

Abstract
The present study selected and characterized a multidrug-resistant HL-60 human acute promyelocytic leukemia cell line, HL-60/RS, by exposure to stepwise incremental doses of doxorubicin. The drug-resistant HL-60/RS cells exhibited 85.68-fold resistance to doxorubicin and were cross-resistant to other chemotherapeutics, including cisplatin, daunorubicin, cytarabine, vincristine and etoposide. The cells over-expressed the transporters P-glycoprotein, multidrug-resistance-related protein 1 and breast-cancer-resistance protein, encoded by the adenosine triphosphate-binding cassette (ABC)B1, ABCC1 and ABCG2 genes, respectively. Unlike other recognized chemoresistant leukemia cell lines, HL-60/RS cells were also strongly cross-resistant to arsenic trioxide. The proportion of leukemia stem cells (LSCs) increased synchronously with increased of drug resistance in the doxorubicin-induced HL-60 cell population. The present study confirmed that doxorubicin-induced HL-60 cells exhibited multidrug-resistance and high arsenic-trioxide resistance. Drug-resistance in these cells may be due to surviving chemoresistant LSCs in the HL-60 population, which have been subjected to long and consecutive selection by doxorubicin.
Keywords: multidrug resistance, arsenic trioxide, leukemia stem cell, acute promyelocytic leukemia, HL-60 cells
Introduction
Chemotherapy serves an important and indispensable role in leukemia therapy. However, chemotherapy-induced multidrug resistance (MDR) frequently induces therapy failure or tumor recurrence (1–3). MDR involves various mechanisms including, adenosine triphosphate-binding cassette (ABC) transporters, P-glycoprotein (P-gp), multidrug-resistance-related protein (MRP) and breast-cancer-resistance protein (BCRP) overexpression, which function to efflux certain molecules out of the cells (4–8). In addition, mechanisms of acquired MDR in leukemia also include abnormal drug metabolism and the presence of leukemia stem cells (LSC). LSCs are particularly of interest and considered to serve an important role in leukemia resistance and relapse (4). Numerous previous studies have confirmed that the rare LSC sub-population in leukemia has self-renewal abilities, uncommitted proliferative capacities, relative dormancy or quiescence, high expression of ABC transporters and good DNA-damage repair ability (9–12). In addition, LSCs have strong resistance to almost all chemotherapeutics (2,5). However, the role of LSCs and the mechanism underlying transmission of super-drug resistance to daughter cells remains unclear. Additionally, the specific markers of LSCs remain uncertain. Previous studies have demonstrated that LSC populations are heterogeneous and may be defined as cluster of differentiation (CD)34+, CD38−, human leukocyte antigen D related, CD90−, CD117− and CD123+ phenotypes (3,13–17). Previous studies demonstrated that CD123 was aberrantly expressed on acute myeloid leukemia CD34+CD38− cells but was not detected on healthy CD34+CD38− cells. Therefore, the level of CD123 expression appears to be leukemic-specific and may be a criterion for identifying LSCs from abnormal hematopoietic stem cells (13,14).
Previous studies have revealed that the majority of MDR leukemia cells have increased sensitivity to As2O3, rather than cross-resistance (3,4). One potential explanation for this is that As2O3 may not be a substrate of P-gp and may inhibit P-gp activity (2,18–22). The present study selected HL-60 human promyelocyte leukemia cells for doxorubicin (ADM)-resistance by long-term exposure to intermittent and continuous stepwise increments of ADM, and characterized the distinguishing features of acquired MDR, particularly in terms of sensitivity to As2O3 and the role of LSCs.
Materials and methods
Materials
ABCB1, ABCC1, ABCG2 and β-actin primers were synthesized by Takara Bio, Inc. (Otsu, Japan). SYBR Premix Ex Taq and Prime Script RT reagents were obtained from Takara Bio, Inc. The following antibodies were used: Mouse anti-β-actin antibody (cat no. 3598-100; dilution, 1:1,000; BioVision, Inc., Milpitas, CA, USA), rabbit anti-P-gp antibody (cat. no. BA1351-2; dilution, 1:500), anti-BCRP antibody (cat. no. BA2307-2; dilution, 1:500), and anti-MRP1 antibody (cat. no. BA0567; dilution, 1:500) from Boster Biological Technology, Wuhai, China, PE-IgG1 (cat. no. GM4993), ECD-IgG1 (cat. no. A99022), PEcy5-IgG1 (cat. no. 85-15-4714-71), PEcy5-CD34 (cat. no. CD3458118) and ECD-CD38 (cat. no. 14-0389-82) and PE-CD123 (cat. no. 12-1239), FITC-BCRP (cat. no. lv1506735) antibodies (eBioscience; Thermo Fisher Scientific, Inc., Waltham, MA, USA), P-gp (MRK16; cat. no. Mc-012; Kamiya Biomedical Co., Tukwila, WA, USA). The chemotherapeutic drugs used were ADM (Shenzhen Wanle Pharmaceutical Co., Ltd., Shenzhen, China), daunorubicin (Zhejiang Haizheng Pharmaceutical Co., Ltd, Zhejiang, China), cisplatin and etoposide (Qilu Pharmaceutical Co., Ltd., Jinan, China), 5-fluorouracil (Haipu Pharmaceutical Co. Ltd., Shanghai, China), vincristine hydrochloride (Guangdong Lingnan Pharmaceutical Co., Ltd. Guangzhou, China), cytarabine (Haipu Pharmaceutical Co., Ltd.) and As2O3 (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany).
Transmission electron microscope observation of cell ultrastructure change
Cells were harvested and washed with 0.1 M PBS. The Cell pellets were immersed in 2.5% glutaraldehyde and incubated at 4°C for 48 h. Following cell fixation, the sample was treated with 1% osmium tetroxide at room temperature for 1 h and then dehydrated in acetone for 30 min. The sample was then embedded in embedding resin, followed by cutting into ultrathin sections with a microtome and lead-uranium double staining and then observed and photographed under a transmission electron microscope.
Scanning electron microscopy analysis
A total of 1×106 cells were washed with PBS and quickly fixed in precooled 2.5% glutaraldehyde (Sigma-Aldrich; Merck KGaA) at 4°C overnight. Following three washes in 0.1 M PBS, the cells were fixed with ice-cold 1% osmium tetroxide (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) for 1.5–2 h, followed by sequential 15 min incubations in 50, 70 and 80% ethanol at 4°C. Subsequently, fixing steps in 90 and 100% ethanol at room temperature, and gradually permeabilizing steps in a series of buffers that consisted of dehydrating agent acetone/isoamyl acetate (1:1) mixed with epoxy resin at a ratio of 2:1, 1:1 and 1:3, followed by 100% resin, for 0.5–1 h each incubation. Finally, the cells were soaked in tert-butyl alcohol twice for 10 min each time, dried with a vacuum pump for 24 h and platinum sputter-coated for 90 sec. The prepared specimen was observed under a scanning electron microscope.
Cell culture and incubation
HL-60 human promyelocyte leukemia cells were purchased from American Type Culture Collection (Manassas, VA, USA). The cells were maintained in RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum (Hyclone; GE Healthcare Life Sciences, Logan, UT, USA) and cultivated at 37°C in a 5% CO2 incubator.
Selection of ADM-resistant cells
HL-60 human leukemia cells were selected for ADM-resistance by long-term exposure to intermittent, repeated and continuous stepwise increments of ADM concentrations from 0.01–40 mg/l. If the cells survived a given ADM concentration and proliferated at a similar rate to parental HL-60 cells, the concentration of treatment was increased. The procedure was repeated intermittently and repeatedly until the cells were stably able to tolerate 40 mg/l ADM. This procedure was used to generate a stable drug-resistant leukemia subline, named HL-60/RS.
In vitro drug sensitivity analysis
HL-60 cells tolerant to various concentrations of ADM were collected for determination of the half-maximal inhibitory concentrations (IC50) of ADM and for cytotoxicity assays. A total of 1×105 cells/ml were plated in 96-well plates and cultured at 37°C with corresponding concentrations of the aforementioned chemotherapeutics (ADM 1, 2, 4, 6, 8, 10 mg/l, cisplatin 1, 5, 10, 20, 40 mg/l, etoposide 0.5, 1, 2, 4, 8, 10 mg/l, 5-fluorouracil 0.5, 1, 2, 4, 8, 16 mg/l, vincristine hydrochloride 0.5, 1, 2, 4, 8, 10 mg/l and cytarabine 0.5, 1, 2, 4, 8, 10 mg/l, all dissolved with 0.9% saline solution and As2O3, dissolved in sodium hydroxide as a storage concentration of 10 mmol/l whose pH value was adjusted with 1 mol/l HCL to 7.2–7.4 and diluted to 2 and 5 µmol/l before using) for 24–72 h. Absorbance was quantified using a Powerwave X plate reader (Omega Bio-Tek, Inc., Norcross, GA, USA) by MTT assay. A total of 5 mg/ml MTT was added to each well and incubated at 37°C for 4 h, then 100 µl 10% SDS was added to each well and incubated at 37°C overnight; absorbance was then detected at 570 nm wavelength.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total cellular RNA was extracted using a TRIzol® kit (Invitrogen; Thermo Fisher Scientific, Inc.). cDNA was derived from the RNA as PCR template using the Prime Script reverse transcriptase kit (Takara Bio, Inc.). Temperature protocol: 70°C for 30 min, 37°C for 15 min, 95°C for 5 min. For PCR, cDNA (2 µl) was mixed with SYBR Premix Ex Taq (Takara Bio, Inc.) and the following relevant primers for PCR were used: β-actin forward, 5′-TGCTCCTCCTGAGCGCAAGTA-3′ and reverse, 5′-CCACATCTGCTGGAAGGTGGA-3′; P-gp forward, 5′CCCATCATTGCAATAGCAGG3′ and reverse, 5′GTTCAAACTTCTGCTCCTGA3′; MRP1 forward, 5′TGCAGAAGGCGGGGAGAACCTC3′ and reverse, 5′GTCGTCCGTTTCCAGGTCCACG3′; BCRP2 forward, 5′GCTGCAAGGAAAGATCCAAGT3′ and reverse, 5′TAGTTGTTGCAAGCCGAAGAG3′, which were designed and synthesized by Takara Bio, Inc. The conditions included an initial denaturing step at 95°C for 10 sec, then 40 cycles of denaturing at 95°C for 5 sec and annealing at 60°C for 30 sec. The relative expression of each mRNA was calculated by comparison to β-actin mRNA using the 2−ΔΔCq method (4).
Western blot analysis
The cells were lysed using a radioimmunoprecipitation assay (RIPA) protein extraction reagent (cat no. P0013B; Beyotime Institute of Biotechnology, Haimen, China), supplemented with phenylmethylsulfonyl fluoride (PMSF) (0.1 mmol/ml PMSF 10 µl in 1 ml RIPA). Total protein concentration was determined using a BCA protein quantitative kit (cat no. P0010s; Beyotime Institute of Biotechnology). Proteins (30 µg per lane) were separated by 10% SDS-PAGE, transferred to a polyvinylidene fluoride membrane and blocked with 5% skimmed milk at room temperature for 1 h. Membranes were washed with PBS-Tween-20 (PBST) three times for 5 min. The membranes were probed with primary antibodies (anti-P-gp, anti-MRP1 or anti-BCRP, with the same conditions as above) at 4°C overnight. The membrane was washed with PBST three times for 5 min. IRDye800CW-(goat anti-mouse; cat. no. 926-32210; dilution, 1:10,000; LI-COR Biosciences, Lincoln, NE, USA) or IRDye680DX-conjugated secondary antibodies (goat anti-rabbit; cat. no. 926-32221; dilution, 1:10,000; LI-COR Biosciences, Lincoln, NE, USA) was added to the membrane and agitated for 1 h at room temperature prior to washing three times with PBST for 5 min, using the anti-β-actin antibody as the control. The blots of antibody-coated protein bands in immunoblots were quantitated and visualized using an Odyssey double-color infrared-laser imaging system (Odyssey v1.2 software; LI-COR Biosciences Inc., Lincoln, NE, USA).
ADM accumulation/efflux assay
In order to determine the intracellular uptake of ADM, the sensitive and resistant HL-60 cells were incubated with 30 mg/l ADM at 37°C for 30 min. Following two washes with PBS and re-suspension in 200 µl PBS, intracellular uptake of ADM in 106 cells was determined immediately using a MoFlo XDP Cell Sorter (Beckman Coulter, Inc., Brea, CA, USA) with excitation at 488 nm and emission at 525 nm wavelengths. The other 106 cells were re-suspended in PBS at 37°C for a further 60 min and assessed by flow cytometry again (18). The intracellular positive rate and fluorescence intensity of ADM were determined using FlowJo v7.6.3 software (FlowJo, LLC, Ashland, OR, USA).
LSC detection
Parental HL-60 and ADM-induced HL-60 cells were co-incubated with PEcy5-CD34 (5 µl), ECD-CD38 (5 µl) and PE-CD123 (10 µl) in 100 µl binding buffer and/or fluorescein isothiocyanate, FITC-BCRP (1 µl) antibodies, with PEcy5-, ECD-, PE- and FITC-murine IgG1 (with the same corresponding volume as aforesaid) as isotype controls, respectively. The relative proportions of LSCs, CD34+CD38−CD123+ and BCRP+CD34+CD38−CD123+ subsets in the cell population were determined by flow cytometry, as aforementioned.
Colony-formation assay
A total of 103 cells/ml were seeded in half-solid RPMI 1640 media with 20% fetal bovine serum and 0.9% methylcellulose. Following 5- and 10-day incubation at 37°C in 5% CO2, the total number of colonies, defined as a mass of >40 cells, within each well was counted under a light microscope (original magnification, ×100). Colony-formation rate=total number of colonies/1,000/well ×100%); three representative fields were imaged.
Statistical analysis
Data were analyzed using SPSS version 17.0 (SPSS, Inc., Chicago, IL, USA). Data are presented as the mean ± standard deviation. Multiple comparisons between the three groups was performed using two-way analysis of variance followed by a Newman-Keuls post hoc test. The intragroup comparisons were performed using paired Student's t-test. All experiments were repeated at least three times. Error bars represent the standard error of the mean and P<0.05 was considered to indicate a statistically significant difference.
Results
Establishment of multidrug-resistant HL-60 cells
Human promyelocytic leukemia HL-60 cells were selected for the MDR phenotype by stepwise increments of ADM treatment until the cells were able to survive and proliferate normally at a concentration of 40 mg/l ADM. The resulting cells were 85.68-fold more resistant to ADM compared with the parental HL-60 cells, as revealed by the IC50 (mg/l) presented in Fig. 1A. Additionally, drug-resistance was maintained following culture in the absence of ADM for 3 months or refrigeration for 6 months. These results demonstrated that following a long induction period imitating clinical chemotherapy, HL-60 cells acquired a stable drug-resistant phenotype, and therefore a stable MDR leukemia subline was established.
Figure 1.

HL-60 cells acquire multidrug resistance. HL-60 cells resistant to 10–40 mg/l ADM were treated with various chemotherapeutic drugs. Inhibitory rate and IC50 values were determined by MTT assay. HL-60 cells resistant to 40 mg/l ADM were defined as HL-60/RS cells. (A) HL-60 cells became increasingly resistant to ADM at increasing concentrations of ADM and increasing exposure times. (B) ADM-resistant HL-60 cells were cross-resistant to other chemotherapeutic drugs, including cisplatin, daunorubicin, cytarabine, vincristine and etoposide, but not 5-fluorouracil. (C) HL-60/RS cells acquired high As2O3 resistance. *P<0.05, **P<0.01 compared with HL-60 cells. IC50, half-maximal inhibitory concentration; ADM, doxorubicin; conc., concentration; 5-FU, 5-fluorouracil.
Resistance and cross-resistance of ADM-selected HL-60 cells
The sensitivity of ADM-induced HL-60 cells to chemotherapeutic agents including ADM was analyzed during and at terminal induction. HL-60 cells became increasingly resistant to ADM at increasing concentrations of ADM and increasing exposure times (Fig. 1A). Additionally, ADM-resistant HL-60 cells were also cross-resistant to other chemotherapeutics including cisplatin, daunorubicin, cytarabine, vincristine, arsenic trioxide and etoposide, but not 5-fluorouracil (Fig. 1B). These results confirmed that HL-60/RS cells possessed high resistance to ADM and other numerous chemotherapeutics.
Sensitivity of ADM-selected HL-60 cells to arsenic trioxide
A previous study revealed that the majority of MDR leukemia cells did not develop cross-resistance, but conversely demonstrated higher sensitivity to As2O3 (4). However, the established HL-60/RS sub-line was strongly cross-resistant to As2O3, with a 12.89-fold increase in resistance compared with HL-60 cells. Therefore, HL-60/RS cells acquired the unique feature of high As2O3-resistance (Fig. 1C).
Morphology and cell cycle distribution of HL-60/RS cells
Drug-resistant HL-60/RS and parental sensitive HL-60 cells were similar in size and had similar morphological phenotypes. The nuclei in HL-60/RS cells were uniformly round or oval (Fig. 2A) and exhibited an increased relative nucleus/cytoplasmic ratio compared with the parental cells (Fig. 2B and C). Sensitive HL-60 cells included more mature, band or polymorphonuclear (segmented)-like cells compared with HL-60/RS cells, indicating that HL-60/RS cells were less mature compared with the parental HL-60 cells. Although both cell lines appeared round, HL-60/RS cells had a smooth surface, whereas the surface of parental HL-60 cells appeared rough and more bulging under scanning electron microscopy (Fig. 2A). Electron microscopy revealed morphologic ultrastructural changes in HL-60/RS cells, including denser cytoplasm and nuclear chromatin and increased heterochromatin, compared with untreated, parental cells (Fig. 2A).
Figure 2.

Morphological and cycles change in drug-resistant HL-60/RS cells. (A) Morphological and cycles change in drug-resistant HL-60/RS cells. (A) Morphological changes were examined by light microscopy in (a) sensitive HL-60 cells and (b) HL-60/RS cells stained with Wright-Giemsa stain (original magnification, ×1,000). The surface micro-structures of (c) HL-60 cells and (d) HL-60/RS cells were observed by scanning electron microscopy (original magnification, ×5,000). The cellular ultrastructures in (e) HL-60 cells and (f) HL-60/RS cells were determined by transmission electron microscopy (original magnification, ×5,000). (B) Diameter of the cytoplasm and nucleus in resistant HL-60/RS and parental HL-60 cells. (C) Mean nuclear-cytoplasmic ratios of HL-60/RS and parental HL-60 cells. (D) Cell-cycle distribution of (a) HL-60 and HL-60/RS cells cultured in doxorubicin-free medium for (b) 7, (c) 15 and (d) 60 days, respectively. *P<0.05 compared with HL-60 cells.
The cell cycle distribution was analyzed in HL-60/RS cells, which were exposed to 40 mg/l ADM and subsequently cultured continuously in ADM-free conditions. At the early stage (7 days), the distribution of total DNA content in G0/G1 was markedly higher compared with the parental HL-60 cells, but lower in S and G2/M phases. The cell cycle distribution became similar in both cell types with increasing incubation times (for 15–60 days) and cell proliferation (Fig. 2D).
Expression of drug transporter genes in ADM-selected resistant HL-60 cells
In cells tolerant to the highest induced dose of 40 mg/l ADM, relative ABCB1, ABCC1 and ABCG2 mRNA expression levels reached 92.5, 45.4 and 75.0-fold of that in sensitive HL-60 cells (Fig. 3B), respectively. The expression of P-gp and BCRP proteins increased simultaneously with increased tolerance to ADM, as determined by flow cytometry. The percentage of P-gp+ cells reached ~100% in cells tolerant to 5 mg/l ADM (Fig. 3A). RT-qPCR demonstrated marked increases in ABCB1, ABCC1 and ABCG2 mRNA expression with increasing tolerance to ADM and extended induction times (Fig. 3Ba). The levels of P-gp, MRP1 and BCRP expression in sensitive parental HL-60 cells and cells tolerant to 30 and 40 mg/l ADM cells (HL-60/RS) was analyzed by western blotting, and the findings were consistent with the gene expression results (Fig. 3Bb). These results revealed that long-term and intermittent ADM exposure of HL-60 cells resulted in overexpression of the drug transporters P-gp, MRP and BCRP, which may represent the major mechanism of drug-resistance in HL-60 cells.
Figure 3.
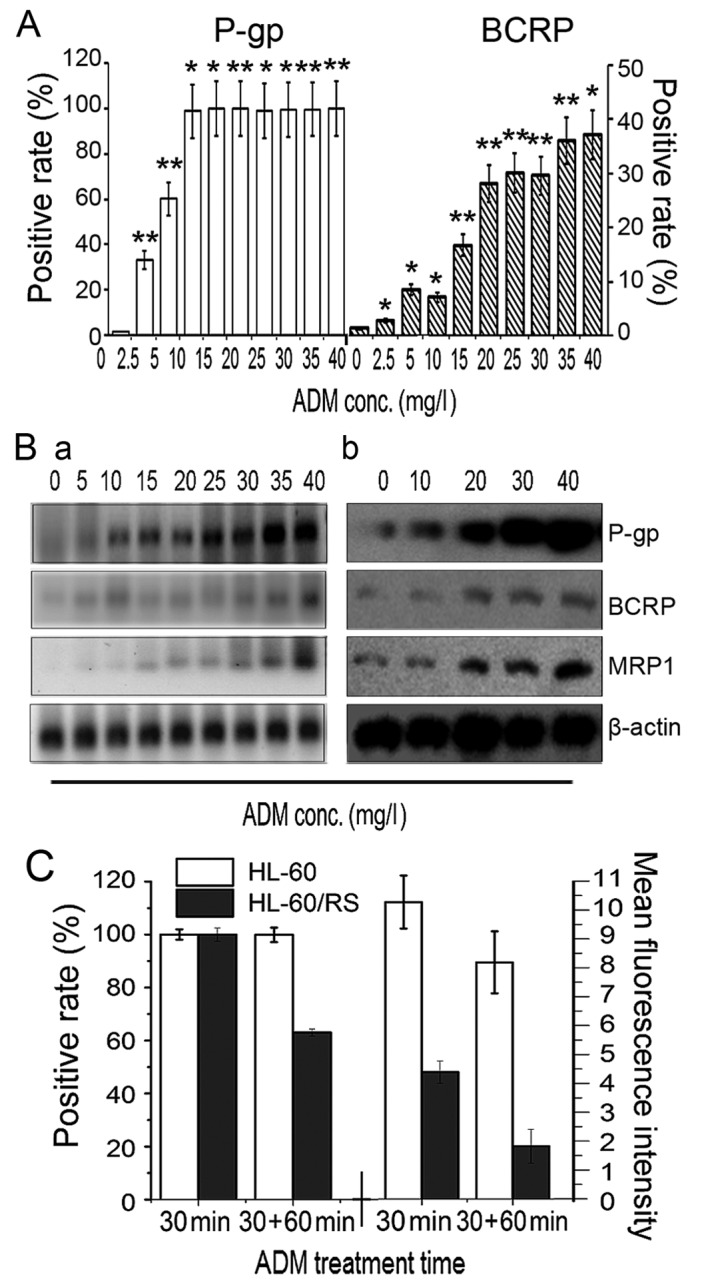
Membrane transporter expression and ADM accumulation-efflux function. (A) The number of P-gp+ and BCRP+ cells increased gradually in ADM-induced HL-60 cells with stepwise increases in ADM doses. (B) Levels of P-gp, MRP1 and BCRP (a) gene mRNA expression were determined by reverse transcription-quantitative polymerase chain reaction, and (b) levels of proteins expression were evaluated by western blotting, in HL-60 parental cells (ADM, 0 mg/l) and in cells tolerant to various doses of ADM. (C) Accumulation and efflux of HL-60 cells and HL-60/RS cells. The cells were incubated in 30 mg/l ADM medium at 37°C for 30 min and ADM content was assessed by the number of fluorescence-positive cells and mean fluorescence intensity. Subsequently, the cells were further incubated for 60 min and re-assessed. *P<0.05, **P<0.01 compared with HL-60 cells. ADM, doxorubicin; conc., concentration; P-gp, P-glycoprotein; BCRP, breast-cancer-resistance protein.
Function of drug transporters in ADM-selected resistant HL-60 cells
ADM has spontaneous fluorescence that can be used to analyze intercellular residual ADM (18). HL-60 cells and HL-60/RS cells were exposed to 30 mg/l ADM-containing medium for 30 min. Almost all cells of both lines demonstrated ADM fluorescence. However, the cellular ADM fluorescence intensity in HL-60/RS cells was only 42.75% of the value of sensitive HL-60 cells. Following re-incubation in ADM-free medium for a further 60 min, only 62.02% of HL-60/RS cells retained ADM fluorescence, compared with ~100% of the HL-60 cells, and the mean fluorescence intensity in HL-60/RS cells remained only 21.01% of that in HL-60 cells (Fig. 3C).
Dynamic changes in LSC proportion during drug-resistance induction of HL-60 cells
Dynamic changes were observed in the proportions of LSCs and BCRP+LSCs in ADM-selected HL-60 cells during the enhancement of drug-resistance, with CD34+CD38−CD123+ and BCRP+CD34+CD38−CD123+ phenotypes, respectively. The relative proportion of LSCs in the HL-60 cell population increased notably along with increments of ADM-inducing concentrations and length of induction time. The proportion of BCRP+LSC increased synchronously (Fig. 4A). In HL-60 cells tolerant to 40 mg/l ADM, the proportions of LSCs and BCRP+LSCs were 24.25 and 23.63-fold higher compared with the sensitive parental HL-60 cells, respectively (Fig. 4B). These levels gradually fell during culture in ADM-free medium, except for P-gp, which remained stable near 100% (Fig. 4B). These findings suggested that LSCs have a pivotal role in the acquisition of drug-resistance in leukemia, and BCRP+ or P-gp+BCRP+ cells may be a specific hallmark of the multidrug-resistant LSC subpopulation.
Figure 4.

Alterations in subsets of LSCs. (A) Proportions of LSC (CD34+CD38−CD123+) and BCRP+LSC cells changed synchronously in HL-60 cells tolerant to 0–40 mg/l ADM as determined by flow cytometry. (B) HL-60 cells were harvested following selection with 10–40 mg/l ADM as they commenced normal proliferation and were then cultured in ADM-free condition for 60 days. Relative proportions of LSC, BCRP+LSC, P-gp- and BCRP-positive cells were determined by flow cytometry. (C) Early colonies (5 days) and terminal colonies (10 days) of HL-60 and HL-60/RS cells were examined by light microscopy (original magnification, ×10). (D) Colony-forming capacities of HL-60 and HL-60/RS cells. LSC, leukemia stem cells; CD, cluster of differentiation; BCRP, breast-cancer-resistance protein; ADM, doxorubicin; P-gp, P-glycoprotein.
Colony-forming capacity
The self-renewal and unrestricted-proliferation potentials of the cells were analyzed by using the methylcellulose half-solid-medium colony-forming-culture (CFC) method. The early and terminal colony-formation ratios of HL-60/RS cells were 3.64 and 7.67-fold than that of HL-60 cells, respectively. The early colonies were morphologically small and loose but became bigger and denser. Additionally, the majority of the late HL-60 colonies were composed of dead and disintegrating cells (Fig. 4C). The colony formation rate in early and late colonies were markedly higher in tolerant HL-60/RS cells compared with the parental HL-60 cells (Fig. 4D).
Discussion
MDR is currently a major and unresolved impediment to leukemia therapy (1–3,10). Although numerous extensive studies have investigated leukemia drug-resistance, regulatory pathways and intervention measures, the underlying mechanisms remain to be fully understood and few effective or clinically practical countermeasures have been identified (23–25). The fundamental role of LSCs in leukemia-MDR has recently attracted widespread attention. LSCs have been suggested to be a core factor underlying leukemia drug resistance and also the key cause of conventional chemotherapy failure and relapse in leukemia (12,26,27). Further studies investigating the characteristics of drug resistance in leukemia and its cellular and molecular mechanisms are required. In the present study, HL-60 human promyelocytic leukemia cells were selected for ADM-resistance by imitating the clinical chemotherapy process. Long-term, intermittent and continuous stepwise increments of ADM concentrations were used to establish a stable MDR leukemia subline (HL-60/RS). The expression of drug-resistance-related genes/proteins and their functions as wells as the LSC composition of the cell lines were examined in relation to the degree of ADM resistance. Following 22 months of ADM induction, the resistance of HL-60/RS cells to ADM had increased 85.68-fold compared with parental HL-60 cells. These ADM-resistant cells were also cross-resistant to other chemically and functionally unrelated chemotherapeutics. In particular, the ADM-induced cells achieved high As2O3-resistance. Additionally, the present study demonstrated that the rate of proliferation, cell morphology and cell cycle distribution all differed between HL-60/RS and parental HL-60 cells.
As2O3 is widely used in chemotherapy for most types of leukemia and solid tumors. As2O3 has good curative effects and little cross-resistance with conventional chemotherapeutics (2,17–21). Previous studies have revealed that conventional MDR leukemia cells exhibit increased sensitivity to As2O3 instead of manifesting cross-resistance. This is possibly because As2O3 is not a P-gp substrate and may even inhibit P-gp expression and activity (2–4,27). However, unlike other ADM-selected P-gp-overexpressing MDR tumor cells, including K562/ADM cells (2,3,27), the HL-60/RS cells established in the present study demonstrated cross-resistance to As2O3, which is a unique feature in MDR cells.
The reason for strong resistance of HL-60/RS cells to arsenic remains unclear, and the possible mechanisms are still being investigated. It was previously reported that the presence of mutations in the arsenic binding domain of promyelocytic leukemia/retinoic acid receptor α (PML-RARA) induced arsenic resistance in patients treated with As2O3 (23–25,28). Previous studies in anti-arsenic organisms indicated that overexpression of arsenic-related transporters and glutathione-S-transferases, which mainly exported arsenic leading to intracellular arsenic load reduction, were responsible for the formation of resistance to arsenic (29–31). Follow-up studies are currently proceeding in the laboratory of the authors to investigate the underlying mechanisms of how MDR cells become sensitive or resistant to As2O3. The preliminary results confirmed that resistance to arsenic in MDR cells was associated with factors, including the overexpression of arsenic transporters.
There is growing evidence demonstrating that LSCs are a main source of leukemia relapse and treatment resistance (13,26,27,32,33). LSCs are naturally resistant to cytotoxic drugs, which kill more mature leukemia cells in the cell proliferation phase, due to their quiescence/dormancy, strong self-DNA-damage-repair capability and overexpression of ABC transporters in LSCs. Therefore, an investigation of the key regulatory pathways of functional ABC family members in drug-resistance of LSCs is required. In the present study, during the process of ADM induction and colony-forming ability, the proportion of LSCs increased with increasing drug-resistance in HL-60 cells. The proportion of LSCs in HL-60 cells gradually decreased when ADM-induced resistant HL-60 cells were cultured in ADM-free medium, stabilizing at ~10%, which was ~10-fold higher compared with the parental HL-60 cells. Notably, ~100% of LSCs expressed P-gp. However, only ~30% expressed BCRP (BCRP+LSCs), which increased (when incubated in ADM-free media for 7 days) or decreased (when incubated in ADM-free media for 15–60 days), but more slowly compared with the total number of LSCs, with increasing resistance to ADM. BCRP in LSCs or leukemic CD34+CD38− stem cells were preferentially expressed and may contribute to their resistant phenotype (26,28,32). Therefore, it was hypothesized that BCRP+ or P-gp+BCRP+LSCs may represent the multi-resistant LSC subset generated by long exposure to ADM, which survived to produce resistant daughter leukemia cells. Repeated stimulation by cytotoxic drugs may drive LSCs to become multi-resistant and develop specific regulatory pathways, which may in turn be transmitted to daughter cells and maintain a drug-resistant leukemia cell population.
In conclusion, the present study established the leukemia HL-60/RS sub-line with MDR characteristics. These characteristics include overexpression of the main ABC transporter members (P-gp, MRP1 and BCRP), an increased proportion of LSCs and a phenotype of high As2O3-resistance, which contrasts with the majority of classical MDR leukemia cells. These findings suggested that the main mechanisms underlying MDR in HL-60/RS cells are mediated by excess LSCs and high expression of ABC transporters. Notably, the leukemia MDR cell line was highly cross-resistant to As2O3. Furthermore, the BCRP+ or P-gp+BCRP+ phenotype may be a specific hallmark of a highly drug-resistant LSC subpopulation. This leukemia MDR cell line may be a good model for further studies that examine the mechanisms underlying As2O3-resistance, particularly the cross-resistance of conventional chemotherapeutics with As2O3, and to investigate strategies to reverse chemotherapy resistance in leukemia.
Acknowledgements
The authors thank Dr David Cushley of International Science Editing (Shannon, Ireland) for language editing and revision of this paper. The present study was supported by the National Natural Science Foundation of China (grant nos. 81541025 and 81141053), the Fundamental Research Funds for the Central Universities (grant no. lzujbky-2016-174), Science and Technology Planning Project from Chengguan District, Lanzhou, Gansu Province, China (grant no. 2015-3-8) and the Natural Science Fund of Gansu (grant no. 1208RJZA183).
References
- 1.Matsumoto T, Jimi S, Hara S, Takamatsu Y, Suzumiya J, Tamura K. Importance of inducible multidrug resistance 1 expression in HL-60 cells resistant to gemtuzumab ozogamicin. Leuk Lymphoma. 2012;53:1399–1405. doi: 10.3109/10428194.2012.656102. [DOI] [PubMed] [Google Scholar]
- 2.Zhang QH, Dou HT, Xu P, Zhuang SC, Liu PS. Tumor recurrence and drug resistance properties of side population cells in high grade ovary cancer. Drug Res. 2015;65:153–157. doi: 10.1055/s-0034-1375609. [DOI] [PubMed] [Google Scholar]
- 3.Gao F, Dong W, Yang W, Liu J, Zheng Z, Sun K. Expression of P-gp in acute myeloid leukemia and the reversal function of As2O3 on drug resistance. Oncol Lett. 2015;9:177–182. doi: 10.3892/ol.2014.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen J, Wei H, Xie B, Wang B, Cheng J, Cheng J. Endoplasmic reticulum stress contributes to arsenic trioxide-induced apoptosis in drug-sensitive and -resistant leukemia cells. Leuk Res. 2012;36:1526–1535. doi: 10.1016/j.leukres.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 5.de Figueiredo-Pontes LL, Pintão MC, Oliveira LC, Dalmazzo LF, Jácomo RH, Garcia AB, Falcão RP, Rego EM. Determination of P-glycoprotein, MDR-related protein 1, breast cancer resistance protein, and lung-resistance protein expression in leukemic stem cells of acute myeloid leukemia. Cytometry B Clin Cytom. 2008;74:163–168. doi: 10.1002/cyto.b.20403. [DOI] [PubMed] [Google Scholar]
- 6.Munić V, Kelnerić Z, Mikac L, Eraković Haber V. Differences in assessment of macrolide interaction with human MDR1 (ABCB1, P-gp) using rhodamine-123 efflux, ATPase activity and cellular accumulation assays. Eur J Pharm Sci. 2010;41:86–95. doi: 10.1016/j.ejps.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 7.Nakanishi T, Ross DD. Breast cancer resistance protein (BCRP/ABCG2): Its role in multidrug resistance and regulation of its gene expression. Chin J Cancer. 2012;31:73–99. doi: 10.5732/cjc.011.10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Laterra J, Pomper MG. Hedgehog pathway inhibitor HhAntag691 is a potent inhibitor of ABCG2/BCRP and ABCB1/Pgp. Neoplasia. 2009;11:96–101. doi: 10.1593/neo.81264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gutiérrez-González A, Belda-Iniesta C, Bargiela-Iparraguirre J, Dominguez G, García Alfonso P, Perona R, Sanchez-Perez I. Targeting Chk2 improves gastric cancer chemotherapy by impairing DNA damage repair. Apoptosis. 2013;18:347–360. doi: 10.1007/s10495-012-0794-2. [DOI] [PubMed] [Google Scholar]
- 10.Ding Q, Gu R, Liang J, Zhang X, Chen Y. PI-103 sensitizes acute myeloid leukemia stem cells to daunorubicin-induced cytotoxicity. Med Oncol. 2013;30:395. doi: 10.1007/s12032-012-0395-5. [DOI] [PubMed] [Google Scholar]
- 11.Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, Uchida N, Suzuki N, Sone A, Najima Y, et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci Transl Med. 2010;2:17ra9. doi: 10.1126/scitranslmed.3000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moore N, Lyle S. Quiescent, slow-cycling stem cell populations in cancer: A review of the evidence and discussion of significance. J Oncol. 2011;2011:pii:396076. doi: 10.1155/2011/396076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pollyea DA, Gutman JA, Gore L, Smith CA, Jordan CT. Targeting acute myeloid leukemia stem cells: A review and principles for the development of clinical trials. Haematologica. 2014;99:1277–1284. doi: 10.3324/haematol.2013.085209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou J, Chng WJ. Identification and targeting leukemia stem cells: The path to the cure for acute myeloid leukemia. World J Stem Cells. 2014;6:473–484. doi: 10.4252/wjsc.v6.i4.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crews LA, Jamieson CH. Selective elimination of leukemia stem cells: Hitting a moving target. Cancer Lett. 2013;338:15–22. doi: 10.1016/j.canlet.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 16.Marques DS, Sandrini JZ, Boyle RT, Marins LF, Trindade GS. Relationships between multidrug resistance (MDR) and stem cell markers in human chronic myeloid leukemia cell lines. Leukemia Res. 2010;34:757–762. doi: 10.1016/j.leukres.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Buda G, Orciuolo E, Maggini V, Galimberti S, Barale R, Ross AM, Petrini M. MDR1 modulates apoptosis in CD34+ leukemic cells. Ann Hematol. 2008;87:1017–1018. doi: 10.1007/s00277-008-0515-7. [DOI] [PubMed] [Google Scholar]
- 18.Zhao D, Jiang Y, Dong X, Liu Z, Qu B, Zhang Y, Ma N, Han Q. Arsenic trioxide reduces drug resistance to adriamycin in leukemic K562/A02 cells via multiple mechanisms. Biomed Pharmacother. 2011;65:354–358. doi: 10.1016/j.biopha.2011.04.016. [DOI] [PubMed] [Google Scholar]
- 19.Perkins C, Kim CN, Fang G, Bhalla KN. Arsenic induces apoptosis of multidrug-resistant human myeloid leukemia cells that express Bcr-Abl or overexpress MDR, MRP, Bcl-2, or Bcl-x(L) Blood. 2000;95:1014–1022. [PubMed] [Google Scholar]
- 20.Zhao H, Guo W, Peng C, Ji T, Lu X. Arsenic trioxide inhibits the growth of adriamycin resistant osteosarcoma cells through inducing apoptosis. Mol Biol Rep. 2010;37:2509–2515. doi: 10.1007/s11033-009-9765-2. [DOI] [PubMed] [Google Scholar]
- 21.Beauchamp EM, Ringer L, Bulut G, Sajwan KP, Hall MD, Lee YC, Peaceman D, Ozdemirli M, Rodriguez O, Macdonald TJ, et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J Clin Invest. 2011;121:148–160. doi: 10.1172/JCI42874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei H, Su H, Bai D, Zhao H, Ge J, Wan B, Yao X, Ma L. Arsenic trioxide inhibits p-glycoprotein expression in multidrug-resistant human leukemia cells that overexpress MDR1 gene. Chin Med J. 2003;116:1644–1648. [PubMed] [Google Scholar]
- 23.Fung TK, So CW. Overcoming treatment resistance in acute promyelocytic leukemia and beyond. Oncotarget. 2013;4:1128–1129. doi: 10.18632/oncotarget.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tomita A, Kiyoi H, Naoe T. Mechanisms of action and resistance to all-trans retinoic acid (ATRA) and arsenic trioxide (As2O3) in acute promyelocytic leukemia. J Hematol. 2013;97:717–725. doi: 10.1007/s12185-013-1354-4. [DOI] [PubMed] [Google Scholar]
- 25.Zhu HH, Qin YZ, Huang XJ. Resistance to arsenic therapy in acute promyelocytic leukemia. N Engl J Med. 2014;370:1864–1866. doi: 10.1056/NEJMc1316382. [DOI] [PubMed] [Google Scholar]
- 26.Felipe Rico J, Hassane DC, Guzman ML. Acute myelogenous leukemia stem cells: From Bench to Bedside. Cancer Lett. 2013;338:4–9. doi: 10.1016/j.canlet.2012.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang F, Wang XK, Shi CJ, Zhang H, Hu YP, Chen YF, Fu LW. Nilotinib enhances the efficacy of conventional chemotherapeutic drugs in CD34+CD38- stem cells and ABC transporter overexpressing leukemia cells. Molecules. 2014;19:3356–3375. doi: 10.3390/molecules19033356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, Ferrara F, Fazi P, Cicconi L, Di Bona E, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111–121. doi: 10.1056/NEJMoa1300874. [DOI] [PubMed] [Google Scholar]
- 29.Xu S, Zhang YF, Carew MW, Hao WH, Loo JF, Naranmandura H, Le XC. Multidrug resistance protein 1 (ABCC1) confers resistance to arsenic compounds in human myeloid leukemic HL-60 cells. Arch Toxicol. 2013;87:1013–1023. doi: 10.1007/s00204-012-0956-6. [DOI] [PubMed] [Google Scholar]
- 30.Hemmingsson O, Nöjd M, Kao G, Naredi P. Increased sensitivity to platinating agents and arsenite in human ovarian cancer by downregulation of ASNA1. Oncol Rep. 2009;22:869–875. doi: 10.3892/or_00000511. [DOI] [PubMed] [Google Scholar]
- 31.Matulis SM, Morales AA, Yehiayan L, Lee KP, Cai Y, Boise LH. Alterations in glutathione levels and apoptotic regulators are associated with acquisition of arsenic trioxide resistance in multiple myeloma. PLoS One. 2012;7:e52662. doi: 10.1371/journal.pone.0052662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yi J, Chen J, Sun J, Wei HL. The relationship between multi-drug resistance and proportion of leukemia stem cells and expression of drug transporters in drug-resistant leukemia K562/ADM cells. Zhonghua Yi Xue Za Zhi. 2009;89:1741–1744. (In Chinese) [PubMed] [Google Scholar]
- 33.Qiu S, Jia Y, Xing H, Yu T, Yu J, Yu P, Tang K, Tian Z, Wang H, Mi Y, et al. N-Cadherin and Tie-positive CD34+CD38−CD123+ leukemic stem cell populations can develop acute myeloid leukemia more effectively in NOD/SCID mice. Leuk Res. 2014;38:632–637. doi: 10.1016/j.leukres.2014.03.007. [DOI] [PubMed] [Google Scholar]