

Abstract
The COPII coat complex, which mediates secretory cargo trafficking from the endoplasmic reticulum, is a key control point for subcellular protein targeting. Because misdirected proteins cannot function, protein sorting by COPII is critical for establishing and maintaining normal cell and tissue homeostasis. Indeed, mutations in COPII genes cause a range of human pathologies, including cranio-lenticulo-sutural dysplasia (CLSD), which is characterized by collagen trafficking defects, craniofacial abnormalities and skeletal dysmorphology. Detailed knowledge of the COPII pathway is required to understand its role in normal cell physiology and to devise new treatments for disorders in which it is disrupted. However, little is known about how vertebrates dynamically regulate COPII activity in response to developmental, metabolic or pathological cues. Several COPII proteins are modified by O-linked β-N-acetylglucosamine (O-GlcNAc), a dynamic form of intracellular protein glycosylation, but the biochemical and functional effects of these modifications remain unclear. Here, we use a combination of chemical, biochemical, cellular and genetic approaches to demonstrate that site-specific O-GlcNAcylation of COPII proteins mediates their protein-protein interactions and modulates cargo secretion. In particular, we show that individual O-GlcNAcylation sites of SEC23A, an essential COPII component, are required for its function in human cells and vertebrate development, because mutation of these sites impairs SEC23A-dependent in vivo collagen trafficking and skeletogenesis in a zebrafish model of CLSD. Our results indicate that O-GlcNAc is a conserved and critical regulatory modification in the vertebrate COPII-dependent trafficking pathway.
Table of Contents figure
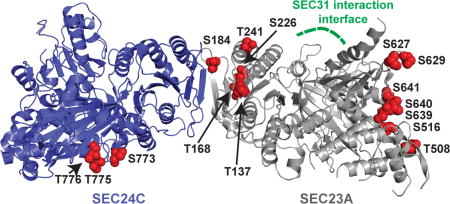
We report sites and functions of the O-linked β-N-acetylglucosamine (O-GlcNAc) modification on components of the vertebrate COPII protein trafficking pathway. Shown is a model of the complex of human SEC23A (silver, PDB: 5VNO) and SEC24C (blue, PDB: 3EH2), with the O-GlcNAc sites discovered in this report indicated in red.
Introduction
One-third of eukaryotic proteins pass through the secretory pathway for targeting to specific locations, including the endoplasmic reticulum (ER), Golgi, plasma membrane and extracellular space1–3. As a result, properly regulated protein secretion is required for cellular and organismal physiology. In particular, the coat protein complex II (COPII), which mediates anterograde trafficking from the ER, is a key component of the early secretory pathway4–9. The formation of a COPII transport carrier begins when the cytosolic GTPase SAR1 binds GTP and inserts into the ER membrane, a process facilitated by the ER-anchored guanine nucleotide exchange factor SEC124–6. Active SAR1-GTP recruits SEC23/SEC24 heterodimers, which effect cargo loading into the nascent carriers via protein-protein interactions among SEC24 and client or adaptor proteins4–6, 10, 11. Then, heterotetramers of SEC13/SEC31 assemble over the SAR1/SEC23/SEC24 pre-budding complex, forming the outer layer of a polyhedral cage that promotes membrane curvature and scission, resulting in a mature COPII transport carrier4–6.
The COPII pathway is required for protein sorting and cell viability in a wide range of organisms4–6, and genetic defects in COPII components cause a variety of human diseases, including cranio-lenticulo-sutural dysplasia (CLSD), hematological disorders, a subtype of osteogenesis imperfecta (OI), and multiple hereditary spastic paraplegias4–6, 12–25. Interestingly, the molecular etiology of these diseases can often be attributed to aberrant trafficking of particular COPII client cargoes. For example, mutations in SEC23A (which cause CLSD) and SEC24D (which cause a subtype of OI) both disrupt collagen trafficking, leading to chondrocyte dysfunction, impaired skeletogenesis, craniofacial disease and bone deformities15, 18, 21–23, 26–30. Despite this broad pathophysiological importance, major aspects of COPII trafficking remain poorly understood. For instance, while the fundamental steps of COPII assembly are relatively well characterized, little is known about how vertebrate cells modulate this activity in response to developmental cues, fluctuating signals, metabolic demands, or stress4–6. The COPII cargo load changes dramatically in both normal (e.g., stimulated B cells or differentiating pancreatic β islets) and pathological (e.g., nutrient, redox, or proteostasis stress) contexts31–37, but the mechanisms through which the COPII machinery adjusts to these changes are largely unclear.
Post-translational modifications (PTMs) represent one likely general mode of COPII pathway regulation. For example, phosphorylation38–44 and ubiquitination45–47 of individual COPII proteins govern particular aspects of vesicular trafficking. Recently, we48 and others49–52 have also shown that multiple COPII components – including SEC23, SEC24 and SEC31 – are modified by O-linked β-N-acetylglucosamine (O-GlcNAc) in mammalian cells. O-GlcNAc is a major intracellular PTM, reversibly decorating serine and threonine side-chains of thousands of nuclear, cytoplasmic and mitochondrial proteins, and is broadly conserved across animals, plants and perhaps some fungi53–56. In animals, O-GlcNAc is added by O-GlcNAc transferase (OGT) and removed by O-GlcNAcase (OGA), both ubiquitous nuclear/cytoplasmic enzymes53–56. O-GlcNAc cycling controls myriad processes, including cell metabolism, cell cycle progression and cell death53–56, and is essential, as genetic ablation of OGT or OGA is lethal in mice57, 58. Because several core COPII proteins are O-GlcNAcylated48–52, we reasoned that this intracellular glycosylation might regulate protein trafficking. Consistent with this notion, SEC24 O-GlcNAcylation is high during interphase but low in mitosis, when COPII-dependent trafficking is suspended through an unknown mechanism50, suggesting that O-GlcNAc may dynamically control COPII function during cell cycle progression. However, the mechanistic and functional effects of O-GlcNAc on the COPII pathway have not been explored.
Here, we address the prevalence, biochemical effects, and physiological importance of O-GlcNAcylation in the COPII pathway. We show that O-GlcNAc is widespread in the COPII system, and that site-specific glycosylation of key COPII proteins impacts protein secretion. Moreover, we use a chemical biology approach to demonstrate that dynamic O-GlcNAcylation mediates the protein-protein interactions of COPII components, identifying a new potential biochemical mechanism for cargo trafficking regulation by this PTM. Using a genome-engineered human cell model, we show that individual glycosylation sites of SEC23A, an essential COPII component, are required to traffic endogenous collagen. Finally, we demonstrate that mutation of specific O-GlcNAc sites impairs in vivo SEC23A-dependent collagen trafficking in a zebrafish model of CLSD. Together, our results establish O-GlcNAc as a conserved and prevalent regulatory modification in the vertebrate COPII pathway.
Materials and Experimental Details
Chemical synthesis
5SGlcNAc was synthesized as described59 and was a gift of Dr. Benjamin M. Swarts (Central Michigan University). Thiamet-G and Ac3GlcNDAz-1-P(Ac-SATE)2 (“GlcNDAz”) were synthesized as described60, 61 by the Duke Small Molecule Synthesis Facility. All other chemicals were purchased from Sigma-Aldrich unless otherwise indicated.
Western blotting
Samples were resolved on Tris-glycine SDS-PAGE gels and electroblotted onto PVDF membrane (88518, ThermoFisher) using standard methods62. Membranes were blocked with Tris-buffered saline with 0.1% Tween (TBST) with 5% bovine serum albumin (BSA). All antibody dilutions were prepared in TBST with 5% BSA. Membranes were incubated with primary antibodies overnight at 4 °C, washed three times in TBST, incubated with secondary antibodies for 1 hour at room temperature, washed three times in TBST, and developed via enhanced chemiluminescence (ECL) according to the manufacturer’s instructions (WesternBright ECL, Advansta). The following primary antibodies were used: rabbit anti-SEC23A (8162, Cell Signaling Technology; 1:2000), rabbit anti-SEC23A serum (gift of Dr. David Ginsburg, University of Michigan, 1:2000), rabbit anti-SEC23B serum (gift of Dr. David Ginsburg, University of Michigan, 1:2000) rabbit anti-SEC24C (8531, Cell Signaling Technology; 1:1000), rabbit anti-SEC24D (9610, Cell Signaling Technology; 1:1000), rabbit anti-SEC24B (7427, Cell Signaling Technology; 1:1000), rabbit anti-SEC31A (A302-336A, Bethyl; 1:2000), rabbit anti-TFG (A302-343A, Bethyl; 1:4000), mouse monoclonal α-tubulin (T6074, Sigma-Aldrich; 1:100000), rabbit monoclonal GAPDH (14C10) (2188, Cell signaling Technology; 1:4000), mouse monoclonal anti-c-myc (9E10) (various vendors), mouse monoclonal anti-O-GlcNAc antibody 9D1 (MA1-039, ThermoFisher; 1:1000), mouse monoclonal anti-O-GlcNAc antibody RL2 (SC-59624, Santa Cruz Biotech; 1:500), rabbit anti-TRAPα (gift of Dr. Chris Nicchitta, Duke University, 1:8000). The following secondary antibodies were used: goat anti-mouse IgG (1030-05, horseradish peroxidase (HRP)-conjugated, SouthernBiotech; 1:10000), goat anti-rabbit IgG (4030-05, HRP-conjugated, SouthernBiotech; 1:10000).
Cell culture
293T, HeLa, HeLa/UAP1(F383G), 293T/UAP1(F383G), COS7(tsVSVG-eGFP) and SW1353 (including all engineered derivative lines) were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, 100 units/ml penicillin, and 100 µg/ml streptomycin in 5% CO2 at 37 °C. Transduced cell lines were also supplemented with puromycin during regular passaging at the following concentrations: 1.5 µg/ml for HeLa/UAP1(F383G); 2 µg/ml for 293T/UAP1(F383G) and COS7(tsVSVG-eGFP).
Mammalian expression vectors
pSin-EF2-UAP1(F383G) was a gift from Dr. Jennifer Kohler, UT Southwestern60. pmTurquoise2-Golgi was a gift from Dr. Dorus Gadella (Addgene plasmid # 36205). mCherry-ER-3 was a gift from Dr. Michael Davidson (Addgene plasmid # 55041). The pMSCV tsVSVG-eGFP was generated by cutting tsVSVG-eGFP from the parent vector (pEGFP-VSVG, Addgene, plasmid #11912) with BglII and NotI and ligating it into pMSCV via standard methods62. pcDNA4-SEC23A-myc-6×His, pcDNA4-SEC31A-myc-6×His, and pcDNA3-myc-6×His-SEC24C, p3×FLAG-SEC24C, pcDNA4-TFG-myc-6×His and p3×FLAG-TFG were generated by amplifying the open reading frames from their respective cDNA and ligating into pcDNA4/myc-6×His, pcDNA3-myc-6×His, or p3×FLAG-CMV-10 using standard methods62. Primers for site-directed mutagenesis were designed using QuikChange Primer Design (http://www.genomics.agilent.com/primerDesignProgram.jsp). Mutagenesis reactions were performed using Phusion polymerase (M0530, New England Biolabs) essentially according to the manufacturer’s instructions, but with an extra initial 30-second 98 °C denaturation step prior to the addition of polymerase. Mutagenesis reactions were digested with DpnI and transformed into One Shot TOP10 chemically competent E. coli (C404010, ThermoFisher). The following primers and their reverse-complements were used for site-directed mutagenesis:
S97 5’-CAGTTCAGATATACCAGCATAAGCAGGTGGAAACTGATTCCTTTGG-3’
S102 5’-AGGCTGATTCAGTTCAGCTATACCAGCATAACTAGGTG-3’
S115A: CATATTCAATGCTAGCAAACTGAGGTAAAAGTTCAGCAGGCT
S116A: CACGCAGAACTACATATTCAATGGCAGAAAACTGAGGTAAAAGTTCAG
T137A: AAATCTTCATCTTCCATGCAAGCATCAACCACATAGAGGAATATC
T168A: CCATTCTCCCAAAAGCAATAAGTCCAACCAAAGCTGTAGGT
S184 5’-CTCTGAAGACATAGCTTTTTGCAATGCCTTCACATCCAAGTTC-3’
S226A: GTAAGAATCTGTTGGCAGGAGGTGGCTGCTGTAC
T241A: GAGTTCTCCCAGAAGATCTGCGAGATTCATGTCTATTTTCT
T508A: TGTTTTGGATTTGAGCTTGAGCATCTGCCCAGTTCC
S516A: TGCCTCCTGGTCAAAAGCTGCAGCAATGTTTTGGA
S627 5’-CCTATCCTGTATGCGTATGCTTTTAGTGGACCACCAG-3’
S629 5’-CTCTGGTGGTCCAGCAAAAGAATACGCATACAGGATAGGC-3’
S639 5’-GCAAGAATGCTACTGGCATCAAGAAGAACCGGCTCTGGT-3’
S640 5’-ACGATCTGCAAGAATGCTAGCGCTATCAAGAAGAACCGGC-3’
S641 5’-GAATACGATCTGCAAGAATGGCACTGCTATCAAGAAGAACCG-3’
Stable cell line creation
UAP1(F383G) and tsVSVG-eGFP retro- and lentiviruses were generated in 293T-17 cells using calcium phosphate transfection and standard methods as described48. Medium containing virus was filtered through pre-wetted 0.45 µm PVDF syringe filters and 1 ml of viral supernatant was added directly to 10 cm plates of 50% confluent cells. HeLa/UAP1(F383G) and 293T/UAP1(F383G) were selected using 1.5 µg/ml and 2 µg/ml puromycin, respectively60. COS7(tsVSVG-eGFP) were selected using 2 µg/ml puromycin until cells exhibited green ER fluorescence.
Immunoprecipitation and tandem affinity purification
Cells transfected with myc-6×His- or 3×FLAG-tagged proteins were lysed in immunoprecipitation (IP) lysis buffer (1% Triton X-100, 0.1% SDS, 150 mM NaCl, 1 mM EDTA, 20 mM Tris-HCl pH 7.4) supplemented with protease inhibitor cocktail (P8340, Sigma), plus 50 µM UDP, and 5 µM PUGNAc (17151, Cayman Chemicals) to inhibit endogenous OGT and hexosaminidases, respectively. Lysates were probe-sonicated, cleared by centrifugation and quantified by BCA protein assay (23225, ThermoFisher) according to the manufacturer’s instructions. IPs were performed on 2–10 mg total protein (for WB) or 50–100 mg total protein (for MS analysis). Cleared lysates were adjusted to final total protein concentration of ~2–5 mg/ml using IP lysis buffer supplemented with protease inhibitor, UDP and PUGNAc, and 2 µg of mouse monoclonal anti-c-myc (9E10) (sc-40, Santa Cruz Biotech) per 1 mg of total protein were added and rotated overnight at 4 °C. 50 µl washed protein A/G UltraLink Resin (53133, ThermoFisher) were added to the lysate and rotated at room temperature for 1 hour. Beads were washed three times with 1 ml of IP lysis buffer and then eluted. For IPs, elution was performed with IP lysis buffer plus 2× SDS-PAGE loading buffer (5× SDS-PAGE loading buffer: 250 mM Tris pH 6.8, 10% SDS, 30% glycerol, 5% β-mercaptoethanol, 0.02% bromophenol blue) and heating at 95 °C for 5 minutes. Eluents were then analyzed by SDS-PAGE and WB. For tandem affinity purification, beads were eluted twice in 500 µl using Ni-NTA wash buffer (8 M urea, 300 mM NaCl, 1% Triton X-100, 5 mM imidazole) with rotation. The two 500 µl elutions were pooled, and 50 µl washed 6×HisPur Ni-NTA resin (88223, ThermoFisher) were added to the eluate and rotated for 2 hours at room temperature. The Ni-NTA resin was washed three times with 1 ml of Ni-NTA wash buffer. Final elution from the Ni-NTA was performed using 8 M urea with 250 mM imidazole.
Digitonin fractionation
Cells were seeded such that they were ~80% confluent in a 10 cm plate on the day of harvest. Cells were washed with 10 ml cold PBS and incubated on ice for 10 minutes. PBS was removed from the plate and 1 ml permeabilization buffer (110 mM KOAc, 25 mM HEPES pH 7.2, 2.5 mM Mg(OAc)2, 1 mM EGTA, 0.015% digitonin, 1 mM DTT, protease inhibitor cocktail) was added, taking care to not dislodge the cells, and incubated on ice for 5 minutes. Soluble material was collected from the plate into a new chilled 1.5 ml centrifuge tube and retained as the cytoplasmic fraction. The remaining cells were washed gently with 5 ml of wash buffer (110 mM KOAc, 25 mM HEPES pH 7.2, 2.5 mM Mg(OAc)2, 1 mM EGTA, 0.004% digitonin, 1 mM DTT). After washing, the cells were incubated on ice in IP lysis buffer supplemented with protease inhibitor cocktail for 5 minutes and then scraped and moved into a chilled 1.5 ml centrifuge tube to obtain the membrane fraction. Both fractions were probe-sonicated and cleared by centrifugation. Equal volumes from each fraction were methanol/chloroform-precipitated to concentrate. Fractions were analyzed by SDS-PAGE and WB.
Densitometry
Western blot band densitometry readings were performed on scanned blot images using the gel analyzer tool within the Fiji software package63. Raw densitometry values were used to calculate cytoplasmic:membrane ratios for each protein within each experimental replicate.
tsVSVG-eGFP imaging
Two days prior to imaging, 200,000 COS7(tsVSVG-eGFP) cells were plated onto 35 mm glass-bottom dishes (P35GCol-1.5-14-C, MatTek) in phenol red-free Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, 10 mM HEPES pH 7.4, 100 units/ml penicillin, and 100 µg/ml streptomycin in 5% CO2. Sixteen hours prior to imaging, cells were shifted to 40 °C and 5% CO2; eight hours prior to imaging cells were treated with either vehicle (DMSO) or 25 µM Thiamet-G. Cells were then shifted to the permissive temperature (32 °C and 5% CO2) and images were collected every 10 seconds for 20 minutes. Imaging was performed on a Zeiss LSM 780 inverted confocal microscope, using 40×/1.4 NA Oil-Plan Apochromat DIC, (UV) VIS-IR (420762-9900), Zen 2011 software (Duke Light Microscopy Core Facility).
Transfections
Cells plated at ~50% confluence (or ~20% confluence, for GlcNDAz labeling experiments – see below) were transfected the following day. 750 µl pre-warmed OPTI-MEM (11058021, ThermoFisher) was placed 1.5 ml tubes with 30 µl TransIT-293 transfection reagent (Mirus), vortexed briefly, and incubated for 15 minutes at room temperature. After incubation, 10 µg of plasmid DNA were added to a 1.5 ml tube, vortexed briefly and incubated for 15 minutes at room temperature. After the final incubation, the transfection mixture was added dropwise to the cells. For GlcNDAz crosslinking experiments (see below), GlcNDAz was added 24 hours after transfection. For other experiments, cells were harvested 24 to 48 hours post-transfection.
HRP secretion assay
293T cells were plated on 10 cm dishes, cultured until 80% confluent, and transfected with 20 µg ssHRP construct using TransIT-LT1 transfect reagent (Mirus) as above. Twenty-four hours post-transfection (and four hours before collecting supernatant), cells were pre-treated with 5SGlcNAc or vehicle only. After four hours, medium was aspirated and cells were gently washed with PBS. Fresh medium containing either 5SGlcNAc or vehicle was re-added. At each time point, 50 µl of medium was removed from the plate and stored at 4 °C until analysis. To quantify ssHRP secretion, 10 µl of medium from each time-point was added to a clear-bottom white 96-well plate with 50 µl of pre-mixed ECL solution (WesternBright ECL, Advansta), and luminescence was measured on a Spectramax M5e plate reader (Molecular Devices) with an integration time of 500 milliseconds.
Cell viability assay
293T cells were transfected with the ssHRP plasmid as above. After 24 hours, cells were trypsinized and 25,000 cells in 100 µl DMEM were seeded into clear-bottom 96-well plates and treated as described above. After an additional 24 hours, the MTS assay was performed according to manufacturer’s protocol (Promega, CellTiter 96 AQueous Proliferation Assay kit, G3581). Reported values are an average of technical triplicate reads minus the absorbance from control wells containing medium only.
GlcNDAz crosslinking
Cells stably expressing UAP1(F383G) were plated at ~50% confluence without puromycin. If transfecting first, cells were plated at ~20% confluence. The next day, 100 µM GlcNDAz60 or vehicle was added to the cells. Medium was changed and fresh GlcNDAz was added every 24 hours. After 48–72 hours of GlcNDAz treatment, plates were placed on ice, lids were removed, and medium was replaced with ice-cold PBS. Cells were irradiated with a 365 nm UV lamp (Blak-Ray XX-20BLB UV Bench Lamp, 95-0045-04) for 20 minutes to induce crosslinking. Cells were collected by scraping into PBS and centrifugation, lysed in IP lysis buffer, and analyzed by BCA assay, SDS-PAGE and WB.
Mass spectrometry (MS) O-GlcNAc site mapping
Eight hours prior to harvest, 293T-17 cells transfected with myc-6×His-tagged COPII components were treated with 25 µM Thiamet-G and 4 mM glucosamine. Tandem affinity-purified SEC23A, SEC24C or SEC31A was separated by SDS-PAGE, visualized by colloidal Coomassie blue, excised, and subjected to a standardized in-gel trypsin digestion (http://www.genome.duke.edu/cores/proteomics/sample-preparation/documents/IngelDigestionProtocolrevised.pdf). Extracted peptides were lyophilized to dryness and resuspended in 20 µl of 0.2% formic acid/2% acetonitrile. Each sample was subjected to chromatographic separation on a Waters NanoAquity UPLC equipped with a 1.7 µm BEH130 C18 75 µm I.D. × 250 mm reversed-phase column. The mobile phase consisted of (A) 0.1% formic acid in water and (B) 0.1% formic acid in acetonitrile. Following a 2 µl injection, peptides were trapped for 3 minutes on a 5 µm Symmetry C18 180 µm I.D. × 20 mm column at 5 µl/minute in 99.9% A. The analytical column was then switched inline and a linear elution gradient of 5% B to 40% B was performed over 90 minutes at 400 nl/minute. The analytical column was connected to a fused silica PicoTip emitter (New Objective, Cambridge, MA) with a 10 µm tip orifice and coupled to a QExactive Plus mass spectrometer (ThermoFisher) through an electrospray interface operating in a data-dependent mode of acquisition. The instrument was set to acquire a precursor MS scan from m/z 375–1675 with MS/MS spectra acquired for the ten most abundant precursor ions. For all experiments, high-energy collisional dissociation (HCD) energy settings were 27 V, and a 120-second dynamic exclusion was employed for previously fragmented precursor ions.
Raw LC-MS/MS data files were processed in Proteome Discoverer (ThermoFisher) and then submitted to independent Mascot searches (Matrix Science) against a SwissProt database (human taxonomy) containing both forward and reverse entries of each protein (20,322 forward entries). Search tolerances were 5 parts per million for precursor ions and 0.02 Da for product ions using semi-trypsin specificity with up to two missed cleavages. Carbamidomethylation (+57.0214 Da on Cys) was set as a fixed modification, whereas oxidation (+15.9949 Da on Met), deamidation (+0.98 Da on Asn/Gln), O-GlcNAc (+203.0793 Da on Ser/Thr), and phosphorylation (+79.9663 Da on Ser/Thr/Tyr) were considered dynamic mass modifications. All searched spectra were imported into Scaffold (v4.7, Proteome Software) and scoring thresholds were set to achieve a peptide false discovery rate below 1% using the PeptideProphet algorithm. Localized O-GlcNAcylated residues were manually verified based on annotated mass spectra generated within Mascot. In the SEC23A sample, peptides assigned by Mascot to SEC23B were assumed to be the cognate peptides in SEC23A, since the SEC23A was purified to homogeneity.
Generation of SEC23A−/− SW1353 cells
LentiCas9 virus was obtained from the Duke Functional Genomics Facility. SW1353 cells were plated in 6-well plates to be ~50% confluent at time of infection. Prior to infection, medium was replaced with fresh medium containing 4 µg/ml polybrene and 50 µl of LentiCas9 virus was added drop-wise to the cells. Plates were then centrifuged at 700 g for 30 minutes and incubated at 37 °C and 5% CO2 overnight. The following morning, medium was replaced and cells were allowed to recover for 48 hours. After recovery, cells were selected using 3 µg/ml blasticidin and passaged until a control, uninfected plate, contained no living cells. These cells were dubbed SW1353/Cas9.
A single-guide RNA (sgRNA) sequence spanning the start codon of the human SEC23A locus was designed and validated via the Surveyor assay64 by the Duke Functional Genomics Facility:
5’ - TTCCAAATAGGTTGTCATTG - 3’
An sgRNA targeting AAVS1, commonly used as a control and deemed the “safe harbor” virus (SHV) locus65, was used a control. Lentiviruses encoding the SEC23A sgRNA and the AAVS1 sgRNA were obtained from the Duke Functional Genomics Facility.
SW1353/Cas9 cells were plated in 6-well plates to be ~50% confluent at time of infection. Prior to infection, medium was replaced with fresh media containing 4 µg/ml polybrene and 50 µl of SEC23A or AAVS1 sgRNA lentivirus was added drop-wise to the cells. Plates were then centrifuged at 700 g for 30 minutes and incubated at 37 °C and 5% CO2 overnight. The following morning, medium was replaced and cells were allowed to recover for 48 hours. After recovery, infected cells were selected using 0.25 µg/ml puromycin and passaged until a control, uninfected plate, contained no living cells. Reduction of SEC23A expression was verified on a mixed population level via WB.
Next, using serial dilution, single cells were plated in 96-well plates for both SEC23A and AAVS1 sgRNA-containing populations, and clones were allowed to recover and expand. Cells were transferred to larger plates under selective pressure of 3 µg/ml blasticidin and 0.25 µg/ml puromycin. WB blot was used to verify loss of SEC23A expression. One SEC23A−/− clone (SW23A-3) and one AAVS1 safe harbor control clone (SHV-23) were used in subsequent reconstitution experiments.
Generation of stable SEC23A-reconstituted SW23A-3
SW23A-3 were plated to be ~30% confluent in 6-well plates the following day. Then, medium was replaced prior to transfections. 150 µl pre-warmed OPTI-MEM was placed 1.5 ml tubes with 9 µl TransIT-LT1 transfection reagent (Mirus), vortexed briefly, and incubated for 15 minutes at room temperature. After incubation, 3 µg of plasmid DNA for either pcDNA4-myc-6×His (empty vector), pcDNA4-GFP-myc-6×His, pcDNA4-SEC23A-myc-6×His (wild type and serine-to-alanine point mutants) were added to a 1.5 ml tube, vortexed briefly and incubated for 15 minutes at room temperature. After the final incubation, the transfection mixture was added dropwise to the cells. Cells recovered at 37 °C and 5% CO2 for 48 hours and then were selected using 200 µg/ml Zeocin. Cells were passaged under selective pressure until a mock-transfected plate no longer contained living cells, the GFP-transfected control plate showed a majority positive cells under fluorescent illumination, and other resistant colonies were expanding. Cell populations were also examined for stable transfectants using myc immunofluorescence.
SEC23B knockdown via siRNA in SW1353
Glass coverslips were dipped in 100% ethanol, flame-sterilized, and placed into 6-well plates with media. Cells were plated to be ~15% the following day. The next morning, two reaction tubes per condition were prepared, both containing 125 µl pre-warmed OPTI-MEM. 100 pmol of a 1:1 mixture of SEC23B siRNA 1 and 2 was added to the first reaction tube (reaction 1) and incubated for 2 minutes. The same was done for the control SEC23B scrambled siRNA 1 and 2. Next, 3 µl per 100 pmol of Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher) was added to the other tube containing 125 µl pre-warmed OPTI-MEM (reaction 2). Then, reaction 1 was added to reaction 2, flicked to mix, and incubated at room temperature for 20 minutes. During the incubation, fresh medium was added to the cells. After 20 minutes, the combined mixture was added drop-wise to the cells. After 72 hours of knockdown, cells were used in IF experiments.
siRNAs sequences targeting SEC23B have been described66 and were ordered from Sigma-Aldrich as duplexes. Scrambled siRNA were generated from parent sequences using Invivogen’s siRNA Wizard scramble tool (http://www.invivogen.com/sirnawizard/scrambled.php). Scrambled sequences were checked using NCBI BLAST to verify no significant homology in the human genome.
SEC23B siRNA 1
5’-CACGUUACAUCAACACGGA[dT][dT]
SEC23B siRNA 2
5’-CACUAUGAGAUGCUUGCUA[dT][dT]
SEC23B scrambled siRNA 1
5’-GAACCCGCACGUAACUAU[dT][dT]
SEC23B scrambled siRNA 2
5’-GCCAAUGCGUCUUGUAAAU[dT][dT]
Collagen immunofluorescence trafficking assay
After SEC23B knockdown via siRNA, cells were treated with 50 µg/ml ascorbate for 7 hours. Then, cells were washed 3 times with PBS and fixed with 4% paraformaldehyde in PBS for 20 minutes. Following fixation, cells were washed 3 times with PBS and permeabilized using 0.1% Triton X-100 in PBS for 10 minutes. After permeabilization, cells were washed 3 times using PBS and then blocked using 0.1% BSA in PBS for 30 minutes. Following blocking, glass coverslips were transferred to microcentrifuge caps inside a humidified 6-well plate. Primary antibody dilutions in 0.1% BSA were added to the coverslips and incubated overnight at 4 °C. The next day, coverslips were transferred back to their original 6-well plate for washing and were washed 3 times with PBS. Coverslips were again transferred to microcentrifuge caps inside a humidified 6-well plate and incubated at room temperature for 1 hour with appropriate Alexa Fluor secondary antibodies in 0.1% BSA. Coverslips were washed 3 times in PBS and transferred to glass slides with ProLong™ Diamond Antifade Mountant with DAPI (Thermo Fisher). Slides were cured at room temperature in a dark cabinet for at least 24 hours before imaging. The following primary antibodies were used: anti-myc (9E10, BioLegend, 626801, 1:500) and anti-collagen I antiserum (LF-68, a gift of Dr. Larry Fisher, National Institute of Dental and Craniofacial Research, 1:2000). The following secondary antibodies were used: goat anti-rabbit Alexa Fluor 488 (Thermo-Fisher, A-11008, 1:2000) and goat anti-mouse Alexa Fluor 594 (Thermo-Fisher, A-11005, 1:2000). At least 200 cells from each cell line were counted per biological replicate. Imaging was performed on a Zeiss LSM 780 inverted confocal microscope, using 40×/1.4 NA Oil-Plan Apochromat DIC, (UV) VIS-IR (420762-9900), Zen 2011 software (Duke Light Microscopy Core Facility).
General immunofluorescence staining
Cells were plated on glass coverslips and treated as indicated. After treatment, cells were fixed, permeabilized, stained, and mounted as above. The following primary antibodies were used: Protein disulfide isomerase (PDI) (C81H6, Cell Signaling Technology, 3501, 1:100), SEC16 (A300-648A, Bethyl, 1:500), human procollagen type I C-peptide (QED Bioscience, 42024, 1:500). The following secondary antibodies were used: goat anti-rabbit Alexa Fluor 488 (Thermo-Fisher, A-11008, 1:2000), goat anti-mouse Alex Fluor 488 (Thermo-Fisher, A-11001; 1:2000), goat anti-mouse Alexa Fluor 594 (Thermo-Fisher, A-11005, 1:2000), goat anti-rabbit 594 (Thermo-Fisher, A-11012).
Construction of Gateway vectors for Tol2kit system
The zebrafish sec23a coding sequence was amplified from pCS2+ zSEC23a vector with primers containing attB1(forward) and attB2 (reverse) recombineering sites. The fragment was purified and cloned into pDONR221 vector using Gateway BP clonase II enzyme mix. After confirming successful cloning by sequencing, this plasmid was used as the middle entry vector for the Multistep Gateway Recombineering system, in combination with p5E_1.7kbCol2a1a promoter67, p3E_v2a-EGFP (a gift of Dr. Josh Gamse, Vanderbilt University), pDestTol2pA2 and incubated with Gateway LR clonase II enzyme mix overnight at room temperature. After transformation and colony screening, a destination vector was obtained that allows expression of the zebrafish sec23a coding sequence fused with a self-cleaving viral 2a peptide-tagged EGFP (v2a-EGFP) under the tissue-specific Col2a1a promoter. Four candidate SEC23A O-GlcNAc sites were sequentially mutated using Q5 site-directed mutagenesis kit (New England Biolabs) with the following primers:
115–116F: TCCACAGTTTgcagctATTGAATATGTTGTC
115–116R: AGTAGCTCAGCAGGCTGG
137F: TGTGGTGGACgCGTGCATGGA
137R: TACAGGAAGTTCAGGGGCATTTG
168F: GGGCCTCATTgCATTTGGACGG
168R: ACTAGGGCAGTAGGGGGC.
Zebrafish microinjection
Zebrafish embryos at the one-cell stage were collected from crusherm299/AB heterozygous crosses, injected with a combination of 50 pg medaka transposase mRNA and 10 pg of sec23a destination vector DNA, and grown for 3 days in embryo medium.
Zebrafish cryosectioning and immunohistochemistry
80 hours post-fertilization (hpf), embryos were fixed with 4% paraformaldehyde at 4 °C overnight and transferred to 30% sucrose in PBS. Embryos were embedded in Cryomatrix (ThermoFisher) and frozen at −80 °C for 15 minutes. Then, 14 µm-thick sections were cut with a cryostat (Leica CM1900) and collected onto Fisherbrand Superfrost Plus slides. Slides were dried and rehydrated in PBS before staining. Following antigen retrieval (20 µg/ml proteinase K at room temperature for 5 minutes), embryos were permeabilized with 0.5% Triton X-100 in PBS for 10 minutes at room temperature. Samples were blocked (2% goat serum and 2 mg/ml BSA) for 30 minutes at room temperature, followed by overnight primary antibody incubation at 4 °C, using 1:250 collagen-II antibody (Rockland) and 1:250 GFP antibody (Vanderbilt University Antibody and Protein Resource). Samples were incubated with secondary antibodies (rabbit AlexaFluor-555 and chicken AlexaFluor-488, LifeTechnologies) for one hour, and 1:4000 DAPI for 15 minutes at room temperature. Slides were mounted in ProlongGold (ThermoFisher).
Zebrafish imaging and quantification
Slides were imaged with Zeiss AxioImager.Z1. Percent collagen area in cytosol was calculated by the following formula: (Collagen-positive intracellular area / [cytosol area − nucleus area marked by DAPI])*100. ImageJ68 was used for intracellular area measurements.
Results
Previous studies have indicated that COPII proteins are O-GlcNAcylated48–52 but the biochemical and functional implications of these observations remain unclear. Indeed, O-GlcNAc sites have not been mapped systematically on any COPII protein, hindering subsequent biochemical and phenotypic experiments. As a first step towards understanding the role of O-GlcNAc cycling in anterograde trafficking, we analyzed the O-GlcNAcylation of three representative human COPII proteins, SEC23A, SEC24C and SEC31A. We expressed and purified each protein to homogeneity from human cells and used mass spectrometry (MS) to map O-GlcNAcylated residues. Using high-stringency criteria, we detected at least twenty-six, eleven and ten O-GlcNAc modifications on SEC23A, SEC24C and SEC31A, respectively, and manual inspection of the MS data permitted unambiguous assignment of many O-GlcNAc moieties to specific residues (Figure 1 and S1). Interestingly, the glycosylation sites were not evenly distributed across each protein, but rather were clustered in specific regions in all three cases. For example, five of the unambiguously assigned SEC23A O-GlcNAc sites (T137, T168, S184, S226, and S241) lie near its site of interaction with SEC24, as determined by prior structural studies69, whereas the others are distal to this interface (Figure 1B). In the case of SEC24C, the three definitively assigned glycosylation sites (S773, T775 and T776) reside in the relatively well-conserved β-sandwich domain. Finally, homology modeling based on structures of the yeast SEC31 ortholog suggests that all four assigned SEC31A glycosylation sites lie in the α-solenoid domain, which is thought to mediate protein-protein interactions and form a flexible hinge, permitting expansion of COPII carriers to accommodate a range of cargo sizes70–73. Therefore, SEC31A glycosylation may modulate protein-protein interactions and/or transport carrier size in the mammalian COPII outer coat. Taken together, these results indicate that site-specific glycosylation is prevalent on COPII proteins and may impact their biochemical functions.
Figure 1.

A. Schematic depicting twelve, three, and four O-GlcNAc sites identified on SEC23A, SEC24C, and SEC31A, respectively. Selected domains of each protein are also illustrated. Each modification shown here was unambiguously assigned to a single residue via manual inspection of MS data. Additional information and spectral data for all candidate O-GlcNAc sites are given in Figure S1.
B. The unambiguously assigned O-GlcNAc sites identified in this report are depicted in red on a model of human SEC23A (silver, PDB: 5VNO) and SEC24C (blue, PDB: 3EH2). The SEC31 interaction interface of SEC23A74 is indicated in green.
C. Representative MS/MS fragmentation spectrum of a SEC23 glycopeptide. Accurate mass measurements of the intact [152]QMSLSLLPPDALVGLITFGR[171] peptide indicated the presence of two O-GlcNAc modifications. HCD fragmentation revealed a y-ion series through y10 (ALVGLITFGR fragment) as well as a fragmentation series y4 (TFGR fragment) plus 203.0794 Da, which is highlighted by y4+203 and expanded in Figure 1D.
D. Fragmentation series with an intact O-GlcNAc modification (magnified from the spectrum in Figure 1C). This fragment ion series localizes one O-GlcNAc modification (denoted +O) unambiguously to Thr168, with either Ser154 or Ser156 as the additional site. All O-GlcNAc sites depicted in Figures 1A and 1B were unambiguously assigned using similar analysis, or because the number of O-GlcNAc moieties detected equaled the number of Ser/Thr residues on a given tryptic peptide.
Next, we asked whether dynamic O-GlcNAcylation regulates the COPII pathway. We used immunoprecipitation (IP) and Western blotting (WB), in combination with specific small molecule inhibitors of OGT (5SGlcNAc) and OGA (Thiamet-G)59, 61, to demonstrate that O-GlcNAc cycles on COPII components, including SEC24C and Trk-fused gene (TFG), a protein required for COPII function in vivo in metazoans (Figure 2A)13, 75–77. These results suggest that dynamic glycosylation may regulate protein secretion. To test this hypothesis, we performed three functional assays to assess the requirement for O-GlcNAc cycling in COPII-dependent trafficking. First, we used a secreted soluble horseradish peroxidase (ssHRP) reporter that traffics through the secretory pathway in a COPII-dependent manner78. ssHRP can be detected in culture supernatants via standard chemiluminescence, providing a quantitative measure of protein secretion78. We found that inhibition of OGT significantly diminished the COPII-dependent secretion of ssHRP without reducing intracellular ssHRP expression or cell viability (Figures 2B & S2). Second, we expressed an enhanced green fluorescent protein (eGFP)-tagged temperature-sensitive mutant of the vesicular stomatitis virus glycoprotein (tsVSVG-eGFP) in mammalian cells79, 80. At the non-permissive temperature, tsVSVG-eGFP is retained in the ER, as indicated by reticular green fluorescence and colocalization with genetically encoded organelle markers (Figures 2C and S3A)79, 80. Upon shifting to the permissive temperature, tsVSVG-eGFP transits to the Golgi in a strictly COPII-dependent manner, displaying characteristic juxtanuclear punctate fluorescence, thereby providing a live-cell assay for pathway function (Figures 2C and S3A)79, 80. We found that inhibition of OGA markedly delayed COPII-dependent tsVSVG-eGFP trafficking (Figures 2C and S3B). Third, we asked whether O-GlcNAcylation affects the subcellular distribution of COPII proteins. All components of the inner and outer COPII coats exist in both soluble cytoplasmic and membrane-associated pools, and reversible membrane association is required for normal COPII function4–6. To determine whether O-GlcNAc regulates the membrane association of COPII components, we used digitonin treatment to permeabilize the plasma membranes of human cells, permitting the separation of soluble cytoplasmic proteins and membrane-associated proteins81, and then performed WBs for COPII components on these subcellular fractions. Interestingly, we found that inhibition of OGA reduced the ratio of membrane-associated to cytoplasmic SEC23A and SEC31A, indicating that O-GlcNAc cycling regulates the subcellular distribution of essential COPII components (Figure 2D and S4). Together, these results indicate that dynamic O-GlcNAcylation is required for normal COPII-dependent secretion.
Figure 2.

A. O-GlcNAc dynamically modifies COPII components. 293T-17 cells were transfected with either myc-6×His-SEC24C or TFG-myc-6×His and treated with vehicle (DMSO), 25 µM Thiamet-G, or 25 µM 5SGlcNAc eight hours prior to harvest. Tandem affinity-purified SEC24C and TFG were analyzed by WB. Both proteins demonstrated O-GlcNAc cycling as indicated by an increase in signal when treated with Thiamet-G, and a decrease in signal with 5SGlcNAc, compared to vehicle.
B. 293T cells were treated with vehicle (DMSO) or 50 µM 5SGlcNAc and transfected with ssHRP. The amount of ssHRP in the medium supernatant was measured after 20 hours via luminescence assay. Inhibition of OGT via 5SGlcNAc significantly diminished secretion. n = 6, ** p = 0.006, Student’s t-test. Error bars are standard error of the mean.
C. COS7 cells stably expressing tsVSVG-eGFP were treated with vehicle (DMSO) or 25 µM Thiamet-G for 8 hours at the non-permissive temperature (40 °C) and then shifted to the permissive temperature (32 °C). Cells were imaged every 10 seconds for 20 minutes, monitoring the movement of the tsVSVG-eGFP from ER (reticular ER fluorescence) to the Golgi (juxtanuclear punctate fluorescence). See also Figure S3.
D. SW1353 cells were incubated with DMSO or 50 µM Thiamet-G for 24 hours. Cytoplasm (C) and endomembrane (M) fractions were prepared by digitonin fractionation, and SEC23A and SEC31A were analyzed by WB. Tubulin and TRAPα serve as loading controls for cytoplasmic and integral ER transmembrane proteins, respectively. One representative experiment is shown. Similar results were obtained in three independent biological replicates (Figure S4).
Like other PTMs, O-GlcNAc can exert diverse biochemical effects on its substrates, including altering protein conformation or subcellular localization, or mediating or disrupting protein-protein interactions53, 54, 57, 58, 82, 83. Elegant biochemical and structural studies have revealed that numerous precise protein-protein interactions are required for COPII coat formation, cargo loading, membrane curvature, and scission4–6. In addition, our MS site-mapping data demonstrate that O-GlcNAcylation occurs on known or predicted protein-protein interaction domains of COPII components (Figures 1 and S1). Therefore, we hypothesized that O-GlcNAc may mediate the protein-protein interactions of COPII components. However, physiologically important O-GlcNAc-mediated protein-protein interactions can be sub-stoichiometric, transient and low-affinity, presenting a significant challenge for their characterization53, 54, 57, 58, 82, 83. To address this challenge, we harnessed a chemical biology method to covalently capture O-GlcNAc-mediated protein-protein interactions in live human cells60. In this strategy, cells are metabolically labeled with a GlcNAc analog bearing a diazirine photocrosslinking moiety, termed “GlcNDAz”60. GlcNDAz is accepted by the GlcNAc salvage pathway, converted to UDP-GlcNDAz, and used by OGT to decorate its native substrates60. Briefly UV-treating GlcNDAz-labeled cells triggers the covalent crosslinking of O-GlcNDAz moieties to any binding partner proteins within ~2–4 Å of the sugar60. Thanks to this short radius, GlcNDAz crosslinking occurs exclusively at sites where the glycan contributes to the interaction interface, without crosslinking to distant or nonspecific proteins60. These covalent crosslinks can be conveniently visualized through the GlcNDAz-dependent appearance of high molecular weight species on a WB for the protein of interest. Therefore, GlcNDAz is a powerful chemical tool for identifying direct, glycosylation-mediated interactions between endogenous proteins in live cells60.
Using GlcNDAz, we found that endogenous SEC23A, SEC24B, SEC24C, SEC31A and TFG all engage in O-GlcNAc-mediated protein-protein interactions in multiple human cell lines, detected as massive crosslinks on a WB (Figure 3A). Importantly, GlcNDAz crosslinking of SEC23A was enhanced when deglycosylation was blocked by brief Thiamet-G treatment (Figure 3B), indicating that these interactions are dynamic and O-GlcNAc-specific. We concluded that O-GlcNAc mediates regulated interactions of COPII proteins.
Figure 3.

A. HeLa/UAP1 or 293T/UAP1 cells were treated with 100 µM GlcNDAz or vehicle (DMSO) and UV light (or not), and lysates were analyzed by WB. Circles indicate uncrosslinked proteins running at their predicted molecular weights. Arrows indicate GlcNDAz- and UV-dependent crosslinked species.
B. HeLa/UAP1 cells were treated with 100 µM GlcNDAz, 50 µM Thiamet-G, vehicle (DMSO), and UV light as indicated for 24 hours and lysates were analyzed by WB. Circle indicates uncrosslinked SEC23A running at its predicted molecular weight. Arrows indicate GlcNDAz- and UV-dependent crosslinked species.
C. Left: 293T/UAP1 cells were transfected with GFP (control), 3×FLAG-TFG only, TFG-myc-6×His only, or co-transfected with both FLAG and myc-6×His constructs (“myc & FLAG”). Then, cells were treated with DMSO (vehicle) only or 100 µM GlcNDAz, and were exposed to UV. Lysates were analyzed by WB, confirming that both myc- and FLAG-tagged TFG crosslinks similarly to endogenous TFG in a GlcNDAz-dependent manner. Covalently crosslinked bands of TFG (~150 kDa) are shown. Right: 293T/UAP1 cells were transfected with GFP (control), 3×FLAG-TFG only, TFG-myc-6×His only, or co-transfected with both FLAG and myc-6×His constructs (“myc & FLAG”). Then, cells were treated with 100 µM GlcNDAz and UV, and lysates were analyzed by IP and WB as indicated. Covalently crosslinked bands of TFG (~150 kDa) are shown.
D. 293T/UAP1 cells were transfected with 3×FLAG-SEC24C or GFP (control) and treated with 100 µM GlcNDAz or vehicle. Lysates were analyzed by anti-FLAG IP and WB. Crosslinked complexes of 3×FLAG-SEC24C were successfully purified (lane 8), but did not contain SEC23A. Notably, endogenous uncrosslinked SEC23A co-IPs with SEC24C (lanes 4 and 8) but endogenous crosslinked SEC23A (lane 5) does not, demonstrating that SEC24C and SEC23A each crosslink, but not to each other. Circles indicate uncrosslinked proteins running at their predicted molecular weights. Arrows indicate GlcNDAz- and UV-dependent crosslinked species.
To test the hypothesis that O-GlcNAc might mediate some of the well-characterized protein-protein interactions in the COPII coat4–6, we examined TFG, SEC23A and SEC24C as model COPII glycoproteins. TFG homo-oligomerizes in vitro and in cells, and this property is required for its subcellular localization and function in COPII cargo trafficking13, 75–77. To determine whether O-GlcNAc participates in homotypic TFG-TFG interactions inside cells, we created constructs of human TFG with different epitope tags. IP/WBs with these constructs revealed that covalent, GlcNDAz-dependent crosslinked complexes of TFG contain both tagged versions of TFG simultaneously (Figure 3C). These results demonstrate that the GlcNDAz-dependent complexes are homo-oligomers, suggesting that O-GlcNAc may participate in TFG-TFG interactions in live cells.
Next, we examined the potential involvement of O-GlcNAc in the heterodimerization of SEC23 and SEC24, an essential step in the formation of the COPII pre-budding complex4–6. We expressed FLAG-SEC24C in human cells and observed GlcNDAz-dependent crosslinking similar to that of endogenous SEC24C (Figure 3A, D). We IP-ed SEC24C and performed WBs for candidate interactors, including SEC23A (Figure 3D), SEC13, SEC31, SAR1 and the scaffolding protein SEC16 (not shown). In all cases, we failed to detect any candidate interactors in the crosslinked SEC24C complexes (Figure 3D and not shown). Notably, endogenous SEC23A again crosslinked in this context, though not to SEC24C (Figure 3D). Similarly, IP-ed GlcNDAz crosslinks of SEC23A did not contain SEC24, SEC13, SEC31 or SAR1 (not shown). We concluded that SEC23A and SEC24C engage in specific but distinct O-GlcNAc-mediated protein-protein interactions, which may influence COPII trafficking. All together, these results demonstrate that dynamic O-GlcNAcylation mediates both known and novel interactions among COPII pathway proteins.
We next focused on SEC23A, because SEC23 is an essential COPII component4–6, 10, 84, and because human SEC23A mutations cause CLSD15, 21, 26. To further elucidate the biochemical effects of SEC23A O-GlcNAcylation, we created unglycosylatable point mutants (Ser/Thr→;Ala) in sixteen of the candidate sites we detected by MS, focusing on residues that are highly conserved across evolution (Figure 1A and S1): S97, S102, S115, S116, T137, T168, S184, S226, T241, S508, S516, S627, S629, S639, S640, and S641. We expressed wild type or point-mutant SEC23A constructs in human cells and performed GlcNDAz crosslinking (Figure 4A). We found that the S115A, S116A, T137A, T168A, and S184A mutants reduced or abrogated GlcNDAz-mediated crosslinking, whereas the others did not (Figure 4A). Interestingly, three of the residues required for SEC23A crosslinking, T137, T168 and S184, lie near its SEC24-binding interface (Figure 1A & B). However, as noted, SEC24 is not the GlcNDAz-mediated crosslinked partner of SEC23 (Figure 3). These observations suggest that the site-specific O-GlcNAcylation of SEC23A near the SEC24 binding interface mediates an interaction distinct from the SEC23/SEC24 pre-budding complex.
Figure 4.


A. 293T/UAP1 cells were transfected with plasmids encoding GFP (control), wild type SEC23A-myc-6×His, or the indicated SEC23A-myc-6×His point mutants. Cells were treated with vehicle (DMSO) or 100 µM GlcNDAz and UV-irradiated. Lysates were analyzed by WB. The S115A, S116A, T137A, T168A and S184A mutants all show a marked reduction in crosslinking. Circles indicate uncrosslinked SEC23A running at its predicted molecular weight. Arrows indicate GlcNDAz- and UV-dependent crosslinked species. White rectangles indicate where irrelevant lanes have been cropped out of a single blot (bottom panels). Duplicate wild type SEC23A crosslinked samples were run on multiple blots due to space constraints. Therefore, apparent variability in wild type crosslinking efficiency is due to blot-to-blot variation in the enhanced chemiluminescence detection.
B. SEC23A−/− SW1353 cells stably reconstituted with empty vector (left), wild type SEC23A-myc-6×His (middle) or S184A SEC23A-myc-6×His (right) were transfected with anti-SEC23B siRNA (or scrambled SEC23B siRNA, not shown) and treated with 50 µg/ml sodium ascorbate for 7 hours to induce collagen translation46, 47. Then, cells were fixed and stained for SEC23A (anti-myc, red), endogenous collagen (LF68 anti-collagen, green) and DNA (DAPI, blue). Cells exhibiting both SEC23A expression and aberrant ER collagen staining (white arrows) were quantified as a fraction of the total number of SEC23A-positive cells. Representative images from eight biological replicates are shown at two magnifications (top and bottom panels).
C. Quantification of IF data presented in Figure 4B. Cells expressing S184A SEC23A were more than twice as likely to exhibit retained collagen, compared to those expressing wild type SEC23A. n = 8, *** p < 0.001, Student’s t-test. Error bars are standard error of the mean.
We next investigated the functional significance of site-specific SEC23A glycosylation in a human cell system. Chondrocytes create and maintain cartilage, largely through the regulated secretion of collagen85, and SEC23A is required for this process in vivo15, 21, 26. To assess the function of SEC23A with a physiologically relevant cell system and model cargo, we used CRISPR/Cas9 methods to delete the endogenous SEC23A gene in SW1353 human chondrosarcoma cells (Figure S5), which secrete endogenous collagen86, 87. Complete loss of SEC23A caused intense collagen accumulation and distended ER morphology (Figures S6 and S7), which resemble the reported previously COPII cargo defects of CLSD patient cells15, 21, 26. These observations confirm that SEC23A−/− SW1353 cells are an appropriate system for testing the function of unglycosylatable SEC23A mutants in trafficking endogenous collagen. Next, we stably re-expressed empty vector, wild type SEC23A, or unglycosylatable single point-mutant SEC23A alleles. To create an appropriately sensitive assay for SEC23A function, we knocked down SEC23B (Figure S8), preventing compensation from this paralog, as described previously66. We then assessed SEC23A function in these reconstituted cell lines by tracking collagen secretion via immunofluorescence (IF) microscopy. Interestingly, SEC23A−/− SW1353 cells reconstituted with the S184A unglycosylatable point mutant exhibited a significantly higher proportion of retained collagen and characteristic dilated ER morphology, as compared to cells reconstituted with wild type SEC23A, despite partial rescue (Figures 4B & C). Importantly, however, wild type and S184A mutant SEC23A co-IPed endogenous SEC24 proteins with similar efficiencies (Figure S9A), indicating that the trafficking defect we observe in the S184A mutant (Figures 4B & C) is not due to gross protein misfolding or lost SEC24 binding. Together, these results demonstrate that the S184 glycosylation site of SEC23A is required for its collagen trafficking function in human chondrosarcoma cells, possibly due to post-translational regulation at the SEC23A/SEC24 interface.
In developing vertebrates, collagen trafficking and skeletogenesis are stringently dependent on SEC23A, because SEC23A partial loss-of-function mutations cause CLSD in humans and similar phenotypes in model organisms15, 18, 21, 26, 27, 88–90. Our biochemical and cellular data (Figure 1–3, 4) suggest that site-specific O-GlcNAcylation might regulate SEC23A function in developing tissues. To test this hypothesis, we took advantage of a vertebrate model of SEC23A dysfunction18, 27. We have shown previously that mutations in zebrafish sec23a underlie the crusher phenotype, exhibiting collagen mis-trafficking, chondrocyte failure and skeletal dysmorphology18, 27 that closely resemble CLSD15, 21, 26. Therefore, crusher fish provide a valuable vertebrate genetic model for studying SEC23A function in vivo18, 27. Importantly, the human and zebrafish SEC23A proteins are 91% identical, and all but one (S629) of the sixteen putative O-GlcNAc sites that we examined (Figure 4A) are conserved between these orthologs.
We tested the functional importance of SEC23A O-GlcNAcylation sites in vivo using the crusher model. First, we confirmed that wild type zebrafish SEC23A crosslinks in our GlcNDAz assay similarly to its human ortholog (Figure 5A). Then, we designed a compound-mutant SEC23A allele, in which four residues required for GlcNDAz crosslinking and conserved from humans to fish (S115, S116, T137, T168) were mutated to alanine (“4A mutant”). We created expression constructs of human and zebrafish SEC23A 4A mutant and confirmed that they are defective in GlcNDAz-mediated crosslinking (Figure 5A). These results indicate that the O-GlcNAc-mediated interactions of SEC23A are biochemically conserved across vertebrates.
Figure 5.

A. 293T/UAP1 cells were transfected with either GFP (control), human wild type SEC23A-myc-6×His, human 4A SEC23A-myc-6×His, zebrafish wild type Sec23A-myc-6×His, or zebrafish 4A Sec23A-myc-6×His. Circle indicates uncrosslinked SEC23A running at its predicted molecular weight. Arrows indicate GlcNDAz- and UV-dependent crosslinked species. The 4A mutations reduce crosslinking in both human and zebrafish SEC23A.
B. Experimental strategy for mosaic expression of Sec23A in zebrafish chondrocytes. Sec23A wild type or 4A mutant was expressed under the zebrafish Col2a1 promoter. Embryos injected with Sec23A constructs were grown until 80 hours post-fertilization (hpf) and cryosectioned for immunohistochemistry.
C. Cells expressing Sec23A constructs are marked by v2a-EGFP. Wild type cells secrete collagen to the extracellular space (arrows, top panel), whereas, crusher (sec23a loss-of-function) mutant chondrocytes accumulate intracellular collagen (arrowhead, middle panel). Overexpression of wild type Sec23A in crusher animals leads to clearance of intracellular collagen, whereas expression of the Sec23A 4A mutant fails to do so (arrowhead, lower panel). Double-headed arrows point to normal levels of collagen in the secretory compartment.
D. Quantification of zebrafish rescue experiments. Percent of cell area occupied by collagen shows that wild type Sec23A expression in crusher chondrocytes restores collagen secretion to normal levels, whereas the Sec23A 4A mutant does not, despite partial rescue. crusher (cru) chondrocytes accumulate the highest level of intracellular collagen. Investigator was blinded to wild type or 4A mutant genotype while performing quantification, per standard practice18, 27, 28. p < 0.05, Student’s t-test. Error bars are standard error of the mean.
Next, we developed an in vivo assay of SEC23A function in skeletogenesis (Figure 5B). We expressed wild type or 4A mutant SEC23A in sec23a-deficient (crusher) zebrafish chondrocytes and tracked COPII-dependent collagen transport by immunohistochemistry (Figure 5B). We measured the cytosolic area occupied by collagen as an established marker of its intracellular retention18, 27, 28. We found that 5% of the cytosolic area of wild type chondrocytes was occupied by collagen at 80 hours post-fertilization (hpf), which corresponds to normal collagen traffic within secretory pathway compartments (Figure 5C, D). In contrast, 50–95% of the cytosolic area of sec23a-deficient chondrocytes was filled with aberrantly retained collagen (Figure 5C, D), consistent with our prior reports18, 27. Interestingly, while the expression of wild type SEC23A in crusher chondrocytes restored collagen secretion to wild type levels in a cell-autonomous fashion, the unglycosylatable SEC23A 4A mutant only partially restored collagen secretion, exhibiting significantly higher collagen retention than wild type (Figure 5C, D). Together, these results suggest that site-specific O-GlcNAcylation of SEC23A may regulate the in vivo COPII-dependent transport of SEC23A-dependent cargoes, including collagen.
Discussion
The COPII trafficking pathway is essential for cell and tissue physiology in vertebrates, and is dysregulated in several human diseases. While the core COPII machinery is relatively well understood, little is known about how trafficking is dynamically regulated in response to physiological or pathological cues. Our results indicate that O-GlcNAcylation of COPII proteins may be one important mode of pathway regulation in vertebrates.
We have shown that O-GlcNAc is a prevalent modification of COPII proteins, including SEC23, SEC24, SEC31 and TFG (Figure 1, 2A, 3A). Like phosphorylation, O-GlcNAcylation can exert a wide range of biochemical effects on its substrates, and it will be important to delineate the effects of COPII protein glycosylation at each individual modification site in order to build an integrated model for how O-GlcNAc affects protein trafficking from the ER. As a first step towards this goal, our GlcNDAz results suggest that O-GlcNAc moieties lie at or very close to the interaction interface between TFG monomers, indicating a potential biochemical function for TFG glycosylation (Figure 3A, C). The N-terminal half of TFG, comprising its PB1 and coiled-coil domains, mediates homo-oligomerization, and this property is required for its trafficking function in vivo13, 75–77. While unglycosylated TFG can homo-oligomerize in vitro, our results suggest that O-GlcNAcylation may facilitate or regulate this process in live cells13, 77. Similarly, we show that both SEC23A and SEC24C also engage in O-GlcNAc-mediated protein-protein interactions, but not with each other, or with several other known binding partners in the COPII system (Figure 3A, B, D). These data suggest that novel O-GlcNAc-mediated protein-protein interactions may govern SEC23 and/or SEC24 function in the COPII system. It will be important to determine in future work how changes in site-specific glycosylation of TFG, SEC23 or SEC24 directly affect their biochemical activities in vivo.
At the cellular level, our results indicate that O-GlcNAc cycling in general (Figure 2), and specific SEC23A glycosylation sites in particular (Figures 4B & C), are required for COPII-dependent protein secretion. Because native PTMs, including O-GlcNAc, are not essential for in vitro COPII vesicle assembly from minimal components91, it is likely that O-GlcNAcylation and other PTMs are instead required to modulate the activity of the pathway in vivo. The precise molecular events affected by O-GlcNAc and the full complement of substrates and modification sites most critical for this regulation remain incompletely understood. Interestingly, however, OGA inhibition both delayed the trafficking of tsVSVG-eGFP (Figure 2C) and decreased the pools of membrane-associated SEC23A and SEC31A (Figure 2D). These results suggest that unfettered O-GlcNAc cycling may be required for the efficient recruitment or recycling of COPII proteins to the ER or Golgi membranes. Other PTMs govern COPII in this way. For example, the Golgi-localized kinase CK1δ and the phosphatase PP6 act reciprocally on SEC23 and SEC24 to ensure orderly COPII carrier budding, fusion and directionality38, 39, and a recent study demonstrated that phosphorylation by CK2 inhibits the membrane association of SEC3144.
Notably, O-GlcNAc and O-phosphate exhibit a complex interplay in cells, frequently competing for nearby or identical residues on a given protein and exerting antagonistic effects54. Indeed, a prior report suggests that the cell cycle-dependent phosphorylation and glycosylation of SEC24 are reciprocal, and may affect its membrane association50. Our data are consistent with this proposed model, but additional cell biological studies with unglycosylatable point mutants of COPII proteins will be required to elucidate the responsible modification sites and potential O-GlcNAc/O-phosphate cross-talk. In the case of SEC23A, none of the glycosylation sites that we examined (Figure 4A) is reported to be phosphorylated, though additional sites of both modifications may yet await discovery. In contrast, phosphorylation of S773 and T776 of SEC24C has been reported92, suggesting that the interplay between O-GlcNAcylation and phosphorylation at these sites could tune COPII activity. Future studies will address the potential cross-talk among PTMs of COPII proteins.
The COPII system of vertebrates is significantly more complex than that of lower eukaryotes. For example, vertebrates possess two paralogs of SEC23 (A and B) and four paralogs of SEC24 (A through D), whereas budding yeast has one essential ortholog of each. The functional significance of vertebrates’ multiple COPII paralogs remains largely unclear. All four SEC24 paralogs exhibit analogous biochemical functions in early COPII coat formation and cargo capture, and yet mouse knockout studies and differences in cargo binding clearly indicate distinct, if overlapping, physiological functions among these proteins17, 28, 93–95. Studies of SEC23A and B revealed analogous results, despite their high level of conservation and similar biochemical functions96–99. Currently, it is not well understood how these COPII components achieve distinct biological roles in vivo, though tissue-specific expression patterns of paralogs and cargoes likely contribute89, 90. Another possibility is that PTMs differentially regulate COPII paralogs. Consistent with this notion, we observed distinct GlcNDAz crosslinks of endogenous SEC24B and C (Figure 3A), suggesting that O-GlcNAcylation may mediate different protein-protein interactions among paralogs. In addition, one glycosylation site that we mapped on human SEC24C (T775) is conserved as a serine or threonine in all four human SEC24 paralogs, whereas the other two localized SEC24C O-GlcNAc sites (Figures 1A & 1B) and several candidate sites (Figure S1) are conserved in only one or none of the other three SEC24 paralogs. Therefore, glycosylation of multiple SEC24 paralogs at the T775-cognate site may impact all COPII traffic, whereas glycosylation of SEC24C at other sites may provide paralog-specific regulation to tune the trafficking of specific cargoes or cell types. In contrast, of the twelve unambiguously localized O-GlcNAc sites we identified on human SEC23A (Figures 1A–B), all except S516 are conserved in SEC23B (with a serine in SEC23B corresponding to SEC23A T508). These results suggest that glycosylation could affect SEC23 paralogs more uniformly than SEC24 paralogs, though this hypothesis remains to be tested.
Finally, our results demonstrate that several evolutionarily conserved glycosylation sites on SEC23A are required for O-GlcNAc-mediated protein-protein interactions in cells, and to support collagen secretion in SW1353 cells and skeletogenesis in developing zebrafish (Figures 4, 5). One of these residues, S184, lies near the SEC23/SEC24 interface, potentially providing a site for a regulatory O-GlcNAc-mediated protein-protein interaction with a protein other than SEC24 (Figures 3D, 4A). In contrast, the residues altered in the SEC23A 4A mutant do not lie exclusively at the SEC23/SEC24 interface, and O-GlcNAcylation may play a biochemically distinct role at those sites. Although our cell- and animal-based experiments (Figures 2, 4, 5) cannot stringently rule out the possibility that the S184A or 4A mutation induces a deleterious conformational change in SEC23A, both mutants co-IP with SEC24 proteins with efficiencies similar to that of wild type SEC23A, arguing against dramatic conformational change or misfolding (Figure S9A and S9B). Similarly, neither SEC23A deletion nor OGT or OGA inhibition disrupted ER exit sites (ERES), as judged by IF staining of the endogenous, canonical ERES marker SEC16 (Figure S10). This result suggests that the functional effects that we observe upon OGT or OGA inhibition (Figure 2) or ablation of SEC23A glycosylation sites (Figure 4) may be due to regulation of SEC23A and/or other COPII proteins downstream of ERES formation. Therefore, we propose that SEC23A O-GlcNAcylation at these sites is required to regulate its interaction with binding partners beyond SEC24. We expect that future GlcNDAz crosslinking and MS proteomic experiments to identify the O-GlcNAc-dependent binding partner(s) of SEC23A will facilitate the elucidation of the molecular mechanisms through which site-specific O-GlcNAcylation of SEC23A influences COPII trafficking.
At the organismal level, our results suggest that dynamic O-GlcNAcylation of SEC23A may govern collagen trafficking during vertebrate development (Figures 4, 5). Consistent with this idea, recent work identified a connection between whole-body O-GlcNAc levels and chondrogenic differentiation in mice100, implicating O-GlcNAcylation in skeletogenesis. We have also shown that O-GlcNAc modifies SEC24C and SEC31A (Figure 1), both of which may participate in efficient in vivo collagen secretion as well46, 47, 101. Therefore, O-GlcNAcylation may be a conserved mode of COPII regulation in chondrocytes during vertebrate development. Testing this hypothesis will be an important goal of future studies.
Supplementary Material
Figure S1: Summary of MS data and identified O-GlcNAc sites
Figure S2: Effect of 5SGlcNAc on ssHRP assay
Figure S3: Organelle marker and Thiamet-G controls for tsVSVG-eGFP assay
Figure S4: Digitonin fractionation replicates and densitometry quantification
Figure S5: Validation of SEC23A deletion by CRISPR/Cas9
Figure S6: Collagen accumulation in SEC23A−/− cells
Figure S7: Distended ER morphology in SEC23A−/− cells
Figure S8: Validation of SEC23B−/− depletion by siRNA
Figure S9: SEC23A S184A and 4A co-IPs with SEC24 paralogs
Figures S10: Lack of effect on ERES by SEC23A deletion or OGT/OGA inhibition
Acknowledgments
We thank Frederic Bard, Larry Fisher, Josh Gamse, David Ginsburg, Rami Khoriaty, Jennifer Kohler, Chris Nicchitta, Randy Schekman and Benjamin Swarts for antibodies or plasmids, and David Gooden of the Duke Small Molecule Synthesis Facility for chemical synthesis. This work was supported by a Scholar Award from the Rita Allen Foundation, a Research Grant from the Mizutani Foundation, and NIH R01GM117473 (to MB), NIH R01HL092217-06 (to EWK), NIH R01GM110567 (to AA), and the Vanderbilt International Scholar Program and an American Heart Association Predoctoral Fellowship 15PRE22940041 (to GU).
Footnotes
Author contributions
NJC, BJB, TRM, BC, PML, TJS and AC performed all experiments conducted in mammalian cells. GU performed the zebrafish experiments. EJS performed MS-based O-GlcNAc site mapping of COPII proteins. MH and AA designed and tested conditions for SEC23B knockdown. NJC, GU, BJB, TRM, BC, AA, EWK and MB designed the experiments, analyzed the data and interpreted the results. EWK supervised the zebrafish experiments. MB supervised all aspects of the work. NJC and MB wrote and revised the manuscript. All authors reviewed and approved the finished manuscript.
References
- 1.Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 2.Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes & development. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 3.Barlowe CK, Miller EA. Secretory protein biogenesis and traffic in the early secretory pathway. Genetics. 2013;193:383–410. doi: 10.1534/genetics.112.142810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller EA, Schekman R. COPII - a flexible vesicle formation system. Current opinion in cell biology. 2013;25:420–427. doi: 10.1016/j.ceb.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Routledge KE, Gupta V, Balch WE. Emergent properties of proteostasis-COPII coupled systems in human health and disease. Molecular membrane biology. 2010;27:385–397. doi: 10.3109/09687688.2010.524894. [DOI] [PubMed] [Google Scholar]
- 6.Brandizzi F, Barlowe C. Organization of the ER-Golgi interface for membrane traffic control. Nature reviews. Molecular cell biology. 2013;14:382–392. doi: 10.1038/nrm3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker D, Hicke L, Rexach M, Schleyer M, Schekman R. Reconstitution of SEC gene product-dependent intercompartmental protein transport. Cell. 1988;54:335–344. doi: 10.1016/0092-8674(88)90196-1. [DOI] [PubMed] [Google Scholar]
- 8.Ruohola H, Kabcenell AK, Ferro-Novick S. Reconstitution of protein transport from the endoplasmic reticulum to the Golgi complex in yeast: the acceptor Golgi compartment is defective in the sec23 mutant. The Journal of cell biology. 1988;107:1465–1476. doi: 10.1083/jcb.107.4.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 1994;77:895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 10.Fromme JC, Orci L, Schekman R. Coordination of COPII vesicle trafficking by Sec23. Trends in cell biology. 2008;18:330–336. doi: 10.1016/j.tcb.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Paczkowski JE, Richardson BC, Fromme JC. Cargo adaptors: structures illuminate mechanisms regulating vesicle biogenesis. Trends in cell biology. 2015;25:408–416. doi: 10.1016/j.tcb.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khoriaty R, Vasievich MP, Ginsburg D. The COPII pathway and hematologic disease. Blood. 2012;120:31–38. doi: 10.1182/blood-2012-01-292086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beetz C, Johnson A, Schuh AL, Thakur S, Varga RE, Fothergill T, Hertel N, Bomba-Warczak E, Thiele H, Nurnberg G, Altmuller J, Saxena R, Chapman ER, Dent EW, Nurnberg P, Audhya A. Inhibition of TFG function causes hereditary axon degeneration by impairing endoplasmic reticulum structure. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:5091–5096. doi: 10.1073/pnas.1217197110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones B, Jones EL, Bonney SA, Patel HN, Mensenkamp AR, Eichenbaum-Voline S, Rudling M, Myrdal U, Annesi G, Naik S, Meadows N, Quattrone A, Islam SA, Naoumova RP, Angelin B, Infante R, Levy E, Roy CC, Freemont PS, Scott J, Shoulders CC. Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nature genetics. 2003;34:29–31. doi: 10.1038/ng1145. [DOI] [PubMed] [Google Scholar]
- 15.Boyadjiev SA, Fromme JC, Ben J, Chong SS, Nauta C, Hur DJ, Zhang G, Hamamoto S, Schekman R, Ravazzola M, Orci L, Eyaid W. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nature genetics. 2006;38:1192–1197. doi: 10.1038/ng1876. [DOI] [PubMed] [Google Scholar]
- 16.Zhang B, Cunningham MA, Nichols WC, Bernat JA, Seligsohn U, Pipe SW, McVey JH, Schulte-Overberg U, de Bosch NB, Ruiz-Saez A, White GC, Tuddenham EG, Kaufman RJ, Ginsburg D. Bleeding due to disruption of a cargo-specific ER-to-Golgi transport complex. Nature genetics. 2003;34:220–225. doi: 10.1038/ng1153. [DOI] [PubMed] [Google Scholar]
- 17.Merte J, Jensen D, Wright K, Sarsfield S, Wang Y, Schekman R, Ginty DD. Sec24b selectively sorts Vangl2 to regulate planar cell polarity during neural tube closure. Nature cell biology. 2010;12:41–46. doi: 10.1038/ncb2002. sup pp 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lang MR, Lapierre LA, Frotscher M, Goldenring JR, Knapik EW. Secretory COPII coat component Sec23a is essential for craniofacial chondrocyte maturation. Nature genetics. 2006;38:1198–1203. doi: 10.1038/ng1880. [DOI] [PubMed] [Google Scholar]
- 19.Schwarz K, Iolascon A, Verissimo F, Trede NS, Horsley W, Chen W, Paw BH, Hopfner KP, Holzmann K, Russo R, Esposito MR, Spano D, De Falco L, Heinrich K, Joggerst B, Rojewski MT, Perrotta S, Denecke J, Pannicke U, Delaunay J, Pepperkok R, Heimpel H. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nature genetics. 2009;41:936–940. doi: 10.1038/ng.405. [DOI] [PubMed] [Google Scholar]
- 20.Wansleeben C, Feitsma H, Montcouquiol M, Kroon C, Cuppen E, Meijlink F. Planar cell polarity defects and defective Vangl2 trafficking in mutants for the COPII gene Sec24b. Development. 2010;137:1067–1073. doi: 10.1242/dev.041434. [DOI] [PubMed] [Google Scholar]
- 21.Fromme JC, Ravazzola M, Hamamoto S, Al-Balwi M, Eyaid W, Boyadjiev SA, Cosson P, Schekman R, Orci L. The genetic basis of a craniofacial disease provides insight into COPII coat assembly. Developmental cell. 2007;13:623–634. doi: 10.1016/j.devcel.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garbes L, Kim K, Riess A, Hoyer-Kuhn H, Beleggia F, Bevot A, Kim MJ, Huh YH, Kweon HS, Savarirayan R, Amor D, Kakadia PM, Lindig T, Kagan KO, Becker J, Boyadjiev SA, Wollnik B, Semler O, Bohlander SK, Kim J, Netzer C. Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta. American journal of human genetics. 2015;96:432–439. doi: 10.1016/j.ajhg.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moosa S, Chung BH, Tung JY, Altmuller J, Thiele H, Nurnberg P, Netzer C, Nishimura G, Wollnik B. Mutations in SEC24D cause autosomal recessive osteogenesis imperfecta. Clin Genet. 2015 doi: 10.1111/cge.12678. [DOI] [PubMed] [Google Scholar]
- 24.Oz-Levi D, Ben-Zeev B, Ruzzo EK, Hitomi Y, Gelman A, Pelak K, Anikster Y, Reznik-Wolf H, Bar-Joseph I, Olender T, Alkelai A, Weiss M, Ben-Asher E, Ge D, Shianna KV, Elazar Z, Goldstein DB, Pras E, Lancet D. Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. American journal of human genetics. 2012;91:1065–1072. doi: 10.1016/j.ajhg.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stadel D, Millarte V, Tillmann KD, Huber J, Tamin-Yecheskel BC, Akutsu M, Demishtein A, Ben-Zeev B, Anikster Y, Perez F, Dotsch V, Elazar Z, Rogov V, Farhan H, Behrends C. TECPR2 Cooperates with LC3C to Regulate COPII-Dependent ER Export. Molecular cell. 2015;60:89–104. doi: 10.1016/j.molcel.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 26.Boyadjiev SA, Kim SD, Hata A, Haldeman-Englert C, Zackai EH, Naydenov C, Hamamoto S, Schekman RW, Kim J. Cranio-lenticulo-sutural dysplasia associated with defects in collagen secretion. Clin Genet. 2011;80:169–176. doi: 10.1111/j.1399-0004.2010.01550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melville DB, Montero-Balaguer M, Levic DS, Bradley K, Smith JR, Hatzopoulos AK, Knapik EW. The feelgood mutation in zebrafish dysregulates COPII-dependent secretion of select extracellular matrix proteins in skeletal morphogenesis. Disease models & mechanisms. 2011;4:763–776. doi: 10.1242/dmm.007625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarmah S, Barrallo-Gimeno A, Melville DB, Topczewski J, Solnica-Krezel L, Knapik EW. Sec24D-dependent transport of extracellular matrix proteins is required for zebrafish skeletal morphogenesis. PLoS ONE. 2010;5:e10367. doi: 10.1371/journal.pone.0010367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malhotra V, Erlmann P. The pathway of collagen secretion. Annual review of cell and developmental biology. 2015;31:109–124. doi: 10.1146/annurev-cellbio-100913-013002. [DOI] [PubMed] [Google Scholar]
- 30.Ohisa S, Inohaya K, Takano Y, Kudo A. sec24d encoding a component of COPII is essential for vertebra formation, revealed by the analysis of the medaka mutant, vbi. Dev Biol. 2010;342:85–95. doi: 10.1016/j.ydbio.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 31.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 32.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 33.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, Hurt EM, Petroulakis E, Sonenberg N, Yewdell JW, Calame K, Glimcher LH, Staudt LM. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 34.Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nature reviews. 2013;12:703–719. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- 35.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. The Journal of cell biology. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farhan H, Weiss M, Tani K, Kaufman RJ, Hauri HP. Adaptation of endoplasmic reticulum exit sites to acute and chronic increases in cargo load. The EMBO journal. 2008;27:2043–2054. doi: 10.1038/emboj.2008.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature reviews. Molecular cell biology. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 38.Lord C, Bhandari D, Menon S, Ghassemian M, Nycz D, Hay J, Ghosh P, Ferro-Novick S. Sequential interactions with Sec23 control the direction of vesicle traffic. Nature. 2011;473:181–186. doi: 10.1038/nature09969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhandari D, Zhang J, Menon S, Lord C, Chen S, Helm JR, Thorsen K, Corbett KD, Hay JC, Ferro-Novick S. Sit4p/PP6 regulates ER-to-Golgi traffic by controlling the dephosphorylation of COPII coat subunits. Molecular biology of the cell. 2013;24:2727–2738. doi: 10.1091/mbc.E13-02-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farhan H, Wendeler MW, Mitrovic S, Fava E, Silberberg Y, Sharan R, Zerial M, Hauri HP. MAPK signaling to the early secretory pathway revealed by kinase/phosphatase functional screening. The Journal of cell biology. 2010;189:997–1011. doi: 10.1083/jcb.200912082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zacharogianni M, Kondylis V, Tang Y, Farhan H, Xanthakis D, Fuchs F, Boutros M, Rabouille C. ERK7 is a negative regulator of protein secretion in response to amino-acid starvation by modulating Sec16 membrane association. The EMBO journal. 2011;30:3684–3700. doi: 10.1038/emboj.2011.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharpe LJ, Luu W, Brown AJ. Akt phosphorylates Sec24: new clues into the regulation of ER-to-Golgi trafficking. Traffic. 2011;12:19–27. doi: 10.1111/j.1600-0854.2010.01133.x. [DOI] [PubMed] [Google Scholar]
- 43.Palmer KJ, Konkel JE, Stephens DJ. PCTAIRE protein kinases interact directly with the COPII complex and modulate secretory cargo transport. Journal of cell science. 2005;118:3839–3847. doi: 10.1242/jcs.02496. [DOI] [PubMed] [Google Scholar]
- 44.Koreishi M, Yu S, Oda M, Honjo Y, Satoh A. CK2 phosphorylates Sec31 and regulates ER-To-Golgi trafficking. PLoS ONE. 2013;8:e54382. doi: 10.1371/journal.pone.0054382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cohen M, Stutz F, Belgareh N, Haguenauer-Tsapis R, Dargemont C. Ubp3 requires a cofactor, Bre5, to specifically de-ubiquitinate the COPII protein, Sec23. Nature cell biology. 2003;5:661–667. doi: 10.1038/ncb1003. [DOI] [PubMed] [Google Scholar]
- 46.Jin L, Pahuja KB, Wickliffe KE, Gorur A, Baumgartel C, Schekman R, Rape M. Ubiquitin-dependent regulation of COPII coat size and function. Nature. 2012;482:495–500. doi: 10.1038/nature10822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGourty CA, Akopian D, Walsh C, Gorur A, Werner A, Schekman R, Bautista D, Rape M. Regulation of the CUL3 Ubiquitin Ligase by a Calcium-Dependent Co-adaptor. Cell. 2016;167:525–538 e514. doi: 10.1016/j.cell.2016.09.026. [DOI] [PubMed] [Google Scholar]
- 48.Boyce M, Carrico IS, Ganguli AS, Yu SH, Hangauer MJ, Hubbard SC, Kohler JJ, Bertozzi CR. Metabolic cross-talk allows labeling of O-linked beta-N-acetylglucosamine-modified proteins via the N-acetylgalactosamine salvage pathway. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3141–3146. doi: 10.1073/pnas.1010045108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zachara NE, Molina H, Wong KY, Pandey A, Hart GW. The dynamic stress-induced "O-GlcNAc-ome" highlights functions for O-GlcNAc in regulating DNA damage/repair and other cellular pathways. Amino acids. 2011;40:793–808. doi: 10.1007/s00726-010-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dudognon P, Maeder-Garavaglia C, Carpentier JL, Paccaud JP. Regulation of a COPII component by cytosolic O-glycosylation during mitosis. FEBS letters. 2004;561:44–50. doi: 10.1016/S0014-5793(04)00109-7. [DOI] [PubMed] [Google Scholar]
- 51.Teo CF, Ingale S, Wolfert MA, Elsayed GA, Not LG, Chatham JC, Wells L, Boons GJ. Glycopeptide-specific monoclonal antibodies suggest new roles for O-GlcNAc. Nature chemical biology. 2010;6:338–343. doi: 10.1038/nchembio.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee A, Miller D, Henry R, Paruchuri VD, O'Meally RN, Boronina T, Cole RN, Zachara NE. Combined Antibody/Lectin Enrichment Identifies Extensive Changes in the O-GlcNAc Sub-proteome upon Oxidative Stress. Journal of proteome research. 2016;15:4318–4336. doi: 10.1021/acs.jproteome.6b00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hanover JA, Krause MW, Love DC. The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochimica et biophysica acta. 2010;1800:80–95. doi: 10.1016/j.bbagen.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annual review of biochemistry. 2011;80:825–858. doi: 10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bond MR, Hanover JA. A little sugar goes a long way: the cell biology of O-GlcNAc. The Journal of cell biology. 2015;208:869–880. doi: 10.1083/jcb.201501101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hart GW. Three Decades of Research on O-GlcNAcylation - A Major Nutrient Sensor That Regulates Signaling, Transcription and Cellular Metabolism. Frontiers in endocrinology. 2014;5:183. doi: 10.3389/fendo.2014.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shafi R, Iyer SP, Ellies LG, O'Donnell N, Marek KW, Chui D, Hart GW, Marth JD. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:5735–5739. doi: 10.1073/pnas.100471497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Keembiyehetty C, Love DC, Harwood KR, Gavrilova O, Comly ME, Hanover JA. Conditional knockout reveals a requirement for O-GlcNAcase in metabolic homeostasis. The Journal of Biological Chemistry. 2015;290:7097–7113. doi: 10.1074/jbc.M114.617779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gloster TM, Zandberg WF, Heinonen JE, Shen DL, Deng L, Vocadlo DJ. Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nature chemical biology. 2011;7:174–181. doi: 10.1038/nchembio.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu SH, Boyce M, Wands AM, Bond MR, Bertozzi CR, Kohler JJ. Metabolic labeling enables selective photocrosslinking of O-GlcNAc-modified proteins to their binding partners. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:4834–4839. doi: 10.1073/pnas.1114356109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, Whitworth GE, Stubbs KA, McEachern EJ, Davies GJ, Vocadlo DJ. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nature chemical biology. 2008;4:483–490. doi: 10.1038/nchembio.96. [DOI] [PubMed] [Google Scholar]
- 62.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning, A Laboratory Manual. Second. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 63.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nature methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature protocols. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sadelain M, Papapetrou EP, Bushman FD. Safe harbours for the integration of new DNA in the human genome. Nature reviews. Cancer. 2011;12:51–58. doi: 10.1038/nrc3179. [DOI] [PubMed] [Google Scholar]
- 66.Cutrona MB, Beznoussenko GV, Fusella A, Martella O, Moral P, Mironov AA. Silencing of mammalian Sar1 isoforms reveals COPII-independent protein sorting and transport. Traffic. 2013;14:691–708. doi: 10.1111/tra.12060. [DOI] [PubMed] [Google Scholar]
- 67.Dale RM, Topczewski J. Identification of an evolutionarily conserved regulatory element of the zebrafish col2a1a gene. Dev Biol. 2011;357:518–531. doi: 10.1016/j.ydbio.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schindelin J, Rueden CT, Hiner MC, Eliceiri KW. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol Reprod Dev. 2015;82:518–529. doi: 10.1002/mrd.22489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bi X, Corpina RA, Goldberg J. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 2002;419:271–277. doi: 10.1038/nature01040. [DOI] [PubMed] [Google Scholar]
- 70.Zanetti G, Prinz S, Daum S, Meister A, Schekman R, Bacia K, Briggs JA. The structure of the COPII transport-vesicle coat assembled on membranes. Elife. 2013;2:e00951. doi: 10.7554/eLife.00951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fath S, Mancias JD, Bi X, Goldberg J. Structure and organization of coat proteins in the COPII cage. Cell. 2007;129:1325–1336. doi: 10.1016/j.cell.2007.05.036. [DOI] [PubMed] [Google Scholar]
- 72.Stagg SM, LaPointe P, Razvi A, Gurkan C, Potter CS, Carragher B, Balch WE. Structural basis for cargo regulation of COPII coat assembly. Cell. 2008;134:474–484. doi: 10.1016/j.cell.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bhattacharya N, J OD, Stagg SM. The structure of the Sec13/31 COPII cage bound to Sec23. Journal of molecular biology. 2012;420:324–334. doi: 10.1016/j.jmb.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bi X, Mancias JD, Goldberg J. Insights into COPII coat nucleation from the structure of Sec23.Sar1 complexed with the active fragment of Sec31. Developmental cell. 2007;13:635–645. doi: 10.1016/j.devcel.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Elsayed LE, Mohammed IN, Hamed AA, Elseed MA, Johnson A, Mairey M, Mohamed HE, Idris MN, Salih MA, El-Sadig SM, Koko ME, Mohamed AY, Raymond L, Coutelier M, Darios F, Siddig RA, Ahmed AK, Babai AM, Malik HM, Omer ZM, Mohamed EO, Eltahir HB, Magboul NA, Bushara EE, Elnour A, Rahim SM, Alattaya A, Elbashir MI, Ibrahim ME, Durr A, Audhya A, Brice A, Ahmed AE, Stevanin G. Hereditary spastic paraplegias: identification of a novel SPG57 variant affecting TFG oligomerization and description of HSP subtypes in Sudan. Eur J Hum Genet. 2016;25:100–110. doi: 10.1038/ejhg.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Witte K, Schuh AL, Hegermann J, Sarkeshik A, Mayers JR, Schwarze K, Yates JR, 3rd, Eimer S, Audhya A. TFG-1 function in protein secretion and oncogenesis. Nature cell biology. 2011;13:550–558. doi: 10.1038/ncb2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Johnson A, Bhattacharya N, Hanna M, Pennington JG, Schuh AL, Wang L, Otegui MS, Stagg SM, Audhya A. TFG clusters COPII-coated transport carriers and promotes early secretory pathway organization. The EMBO journal. 2015;34:811–827. doi: 10.15252/embj.201489032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bard F, Casano L, Mallabiabarrena A, Wallace E, Saito K, Kitayama H, Guizzunti G, Hu Y, Wendler F, Dasgupta R, Perrimon N, Malhotra V. Functional genomics reveals genes involved in protein secretion and Golgi organization. Nature. 2006;439:604–607. doi: 10.1038/nature04377. [DOI] [PubMed] [Google Scholar]
- 79.Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- 80.Scales SJ, Pepperkok R, Kreis TE. Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell. 1997;90:1137–1148. doi: 10.1016/s0092-8674(00)80379-7. [DOI] [PubMed] [Google Scholar]
- 81.Reid DW, Chen Q, Tay AS, Shenolikar S, Nicchitta CV. The unfolded protein response triggers selective mRNA release from the endoplasmic reticulum. Cell. 2014;158:1362–1374. doi: 10.1016/j.cell.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mondoux MA, Love DC, Ghosh SK, Fukushige T, Bond M, Weerasinghe GR, Hanover JA, Krause MW. O-linked-N-acetylglucosamine cycling and insulin signaling are required for the glucose stress response in Caenorhabditis elegans. Genetics. 2011;188:369–382. doi: 10.1534/genetics.111.126490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bond MR, Hanover JA. O-GlcNAc cycling: a link between metabolism and chronic disease. Annual review of nutrition. 2013;33:205–229. doi: 10.1146/annurev-nutr-071812-161240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barlowe C, Helenius A. Cargo Capture and Bulk Flow in the Early Secretory Pathway. Annual review of cell and developmental biology. 2016;32:197–222. doi: 10.1146/annurev-cellbio-111315-125016. [DOI] [PubMed] [Google Scholar]
- 85.Malhotra V, Erlmann P, Nogueira C. Procollagen export from the endoplasmic reticulum. Biochemical Society transactions. 2015;43:104–107. doi: 10.1042/BST20140286. [DOI] [PubMed] [Google Scholar]
- 86.Vincourt JB, Etienne S, Cottet J, Delaunay C, Malanda CB, Lionneton F, Sirveaux F, Netter P, Plenat F, Mainard D, Vignaud JM, Magdalou J. C-propeptides of procollagens I alpha 1 and II that differentially accumulate in enchondromas versus chondrosarcomas regulate tumor cell survival and migration. Cancer research. 2010;70:4739–4748. doi: 10.1158/0008-5472.CAN-10-0046. [DOI] [PubMed] [Google Scholar]
- 87.Aigner T. Towards a new understanding and classification of chondrogenic neoplasias of the skeleton--biochemistry and cell biology of chondrosarcoma and its variants. Virchows Arch. 2002;441:219–230. doi: 10.1007/s00428-002-0641-x. [DOI] [PubMed] [Google Scholar]
- 88.Kim SD, Pahuja KB, Ravazzola M, Yoon J, Boyadjiev SA, Hammamoto S, Schekman R, Orci L, Kim J. The [corrected] SEC23-SEC31 [corrected] interface plays critical role for export of procollagen from the endoplasmic reticulum. The Journal of Biological Chemistry. 2012;287:10134–10144. doi: 10.1074/jbc.M111.283382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Melville DB, Knapik EW. Traffic jams in fish bones: ER-to-Golgi protein transport during zebrafish development. Cell Adh Migr. 2011;5:114–118. doi: 10.4161/cam.5.2.14377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Unlu G, Levic DS, Melville DB, Knapik EW. Trafficking mechanisms of extracellular matrix macromolecules: insights from vertebrate development and human diseases. The international journal of biochemistry & cell biology. 2014;47:57–67. doi: 10.1016/j.biocel.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Matsuoka K, Orci L, Amherdt M, Bednarek SY, Hamamoto S, Schekman R, Yeung T. COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell. 1998;93:263–275. doi: 10.1016/s0092-8674(00)81577-9. [DOI] [PubMed] [Google Scholar]
- 92.Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Science signaling. 2011;4:rs5. doi: 10.1126/scisignal.2001497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen XW, Wang H, Bajaj K, Zhang P, Meng ZX, Ma D, Bai Y, Liu HH, Adams E, Baines A, Yu G, Sartor MA, Zhang B, Yi Z, Lin J, Young SG, Schekman R, Ginsburg D. SEC24A deficiency lowers plasma cholesterol through reduced PCSK9 secretion. Elife. 2013;2:e00444. doi: 10.7554/eLife.00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Adams EJ, Chen XW, O'Shea KS, Ginsburg D. Mammalian COPII coat component SEC24C is required for embryonic development in mice. The Journal of Biological Chemistry. 2014;289:20858–20870. doi: 10.1074/jbc.M114.566687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Baines AC, Adams EJ, Zhang B, Ginsburg D. Disruption of the Sec24d gene results in early embryonic lethality in the mouse. PLoS ONE. 2013;8:e61114. doi: 10.1371/journal.pone.0061114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Khoriaty R, Everett L, Chase J, Zhu G, Hoenerhoff M, McKnight B, Vasievich MP, Zhang B, Tomberg K, Williams J, Maillard I, Ginsburg D. Pancreatic SEC23B deficiency is sufficient to explain the perinatal lethality of germline SEC23B deficiency in mice. Sci Rep. 2016;6:27802. doi: 10.1038/srep27802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Khoriaty R, Vasievich MP, Jones M, Everett L, Chase J, Tao J, Siemieniak D, Zhang B, Maillard I, Ginsburg D. Absence of a red blood cell phenotype in mice with hematopoietic deficiency of SEC23B. Molecular and cellular biology. 2014;34:3721–3734. doi: 10.1128/MCB.00287-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tao J, Zhu M, Wang H, Afelik S, Vasievich MP, Chen XW, Zhu G, Jensen J, Ginsburg D, Zhang B. SEC23B is required for the maintenance of murine professional secretory tissues. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E2001–2009. doi: 10.1073/pnas.1209207109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhu M, Tao J, Vasievich MP, Wei W, Zhu G, Khoriaty RN, Zhang B. Neural tube opening and abnormal extraembryonic membrane development in SEC23A deficient mice. Sci Rep. 2015;5:15471. doi: 10.1038/srep15471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Andres-Bergos J, Tardio L, Larranaga-Vera A, Gomez R, Herrero-Beaumont G, Largo R. The increase in O-linked N-acetylglucosamine protein modification stimulates chondrogenic differentiation both in vitro and in vivo. The Journal of Biological Chemistry. 2012;287:33615–33628. doi: 10.1074/jbc.M112.354241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Saito K, Yamashiro K, Ichikawa Y, Erlmann P, Kontani K, Malhotra V, Katada T. cTAGE5 mediates collagen secretion through interaction with TANGO1 at endoplasmic reticulum exit sites. Molecular biology of the cell. 2011;22:2301–2308. doi: 10.1091/mbc.E11-02-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Summary of MS data and identified O-GlcNAc sites
Figure S2: Effect of 5SGlcNAc on ssHRP assay
Figure S3: Organelle marker and Thiamet-G controls for tsVSVG-eGFP assay
Figure S4: Digitonin fractionation replicates and densitometry quantification
Figure S5: Validation of SEC23A deletion by CRISPR/Cas9
Figure S6: Collagen accumulation in SEC23A−/− cells
Figure S7: Distended ER morphology in SEC23A−/− cells
Figure S8: Validation of SEC23B−/− depletion by siRNA
Figure S9: SEC23A S184A and 4A co-IPs with SEC24 paralogs
Figures S10: Lack of effect on ERES by SEC23A deletion or OGT/OGA inhibition