

Abstract
Disruption of the endothelial barrier in response to Gram positive (G+) bacterial toxins is a major complication of acute lung injury (ALI) and can be further aggravated by antibiotics which stimulate toxin release. The integrity of the pulmonary endothelial barrier is mediated by the balance of disruptive forces such as the small GTPase RhoA, and protective forces including endothelium-derived nitric oxide (NO). How NO protects against the barrier dysfunction is incompletely understood and our goal was to determine whether NO and S-nitrosylation can modulate RhoA activity and whether this mechanism is important for G+ toxin-induced microvascular permeability. We found that the G+ toxin listeriolysin-O (LLO) increased RhoA activity and that NO and S-NO donors inhibit RhoA activity. RhoA was robustly S-nitrosylated as determined by biotin-switch and mercury column analysis. MS revealed that three primary cysteine residues are S-nitrosylated including cys16, cys20 and cys159. Mutation of these residues to serine diminished S-nitrosylation to endogenous NO and mutant RhoA was less sensitive to inhibition by S-NO. G+-toxins stimulated the denitrosylation of RhoA which was not mediated by S-nitrosoglutathione reductase (GSNOR), thioredoxin (TRX) or thiol-dependent enzyme activity but was instead stimulated directly by elevated calcium levels. Calcium-promoted the direct denitrosylation of WT but not mutant RhoA and mutant RhoA adenovirus was more effective than WT in disrupting the barrier integrity of human lung microvascular endothelial cells. In conclusion, we reveal a novel mechanism by which NO and S-nitrosylation reduces RhoA activity which may be of significance in the management of pulmonary endothelial permeability induced by G+-toxins.
Keywords: Nitric oxide, S-nitrosylation, RhoA, G+-toxins, Endothelial, Permeability
1. Introduction
Acute lung injury (or ALI) is a serious medical condition with a high mortality rate that can result in non-cardiogenic pulmonary edema and respiratory failure. A major cause of ALI is Gram positive (G+) bacterial infections [1,2] with Streptococcus pneumonia accounting for up to 45% of all community-acquired pneumonia (CAP) cases in the US. In the United States alone, there are greater than 500,000 yearly cases of pneumonia and 40,000 pneumococcal-related deaths, which results in a health care burden exceeding 5 billion dollars [3]. Currently, there are no effective treatments that specifically target the pulmonary barrier dysfunction in ALI and a greater understanding of the cellular and molecular foundations of G+-toxin induced pulmonary barrier dysfunction is needed.
The vascular endothelium is a monolayer of tightly connected endothelial cells that forms a barrier between the blood and the underlying interstitium which regulates the passage of fluid and cells. Dysfunction of the endothelial barrier arises from changes in cell behavior in response to signaling events that promote contraction, dissolution of adhesive complexes between cells and cell death. Many signaling pathways are involved including calcium, MLC (myosin light chain)-dependent mechanisms [4], cytoskeletal rearrangements [5], disassembly of junctional proteins between cells [6], activation of PKC (protein kinase C) [7], alteration of nitric oxide (NO) signaling [8] and numerous others [9,10]. Unresolved disruption of the endothelial barrier results in the inappropriate loss of fluid from the vasculature and extensive pulmonary edema which are the major complications of acute respiratory distress syndrome (ARDS) and CAP [11].
The mortality rate of ALI remains unacceptably high despite a high level of support and the extensive use of antibiotics. A complicating factor in the use of bactericidal antibiotics is the release of toxins from G+ bacteria, including LLO (listeriolysin-O, Listeria monocytogenes) and pneumolysin (PLY, S. pneumoniae). Both LLO and PLY belong to a family of cholesterol-dependent poreforming cytolysins which form plasma membrane pores in the presence of calcium that stimulate the entry of calcium and other ions as well as stimulate intracellular signaling [7,8]. The mechanisms by which G+ toxins promote endothelial permeability are not fully understood.
Nitric Oxide (NO) is an important regulator of vascular homeostasis and pathophysiology. In blood vessels, the primary source of NO is the enzyme endothelial nitric oxide synthase (eNOS), which is found almost exclusively in the endothelium, and has been shown to regulate endothelial permeability [12,13]. Previously we showed that G+ toxins induce eNOS uncoupling which results in increased superoxide production. This occurs in part through an increase in PKCα-dependent eNOS T495 phosphorylation which alters enzymatic fidelity to reduce NO and increase superoxide synthesis [8]. However, the particular role of NO in regulating endothelial permeability is poorly understood and complicated by the amount of NO, the vascular bed and the agonist [14]. NO signaling is primarily mediated by soluble guanylate cyclase and cGMP-dependent signaling in smooth muscle. In endothelial cells, there are higher concentrations of NO and lower levels of soluble guanylate cyclase and NO-dependent signaling can be mediated via an alternative pathway. Sulfhydryl groups on substrate proteins can be covalently modified by NO and its metabolites in a process called S-nitrosylation that can modulate the function of numerous substrate proteins [12,15–18]. S-nitrosylation is a reversible modification and the removal of S-NO is primarily mediated by two enzymes, the GSNOR (S-nitrosoglutathione reductase) and thioredoxin 1(Trx1) [19].
RhoA belongs to the family of small GTPases which are activated by the binding of GTP versus GDP [20]. RhoA is a key regulator of numerous cellular responses involved in barrier function including cell adhesion and contraction. Upon activation, RhoA translocates to the plasma membrane where it contributes to the loss of barrier function by destabilizing endothelial junction proteins and increasing myosin light chain phosphorylation to promote cell contraction [21]. The actions of RhoA are opposed by another GTPase, Rac1 and the loss of pulmonary barrier function in ALI is partly due to a shift in the balance of RhoA and Rac1 signaling where RhoA becomes dominant [22,23]. Several post-translational modifications have been shown to modulate the activity of Rho family proteins, including transglutamination [24], deamidation [25], glycosylation [26], ADP-ribosylation [26], adenylylation [27], and phosphorylation [27]. Our group has previously demonstrated the nitration of RhoA in response to LPS which induces enhanced activity through the changes in nucleotide cycling [23] but the role of other nitrosative modifications are not well understood.
However, the ability of NO (as opposed to ONOO-) to regulate the activity of RhoA in human lung microvascular endothelial cells (HLMVECs), i.e. whether NO directly S-nitrosylates RhoA and whether this pathway is important for G+ toxin-mediated disrupted endothelial barrier function are not well described. Therefore, the goal of the current study was to determine the role of S-nitrosylation in the LLO-induced activation of RhoA. We found that RhoA activity is decreased by NO, that RhoA is an S-nitrosylated protein and identify a novel calcium-dependent mechanism by which G+-toxins stimulate the denitrosylation of RhoA. We also identify three sites of S-nitrosylation on RhoA (C16, C20 and C159) and show that mutation of these cysteine residues to serine provides resistance against the inhibitory effects of NO and results in a form of RhoA that is more effective at promoting endothelial barrier disruption compared to WT.
2. Materials and methods
2.1. Cell culture and transfection
COS-7 and HEK-293 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 100 U/ml penicillin, 100 mg/ ml streptomycin, and 10% FBS [28,29]. Human Lung Microvascular Endothelial Cells (HLMVECs) were isolated and grown in house as previously described [8,23] or purchased from Lonza and grown in Endothelial Growth Medium-2-Microvessel (EGM-2MV) containing the requisite growth factors and 5%FBS (Lonza, Allendale, NJ) and used below passage 8. Cells were grown in a 5% CO2 incubator at 37 °C and used from passage 2–8. For transfection, COS-7 and HEK-293 cells were transfected using Lipofectamine 2000 (Invitrogen, Grand Island, NY) according to the manufacturer’s instructions. HEK 293 cells stably expressing eNOS were generated using the Flp-In System (Invitrogen, Grand Island, NY) [30].
2.2. DNA constructs, antibodies and reagents
Plasmid DNA coding RhoA have been described previously [23]. The C16, 20, 159S triple mutant RhoA was generated by Q5® Site- Directed Mutagenesis Kit (NEB, Ipswich, MA) and confirmed by DNA sequencing. The antibody against RhoA was purchased from Cell Signaling. The G+-bacterial toxin, listeriolysin (LLO), was purified as described previously [7].
GSNOR/TRX genes were transiently silenced using siRNA. The siRNA targeting GSNOR (siRNA ID: s1071) and TRX (siRNA ID: s2) as well as validated non-targeting controls were purchased from Applied Biosystems (Carlsbad, California). Lipofectamine® RNAi- MAX transfection reagent was used to deliver the siRNA in HLMVECs (Invitrogen, Grand Island, NY).
2.3. Measurement of nitric oxide and cell viability
Confluent HLMVECs were treated with or without LLO in serum free medium for 1 h, and the accumulation of in the medium was measured by NO-specific chemiluminescence as previously described [8,31]. Cell viability was assessed by using the Cell Titer-Glo® Luminescent cell viability assay per the manufacturer’s instructions (Promega, Madison, WI).
2.4. Measurement of RhoA activity
RhoA activity was measured using the RhoA G-LISA assay kit (Colorimetric format, Cat: # BK124) (Cytoskeleton, Denver, CO) following the manufacturer’s instruction. In brief, approximately 1– 2 × 106 HLMVECs were plated into 10 cm dishes and incubated overnight in EGM-2MV media. Following the indicated treatments, cells were lysed and the level of GTP-bound RhoA determined by the absorbance at 490 nm using a PolarSTAR luminometer (BMG Labtech, Cary, NC).
2.5. Rapid kinetic analysis of nucleotide binding to RhoA
RhoA was treated with MAHMA NONOate (100 μM) (Sigma, St. Louis, MO) for 30 min in buffered solution, and then 2 μM of mant- GTP was mixed with RhoA (0.1 μM) in a stopped flow instrument (SX-20, Applied Biophysics, Carlsbad, California). The fluorescent intensity of mant-nucleotide binding to RhoA was then detected using excitation at 350 nm and a cutoff filter of 395 nm for 0.2– 1 s. The kinetic curves acquired from at least three experiments were averaged, and the Pro-kineticist software was used to calculate binding constants.
2.6. Endothelial permeability
The trans-endothelial resistance (TER) was measured in confluent HLMVECs. Previous studies have shown that acute changes in TER reflect changes in cell permeability [7,8]. In brief, approximately 60,000 HLMVECs/well were seeded into 8W10E arrays. Media was replaced with fresh media at 24 h and then again to serum free media at 48 h prior to the addition of LLO. ECIS Zh model (Applied Biophysics, Carlsbad, California) was used to monitor resistance values overtime and then normalized to time = 0. Different concentrations of LLO or vehicle were added after the resistance was stable around 1200 O at a frequency of 4000 Hz and the capacitance was between 22 and 29 nanoFarads.
2.7. Measurement of protein S-nitrosylation and Western blotting analysis
Cells, cell lysates or recombinant RhoA were treated with either vehicle or different concentrations of LLO (60–250 ng/ml), Cys-NO (100–300 μM), Calcium (3 mM), ionomycin (1 μM), EGTA (3 mM) and NEM (40 mM). The level of S-nitrosylated RhoA was determined using the biotin-switch assay as shown previously [12,32]. Briefly, cells were washed two times with cold phosphate-buffered saline and then lysed with HEN buffer (250 mM Hepes, 0.1 mM neocuproine (pH 7.7), 1 mM EDTA, SDS (2.5%) and 20 mM methyl methane thiosulfonate (Sigma, St. Louis, MO)) at 50 °C for 20 min. Proteins were precipitated with acetone, washed three times with 70% acetone, and mixed with 0.2 mM N-(6-(bioti namido)hexyl)-3′-(2′-pyridyldithio)propionamide (HPDP-biotin, Thermo Fisher Scientific, Grand Island, NY) in the absence or presence of 50 mM ascorbate for 1 h. Streptavidin–agarose beads (Sigma, St. Louis, MO) adopted to pull down the biotinylated proteins, which was then separated by 12% SDS–PAGE, and immunoblotted with anti-RhoA antibody (Cell Signaling, Danvers, MA) [33,34]. While, biotinylated proteins were detected under non reducing condition using anti-biotin antibody (Cell Signaling, Danvers, MA). Alternatively, protein S-nitrosylation was determined using organic mercury assisted enrichment assay which has been described previously [35].
2.8. Measurements of intracellular Ca2+
Intracellular calcium levels were monitored using the calcium-activated photoprotein, aequorin, as described previously [31]. In brief, HLMVECs were transduced with an adenovirus encoding aequorin at 30MOI, and 24 h later the aequorin was reconstituted by treating cells with 5 μM coelenterazine (Sigma, St. Louis, MO) for 30 min in serum-free and phenol free DMEM. The cells were then exposed to different concentrations of LLO, and luminescence was recorded using a PolarSTAR luminometer (BMG Labtech, Cary, NC).
2.9. MS analysis
To identify sites of S-nitrosylation, the biotin-switch assay was performed as described above on 1 mg of recombinant RhoA incubated with 100 μM of the S-NO donor, nitrosocysteine for 30 min. RhoA was precipitated, washed and then trypsinized overnight using 5 μg of sequencing-grade trypsin (Promega, Madison, WI) at 37 °C. Trypsin was inactivated by addition of 1 mM 4-(2- Aminomethyl) benzenesulfonyl fluoride hydrochloride (AEBSF, Sigma, St. Louis, MO), and biotinylated peptides of RhoA were precipitated with streptavidin–agarose (Sigma, St. Louis, MO) and washed four times with 50 mM ammonium bicarbonate. Peptide digests were analyzed on an Orbitrap Fusion tribrid mass spectrometer (Thermo Fisher Scientific, Grand Island, NY) coupled with an Ultimate 3000 nano-UPLC system (Thermo Fisher Scientific, Grand Island, NY). Two microliters of reconstituted peptide was first trapped and washed on a Pepmap100 C18 trap (5 μm, 0.3 × 5 mm) at 20 μl/min using 2% acetonitrile in water (with 0.1% formic acid) for 10 min and then separated on a Pepman 100 RSLC C18 column (2.0 μm, 75-μm × 150-mm) using a gradient of 2–40% acetonitrile with 0.1% formic acid over 40 min at a flow rate of 300 nl/min and a column temperature of 40 °C. Samples were analyzed by data-dependent acquisition in positive mode using Orbitrap MS analyzer for precursor scan at 120,000 FWHM from 300 to 1500 m/z and ion-trap MS analyzer for MS/MS scans at top speed mode (3-s cycle time). Collision-induced dissociation (CID) was used as fragmentation method. Raw data were processed using Proteome Discoverer (v1.4, Thermo Fisher Scientific, Grand Island, NY) and submitted for Sequest HT search against the Uniprot human database. Fixed value PSM validator algorithm was used for peptide spectrum matching validation. Sequest HT search parameters were 10 ppm precursor and 0.6 Da product ion tolerance, with dynamic carbidomethylation (+57.021 Da) and Biotin- BMCC modification (+533.267 Da).
2.10. Statistical analysis
Data are expressed as means ± SE and statistical analyses performed using Instat software (GraphPad Software Inc., San Diego, CA) with a two-tailed student t-test or ANOVA with a post hoc test where appropriate. Statistical significances were considered as p < 0.05.
3. Results
3.1. NO signaling inhibits G+ toxin-induced RhoA activation and prevents G+ toxin-induced microvascular permeability
We have previously shown that NO signaling plays an important role in G+ toxin-induced endothelial barrier dysfunction [8], however the mechanisms involved are poorly understood. RhoA is a key regulator of endothelial barrier dysfunction, but its regulation by G+-toxins and NO is not completely understood. To assess whether G+ toxins can influence RhoA activity in HLMVECs, cells were exposed to either vehicle or LLO (250 ng/ml) in serum-free conditions. In agreement with our previous findings [46], LLO significantly increased RhoA activity (Fig. 1). We next examined whether NO is important in regulating RhoA activity. Confluent HLMVECs incubated with DETA NONOate or Cys-NO for 30 min and RhoA activity was measured by the G-LISA assay. We found that both NONOate and Cys-NO reduced RhoA activity (Fig. 2A). We then performed kinetic studies to assess the relative affinity of RhoA for GTP using fluorescently labeled guanine nucleotides. The exposure of recombinant RhoA to NO markedly decreased the GTP binding constant (Fig. 2B), indicating that the ability of LLO to activate RhoA can be opposed by NO signaling. To determine the role NO signaling in G+-toxin-induced microvascular permeability, we pretreated the HLMVECs with or without Cys-NO for 30 min and then challenged the cells with 60 and 100 ng of LLO. Transendothelial resistance (TER) was monitored overtime on an ECIS array. As seen in Fig. 2C, LLO dose dependently decreased transendothelial resistance (TER) in HLMVECs which was significantly attenuated in cells pretreated with Cys-NO. Over the duration studied (1hr), we did not detect any significant toxicity of LLO (15–120 ng, Fig. 2D).
Fig. 1.
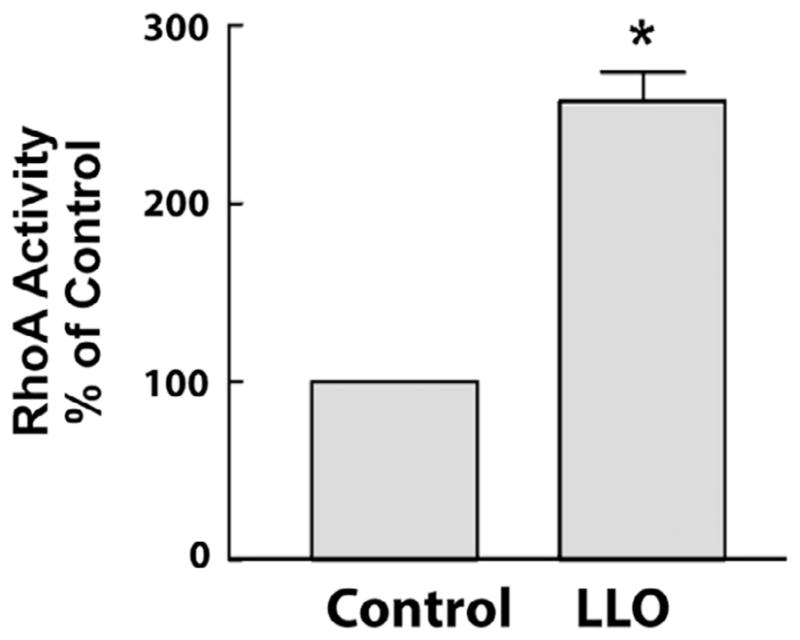
G+-toxins activate RhoA in HLMVECs. Confluent HLMVECs grown in 100-mm dishes were treated with or without the gram positive bacterial (G+) toxin, listeriolysin (LLO, 250 ng/ml) for 30 min. Cells were then lysed and RhoA activity was determined by using the G-LISA RhoA activation assay. Data are expressed as means ± S.E., *P < 0.05 versus control. (n = 6).
Fig. 2.

Nitric oxide is an inhibitor of RhoA activity and G+-toxin-induced microvascular permeability. (A) HLMVEC cells were grown to confluency in 100-mm dishes, treated with NONOate (100 μM) or Cys-NO (100 μM) for 30 min. Cells were then lysed and RhoA activity was determined by using the G-LISA RhoA activation assay. Data are expressed as means ± S.E., *P < 0.05 versus control. (n = 4–6). (B) The kinetics of binding fluorescently labeled GTP to RhoA was performed using stopped flow analysis. Recombinant RhoA (0.1 μM) was mixed with 2 μM of mant-GTP, excitation set to 350 nm and the cutoff filter for emission was 395 nm. Increased fluorescence upon binding mant-GTP was monitored over a 0.5 s time scale. RhoA nitrosylation was achieved by incubating with 100 μM MAHMA NONOate for 30 min prior to mixing with mant-GTP. Three measurements were averaged and fitted with bimolecular reaction equation. Nitric oxide decreased the binding constant of GTP to RhoA from 19.78 to 5.11. (C) HLMVECs were treated with or without Cys-NO (100 μM) for 30 min and then challenged with LLO (60 ng/ml and 120 ng/ml). Transendothelial resistance (TER) was monitored overtime using ECIS. Resistance was normalized to time = 0 and plotted as a function of time. Data are expressed as means ± S.E., *P < 0.05 versus control. #P < 0.05 versus LLO (n = 3–6). (D) LLO does not influence cell viability. Confluent HLMVECs were treated with LLO (0–120 ng/ml) for 1 h, and cell viability was measured using the CellTiter-Glo® Luminescent Cell Viability Assay. Data are expressed as means ± S.E. (n = 3–6).
3.2. RhoA activation is regulated by S-nitrosylation
To investigate whether RhoA is a substrate for S-nitrosylation, the biotin switch assay was performed in HLMVECs treated with or without the S-NO donor, Cys-NO (S-nitrosocysteine). In cells incubated with Cys-NO, there was significantly more S-nitrosylated RhoA compared to control cells (Fig. 3A). To confirm the specificity of the biotin switch assay, we also detected the S-nitrosylation of RhoA using organomercury columns. Organic mercury reacts with S-nitrosothiols (Saville reaction) and displaces the thiol-bound NO to form a covalent bond with the thiol. S-nitrosylated proteins bound to the column were eluted with 50 mM β-mercaptoethanol. Using both methods, our data indicate that RhoA is a substrate for S-nitrosylation (Fig. 3B). To identify the sites of S-nitrosylation on RhoA, recombinant RhoA was treated with Cys-NO, followed by the biotin-switch procedure and MS/MS analysis (Fig. 4A). RhoA peptides containing three modified cysteine residues (C16, C20 and C159) were identified from Cys- NO-treated RhoA (Fig. 4B–D). The residues are highly conserved among different species, suggesting the potential for functional significance. Site-directed mutagenesis was used to make individual cysteine to serine mutants in RhoA (C16S, C20S, C83S, C107S, C159S, C190S) but failed to alter the ability of Cys-NO to induce S-nitrosylation (data not shown). Combined mutation of 3 sites (C16, 20 and 159S) did not prevent Cys-NO induced S-nitrosylation of RhoA in COS-7 cells (Fig. 5A), possibly due to the increased S-nitrosylation of RhoA on other sites in the presence of an exogenous S-NO donor. To better approximate the levels of NO in cells, we generated HEK293-eNOS cells using the Flp-In System to provide a source of endogenous NO. We found that the RhoA triple mutant exhibited a substantial reduction in eNOS-dependent S-nitrosylation compared to wild type (Fig. 5B). To assess the functional consequences of the loss of S-nitrosylation of these 3 sites, we next measured activity in the presence and absence of Cys-NO. We found that the triple C16, 20, 159S mutant of RhoA retains more activity in the presence of increasing concentrations of Cys-NO (Fig. 5C). These data indicate that RhoA is S-nitrosylated on many cysteine residues, with C16, 20 and 159 being the most prominent and functionally important for inactivation of RhoA in the presence of NO.
Fig. 3.

RhoA is a substrate for S-nitrosylation. (A) HLMVECs were treated with either vehicle or Cys-NO (100 μM) for 30 min, and the S-nitrosylation of proteins was determined by the biotin-switch assay in the presence of ascorbate and trace levels of copper. Biotinylated proteins were concentrated using streptavidin–agarose beads, and immunoblotted for RhoA (SNO-RhoA, top panel) versus total RhoA in cell lysates (total RhoA, bottom panel). (B) HLMVECs were treated with or without Cys-NO (100 μM) for 30 min, and S-nitrosylated proteins were selected using organomercury columns followed by immunoblotting for RhoA (SNO-RhoA, top panel) versus total RhoA in cell lysates (total RhoA, bottom panel). The relative densitometry of SNO-RhoA vs total-RhoA is expressed as means ± S.E., *P < 0.05 versus control. (n = 3–6).
Fig. 4.

Identification of C16, C20 and C159 as sites of S-nitrosylation on RhoA. (A) Recombinant RhoA was treated with or without Cys-NO (100 μM) for 30 min and the S-nitrosylation of RhoA was confirmed using the biotin-switch assay and Western blotting. (B–D) Biotinylated RhoA was analyzed by MS/MS and the spectrum of C16, C20 and C159-carbido-Biotin-BMCC tryptic peptides is shown with a summary of the molecular weights of these peptides (top right).
Fig. 5.

Mutation of RhoA on C16, 20, 159S reduces the eNOS-dependent S-nitrosylation of RhoA and protects RhoA from the inhibitory effects of NO. (A) COS-7 cells transfected with WT or mutant C16, 20, 159S RhoA constructs were treated with or without Cys-NO (100 μM) for 30 min. Cells were then lysed, the biotin-switch assay performed and biotinylated proteins concentrated using streptavidin agarose. Total S-nitrosylated proteins were identified using an anti-biotin antibody (top panel) and S-nitrosylated RhoA using a RhoA antibody (lower panel). (B) HEK293-eNOS cells were transfected with RhoA WT or the RhoAC16, 20, 159S mutant, and the degree of S-nitrosylation of RhoA was determined using the biotin-switch assay and immunoblotted for RhoA (SNO-RhoA, top panel) versus total RhoA in cell lysates (total RhoA, bottom panel). (C) COS-7 cells were transfected with WT or mutant C16, 20, 159S RhoA and exposed to the indicated concentrations of Cys-NO for 30 min. Cells were then lysed and RhoA activity determined using the G-LISA RhoA activation assay. Data are expressed as means ± S.E., *P < 0.05 versus control. (n = 4–6).
3.3. G+-toxin stimulated calcium entry mediates RhoA denitrosylation
To determine the effect of G+-toxins on RhoA S-nitrosylation, we exposed HLMVECs to LLO and measured S-nitrosylation using the biotin-switch assay. We found that LLO potently decreased RhoA S-nitrosylation (Fig. 6A). Next, we incubated HLMVECs with different amounts of LLO for 1 h and measured NO production using NO-specific chemiluminescent. We found no significant change in the level of NO following the short term treatment with LLO (Fig. 6B). To further elucidate the mechanisms underlying RhoA denitrosylation in response to G+-toxins, we measured the level of intracellular calcium in response to LLO using the genetically encoded calcium-sensor, aequorin. LLO robustly stimulated calcium entry in HLMVECs (Fig. 6C). The ability of LLO to promote calcium entry results from actions on plasma membrane calcium channels [36]. To determine the importance of calcium entry to LLO stimulated endothelial barrier disruption, we employed a non-selective steric inhibitor of plasma membrane calcium-channels, lanthanum chloride (LaCl3). LLO potently decreased transendothelial resistance (TER) in HLMVECs and this ability was significantly reduced in cells pretreated with the LaCl3 (Fig. 6D). We next investigated whether calcium entry influences RhoA S-nitrosylation. We found that EGTA prevented the loss of RhoA S-nitrosylation following stimulation with LLO (Fig. 6E). We also investigated whether RhoA can be nitrated in response to G+-toxins as has been shown for the G− toxin, LPS [23]. Using anti-nitrotyrosine antibodies we did not observe an increase in RhoA nitration (data not shown).
Fig. 6.

G+-toxin stimulated calcium entry is necessary for endothelial barrier disruption and mediates RhoA denitrosylation. (A) Confluent HLMVECs were treated with or without the G+ toxin, listeriolysin (LLO, 250 ng/ml) for 30 min. The levels of S-nitrosylated RhoA were determined using the biotin-switch assay, and the relative densitometry of SNO-RhoA vs total-RhoA is shown. (B) Acute treatment of G +-toxin LLO does not significantly change the level of NO in HLMVECs. Confluent HLMVECs were treated with listeriolysin (LLO, 0, 125 and 250 ng/ml) for 1 h, and NO release was measured by chemiluminescence. (C) Intracellular calcium was monitored in HLMVEC cells transduced with an adenovirus encoding the calcium-sensor aequorin (30MOI) in response to the indicated amounts of LLO. (D) HLMVECs were pretreated with LaCl3, a non-selective inhibitor of calcium channels for 30 min and then challenged with LLO (120 ng/ml). Transendothelial resistance (TER) values were monitored overtime on an ECIS array. Resistance was normalized to time = 0 and plotted as a function of time. (E) Confluent HLMVECs were treated with or without LLO (LLO, 250 ng/ml) in the presence or absence of EGTA (3 mM). The level of S-nitrosylated RhoA was determined using the biotin-switch assay and immunoblotting for RhoA (SNO-RhoA, top panel) versus total RhoA in cell lysates (total RhoA, bottom panel). Data are expressed as means ± S.E., *P < 0.05 versus control. (n = 3–6).
Protein denitrosylation can be catalyzed by a number of enzymes including GSNOR and Trx1 which utilize a catalytic, sulfhydryl based mechanism to remove NO [12,37]. We next explored the mechanisms of RhoA denitrosylation and the role of calcium in HEK293-eNOS cells. Consistent with results in HLMVECs, LLO robustly decreased the S-nitrosylation of RhoA (Fig. 7A). In addition, the selective and potent calcium ionophore, ionomycin, also efficiently decreased RhoA S-nitrosylation (Fig. 7B). However, the genetic silencing of either GSNOR or Trx1 expression did not influence the ability of ionomycin to stimulate RhoA denitrosylation (Fig. 7C). Furthermore, the thiol-blocking compound N-ethylmaleimide (NEM) also failed to prevent the calcium-induced denitrosylation of RhoA (Fig. 7D) suggesting a non-enzymatic mechanism. To test this directly, we incubated S-nitrosylated, recombinant RhoA with CaCl2 and observed reduced S-nitrosylation in the absence of other factors (Fig. 7E). To determine which sites on RhoA are denitrosylated by calcium, we expressed WT and triple mutant C16, C20, C159S RhoA in HLMVECs using adenovirus, lysed cells and incubated with calcium for 1hr. We found that exposure to calcium promoted the denitrosylation of the WT but not the triple mutant RhoA (Fig. 8A) suggesting these sites are preferentially S-nitrosylated and denitrosylated. To assess functional significance, HLMVECs were transduced with adenoviruses encoding WT and triple mutant RhoA and barrier function measured using TER. Mutant RhoA was more effective in promoting endothelial barrier disruption than WT RhoA (Fig. 8B).
Fig. 7.

Denitrosylation of RhoA is mediated by calcium and not the denitrosylates GSNOR and TRX. (A-B) HEK293-eNOS cells were transfected with WT RhoA for 48 h and then treated with or without LLO (250 ng/ml), the selective calcium ionophore, ionomycin (1 μM) for 30 min. The degree of RhoA S-nitrosylation was determined by the biotin-switch assay, and the relative densitometric ratio of SNO-RhoA (top panel) vs total-RhoA (bottom panel) determined. (C) HEK293-eNOS cells expressing RhoA were transfected with a scrambled negative control siRNA or siRNAs targeting GSNOR or TRX, exposed to ionomycin (1 μM) for 30 min and the level of S-nitrosylated RhoA determined by the biotin-switch assay and shown by the relative densitometry of SNO-RhoA (top panel) vs total-RhoA (lower panel). (D) HEK293-eNOS cells transfected with WT RhoA for 48 h were treated with NEM (40 mM) to block free thiol groups, and then stimulated with or without ionomycin (1 μM) for 30 min. S-nitrosylated RhoA was determined by the biotin-switch assay, and the relative densitometry of SNO-RhoA (top panel) vs total-RhoA (lower panel) determined. (E) Recombinant RhoA was nitrosylated with 100 μM Cys-NO for 30 min and then exposed to 3 mM CaCl2 for 1 h. The level of S-nitrosylated RhoA was determined by the biotin-switch assay, and shown by the relative densitometry of SNO-RhoA (top panel) vs total-RhoA (lower panel). Data are expressed as means ± S.E., *P < 0.05 versus control. (n = 3–4).
Fig. 8.

Calcium promotes the denitrosylation of WT but not C16, 20, 159S mutant RhoA, and mutant RhoA is more effective at promoting barrier disruption in HLMVECs than WT. (A) HLMVECs were transduced with adenoviruses encoding WT or mutant C16, 20, 159S RhoA (30MOI), lysed and then incubated with or without 3 mM CaCl2. The level of S-nitrosylated RhoA was determined by the biotin-switch assay, and shown by the relative densitometry of SNO-RhoA (top panel) vs total-RhoA (lower panel). (B) HLMVECs were transduced with WT or mutant C16, 20, 159S RhoA viruses (30MOI), and transendothelial resistance (TER) values were monitored overtime on an ECIS array. Resistance was normalized to time = 0 and plotted as a function of time. Data are expressed as means ± S.E., *P < 0.05 versus control. (n = 3–4).
4. Discussion
Our previous studies have shown that G+-toxins stimulate endothelial barrier dysfunction through the activation of PKCα and increased eNOS T495 phosphorylation, as well as through the activation of arginase 1, both events lead to the uncoupling of eNOS activity and decreased NO and increased superoxide production [7,8,23,38]. However, the mechanisms by which the diminished NO production in endothelial cells contributes to barrier dysfunction are poorly understood. The activation of RhoA plays an important role in disrupting endothelial barrier function in the lung [39]. In the present study we found that LLO increased the activity of RhoA which is consistent with the actions of other G+-toxins [40]. In endothelial cells, a major pathway of NO signaling is the S-nitrosylation of cysteine residues on target proteins. Using multiple approaches, we found that RhoA is an efficient substrate for S-nitrosylation and that the addition of NO reduces RhoA activity. MS analysis identified multiple cysteine residues that are modified by S-nitrosylation, including C16, C20 and C159. Mutation of these residues to serine prevents S-nitrosylation and reduces sensitivity to the inhibitory actions of NO on RhoA activity. We found that G+-toxins stimulate the robust denitrosylation of RhoA. The denitrosylation of RhoA is not mediated by GSNOR or TRX or thiol based mechanisms and instead appears to be directly mediated by the elevated calcium in response to LLO. Collectively, these data add significantly to our knowledge of RhoA activation by G+-toxins and elucidate a new regulatory mechanism that includes inhibitory S-nitrosylation and the calcium-dependent denitrosylation at three cysteine residues (C16, C20, and C159).
The best known pathway of NO signaling is through the activation of soluble guanylate cyclase (SGC) and production of cGMP [41,42]. The activation of the sGC-cGMP-PKG signaling pathway signaling accounts for many but not all of the biological effects of NO. An alternative pathway of NO signaling involves S-nitrosylation [15,43] which is prominent in endothelial cells where NO concentrations are higher. A previous study has shown that NO inhibits RhoA activity in smooth muscle cells in a cGMP-independent manner and is instead mediated through S-nitrosylation [44]. However, the sites of S-nitrosylation on RhoA and the impact of RhoA S-nitrosylation on microvascular endothelial function have not, to the best of our knowledge, been investigated. In the endothelium of caveolin-1 knockout mice, which synthesize greater amounts of NO, there was no difference in overall S-nitrosylation, but increased nitration of the GTPase-activating protein (GAP) p190RhoGAP-A [45]. Others have shown that the S-nitrosylation of beta-catenin mediates VEGF-induced endothelial cell permeability in aortic endothelial cells [46]. In our study, we found that S-NO and nitric oxide donors can inhibit RhoA activity. However, a limitation of our study is that we have not directly shown that Cys-NO can reduce the activity of LLO-stimulated RhoA. However, we consider this to be likely given the potent ability of Cys-NO to attenuate LLO-induced disruption of the endothelial barrier.
The mechanisms by which NO and S-nitrosylation inhibit RhoA activity are not fully elucidated. We used both the biotin-switch procedure and organomercury columns to confirm that RhoA is a substrate for S-nitrosylation. MS analysis was then used to determine the sites of S-nitrosylation on RhoA. We found that RhoA is S-nitrosylated on C16, C20, and C159, which are highly conserved residues in the amino acid sequences of RhoA from different species [47]. C16, C20, and C159 are predicted to lie within the GTP/ Mg2+ binding site and could possibly interfere with GTP binding once S-nitrosylated. To test this, we performed kinetic studies to assess the relative affinity of RhoA for GTP using fluorescently labeled guanine nucleotides. The exposure of recombinant RhoA to NO markedly reduced time to equilibrium and decreased the GTP binding constant. These data suggest that S-nitrosylation of RhoA has altered the conformation of the nucleotide binding pocket making it harder for the GTP to gain entry and also that when the GTP gains entry to the nucleotide binding site it binds more avidly. We also employed a site-directed mutagenesis approach to confirm the loss of S-nitrosylation in mutants lacking select cysteine residues. However, there was no change in S-nitrosylation in mutants exposed to NO donors. This was a surprising observation and suggests that the labeling of other available cysteine residues occurs at higher NO concentrations. To determine the effect of lower amounts of NO, we employed HEK293-eNOS cells as a source of endogenous NO and found that there was a substantial reduction in S-nitrosylation of the RhoA triple C16, 20, 159S mutant compared to wild type. To confirm that the S-nitrosylation of these residues impacts RhoA function we compared the activity of WT and mutant RhoA in activity assays that recognize the GTP-bound form of RhoA. We found that a NO donor decreased the levels of GTP-bound RhoA and this was reduced in a mutant harboring the combined mutation of C16, 20 and C159S. These results are in agreement with the stopped flow analysis of recombinant RhoA in that the S-nitrosylation of these residues alters the normal operation of the guanine nucleotide binding pocket.
LLO and other members of the family of cholesterol-dependent pore-forming cytolysins from G+-bacteria have been shown to form plasma membrane pores that stimulate calcium entry and promote disruption of the endothelial barrier [7,8,36]. In HLMVECs, we found that LLO robustly increased intracellular calcium and that calcium entry was necessary for disruption of the endothelial barrier. Surprisingly, we also found that LLO stimulated the robust denitrosylation of RhoA and that the loss of S-nitrosylation was calcium-dependent. Similar results were found with PLY (data not shown). This result was not restricted to HLMVEC and in HEK293-eNOS cells, RhoA denitrosylation was also observed with the calcium selective ionophore, ionomycin.
Analogous to phosphorylation, the S-nitrosylation of substrate proteins is a reversible posttranslational modification. GSNOR (also known as alcohol dehydrogenase 5 or ADH5) was one of the earliest identified denitrosylases and has been shown to facilitate the denitrosylation of numerous S-nitrosylated proteins [48]. Additionally, thioredoxin1 (Trx1) is another enzyme capable of removing NO from a wide variety of S-nitrosylated substrates [19,49,50]. Surprisingly, we found that the silencing of either GSNOR or Trx1 expression failed to alter the level of RhoA S-nitrosylation in ionomycin stimulated cells, indicating that denitrosylation of RhoA is not mediated by GSNOR or TRX. We also used the thiol blocking agent, NEM to inhibit other enzymatic pathways of denitrosylation but there was no diminution in the ability of ionomycin to stimulate reduced RhoA s-nitrosylation. The mechanisms by which calcium promotes the denitrosylation of RhoA in the presence of LLO remain unknown. Others have shown that calcium can directly promote protein denitrosylation [51,52]. Calcium-induced denitrosylation has been shown to be independent of trace metals, but may depend on the activation of calpain as calpain inhibitors block calcium-dependent denitrosylation [52]. Alternatively, calcium may allosterically regulate RhoA S-nitrosylation as has been shown for hemoglobin where oxygen binding can alter the stability of distal S-NO modifications [53]. Calcium-dependent denitrosylation also provides an explanation for the paradoxical denitrosylation of eNOS following stimulation with a calcium-mobilizing agonist [54].
To assess the functional role of RhoA denitrosylation we generated adenoviruses encoding WT and triple mutant (C16, 20,159S) RhoA and transduced HLMVECs. The direct addition of calcium to cell lysates resulted in the denitrosylation of WT RhoA but not mutant RhoA. Furthermore, in HLMVECs expressing both RhoA constructs, the RhoA triple mutant which is resistant to the inhibitory effects of NO, was more effective in promoting endothelial barrier dysfunction than the WT.
In conclusion, our study reveals that RhoA is directly modified by S-nitrosylation on C16, C20 and C159 which inhibits RhoA activity and that S-nitrosylation can be reversed through a direct calcium-dependent mechanism. Current therapies to treat G+-toxin induced endothelial barrier dysfunction are not effective, and a deeper understanding of the molecular mechanisms of lung microvascular permeability is needed. The findings in this study not only advance our understanding of how G+-toxins promote hyperpermeability but will facilitate the development of more effective therapeutic approaches that preserve or increase the level of S-nitrosylation of RhoA which may be of clinical significance in the management of ALI-related pulmonary barrier dysfunction.
Acknowledgments
This work was supported by NIH grants R01HL092446, P01 HL101902-01A1, by a Transregio 84/2 SFB grant from the German Research Foundation (TC and RL) “Innate Immunity of the Lung” (TC and RL) and by awards from the AHA (FC and YW) and NSFC 81570378(FC).
Footnotes
Conflict of interest statement
This study was funded by the NIH and there are no conflicts to report.
References
- 1.Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, Gattinoni L, van Haren F, Larsson A, McAuley DF, Ranieri M, Rubenfeld G, Thompson BT, Wrigge H, Slutsky AS, Pesenti A Investigators LS, Group ET. Epidemiology. JAMA. 2016;315(8):788–800. doi: 10.1001/jama.2016.0291. [DOI] [PubMed] [Google Scholar]
- 2.Mansur A, Steinau M, Popov AF, Ghadimi M, Beissbarth T, Bauer M, Hinz J. Impact of statin therapy on mortality in patients with sepsis-associated acute respiratory distress syndrome (ARDS) depends on ARDS severity: a prospective observational cohort study. BMC Med. 2015;13:128. doi: 10.1186/s12916-015-0368-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weycker D, Strutton D, Edelsberg J, Sato R, Jackson LA. Clinical and economic burden of pneumococcal disease in older US adults. Vaccine. 2010;28(31):4955–4960. doi: 10.1016/j.vaccine.2010.05.030. [DOI] [PubMed] [Google Scholar]
- 4.Han B, Sheng B, Zhang Z, Pu A, Yin J, Wang Q, Yang K, Sun L, Yu M, Qiu Y, Xiao W, Yang H. Aryl Hydrocarbon receptor activation in intestinal obstruction ameliorates intestinal barrier dysfunction via suppression of MLCK-MLC phosphorylation pathway. Shock. 2016 doi: 10.1097/SHK.0000000000000594. [DOI] [PubMed] [Google Scholar]
- 5.Chistiakov DA, Orekhov AN, Bobryshev YV. Endothelial barrier and its abnormalities in cardiovascular disease. Front Physiol. 2015;6:365. doi: 10.3389/fphys.2015.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao F, Artham S, Sabbineni H, Al-Azayzih A, Peng XD, Hay N, Adams RH, Byzova TV, Somanath PR. Akt1 promotes stimuli-induced endothelial-barrier protection through FoxO-mediated tight-junction protein turnover. Cell Mol Life Sci. 2016 doi: 10.1007/s00018-016-2232-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lucas R, Yang G, Gorshkov BA, Zemskov EA, Sridhar S, Umapathy NS, Jezierska-Drutel A, Alieva IB, Leustik M, Hossain H, Fischer B, Catravas JD, Verin AD, Pittet JF, Caldwell RB, Mitchell TJ, Cederbaum SD, Fulton DJ, Matthay MA, Caldwell RW, Romero MJ, Chakraborty T. Protein kinase C-alpha and arginase I mediate pneumolysin-induced pulmonary endothelial hyperpermeability. Am J Respir Cell Mol Biol. 2012;47(4):445–453. doi: 10.1165/rcmb.2011-0332OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen F, Kumar S, Yu Y, Aggarwal S, Gross C, Wang Y, Chakraborty T, Verin AD, Catravas JD, Lucas R, Black SM, Fulton DJ. PKC-dependent phosphorylation of eNOS at T495 regulates eNOS coupling and endothelial barrier function in response to G+-toxins. PLoS ONE. 2014;9(7):e99823. doi: 10.1371/journal.pone.0099823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen F, Haigh S, Barman S, Fulton DJ. From form to function: the role of Nox4 in the cardiovascular system. Front Physiol. 2012;3:412. doi: 10.3389/fphys.2012.00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng B, Anea CB, Yao L, Chen F, Patel V, Merloiu A, Pati P, Caldwell RW, Fulton DJ, Rudic RD. Tissue-intrinsic dysfunction of circadian clock confers transplant arteriosclerosis. Proc Natl Acad Sci USA. 2011;108(41):17147– 17152. doi: 10.1073/pnas.1112998108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Witzenrath M, Gutbier B, Hocke AC, Schmeck B, Hippenstiel S, Berger K, Mitchell TJ, de los Toyos TJ, Rosseau S, Suttorp N, Schutte H. Role of pneumolysin for the development of acute lung injury in pneumococcal pneumonia. Crit Care Med. 2006;34(7):1947–1954. doi: 10.1097/01.CCM.0000220496.48295.A9. [DOI] [PubMed] [Google Scholar]
- 12.Qian J, Chen F, Kovalenkov Y, Pandey D, Moseley MA, Foster MW, Black SM, Venema RC, Stepp DW, Fulton DJ. Nitric oxide reduces NADPH oxidase 5 (Nox5) activity by reversible S-nitrosylation. Free Radic Biol Med. 2012;52(9):1806–1819. doi: 10.1016/j.freeradbiomed.2012.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Predescu D, Predescu S, Shimizu J, Miyawaki-Shimizu K, Malik AB. Constitutive eNOS-derived nitric oxide is a determinant of endothelial junctional integrity. Am J Physiol Lung Cell Mol Physiol. 2005;289(3):L371–L381. doi: 10.1152/ajplung.00175.2004. [DOI] [PubMed] [Google Scholar]
- 14.Duran WN, Breslin JW, Sanchez FA. The NO cascade, eNOS location, and microvascular permeability. Cardiovasc Res. 2010;87(2):254–261. doi: 10.1093/cvr/cvq139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stamler JS, Lamas S, Fang FC. Nitrosylation. the prototypic redox-based signaling mechanism. Cell. 2001;106(6):675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 16.Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science (New York, NY) 1998;279(5348):234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 17.Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, Hara MR, Quick RA, Cao W, O’Rourke B, Lowenstein JM, Pevsner J, Wagner DD, Lowenstein CJ. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide- sensitive factor. Cell. 2003;115(2):139–150. doi: 10.1016/s0092-8674(03)00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalez DR, Beigi F, Treuer AV, Hare JM. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci USA. 2007;104(51):20612–20617. doi: 10.1073/pnas.0706796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitchell DA, Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat Chem Biol. 2005;1(3):154–158. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- 20.Birukov KG. Small GTPases in mechanosensitive regulation of endothelial barrier. Microvasc Res. 2009;77(1):46–52. doi: 10.1016/j.mvr.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bogatcheva NV, Zemskova MA, Poirier C, Mirzapoiazova T, Kolosova I, Bresnick AR, Verin AD. The suppression of myosin light chain (MLC) phosphorylation during the response to lipopolysaccharide (LPS): beneficial or detrimental to endothelial barrier? J Cell Physiol. 2011;226(12):3132– 3146. doi: 10.1002/jcp.22669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goeckeler ZM, Wysolmerski RB. Myosin phosphatase and cofilin mediate cAMP/cAMP-dependent protein kinase-induced decline in endothelial cell isometric tension and myosin II regulatory light chain phosphorylation. J Biol Chem. 2005;280(38):33083–33095. doi: 10.1074/jbc.M503173200. [DOI] [PubMed] [Google Scholar]
- 23.Rafikov R, Dimitropoulou C, Aggarwal S, Kangath A, Gross C, Pardo D, Sharma S, Jezierska-Drutel A, Patel V, Snead C, Lucas R, Verin A, Fulton D, Catravas JD, Black SM. Lipopolysaccharide-induced lung injury involves the nitration-mediated activation of RhoA. J Biol Chem. 2014;289(8):4710– 4722. doi: 10.1074/jbc.M114.547596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McNichol BA, Rasmussen SB, Meysick KC, O’Brien AD. A single amino acid substitution in the enzymatic domain of cytotoxic necrotizing factor type 1 of Escherichia coli alters the tissue culture phenotype to that of the dermonecrotic toxin of Bordetella spp. Mol Microbiol. 2006;60(4):939–950. doi: 10.1111/j.1365-2958.2006.05157.x. [DOI] [PubMed] [Google Scholar]
- 25.Lerm M, Selzer J, Hoffmeyer A, Rapp UR, Aktories K, Schmidt G. Deamidation of Cdc42 and Rac by Escherichia coli cytotoxic necrotizing factor 1: activation of c-Jun N-terminal kinase in HeLa cells. Infect Immun. 1999;67(2):496–503. doi: 10.1128/iai.67.2.496-503.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sehr P, Joseph G, Genth H, Just I, Pick E, Aktories K. Glucosylation and ADP ribosylation of rho proteins: effects on nucleotide binding, GTPase activity, and effector coupling. Biochemistry. 1998;37(15):5296–5304. doi: 10.1021/bi972592c. [DOI] [PubMed] [Google Scholar]
- 27.Visvikis O, Maddugoda MP, Lemichez E. Direct modifications of Rho proteins: deconstructing GTPase regulation. Biol Cell. 2010;102(7):377–389. doi: 10.1042/BC20090151. [DOI] [PubMed] [Google Scholar]
- 28.Chen F, Wang Y, Barman S, Fulton DJ. Enzymatic regulation and functional relevance of NOX5. Curr Pharm Des. 2015;21(41):5999–6008. doi: 10.2174/1381612821666151029111528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen F, Haigh S, Yu Y, Benson T, Wang Y, Li X, Dou H, Bagi Z, Verin AD, Stepp DW, Csanyi G, Chadli A, Weintraub NL, Smith SM, Fulton DJ. Nox5 stability and superoxide production is regulated by C-terminal binding of Hsp90 and CO-chaperones. Free Radic Biol Med. 2015;89:793–805. doi: 10.1016/j.freeradbiomed.2015.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen F, Yu Y, Qian J, Wang Y, Cheng B, Dimitropoulou C, Patel V, Chadli A, Rudic RD, Stepp DW, Catravas JD, Fulton DJ. Opposing actions of heat shock protein 90 and 70 regulate nicotinamide adenine dinucleotide phosphate oxidase stability and reactive oxygen species production. Arterioscler Thromb Vasc Biol. 2012;32(12):2989–2999. doi: 10.1161/ATVBAHA.112.300361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen F, Fulton DJ. An inhibitor of protein arginine methyltransferases, 7,7′-carbonylbis(azanediyl)bis(4-hydroxynaphthalene-2-sulfonic acid (AMI-1), is a potent scavenger of NADPH-oxidase-derived superoxide. Mol Pharmacol. 2010;77(2):280–287. doi: 10.1124/mol.109.061077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forrester MT, Foster MW, Benhar M, Stamler JS. Detection of protein S-nitrosylation with the biotin-switch technique. Free Radic Biol Med. 2009;46(2):119–126. doi: 10.1016/j.freeradbiomed.2008.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen F, Barman S, Yu Y, Haigh S, Wang Y, Black SM, Rafikov R, Dou H, Bagi Z, Han W, Su Y, Fulton DJ. Caveolin-1 is a negative regulator of NADPH oxidase-derived reactive oxygen species. Free Radic Biol Med. 2014;73:201– 213. doi: 10.1016/j.freeradbiomed.2014.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barman SA, Chen F, Su Y, Dimitropoulou C, Wang Y, Catravas JD, Han W, Orfi L, Szantai-Kis C, Keri G, Szabadkai I, Barabutis N, Rafikova O, Rafikov R, Black SM, Jonigk D, Giannis A, Asmis R, Stepp DW, Ramesh G, Fulton DJ. NADPH oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling. Arterioscler Thromb Vasc Biol. 2014;34(8):1704–1715. doi: 10.1161/ATVBAHA.114.303848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doulias PT, Raju K, Greene JL, Tenopoulou M, Ischiropoulos H. Mass spectrometry-based identification of S-nitrosocysteine in vivo using organic mercury assisted enrichment. Methods. 2013;62(2):165–170. doi: 10.1016/j.ymeth.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiong C, Yang G, Kumar S, Aggarwal S, Leustik M, Snead C, Hamacher J, Fischer B, Umapathy NS, Hossain H, Wendel A, Catravas JD, Verin AD, Fulton D, Black SM, Chakraborty T, Lucas R. The lectin-like domain of TNF protects from listeriolysin-induced hyperpermeability in human pulmonary microvascular endothelial cells - a crucial role for protein kinase C-alpha inhibition. Vascul Pharmacol. 2010;52(5–6):207–213. doi: 10.1016/j.vph.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol. 2009;10(10):721–732. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- 38.Joshi AD, Dimitropoulou C, Thangjam G, Snead C, Feldman S, Barabutis N, Fulton D, Hou Y, Kumar S, Patel V, Gorshkov B, Verin AD, Black SM, Catravas JD. Heat shock protein 90 inhibitors prevent LPS-induced endothelial barrier dysfunction by disrupting RhoA signaling. Am J Respir Cell Mol Biol. 2014;50(1):170–179. doi: 10.1165/rcmb.2012-0496OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrington EO, Newton J, Morin N, Rounds S. Barrier dysfunction and RhoA activation are blunted by homocysteine and adenosine in pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol. 2004;287(6):L1091– L1097. doi: 10.1152/ajplung.00421.2003. [DOI] [PubMed] [Google Scholar]
- 40.Iliev AI, Djannatian JR, Nau R, Mitchell TJ, Wouters FS. Cholesterol-dependent actin remodeling via RhoA and Rac1 activation by the Streptococcus pneumoniae toxin pneumolysin. Proc Natl Acad Sci USA. 2007;104(8):2897–2902. doi: 10.1073/pnas.0608213104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Friebe A, Koesling D. Regulation of nitric oxide-sensitive guanylyl cyclase. Circ Res. 2003;93(2):96–105. doi: 10.1161/01.RES.0000082524.34487.31. [DOI] [PubMed] [Google Scholar]
- 42.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43(2):109–142. [PubMed] [Google Scholar]
- 43.Foster MW, Forrester MT, Stamler JS. A protein microarray-based analysis of S-nitrosylation. Proc Natl Acad Sci USA. 2009;106(45):18948–18953. doi: 10.1073/pnas.0900729106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuckerbraun BS, Stoyanovsky DA, Sengupta R, Shapiro RA, Ozanich BA, Rao J, Barbato JE, Tzeng E. Nitric oxide-induced inhibition of smooth muscle cell proliferation involves S-nitrosation and inactivation of RhoA. Am J Physiol Cell Physiol. 2007;292(2):C824–C831. doi: 10.1152/ajpcell.00592.2005. [DOI] [PubMed] [Google Scholar]
- 45.Siddiqui MR, Komarova YA, Vogel SM, Gao X, Bonini MG, Rajasingh J, Zhao YY, Brovkovych V, Malik AB. Caveolin-1-eNOS signaling promotes p190RhoGAP-A nitration and endothelial permeability. J Cell Biol. 2011;193(5):841–850. doi: 10.1083/jcb.201012129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thibeault S, Rautureau Y, Oubaha M, Faubert D, Wilkes BC, Delisle C, Gratton JP. S-nitrosylation of beta-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Mol Cell. 2010;39(3):468–476. doi: 10.1016/j.molcel.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 47.Kakiuchi M, Nishizawa T, Ueda H, Gotoh K, Tanaka A, Hayashi A, Yamamoto S, Tatsuno K, Katoh H, Watanabe Y, Ichimura T, Ushiku T, Funahashi S, Tateishi K, Wada I, Shimizu N, Nomura S, Koike K, Seto Y, Fukayama M, Aburatani H, Ishikawa S. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat Genet. 2014;46(6):583–587. doi: 10.1038/ng.2984. [DOI] [PubMed] [Google Scholar]
- 48.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410(6827):490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 49.Benhar M, Thompson JW, Moseley MA, Stamler JS. Identification of S-nitrosylated targets of thioredoxin using a quantitative proteomic approach. Biochemistry. 2010;49(32):6963–6969. doi: 10.1021/bi100619k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kahlos K, Zhang J, Block ER, Patel JM. Thioredoxin restores nitric oxide-induced inhibition of protein kinase C activity in lung endothelial cells. Mol Cell Biochem. 2003;254(1–2):47–54. doi: 10.1023/a:1027380828645. [DOI] [PubMed] [Google Scholar]
- 51.Lai TS, Hausladen A, Slaughter TF, Eu JP, Stamler JS, Greenberg CS. Calcium regulates S-nitrosylation, denitrosylation, and activity of tissue transglutaminase. Biochemistry. 2001;40(16):4904–4910. doi: 10.1021/bi002321t. [DOI] [PubMed] [Google Scholar]
- 52.Chvanov M, Gerasimenko OV, Petersen OH, Tepikin AV. Calcium-dependent release of NO from intracellular S-nitrosothiols. EMBO J. 2006;25(13):3024–3032. doi: 10.1038/sj.emboj.7601207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jia L, Bonaventura C, Bonaventura J, Stamler JS. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature. 1996;380(6571):221–226. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 54.Erwin PA, Lin AJ, Golan DE, Michel T. Receptor-regulated dynamic S-nitrosylation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem. 2005;280(20):19888–19894. doi: 10.1074/jbc.M413058200. [DOI] [PubMed] [Google Scholar]