y, Faurot KF, Macintosh B, Majchrzak-Hong SF, Mann JD, et al. Dietary omega-6 fatty acid lowering increases bioavailability of omega-3 polyunsaturated fatty acids in human plasma lipid pools. Prostaglandins Leukot Essent Fatty Acids. 2014;90:151–7. doi: 10.1016/j.plefa.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Madsen L, Kristiansen K. Of mice and men: factors abrogating the anti-obesity effect of omega-3 fatty acids. Adipocyte. 2012;1:173–6. doi: 10.4161/adip.20689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang X, Margolis KL, Hendryx M, Rohan TE, Groessl EJ, Thomson CA, et al. Metabolic phenotype and risk of colorectal cancer in normal-weight postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2017;26:1–7. doi: 10.1158/1055-9965.EPI-16-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kyrgiou M, Kalliala I, Markozannes G, Gunter MJ, Paraskevaidis E, Gabra H, et al. Adiposity and cancer at major anatomical sites: umbrella review of the literature. BMJ. 2017;356:j477. doi: 10.1136/bmj.j477. [DOI] [PMC free article] [PubMed] [Google Scholar]