

Abstract
The properties of graphene nanoribbons are highly dependent on structural variables such as width, length, edge structure, and heteroatom doping. Therefore, atomic precision over all these variables is necessary for establishing their fundamental properties and exploring their potential applications. An iterative approach is presented that assembles a small and carefully designed molecular building block into monodisperse N‐doped graphene nanoribbons with different lengths. To showcase this approach, the synthesis and characterisation of a series of nanoribbons constituted of 10, 20 and 30 conjugated linearly‐fused rings (2.9, 5.3, and 7.7 nm in length, respectively) is presented.
Keywords: graphene, nanoribbons, polycyclic aromatic hydrocarbons
The discovery of fullerenes, nanotubes, and graphene has stimulated the exploration of synthetic low‐dimensional carbon nanostructures. Among these, quasi‐one‐dimensional atomically precise substructures of graphene, known as graphene nanoribbons (NRs),1 combine the one‐atom thickness of graphene with the structure‐dependent metallicity of carbon nanotubes. NRs have unique electronic, optical and mechanical properties and are considered promising candidates to develop new technologies for electronics,2 photonics,3 and energy conversion,4 among others. The properties of NRs are highly dependent on several structural variables such as width, length, edge structure, and heteroatom doping. Therefore, atomic precision over these variables is necessary for establishing their fundamental properties and exploring their potential applications. The edge structure of NRs influences their metallicity5 and their photonic properties.3 The size of the energy gap of NRs is strongly influenced by the width.5g For example, energy gaps >1.4 eV are expected for NRs with sub‐nm widths. The length is also an important variable in NRs, as the size of the energy gap decreases with increasing length until saturation. Also, lengths of more than 5 nm constitute a structural prerequisite to explore the potential of NRs in single NR field‐effect transistors.6
Even if there have been enormous advances in the synthesis of NRs,7 current approaches do not allow the attainment of atomic precision over width, length, and edge structure simultaneously on NRs of more than 5 nm in length. Top‐down methods such as cutting graphene or unzipping carbon nanotubes by means of lithography or etching have been applied to prepare NRs,8 but they do not provide atomic precision over any structural variable. Bottom‐up on‐surface synthesis,5h, 9 in‐nanotube synthesis,10 and solution polymerisation methods11 provide atomically precise control over the edge and width of the NRs, but do not provide atomic precision over the length.
A promising approach that can provide simultaneously atomic precision over edge, width, and length is multistep organic synthesis in solution. In fact, several families of monodisperse NRs with more than 2 nm in length have been reported2c, 4c, 11h, 12 that evolve from acenes, naphthalene, pyrene, perylene, coronene, and rylene derivatives, among others. However, until now, only NRs with lengths approaching 5 nm have been obtained by this approach.4c, 12b This is up to 18 fused aromatic rings in a linear arrangement4c and up to 23 fused aromatic rings in an armchair arrangement.12b Approaching the synthesis of NRs more than 5 nm in length from an organic chemistry perspective is very challenging because of the large number of different synthetic and purification steps that have to be individually optimised and also because of the high tendency of large aromatic systems to aggregate in solution, which makes difficult, and in some cases even hamper, their synthesis, purification, characterisation, and processing.
Herein we report an iterative approach that assembles a small molecular building block into NRs of different lengths, opening up a new route for the preparation of monodisperse NRs. To showcase this approach, we describe the synthesis of a series of NRs constituted of 10, 20, and 30 linearly‐fused aromatic rings (with 2.9, 5.3, and 7.7 nm in length, respectively), which include the longest monodisperse NRs reported to date (Scheme 1; Supporting Information, Figure S1). Remarkably, the whole NR series is soluble in chlorinated solvents, which has allowed their purification by flash chromatography and their characterisation by 1H and 13C NMR spectroscopy, high‐resolution mass spectrometry, absorption and photoluminescence spectroscopy, cyclic voltammetry, and time‐resolved microwave conductivity measurements. These studies, besides confirming unequivocally the structure of the NRs, provide a detailed picture of their structure‐property relationship.
Scheme 1.
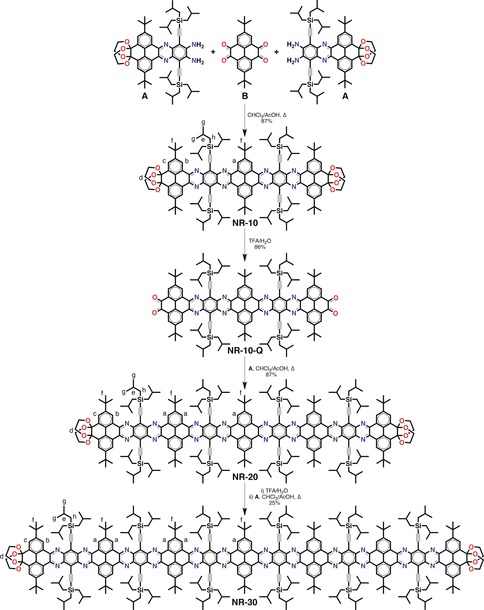
Iterative synthesis of NR‐10, NR‐20, and NR‐30.
Our approach is based on the iterative assembly molecular building block A into NRs of different lengths. Building block A consists of a dibenzodiazatetracene core equipped with o‐diamines at one end and diacetal‐protected o‐dione functionalities at the other end (Scheme 1). o‐Diamines and o‐diones have been selected as they converge through the formation of a pyrazine ring by an imine‐type cyclocondensation reaction, providing an efficient mean to interconnect building blocks. The o‐dione functionalities have been protected to avoid the self‐condensation of building block A, and thus enable a stepwise condensation of one building block after the other. The presence of lateral rings increases the number of aromatic sextets (indicated by Robinson's circle notation) on the electronic structure of the resulting NRs, providing them with a high stability.11b,11d Importantly, carefully selected solubilising groups have been introduced in key positions to ensure the dispersibility of the resulting NRs. We selected tert‐butyl and tri‐isobutylsilyl groups because of their relatively small size and large solubilising power.13 The synthesis of building block A has been achieved in twelve steps and is described in the Supporting Information.
2,7‐Di‐tert‐butylpyrene‐4,5,9,10‐tetraone B14 has been condensed with 2.5 equivalents of building block A by a double cyclocondensation reaction, providing NR‐10, a hexabenzododecacene with 10 linearly‐fused aromatic rings in one single step in an 87 % yield (Scheme 1). This process is followed by the deprotection of the terminal ketones in the presence of TFA/H2O yielding NR‐10‐Q, which was isolated in a 86 % yield and characterised. Then, NR‐10‐Q was subsequently cyclocondensed with 4 equivalents of building block A to provide NR‐20, a decabenzodocosacene with 20 linearly fused aromatic rings in a 87 % yield. This condensation/deprotection process has been repeated over NR‐20 without the complete isolation of the tetraone intermediate to provide NR‐30, a tetradecabenzodotriacontacene with 30 linearly‐fused aromatic rings in a 25 % yield over the two steps. The complete synthetic procedures for NR‐10, NR‐20, and NR‐30 are described in the Supporting Information. NR‐10, NR‐20, and NR‐30 were soluble in common chlorinated solvents, such as CHCl3, CH2Cl2, and o‐dichlorobenzene (ODCB), providing bright magenta solutions similar to the colour of the powders.
The structure of the NRs was unambiguously confirmed by a combination of 1H NMR, 13C NMR, and high‐resolution mass spectrometry (HRMS), and is consistent with UV/Vis steady‐state absorption and photoluminescence spectroscopy (see below). The high solubility made the structural characterisation of the NRs very easy as resolved 1H NMR and 13C NMR spectra could be recorded at room temperature in CDCl3 for the whole NR series (Figure 1 and Supporting Information). The sharp NMR signals in the aromatic and aliphatic region are consistent with the structure of the NRs. For instance, the integration in 1H NMR correlates with the length of the NRs, as signal a (the assignments correspond to the lettering shown in Scheme 1) integrates 4, 12, and 20 respectively for NR‐10, NR‐20, and NR‐30, while protons b, c, and d integrate 4, 4, and 8 independently of the length of the NRs, as expected (Figure 1). Matrix‐assisted laser desorption/ionization time of flight high‐resolution mass spectrometry (MALDI‐TOF HRMS) show both the expected molecular ion peaks (M+Ag)+ and the isotopic distributions for NR‐10, NR‐20, and NR‐30 (Supporting Information, Figure S2), which are in agreement with the calculated spectra.
Figure 1.
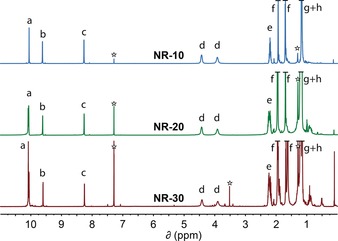
NMR spectra of NR‐10, NR‐20, and NR‐30 in CDCl3. The assignments correspond to the lettering in Scheme 1. The stars indicate residual solvent peaks.
After several attempts, we were unable to grow crystals suitable for X‐ray diffraction, so we relied on calculations to gain insight into the structure of the NRs. The GFN‐xTB (geometry, frequency, non‐covalent, extended tight‐binding) Hamiltonian15 was used to generate and screen different conformers that were optimized at the B3LYP‐6–31g(d,p) level. DFT models show that the bulky solubilising groups possess some, but not large, intramolecular steric hindrance, which gives rise to a wealth of non‐flat structures (Supporting Information, Figure S1). To further understand the flexibility of the aromatic framework, we computed the energy needed to progressively twist the aromatic backbone of NR‐10‐H, in which the tri‐isobutylsilyl groups have been exchanged for a H atom (Supporting Information, Figure S3), from 0° to 90° (Supporting Information, Figure S4). It was found that, for instance, a 30° end‐to‐end twist only costs about 1 kcal mol−1. Therefore, a large number of different longitudinally twisted conformations and of alternated twisted conformations are possible at room temperature. The models allow us to compute the dimensions of the NR series yielding 2.9, 5.3, and 7.7 nm in length, respectively for NR‐10, NR‐20, and NR‐30, and 0.7 nm in width across the pyrene moiety (Supporting Information, Figure S1).
The electronic absorption features of the NR series in CHCl3 are consistent with their colour and their structure. The spectra show a small band at about 600 nm, a set of bands with clear vibronic features between 400 and 600 nm and another set in the UV region, which were assigned to the α, ρ, and β bands16 from longer to shorter wavelengths, respectively (Figure 2 a). The absorption features are consistent with the electronic structure of previously reported dibenzohexacenes17 and dibenzooctacenes,17 but bathocromically shifted as expected for the more extended π‐system of the NRs. The α bands (with maxima at 604, 605, and 605 nm, respectively for NR‐10, NR‐20, and NR‐30) are nearly independent of the length of the NRs, while the ρ (with maxima at 528, 544, and 549 nm, respectively for NR‐10, NR‐20, and NR‐30) and the β bands (with maxima at 378, 385, and 387 nm, respectively for NR‐10, NR‐20, and NR‐30) show a clear bathochromic shift with increasing lengths. Importantly, the molar attenuation coefficients of NR‐10 (185 527 L mol−1 cm−1), NR‐20 (426 294 L mol−1 cm−1), and NR‐30 (730 137 L mol−1 cm−1) increase with increasing lengths as an effect of the more extended π‐system.
Figure 2.
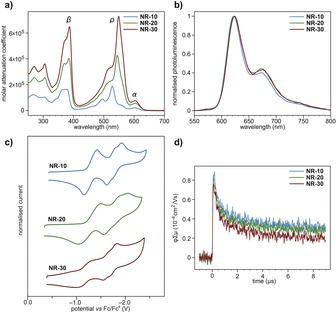
a) UV/Vis electronic absorption and b) photoluminescence spectra in CHCl3. c) Cyclic voltammograms in an Ar‐saturated 0.1 m solution of nBu4NPF6 in ODCB. Potentials versus Fc/Fc+. d) TRMC (λ ex=355 nm, I 0=9.1×1015 photons cm−2).
The electronic absorption spectra of NR‐10, NR‐20, and NR‐30 were simulated to provide an insight into the nature of the lowest‐energy electronic transitions. TD‐DFT was computed with the 6–31g(d,p) basis set with the B3LYP Hamiltonian. The simulated spectra of the lowest‐energy conformations of NR‐10, NR‐20, and NR‐30 correlate well with the experimental electronic spectra (Supporting Information, Figure S5). Similar to the experiments, two bands were obtained in all cases: a band with a smaller oscillator strength that corresponds to the experimental α band, which is the result of the transitions between the quasi‐degenerate HOMOs and LUMOs as the eigenvalues of the frontier orbitals show quasi‐degeneration of 2, 4, and 6 for NR‐10, NR‐20, and NR‐30, respectively (Supporting Information, Table S1); and a band with a larger oscillator strength that corresponds to the experimental ρ band, which is the result of transitions from the lower occupied molecular orbitals underneath the quasidegenerate HOMOs to the quasidegenerate LUMO levels. Additionally, we explicitly modelled the effect of the NR twisting on the absorption spectra by computing the TD‐DFT spectra for different twisted geometries of NR‐10‐H and found it to be negligible (Supporting Information, Figure S6).
The photoluminescence spectra in CHCl3 are almost superimposable over the whole series of NRs and show a clear emission band with vibrational features that span from 600 to 800 nm (Figure 2 b). This is because the emission emerges from the longest wavelength α absorption band, which remains nearly invariable as the length of the NRs increases. Only small increasing bathochromic shifts of <1 nm are observed with increasing length. On the other hand the quantum yields (Φ)18 of NR‐10 (0.27), NR‐20 (0.14), and NR‐30 (0.11) decrease with increasing lengths.
The electrochemical properties of the complete NR series were investigated by cyclic voltammetry in an Ar‐saturated 0.1 m solution of nBu4NPF6 in ODCB using the ferrocene/ferrocinium redox couple (Fc/Fc+) as an internal standard. The voltammograms show three reduction processes and no oxidation processes in the potential window of the solvent‐supported electrolyte system used (Figure 2 c). The voltammograms are similar and show a first reduction wave, followed by two reduction waves grouped at more cathodic potentials. The half‐wave potentials of the first reduction wave (E 1/2 I) are similar for NR‐10 (−1.28 V) and NR‐20 (−1.27 V), while in the case of NR‐30 (−1.18 V) the potential is more anodic. The same trend was also observed on the half‐wave potentials of the second (E 1/2 II) and third (E 1/2 III) reduction waves. Similar E 1/2 II and E 1/2 III potentials were observed for NR‐10 (−1.72 V and −1.82 V, respectively) and NR‐20 (−1.72 V and −1.89 V, respectively), while in the case of NR‐30 (−1.62 V and −1.75 V, respectively) the E 1/2 II and E 1/2 III potentials are again more anodic. All this confirms that the NR‐10, NR‐20, and NR‐30 are highly electron‐deficient materials and that the stability of their electrogenerated radical‐anions increase with increasing lengths.
The optical HOMO–LUMO gaps (E gap) have been estimated from the absorption onset of the longest wavelength transition. The E gap of NR‐10 (1.97 eV), NR‐20 (1.96 eV), and NR‐30 (1.96 eV) are in a similar range of inorganic semiconductors, such as BP, AlAs, AlSb, CdSe, Cu2O, GaSe, SiC, SnS2, and ZnTe. A comparison of the E gap of the NR series with those of previously reported dibenzohexacenes17 (2.8 eV) and dibenzooctacenes17 (2.4 eV), with 6 and 8 linearly‐fused rings, respectively, reveals that the E gap for this NR family decreases rapidly with increasing lengths until NR‐10, from where the E gap remains almost invariable. This is in line with the computed E gap values at the B3LYP 6–31g(d,p) level in vacuum for NR‐10 (2.23 eV), NR‐20 (2.13 eV), and NR‐30 (2.13 eV). The electron affinities or electrochemical LUMO levels (E LUMO) have been estimated from the onset potential of the first reduction wave of the cyclic voltammograms.19 The E LUMO values of NR‐10 (−3.60 eV), NR‐20 (−3.63 eV), and NR‐30 (−3.69 eV) decrease with increasing lengths. This is again consistent with the computed E LUMO values at the B3LYP‐6–31g(d,p) level in vacuum for NR‐10 (−3.12 eV), NR‐20 (−3.23 eV), and NR‐30 (−3.26 eV). The eigenvalues of both frontier orbitals, HOMO and LUMO, show quasidegeneration of 2, 4 and 6 for NR‐10, NR‐20, and NR‐30, respectively (Supporting Information, Table S1). The quasidegenerate HOMOs and LUMOs show electron densities mostly localised in specific residues across the different NRs, as seen on the 6 quasidegenerate HOMOs and 6 quasidegenerate LUMOs of NR‐30 (Supporting Information, Figure S7).
To shed light on the conducting properties of the NRs, time‐resolved microwave conductivity (TRMC)20 measurements have been carried out (Figure 2 d). TRMC measures the pseudo‐photoconductivity (ϕΣμ, where ϕ is the product of the quantum yield, and Σμ is the sum of the charge carrier mobilities) under an oscillating microwave electric field directly from powder samples with no metal contacts. The measurements show nearly invariable ϕΣμ maxima for NR‐10 (0.85×10−4 cm2 V−1 s−1), NR‐20 (0.82×10−4 cm2 V−1 s−1), and NR‐30 (0.75×10−4 cm2 V −1 s−1), and also nearly invariable half lifetimes (τ 1/2) for NR‐10 (1.4 μs), NR‐20 (1.1 μs), and NR‐30 (0.9 μs). These ϕΣμ values are in line with those observed on conjugated polymers such as P3HT and polyfluorene.20 The insignificant differences found in ϕΣμ maxima and τ 1/2 with respect to NR length are consistent with the localized HOMO/LUMO densities.
To conclude, we have successfully synthesised a series of monodisperse NRs constituted of 10, 20, and 30 linearly‐fused conjugated rings (2.9, 5.3, and 7.7 nm in length, respectively), by assembling a small and carefully designed molecular building block through a new iterative approach. Remarkably, the resulting NRs are soluble in chlorinated solvents, which has allowed their purification by flash chromatography and their characterisation by 1H and 13C NMR spectroscopy, HRMS, absorption and photoluminescence spectroscopy, cyclic voltammetry, and TRMC measurements. These studies, besides confirming unequivocally the structure of the NRs, show increasing molar attenuation coefficients, decreasing photoluminescence Φ, and decreasing LUMO levels with increasing lengths. Conversely, a series of nearly length‐invariable properties such as optical energy gaps, photoluminescence spectra and pseudo‐photoconductivities are also revealed for this family of NRs. Overall, the approach reported herein has not only allowed the preparation of the longest monodisperse NRs reported to date, but most importantly paves the way for the synthesis of longer monodisperse NRs, for validating their predicted theoretical properties and for exploring their properties in field‐effect transistors,2c photodetectors,21 solar cells4, 22 and molecular wires,23 in which NRs have shown a lot of promise. Excitingly, the limits of this iterative approach are yet to be seen.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful to the Basque Science Foundation for Science (Ikerbasque), POLYMAT, the University of the Basque Country (SGIker), the Deutsche Forschungsgemeinschaft (MA 5215/4‐1), Gobierno de España (Ministerio de Economía y Competitividad CTQ2016‐77970‐R and CTQ2015‐71936‐REDT), Gobierno Vasco (BERC program and PC2015‐1‐01(0637)), Diputación Foral de Guipúzcoa (OF215/2016(ES)), CICECO—Aveiro Institute of Materials, POCI‐01‐0145‐FEDER‐007679 (FCT ref. UID/CTM/50011/ 2013), ON2 (NORTE‐07‐0162‐FEDER‐000086), and the FP7 framework program of the European Union (ERA Chemistry, Marie Curie Career Integration Grant No. 618247 (NIRVANA)). This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 664878. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 722951).
D. Cortizo-Lacalle, J. P. Mora-Fuentes, K. Strutyński, A. Saeki, M. Melle-Franco, A. Mateo-Alonso, Angew. Chem. Int. Ed. 2018, 57, 703.
Contributor Information
Prof. Dr. Manuel Melle‐Franco, Email: manuelmelle.research@gmail.com
Prof. Dr. Aurelio Mateo‐Alonso, Email: amateo@polymat.eu.
References
- 1. Fujita M., Wakabayashi K., Nakada K., Kusakabe K., J. Phys. Soc. Jpn. 1996, 65, 1920–1923. [Google Scholar]
- 2.
- 2a. Jiao L., Wang X., Diankov G., Wang H., Dai H., Nat. Nanotechnol. 2010, 5, 321–325; [DOI] [PubMed] [Google Scholar]
- 2b. Wei D., Xie L., Lee K. K., Hu Z., Tan S., Chen W., Sow C. H., Chen K., Liu Y., Wee A. T. S., Nat. Commun. 2013, 4, 1374; [DOI] [PubMed] [Google Scholar]
- 2c. Zhong Y., Kumar B., Oh S., Trinh M. T., Wu Y., Elbert K., Li P., Zhu X., Xiao S., Ng F., Steigerwald M. L., Nuckolls C., J. Am. Chem. Soc. 2014, 136, 8122–8130. [DOI] [PubMed] [Google Scholar]
- 3. Fei Z., Goldflam M. D., Wu J. S., Dai S., Wagner M., McLeod A. S., Liu M. K., Post K. W., Zhu S., Janssen G. C. A. M., Fogler M. M., Basov D. N., Nano Lett. 2015, 15, 8271–8276. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Meng X., Yu C., Song X., Liu Y., Liang S., Liu Z., Hao C., Qiu J., Adv. Energy Mater. 2015, 5, 1500180; [Google Scholar]
- 4b. Zhong Y., Trinh M. T., Chen R., Purdum G. E., Khlyabich P. P., Sezen M., Oh S., Zhu H., Fowler B., Zhang B., Wang W., Nam C.-Y., Sfeir M. Y., Black C. T., Steigerwald M. L., Loo Y.-L., Ng F., Zhu X. Y., Nuckolls C., Nat. Commun. 2015, 6, 8242; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Sisto T. J., Zhong Y., Zhang B., Trinh M. T., Miyata K., Zhong X., Zhu X. Y., Steigerwald M. L., Ng F., Nuckolls C., J. Am. Chem. Soc. 2017, 139, 5648–5651. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Son Y.-W., Cohen M. L., Louie S. G., Nature 2006, 444, 347–349; [DOI] [PubMed] [Google Scholar]
- 5b. Wang X., Ouyang Y., Jiao L., Wang H., Xie L., Wu J., Guo J., Dai H., Nat. Nanotechnol. 2011, 6, 563–567; [DOI] [PubMed] [Google Scholar]
- 5c. Ritter K. A., Lyding J. W., Nat. Mater. 2009, 8, 235–242; [DOI] [PubMed] [Google Scholar]
- 5d. Kimouche A., Ervasti M. M., Drost R., Halonen S., Harju A., Joensuu P. M., Sainio J., Liljeroth P., Nat. Commun. 2015, 6, 10177; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. Chen Y.-C., Cao T., Chen C., Pedramrazi Z., Haberer D., de OteyzaDimas G., Fischer F. R., Louie S. G., Crommie M. F., Nat. Nanotechnol. 2015, 10, 156–160; [DOI] [PubMed] [Google Scholar]
- 5f. Magda G. Z., Jin X., Hagymasi I., Vancso P., Osvath Z., Nemes-Incze P., Hwang C., Biro L. P., Tapaszto L., Nature 2014, 514, 608–611; [DOI] [PubMed] [Google Scholar]
- 5g. Barone V., Hod O., Scuseria G. E., Nano Lett. 2006, 6, 2748–2754; [DOI] [PubMed] [Google Scholar]
- 5h. Cai J., Ruffieux P., Jaafar R., Bieri M., Braun T., Blankenburg S., Muoth M., Seitsonen A. P., Saleh M., Feng X., Mullen K., Fasel R., Nature 2010, 466, 470–473. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Qiu C., Zhang Z., Xiao M., Yang Y., Zhong D., Peng L.-M., Science 2017, 355, 271–276; [DOI] [PubMed] [Google Scholar]
- 6b. Narita A., Feng X., Müllen K., Chem. Rec. 2015, 15, 295–309. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Chen L., Hernandez Y., Feng X., Müllen K., Angew. Chem. Int. Ed. 2012, 51, 7640–7654; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7758–7773; [Google Scholar]
- 7b. Segawa Y., Ito H., Itami K., Nat. Rev. Mater. 2016, 1, 15002; [Google Scholar]
- 7c. Narita A., Wang X.-Y., Feng X., Mullen K., Chem. Soc. Rev. 2015, 44, 6616–6643; [DOI] [PubMed] [Google Scholar]
- 7d. Mateo-Alonso A., Chem. Soc. Rev. 2014, 43, 6311–6324. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Jia X., Hofmann M., Meunier V., Sumpter B. G., Campos-Delgado J., Romo-Herrera J. M., Son H., Hsieh Y.-P., Reina A., Kong J., Terrones M., Dresselhaus M. S., Science 2009, 323, 1701–1705; [DOI] [PubMed] [Google Scholar]
- 8b. Jiao L., Zhang L., Wang X., Diankov G., Dai H., Nature 2009, 458, 877–880; [DOI] [PubMed] [Google Scholar]
- 8c. Kosynkin D. V., Higginbotham A. L., Sinitskii A., Lomeda J. R., Dimiev A., Price B. K., Tour J. M., Nature 2009, 458, 872–876. [DOI] [PubMed] [Google Scholar]
- 9. Jiang L., Papageorgiou A. C., Oh S. C., Sağlam Ö., Reichert J., Duncan D. A., Zhang Y.-Q., Klappenberger F., Guo Y., Allegretti F., More S., Bhosale R., Mateo-Alonso A., Barth J. V., ACS Nano 2016, 10, 1033–1041. [DOI] [PubMed] [Google Scholar]
- 10. Chuvilin A., Bichoutskaia E., Gimenez-Lopez M. C., Chamberlain T. W., Rance G. A., Kuganathan N., Biskupek J., Kaiser U., Khlobystov A. N., Nat. Mater. 2011, 10, 687–692. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Stille J. K., Mainen E. L., J. Polym. Sci. B 1966, 4, 39–41; [Google Scholar]
- 11b. Stille J. K., Mainen E. L., Macromolecules 1968, 1, 36–42; [Google Scholar]
- 11c. Stille J. K., Noren G. K., Green L., J. Polym. Sci. A-1 1970, 8, 2245–2254; [Google Scholar]
- 11d. Imai K., Kuhihara M., Mathias L., Wittmann J., Alston W. B., Stille J. K., Macromolecules 1973, 6, 158–162; [Google Scholar]
- 11e. Fogel Y., Zhi L., Rouhanipour A., Andrienko D., Räder H. J., Müllen K., Macromolecules 2009, 42, 6878–6884; [Google Scholar]
- 11f. Tan Y.-Z., Yang B., Parvez K., Narita A., Osella S., Beljonne D., Feng X., Müllen K., Nat. Commun. 2013, 4, 2646; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11g. Narita A., Feng X., Hernandez Y., Jensen S. A., Bonn M., Yang H., Verzhbitskiy I. A., Casiraghi C., Hansen M. R., Koch A. H. R., Fytas G., Ivasenko O., Li B., Mali K. S., Balandina T., Mahesh S., De Feyter S., Müllen K., Nat. Chem. 2014, 6, 126–132; [DOI] [PubMed] [Google Scholar]
- 11h. Liu J., Li B.-W., Tan Y.-Z., Giannakopoulos A., Sanchez-Sanchez C., Beljonne D., Ruffieux P., Fasel R., Feng X., Müllen K., J. Am. Chem. Soc. 2015, 137, 6097–6103; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11i. Lehnherr D., Chen C., Pedramrazi Z., DeBlase C. R., Alzola J. M., Keresztes I., Lobkovsky E. B., Crommie M. F., Dichtel W. R., Chem. Sci. 2016, 7, 6357–6364; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11j. Jordan R. S., Wang Y., McCurdy R. D., Yeung M. T., Marsh K. L., Khan S. I., Kaner R. B., Rubin Y., Chem 2016, 1, 78–90; [Google Scholar]
- 11k. Yang W., Lucotti A., Tommasini M., Chalifoux W. A., J. Am. Chem. Soc. 2016, 138, 9137–9144; [DOI] [PubMed] [Google Scholar]
- 11l. Huang Y., Mai Y., Beser U., Teyssandier J., Velpula G., van Gorp H., Straasø L. A., Hansen M. R., Rizzo D., Casiraghi C., Yang R., Zhang G., Wu D., Zhang F., Yan D., De Feyter S., Müllen K., Feng X., J. Am. Chem. Soc. 2016, 138, 10136–10139; [DOI] [PubMed] [Google Scholar]
- 11m. Daigle M., Miao D., Lucotti A., Tommasini M., Morin J.-F., Angew. Chem. Int. Ed. 2017, 56, 6213–6217; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 6309–6313. [Google Scholar]
- 12.
- 12a. Schlicke B., Schlüter A. D., Hauser P., Heinze J., Angew. Chem. Int. Ed. Engl. 1997, 36, 1996–1998; [Google Scholar]; Angew. Chem. 1997, 109, 2091–2093; [Google Scholar]
- 12b. Zeng W., Phan H., Herng T. S., Gopalakrishna T. Y., Aratani N., Zeng Z., Yamada H., Ding J., Wu J., Chem 2017, 2, 81–92; [Google Scholar]
- 12c. Ozaki K., Kawasumi K., Shibata M., Ito H., Itami K., Nat. Commun. 2015, 6, 6251; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12d. Endres A. H., Schaffroth M., Paulus F., Reiss H., Wadepohl H., Rominger F., Krämer R., Bunz U. H. F., J. Am. Chem. Soc. 2016, 138, 1792–1795; [DOI] [PubMed] [Google Scholar]
- 12e. Gao B., Wang M., Cheng Y., Wang L., Jing X., Wang F., J. Am. Chem. Soc. 2008, 130, 8297–8306; [DOI] [PubMed] [Google Scholar]
- 12f. Wang Z., Gu P., Liu G., Yao H., Wu Y., Li Y., Rakesh G., Zhu J., Fu H., Zhang Q., Chem. Commun. 2017, 53, 7772–7775; [DOI] [PubMed] [Google Scholar]
- 12g. Purushothaman B., Bruzek M., Parkin S. R., Miller A.-F., Anthony J. E., Angew. Chem. Int. Ed. 2011, 50, 7013–7017; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7151–7155; [Google Scholar]
- 12h. Kaur I., Jazdzyk M., Stein N. N., Prusevich P., Miller G. P., J. Am. Chem. Soc. 2010, 132, 1261–1263; [DOI] [PubMed] [Google Scholar]
- 12i. Huang R., Phan H., Herng T. S., Hu P., Zeng W., Dong S.-q., Das S., Shen Y., Ding J., Casanova D., Wu J., J. Am. Chem. Soc. 2016, 138, 10323–10330; [DOI] [PubMed] [Google Scholar]
- 12j. Gu P. Y., Wang N., Wu A., Wang Z., Tian M., Fu Z., Sun X. W., Zhang Q., Chem. Asian J. 2016, 11, 2135–2138; [DOI] [PubMed] [Google Scholar]
- 12k. Gu P.-Y., Wang Z., Xiao F.-X., Lin Z., Song R., Xu Q.-F., Lu J.-M., Liu B., Zhang Q., Mater. Chem. Front. 2017, 1, 495–498. [Google Scholar]
- 13.
- 13a. More S., Choudhary S., Higelin A., Krossing I., Melle-Franco M., Mateo-Alonso A., Chem. Commun. 2014, 50, 1976–1979; [DOI] [PubMed] [Google Scholar]
- 13b. Choudhary S., Gozalvez C., Higelin A., Krossing I., Melle-Franco M., Mateo-Alonso A., Chem. Eur. J. 2014, 20, 1525–1528. [DOI] [PubMed] [Google Scholar]
- 14. Hu J., Zhang D., Harris F. W., J. Org. Chem. 2005, 70, 707–708. [DOI] [PubMed] [Google Scholar]
- 15. Grimme S., Bannwarth C., Shushkov P., J. Chem. Theory Comput. 2017, 13, 1989–2009. [DOI] [PubMed] [Google Scholar]
- 16. Clar E., The Aromatic Sextet, Wiley, London, 1972. [Google Scholar]
- 17. More S., Bhosale R., Choudhary S., Mateo-Alonso A., Org. Lett. 2012, 14, 4170–4173. [DOI] [PubMed] [Google Scholar]
- 18.Estimated using Rhodamine 6G in MeOH as a reference.
- 19. E LUMO=−4.8−e(E ONSET−E 1/2 Fc).
- 20. Saeki A., Koizumi Y., Aida T., Seki S., Acc. Chem. Res. 2012, 45, 1193–1202. [DOI] [PubMed] [Google Scholar]
- 21. Zhong Y., Sisto T. J., Zhang B., Miyata K., Zhu X. Y., Steigerwald M. L., Ng F., Nuckolls C., J. Am. Chem. Soc. 2017, 139, 5644–5647. [DOI] [PubMed] [Google Scholar]
- 22. Castro E., Sisto T. J., Romero E. L., Liu F., Peurifoy S. R., Wang J., Zhu X., Nuckolls C., Echegoyen L., Angew. Chem. Int. Ed. 2017, 56, 14648–14652; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14840–14844. [Google Scholar]
- 23. Koch M., Ample F., Joachim C., Grill L., Nat. Nanotechnol. 2012, 7, 713. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary