

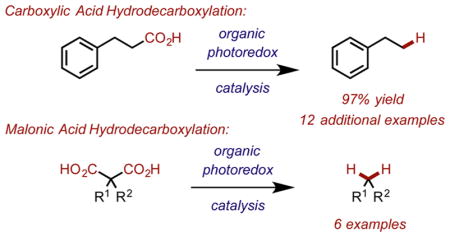
Abstract
A direct, catalytic hydrodecarboxylation of primary, secondary, and tertiary carboxylic acids is reported. The catalytic system consists of a Fukuzumi acridinium photooxidant with phenyldisulfide acting as a redox-active cocatalyst. Substoichiometric quantities of Hünig’s base are used to reveal the carboxylate. Use of trifluoroethanol as a solvent allowed for significant improvements in substrate compatibilities, as the method reported is not limited to carboxylic acids bearing α heteroatoms or phenyl substitution. This method has been applied to the direct double decarboxylation of malonic acid derivatives, which allows for the convenient use of dimethyl malonate as a methylene synthon. Kinetic analysis of the reaction is presented showing a lack of a kinetic isotope effect when generating deuterothiophenol in situ as a hydrogen atom donor. Further kinetic analysis demonstrated first-order kinetics with respect to the carboxylate, while the reaction is zero-order in acridinium catalyst, consistent with another finding suggesting the reaction is light limiting and carboxylate oxidation is likely turnover limiting. Stern–Volmer analysis was carried out in order to determine the efficiency for the carboxylates to quench the acridinium excited state.
Graphical Abstract
INTRODUCTION
The utility of carboxylic acids and esters as functional handles and activating groups is vital to the strategic deployment of classical C–C bond forming reactions in complex synthetic sequences via enolate and Michael reactivity.1,2 Furthermore, carboxylic acids and esters are also commonly used to activate dienophiles for Diels–Alder cycloadditions,3 a reaction that is ubiquitous in complex molecule synthesis. Though carbonyls are valuable for their ability to facilitate carbon–carbon bond formation, the carboxylic acid functionality is not always desired in downstream adducts which would necessitate removal. Excising carboxylic acid functionality via a hydrodecarboxylation strategy allows for the use of carbonyls as traceless functional handles for assembling molecular complexity.
The Barton decarboxylation4–7 is perhaps the most commonly utilized method for the reduction of carboxylic acids to alkanes via a hydrodecarboxylation mechanism;8–10 however, it requires prefunctionalization of the carboxylic acid (PTOC ester formation) and utilizes stoichiometric amounts of toxic tin hydrides as the source of hydrogen atoms (Scheme 1). Modifications of the Barton decarboxylation employ thiols,11,12 silanes,13 or chloroform14 as H atom donors, but necessitate superstoichiometric quantities of H atom donor or produce significant amounts of unwanted byproducts. Electrochemical methods for decarboxylation have also been employed, such as the Kolbe electrolysis which proceeds through the single electron oxidation of a carboxylate to form an acyloxyl radical.15,16 These radicals are known to rapidly rearrange to expel CO2 and form carbon-centered radicals.17,18 On the surface of an electrode, these radicals can be successively oxidized to the corresponding cation, known as the non-Kolbe pathway.19
Scheme 1.

Progression of Hydrodecarboxylation Strategies
To avoid dimerization of radicals and successive radical oxidation, several methods for the catalytic hydrodecarboxylation and decarboxylative coupling reactions have been developed using both ground state,20,21 and photochemical oxidants.22–24 These methods often utilize stoichiometric amounts of a terminal oxidant and hydrogen atom donor.22,23
Several methods for hydrodecarboxylation and decarboxylative coupling25 have been developed using Pd,26–31 Cu,32–35 Ag33,36,37 and Rh,38 but these methods are limited to aryl and alkynyl carboxylic acids. Photoredox catalysis has been utilized to implement decarboxylative functionalizations, including additions to arenes39–42 and alkenes,43–50 as well as decarboxylative fluorinations51–53 and decarboxylation of keto carboxylic acids to form ketones.54 Wallentin and co-workers recently reported a photoredox method for the hydrodecarboxylation of stabilized carboxylic acids, such as protected amino acid derivatives, and phenyl acetic acid derivatives using a similar catalyst system to our own; however, aliphatic carboxylic acids were not found to be viable substrates using this method.24 Thus, a method for the direct catalytic hydrodecarboxylation of unstabilized aliphatic carboxylic acids has remained elusive.
A direct catalytic hydrodecarboxylation of aliphatic carboxylic acids and malonic acid derivatives would be complementary to these methods. Hydrodecarboxylation of malonic acid derivatives could be particularly interesting from a synthetic standpoint because it would allow for the use of malonate as a “(−)CH2(−)” synthon by directly reducing the corresponding malonic acid. This would have the advantage over conventional methods, which require several additional steps to reach the desired product. There are also numerous examples of reactions that rely on malonates to increase the rate of intramolecular reactions such as intramolecular Diels-Alder reactions or olefin metathesis.55 Although, malonates are useful for facilitating Thorpe-Ingold effects and as functional handles, they are not always desirable in the final product. Traditional methods for removing both carboxylates would require several steps including thermal decomposition of the malonic acid at high temperatures, then formation of the Barton ester and decomposition thereof to remove the second carboxylate. Thus, we set out to develop a general method for decarboxylating unstabilized carboxylic acids and malonic acid derivatives.
Due to the low oxidation potential of aliphatic carboxylates (vide infra), we believed that the direct, catalytic, photoredox hydrodecarboxylation of aliphatic carboxylic acids should not, in theory, be limited to those acids stabilized by either heteroatomic functionality or aryl substitution. Thus, we found it surprising that the photoredox hydrodecarboxylation of aliphatic carboxylic acids was apparently more difficult to achieve. We hoped to provide mechanistic insight into the factors contributing to this problem, as we believed it could lead to the transformation being developed further.
We envisioned using Fukuzumi acridinium photooxidants,56 which have recently been employed in photoredox systems in our lab,57–65 as they are cited to have excited state reduction potentials of greater than +2.0 V.57 Carboxylic acids have been used in various other transformations in our lab, indicating that the carboxylic acid is not itself oxidizable;57,60,65 however, we believed that deprotonation by a strong, noncoordinating base could reveal the carboxylate and render them more susceptible to oxidation. This is further evidenced by the fact that carboxylic acids have oxidation potentials higher than the solvent window in acetonitrile, whereas tetrabutylammonium carboxylates are cited to have oxidation potentials close to +1.2 V.66 This indicates that base selection would be critical to the success of the proposed oxidative decarboxylation.
RESULTS AND DISCUSSION
Aware that solvent could play an important role in the success of the transformation, we began by conducting a solvent screen using both protic and aprotic solvents with a range of polarity. We began optimization with conditions similar to those previously reported by our lab63 and with the aliphatic tertiary acid 1a, as we believed the formation of the tertiary carbon centered radical would be relatively facile and the substrate could represent a feasible expansion of previous substrate limitations. After some initial screening, Mes-Acr-Ph was found to be the catalyst of choice, potentially because it is less susceptible to deactivation via dealkylation than the more widely used Mes-Acr-Me.64 Under these conditions, we did observe product formation, albeit in very low yields (Table 1, entry 1). Changing the solvent to more polar solvents such as acetonitrile and methanol seemed to have no effect (entries 2 and 3). When a 9:1 MeOH/H2O solvent system was used, gains in yields were observed (entry 4). This indicates that increasing the equilibrium concentration of carboxylate relative to the carboxylic acid was important. The pKa of carboxylic acids can be up to five units greater in methanol than in water (the pKa of acetic acid in water is 4.76 vs 9.63 in methanol), while the pKa of protonated amines are similar in both solvents (triethylammonium is 10.75 in water and 10.78 in methanol).67 This prompted us to conduct a thorough examination of the base employed in the reaction. The more strongly basic 2,4,6-collidine gave improved yields (entry 5), as did the even more basic, N,N-diisopropylethylamine (DIPEA, entry 6). Although it may be somewhat surprising that oxidizable amine bases are compatible in these reactions as they are known to form aminium radical cations in similar photoredox systems,68,69 their use can be rationalized by noting that they likely predominately exist in solution as the ammonium salts which are insulated from oxidation. Due to the success with the MeOH/H2O system, we considered other polar alcohol solvents, and found that trifluoroethanol (TFE, entry 8) gave a marked improvement. Primary aliphatic carboxylic acids such as hydrocinnamic acid were poor substrates when using 9:1 MeOH/H2O as solvent (entry 9); however, using TFE as a solvent dramatically improved their reactivity (entry 10). Control experiments revealed that base was necessary for reactivity (entry 7) as was phenyl disulfide (entry 11).
Table 1.
Optimization of Reaction Conditionsa

| ||||
|---|---|---|---|---|
| Entry | Substrate | Solvent | Baseb | Yieldc |
| 1 | 1a | CHCl3 | 2,6-lutidine | <10% |
| 2 | 1a | MeCN | 2,6-lutidine | <10% |
| 3 | 1a | MeOH | 2,6-lutidine | <10% |
| 4 | 1a | MeOH/H2O (9:1) | 2,6-lutidine | 21% |
| 5 | 1a | MeOH/H2O (9:1) | collidine | 51% |
| 6 | 1a | MeOH/H2O (9:1) | i-Pr2NEt | 81% |
| 7 | 1a | MeOH/H2O (9:1) | – | No rxn |
| 8 | 1a | CF3CH2OH | i-Pr2NEt | 77% |
| 9d | 1b | MeOH/H2O (9:1) | i-Pr2NEt | 14% |
| 10d | 1b | CF3CH2OH | i-Pr2NEt | 97% |
| 11d,e | 1b | CF3CH2OH | i-Pr2NEt | <10% |
Reactions carried out on a 0.3 mmol scale in N2-sparged solvents [0.5 M] at ambient temperature.
Twenty mole percent base loading.
Yields determined by 1H NMR analysis of crude reactions.
Reaction run for 72 h.
Reaction without phenyl disulfide.
With these optimized conditions, we decided to explore the scope of this reaction (Chart 1). Primary (2a–c), secondary (2d), tertiary (2e) alkyl substituted carboxylic acids were all competent substrates. Further investigation of the scope of this reaction revealed that electron deficient (2b) and moderately electron rich arenes (2c) were tolerated under the standard reaction conditions, while electron rich arenes, such as p-methoxyhydrocinnamic acid, were not viable substrates presumably due to competitive oxidation of the aromatic ring with the carboxylate functional group. Substrates bearing bi-(2f) and monoaryl (2g) substitution adjacent to the carboxylate were found to be excellent substrates. Protected amino acids (2h) and other protected amine-containing substrates (2i and 2j) were also tolerated using this method. Substrates bearing α-esters (2k) could be efficiently decarboxylated under these conditions. Fatty acid tridecanoic acid initially gave only trace amounts of dodecane (2l). Tridecanoic acid was only sparingly soluble in TFE; therefore, an additional solvent screen was conducted, and revealed that using 4:1 TFE/EtOAc [0.3 M] improved the reactivity substantially. Increasing disulfide loading from 10 to 20 mol % was also found to be optimal for this substrate. Remarkably, the highly functionalized natural product Enoxolone (2m) underwent hydrodecarboxylation in good yield as a mixture of diastereomers (3:1), albeit with an extended reaction time (96 h, 85%). The increased reaction time required for this substrate is most likely due to the limited solubility of the substrate in TFE, even at lower concentrations. However, with the use of ethyl acetate as a cosolvent, the reaction time could be reduced to 24 h, with an improved yield. Using ethyl acetate as a cosolvent also improved reactivity for substrate 2j, which also exhibited low solubility in TFE.
Chart 1. Hydrodecarboxylation Reaction Scopea.

aReactions carried out in N2-sparged TFE [0.5 M]. bYields for volatile compounds were determined by GC. cAverage of two isolated yields on >100 mg scale. d[0.3 M] in TFE/EtOAc (4:1). eTwenty mole percent Ph2S2.
We propose a mechanism for this reaction in which the carboxylic acid (1) is deprotonated, then single electron oxidation by Mes-Acr-Ph* results in the formation of an acyloxyl radical (2), which then rapidly rearranges to form carbon dioxide and a carbon-centered radical (3). Hydrogen atom abstraction from thiophenol by 3 furnishes the final hydrodecarboxylation adduct. In prior work, we have mechanistic evidence that supports the generation of phenylthiyl radical via photoinduced S–S bond homolysis.70 The thiyl radical then reoxidizes the reduced catalyst and is protonated to furnish the active hydrogen atom donor (Scheme 2).
Scheme 2.

Proposed Mechanism for Decarboxylation
Next, we elected to extend this method to the decarboxylation of malonic acid derivatives. We found that our previously optimized conditions did not result in the doubly decarboxylated product and only small amounts of monodecarboxylated product. We posited that this was due to hydrogen bonding events from the second acid moiety on the malonic acid increasing the oxidation potential of the carboxylate. A comparison of benzyl malonic acid and substrate 2k supports this hypothesis (Figure 1). Almost no reactivity was observed for the malonic acid under standard conditions, while the monoacid can be efficiently decarboxylated.
Figure 1.

Comparison between efficiency of decarboxylation of (a) malonic acids and (b) malonate monoesters. Control experiments were done without catalyst to ensure a thermal decomposition pathway was not active.
We presumed that using increased base loading could improve the reactivity, and indeed using 1.2 equiv of DIPEA instead of 0.2 equiv resulted in small amounts of toluene from phenyl malonic acid; however, we believed that the reactivity and scope could be improved through the use of a stronger base. We were pleased to see that when using 1.0 equiv of KOt-Bu instead of DIPEA under otherwise standard reactions conditions, we were able to isolate the corresponding doubly decarboxylated products. These reactions were found to require longer reactions times, due to the increased amount of carboxylate relative to the catalyst. Not surprisingly, increased catalyst (7.5 mol %) and disulfide (15 mol %) loading was found to improve the effciency of the reaction. In most cases the mass balance consisted of the monodecarboxylated and doubly decarboxylated products, with a small amount of unreacted starting material. Importantly, a control experiment revealed no reaction occurred without the inclusion of the Mes-Acr-Ph photocatalyst, excluding thermal decomposition as a potential mechanism.
During our investigation of the reaction scope, we observed that aryl malonic acid derivatives were particularly prone to hydrodecarboxylation, potentially because of an ability to stabilize the resulting radical (3a and 3b). We were also able to demonstrate that alkyl-substituted malonic acid derivatives were viable substrates for decarboxylation in this system, although they required prolonged reaction times. Dialkyl substituted malonic acids 2-benzyl-2-methylmalonic acid, indan-2,2-dicarboxylic acid, and 2-benzyl-2-(3-oxobutyl)-malonic acid were able to be decarboxylated to give products 3c–e, respectively. Unfortunately, monoalkyl substituted benzyl malonic acid gave poor yields of the doubly decarboxylated product 3f (Chart 2).
Chart 2. Malonic Acid Derivative Decarboxylationa.

aReactions carried out in N2-sparged TFE [0.5 M]. bYields for volatile compounds were determined by GC. c1.1 equiv of KOH used in place of KOt-Bu. dAverage of two isolated yields on >100 mg scale.
Several mechanistic studies were conducted to understand the role of TFE as very large gains in yields were observed, particularly for the primary carboxylic acid substrates, when using TFE as a solvent. This behavior would not be solely explained by polarity, as the MeOH/H2O system is more polar (the dielectric constants of 9:1 MeOH/H2O and TFE are 36.8 and 27.1 F/m, respectively).71,72 We considered the possibility of TFE supplementing thiophenol as a hydrogen atom donor in this reaction as our lab and others have shown that alcohols (αC-H bonds) can act as hydrogen atom donors.61 However, exclusion of disulfide in the reaction led to only trace amounts of product formation, indicating either that TFE is not a competitive H atom donor in this reaction or that the corresponding radical formed is unable to reoxidize the catalyst (Table 1, entry 11). This was also confirmed with a deuterium labeling study in which 2,2,2-trifluoroethanol-d2 (d2-TFE) was used as a solvent under otherwise standard conditions: no deuterium incorporation in the product was observed at full reaction conversion (Figure 2a). When using 2,2,2-Trifluoroethanol-d1 (d1-TFE) as a solvent, 63% deuterium incorporation was observed at reaction completion, supporting a mechanism in which thiophenol is generated in situ and acts as a hydrogen atom donor, as thiolate being formed in the reaction could deprotonate an equivalent of carboxylic acid, regenerating the H atom donor (Figure 2b).
Figure 2.

Deuterium labeling experiments (a) decarboxylation of 2,2-dimethyl 3-phenyl propanoic acid run in d2-TFE to determine if TFE was a catalytically active hydrogen atom donor. (b) Decarboxylation in d1-TFE showing that the proton from the carboxylic acid starting material is incorporated in the product.
Since it was evident that TFE was not contributing significantly as a hydrogen atom donor, further investigations were undertaken to elucidate its role. We measured the fluorescence lifetime of Mes-Acr-Ph using time-correlated single photon counting (TCSEC) in both methanol and TFE. The fluorescence lifetime was found to be 10.8 ns in TFE. In methanol, the catalyst displayed two fluorescence decay components having lifetimes of 0.49 and 5.5 ns (see Figure S4), both significantly shorter than in TFE. We also found that significant catalyst decomposition occurred upon standing in a solution of 9:1 MeOH/H2O overnight with blue LED irradiation. This suggests that both the increased catalyst excited state lifetime of Mes-Acr-Ph in TFE and the decreased nucleophilicity73 of TFE relative to methanol makes TFE an ideal solvent for this reaction. Though other solvents have been shown to demonstrate relatively long excited state lifetimes for acridinium catalysts, TFE meets the requirements of being a polar protic solvent, which is apparently necessary to stabilize carboxylate formation in this reaction.
To determine if a hydrogen atom transfer step could be rate limiting, the rate of decarboxylation was measured for a tertiary carboxylic acid under standard conditions and for the deuterated analogue in d1-TFE (Figure 3). This would generate d1-thiophenol in situ and, with no other sources of exchangeable protons, should allow for the determination of a KIE. The kinetics were determined using the initial rates method with good mass balance observed. A very short induction period was observed in some cases, most likely due to the limited solubility of the disulfide at the beginning of the reaction, which was not found to significantly affect the reaction kinetics after multiple trials with each set of conditions. A KIE near unity was observed (1.01) when rates were measured in separate vessels. A competition experiment in which a mixture of 1:1 proteo and deutero acid in a 1:1 mixture of TFE/d1-TFE resulted in greater than 20:1 proton incorporation in the final product observed.
Figure 3.

Initial rates of decarboxylation for (a) 2,2-dimethyl 3-phenyl propanoic acid and (b) the deuterated analogue. The rate of decarboxylation of the deuterated analogue was determined using d1-TFE as a solvent; 1H NMR analysis shows the complete deuterium incorporation in the product. Each initial rate was calculated based on 3 trials, giving an average KIE of 1.01.
The lack of KIE observed when reactions were run in separate vessels indicates that no proton transfer or hydrogen atom transfer step is likely to be rate-limiting in the reaction. This is consistent with the observation that no products resulting from dimerization were observed in these reactions. The results of the competition experiment, while indicating a kinetic preference for hydrogen atom transfer over deuterium atom transfer, cannot give any information about the rate-limiting step in the reaction. The large KIE observed in the competition experiment could be indicative of an equilibrium isotope effect in which thiophenol is formed in higher concentrations than deuterothiophenol, via deprotonation of carboxylic acid or exchange with the solvent (this would be expected considering the relevant bond dissociation energies). This could also be compounded by a faster rate of H atom transfer than D atom transfer; however, this does not indicate that H atom transfer is rate limiting, as this experiment only indicates that H atom transfer is irreversible and product determining. Our lab has previously discovered that thiyl radical oxidation of the acridine radical intermediate is essentially diffusion controlled,70 and as previously mentioned, others have determined that the rate of acyloxyl radical rearrangement to lose carbon dioxide is also very rapid. Therefore, the lack of a kinetic isotope effect could be indicative of carboxylate oxidation being rate limiting.
Further kinetic analysis showed that the reaction is first-order with respect to the carboxylate, while being zero-order with respect to Mes-Acr-Ph; this finding is suggestive of a light limiting reaction. (Table S4). Indeed, the reaction was found to be very light-dependent. Reactions were normally irradiated with two LED lamps over the course of the reaction, as there is a dramatic decrease in the initial rate of the reaction when only one lamp is used to irradiate the reaction vessel (Figure 4). The light dependence of the reaction suggests that the method could be improved through the use of a flow reactor setup, which could allow for a greater absorbance of light because of an increased surface area.
Figure 4.

Comparison of initial rate of reaction for the decarboxylation of 2,2-dimethyl 3-phenyl propanoic acid under irradiation with 2 blue LED lamps and 1 blue LED lamp.
During the course of optimization and investigating the scope of the reaction, it became apparent that substrates were decarboxylated much more readily as alkyl substitution at the α position increased when using a methanol/water system, as only very small amounts of product were produced for primary carboxylic acids in this system. It seemed possible that a difference in the oxidation potential of substrates based on the degree of substitution alpha to the carboxylic acid could explain this trend. To probe these reactivity differences, redox data was collected for three carboxylates of increasing amounts of alpha substitution. To our surprise, there was not a significant difference in oxidation potential among these substrates (Figure 5). The relatively low oxidation potentials for carboxylates indicate that electron transfer from the carboxylate to the excited Mes-Acr-Ph should be very thermodynamically favorable in each case. With the oxidation potentials of the three representative carboxylates being very close to one another, it seems that oxidation potential alone is not sufficient to describe differences in reactivity.
Figure 5.

Oxidation potentials of the tetrabutylammonium salts of three representative carboxylic acids were measured in a 0.1 M solution of tetrabutylammonium hexafluorophosphate in acetonitrile, vs SCE.
Electrochemical oxidation potentials suggest electron transfer should be thermodynamically favorable, however it seemed reasonable that there could be differences in the kinetic barrier for oxidation between carboxylates bearing differing α substitution causing a difference in their apparent reactivity. Therefore, comparisons were also made between potassium salts of three carboxylic acids with increasing alkyl substitution at the α position, in their ability to quench the excited state of the catalyst in TFE. The quenching constants were determined by Stern-Volmer analysis of fluorescence quenching, and the quenching constants are reported in Figure 6. The observed quenching constants (kq) indicate that there is only a small difference in the rate of quenching of the acridinium excited singlet state in TFE, with the primary carboxylate possessing the largest kq, albeit by a narrow margin.
Figure 6.

Bimolecular quenching constants measured for the potassium salts of each carboxylic acid in TFE.
Indeed, the rate of the reaction in TFE seems to be nearly independent of substitution at the α position, as shown by a competition experiment in which equimolar amounts of each carboxylate were added to the same reaction vial (Figure 7 top). The experiment revealed that the primary carboxylic acid was actually decarboxylated most rapidly in TFE. This observation was counter to what was previously observed in other solvent systems, as seen from an analogous competition experiment in 9:1 MeOH/H2O in which the selectivity is reversed (Figure 7 bottom). It should also be noted that the overall rate of conversion in MeOH/H2O was much slower than what was expected based on previous observations with the tertiary substrate (Table 1, entry 6), as 24 h of irradiation was required to reach about 30% total conversion.
Figure 7.

Competition experiments in (a) TFE and (b) 9:1 MeOH/H2O in which equimolar amounts (0.25 mmol) of each substrate were in the same reaction vessel. Other reagents were added in their respective quantities relative to the total amount of carboxylate in the reaction. The reactions were stopped at about 30% conversion. Yields were measured by analysis of crude 1H NMR spectra.
Fukuzumi has demonstrated in a similar system that in an acetonitrile/water mixture there are significant differences in the ability of a series of primary, secondary, and tertiary alkyl substituted carboxylates to quench the photoexcited state of a 10-methyl acridinium catalyst via an electron transfer mechanism.74 This could potentially indicate a different mechanism in different solvent systems and highlights the importance of solvent in these systems. We noted in early optimization of this reaction (Tables S1–S3) that substrates bearing α-phenyl groups could be efficiently decarboxylated in chloroform, whereas alkyl substituted carboxylic acids were sluggish using this solvent. Wallentin et al. have also demonstrated the ability for an acridinium photooxidant to decarboxylate protected amino acids and phenyl acetic acid derivatives in dichloroethane, but alkyl-substituted acids were not possible.24
It is possible that a back electron transfer process occurring from the acridine radical to the acyloxyl radical is faster than CO2 loss for primary carboxylic acids in MeOH/H2O, as this electron transfer is thermodynamically favorable and probably rapid. This would suggest that for tertiary carboxylic acids CO2 loss is competitive with back electron transfer. However, the competition experiment in Figure 7 in MeOH/H2O seems to suggest that at early conversions the rate of product formation is similar for primary, secondary, and tertiary carboxylic acids. As previously mentioned, the overall rate of conversion for the tertiary acid was slower than expected in the competition experiment. Since there is only a slight rate enhancement for the tertiary substrate in MeOH/H2O, it seems plausible that catalyst deactivation is an issue with the primary substituted acids, consistent with a slower than expected rate for the tertiary acid in the competition experiment. Thus, while TFE seems to have a role in accelerating these reactions in the case of the primary alkyl substituted carboxylic acid, its exact role is unclear.
It is also of interest to note the magnitude of fluorescence quenching of the excited state. Although the quenching constants derived from Stern-Volmer experiments indicate a rapid rate of oxidation, the quenching efficiency is very low; for potassium hydrocinnamate (5 mM), only 2% of Mes-Acr-Ph fluorescence is quenched. This reflects that bimolecular quenching is competitive with fast decay of the excited state by fluorescence (kF = 9.3 × 107 s−1 in TFE) and is consistent with the light dependence shown in Figure 4. This shows that even though the rate constant for carboxylate oxidation is very large, it is still possible for oxidation to be turnover limiting in the reaction.
Previous results from our lab indicated that a donor–acceptor complex could exist between ground state acridinium catalyst and alkenes, resulting in a preassociation equilibrium prior to oxidation.70 Thus, it seemed plausible that a ground state Donor–Acceptor complex could form between carboxylates and the ground state Mes-Acr-Ph. Figure 8 shows the absorption spectra of Mes-Acr-Ph (25 μM) before and after the addition of potassium 3-phenyl propanoate (up to 100 mM). After subtraction of the absorbance to the carboxylate, the UV/vis spectrum of the catalyst was unchanged. Therefore, we found no evidence of the formation of a ground state charge-transfer complex.
Figure 8.

UV/vis absorption spectra of the catalyst before and after adding carboxylate salt. The red line shows Mes-Acr-Ph before the addition of carboxylate. The yellow line shows the absorption spectrum of the catalyst after adding the carboxylate. The blue line is the absorption spectrum of the carboxylate and the dashed black line is the subtraction of the carboxylate from the absorption spectrum of the catalyst with added quencher (yellow-blue).
We found it likely that some sort of preassociation complex was formed between the catalyst and the carboxylate salt. 1H NMR spectra of Mes-Acr-Ph in CD3OD show that adding increasing amounts of tetrabutylammonium hydrocinnamate cause an upfield shift in the proton signals of the acridinium, which was found to be linear with respect to carboxylate concentration (Figure 9a and Figure S7). The peaks were also found to broaden out significantly at higher concentrations of carboxylate, potentially indicating a rapid exchange of BF4− counterion with carboxylate counterion. This can also be observed by 19F NMR in which the BF4− counterion can be observed to shift upfield upon the addition of increasing amounts of carboxylate (Figure 9b). Again significant broadening of the signals is observed upon addition of large amounts of carboxylate suggesting an exchange process.
Figure 9.

(a) 1H NMR spectra of Mes-Acr-Ph BF4 [25 mM] in CD3OD. Residual methanol solvent peak was set to 3.31 ppm in each 1H NMR. (b) 19F NMR spectra of Mes-Acr-Ph BF4 [25 mM] in CD3OD. Samples were spiked with 20 μL of TFE before taking 19F NMRs and the corresponding peak was set to −78.82 ppm in each spectrum. TBA+ RCOO− = tetrabutylammonium hydrocinnamate.
Thus, it is likely that a ground state preassociation complex is present between the carboxylate and acridinium catalyst. This could help to explain the slight rate enhancement of primary alkyl substituted substrates over secondary and tertiary alkyl substituted substrates (Figure 7 top). When exploring the scope of the primary carboxylic acids, we found that there were some apparent rate differences among primary carboxylic acids, even in TFE. A competition experiment between tridecanoic acid and hydrocinnamic acid to give alkane products 2a and 2l shows that there is a rate difference of about 4.7:1, in favor of hydrocinnamic acid, at early levels of conversion (see Supporting Information for details). If a preassociation interaction occurs prior to electron transfer, the steric environment around the carboxylate could have an impact on the reaction rate.
CONCLUSION
In summary, we have described a direct organocatalytic protocol for the decarboxylation of carboxylic acids to alkanes, including carboxylic acid substrates previously inaccessible through other methods. We have also extended this method to malonic acid derivatives as we believe this provides an efficient route to the doubly decarboxylated alkyl products. Finally, we have provided insight into the mechanism of this reaction through fluorescence quenching studies, kinetic data, and NMR analysis. We have found that choice of solvent has a major impact on substrate compatibility for this transformation.
Supplementary Material
Acknowledgments
This work was supported in part by Award No. R01 GM098340 from the National Institute of General Medical Sciences, a grant from Eastman Chemical and the David and Lucile Packard Foundation. The authors would like to thank Nathan Romero for his assistance with fluorescence quenching experiments.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b07770.
Experimental procedures and supporting data (PDF) 1H and 13C NMR spectra (PDF)
References
- 1.Rossiter BE, Swingle NM. Chem Rev. 1992;92:771–806. [Google Scholar]
- 2.Alexakis A, Bäckvall JE, Krause N, Pàmies O, Diéguez M. Chem Rev. 2008;108:2796–2823. doi: 10.1021/cr0683515. [DOI] [PubMed] [Google Scholar]
- 3.Kagan HB, Riant O. Chem Rev. 1992;92:1007–1019. [Google Scholar]
- 4.Barton DHR, Dowlatshahi HA, Motherwell WB, Villemin D. J Chem Soc, Chem Commun. 1980:732–733. [Google Scholar]
- 5.Barton DHR, Crich D, Motherwell WB. Tetrahedron Lett. 1983;24:4979–4982. [Google Scholar]
- 6.Barton DHR, Crich D, Motherwell WB. J Chem Soc, Chem Commun. 1983:939–941. [Google Scholar]
- 7.Saraiva MF, Couri MRC, Le Hyaric M, de Almeida MV. Tetrahedron. 2009;65:3563–3572. [Google Scholar]
- 8.Ihara M, Suzuki M, Fukumoto K, Kametani T, Kabuto C. J Am Chem Soc. 1988;110:1963–1964. [Google Scholar]
- 9.Ling T, Poupon E, Rueden EJ, Theodorakis EA. Org Lett. 2002;4:819–822. doi: 10.1021/ol025501z. [DOI] [PubMed] [Google Scholar]
- 10.Ito H, Takeguchi S, Kawagishi T, Iguchi K. Org Lett. 2006;8:4883–4885. doi: 10.1021/ol061947u. [DOI] [PubMed] [Google Scholar]
- 11.Barton DHR, Zard SZ. Pure Appl Chem. 1986;58:675–684. [Google Scholar]
- 12.Fang C, Shanahan CS, Paull DH, Martin SF. Angew Chem, Int Ed. 2012;51:10596–10599. doi: 10.1002/anie.201205274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamaguchi K, Kazuta Y, Abe H, Matsuda A, Shuto S. J Org Chem. 2003;68:9255–9262. doi: 10.1021/jo0302206. [DOI] [PubMed] [Google Scholar]
- 14.Ho J, Zheng J, Meana-Pañeda R, Truhlar DG, Ko EJ, Savage GP, Williams CM, Coote ML, Tsanaktsidis J. J Org Chem. 2013;78:6677–6687. doi: 10.1021/jo400927y. [DOI] [PubMed] [Google Scholar]
- 15.Andrieux CP, Gonzalez F, Savéant JM. J Electroanal Chem. 2001;498:171–180. [Google Scholar]
- 16.Vijh AK, Conway BE. Chem Rev. 1967;67:623–664. [Google Scholar]
- 17.Hilborn JW, Pincock JA. J Am Chem Soc. 1991;113:2683–2686. [Google Scholar]
- 18.Fraind A, Turncliff R, Fox T, Sodano J, Ryzhkov LR. J Phys Org Chem. 2011;24:809–820. [Google Scholar]
- 19.Perkins RJ, Xu HC, Campbell JM, Moeller KD. Beilstein J Org Chem. 2013;9:1630–1636. doi: 10.3762/bjoc.9.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson JM, Kochi JK. J Am Chem Soc. 1970;92:1651–1659. [Google Scholar]
- 21.Minisci F, Bernardi R, Bertini F, Galli R, Perchinummo M. Tetrahedron. 1971;27:3575–3579. [Google Scholar]
- 22.Davidson RS, Steiner PR. J Chem Soc C. 1971:1682–1689. [Google Scholar]
- 23.Yoshimi Y, Itou T, Hatanaka M. Chem Commun. 2007:5244–5246. doi: 10.1039/b714526h. [DOI] [PubMed] [Google Scholar]
- 24.Cassani C, Bergonzini G, Wallentin CJ. Org Lett. 2014;16:4228–4231. doi: 10.1021/ol5019294. [DOI] [PubMed] [Google Scholar]
- 25.Rodríguez N, Goossen LJ. Chem Soc Rev. 2011;40:5030–5048. doi: 10.1039/c1cs15093f. [DOI] [PubMed] [Google Scholar]
- 26.Myers AG, Tanaka D, Mannion MR. J Am Chem Soc. 2002;124:11250–11251. doi: 10.1021/ja027523m. [DOI] [PubMed] [Google Scholar]
- 27.Tanaka D, Myers AG. Org Lett. 2004;6:433–436. doi: 10.1021/ol0363467. [DOI] [PubMed] [Google Scholar]
- 28.Matsubara S, Yokota Y, Oshima K. Org Lett. 2004;6:2071–2073. doi: 10.1021/ol0492602. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka D, Romeril SP, Myers AG. J Am Chem Soc. 2005;127:10323–10333. doi: 10.1021/ja052099l. [DOI] [PubMed] [Google Scholar]
- 30.Dickstein JS, Mulrooney CA, O’Brien EM, Morgan BJ, Kozlowski MC. Org Lett. 2007;9:2441–2444. doi: 10.1021/ol070749f. [DOI] [PubMed] [Google Scholar]
- 31.Cornella J, Righi M, Larrosa I. Angew Chem, Int Ed. 2011;50:9429–9432. doi: 10.1002/anie.201103720. [DOI] [PubMed] [Google Scholar]
- 32.Gooßen LJ, Thiel WR, Rodríguez N, Linder C, Melzer B. Adv Synth Catal. 2007;349:2241–2246. [Google Scholar]
- 33.Gooßen LJ, Rodríguez N, Linder C, Lange PP, Fromm A. ChemCatChem. 2010;2:430–442. [Google Scholar]
- 34.Gooßen LJ, Deng G, Levy LM. Science. 2006;313:662–664. doi: 10.1126/science.1128684. [DOI] [PubMed] [Google Scholar]
- 35.Kolarovič A, Fáberová Z. J Org Chem. 2009;74:7199–7202. doi: 10.1021/jo901377b. [DOI] [PubMed] [Google Scholar]
- 36.Gooßen LJ, Linder C, Rodríguez N, Lange PP, Fromm A. Chem Commun. 2009:7173–7175. doi: 10.1039/b912509d. [DOI] [PubMed] [Google Scholar]
- 37.Wang PF, Wang XQ, Dai JJ, Feng YS, Xu HJ. Org Lett. 2014;16:4586–4589. doi: 10.1021/ol502144c. [DOI] [PubMed] [Google Scholar]
- 38.Sun ZM, Zhang J, Zhao P. Org Lett. 2010;12:992–995. doi: 10.1021/ol100001b. [DOI] [PubMed] [Google Scholar]
- 39.Itou T, Yoshimi Y, Morita T, Tokunaga Y, Hatanaka M. Tetrahedron. 2009;65:263–269. [Google Scholar]
- 40.Kachkovskyi G, Faderl C, Reiser O. Adv Synth Catal. 2013;355:2240–2248. [Google Scholar]
- 41.Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC. Science. 2014;345:437–440. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zuo Z, MacMillan DWC. J Am Chem Soc. 2014;136:5257–5260. doi: 10.1021/ja501621q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okada K, Okamoto K, Morita N, Okubo K, Oda M. J Am Chem Soc. 1991;113:9401–9402. [Google Scholar]
- 44.Yoshimi Y, Masuda M, Mizunashi T, Nishikawa K, Maeda K, Koshida N, Itou T, Morita T, Hatanaka M. Org Lett. 2009;11:4652–4655. doi: 10.1021/ol9019277. [DOI] [PubMed] [Google Scholar]
- 45.Schnermann MJ, Overman LE. Angew Chem, Int Ed. 2012;51:9576–9580. doi: 10.1002/anie.201204977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lackner GL, Quasdorf KW, Overman LE. J Am Chem Soc. 2013;135:15342–15345. doi: 10.1021/ja408971t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshimi Y, Washida S, Okita Y, Nishikawa K, Maeda K, Hayashi S, Morita T. Tetrahedron Lett. 2013;54:4324–4326. [Google Scholar]
- 48.Chu L, Ohta C, Zuo Z, MacMillan DWC. J Am Chem Soc. 2014;136:10886–10889. doi: 10.1021/ja505964r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Noble A, MacMillan DWC. J Am Chem Soc. 2014;136:11602–11605. doi: 10.1021/ja506094d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Noble A, McCarver SJ, MacMillan DWC. J Am Chem Soc. 2015;137:624–627. doi: 10.1021/ja511913h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rueda-Becerril M, Mahé O, Drouin M, Majewski MB, West JG, Wolf MO, Sammis GM, Paquin JF. J Am Chem Soc. 2014;136:2637–2641. doi: 10.1021/ja412083f. [DOI] [PubMed] [Google Scholar]
- 52.Ventre S, Petronijevic FR, MacMillan DWC. J Am Chem Soc. 2015;137:5654–5657. doi: 10.1021/jacs.5b02244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu X, Meng C, Yuan X, Jia X, Qian X, Ye J. Chem Commun. 2015;51:11864–11867. doi: 10.1039/c5cc04527d. [DOI] [PubMed] [Google Scholar]
- 54.Chu L, Lipshultz JM, MacMillan DWC. Angew Chem, Int Ed. 2015;54:7929–7933. doi: 10.1002/anie.201501908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jung ME, Piizzi G. Chem Rev. 2005;105:1735–1766. doi: 10.1021/cr940337h. [DOI] [PubMed] [Google Scholar]
- 56.Fukuzumi S, Kotani H, Ohkubo K, Ogo S, Tkachenko NV, Lemmetyinen H. J Am Chem Soc. 2004;126:1600–1601. doi: 10.1021/ja038656q. [DOI] [PubMed] [Google Scholar]
- 57.Hamilton DS, Nicewicz DA. J Am Chem Soc. 2012;134:18577–18580. doi: 10.1021/ja309635w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grandjean JMM, Nicewicz DA. Angew Chem, Int Ed. 2013;52:3967–3971. doi: 10.1002/anie.201210111. [DOI] [PubMed] [Google Scholar]
- 59.Nguyen TM, Nicewicz DA. J Am Chem Soc. 2013;135:9588–9591. doi: 10.1021/ja4031616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perkowski AJ, Nicewicz DA. J Am Chem Soc. 2013;135:10334–10337. doi: 10.1021/ja4057294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilger DJ, Gesmundo NJ, Nicewicz DA. Chem Sci. 2013;4:3160–3165. [Google Scholar]
- 62.Nicewicz DA, Nguyen TM. ACS Catal. 2014;4:355–360. [Google Scholar]
- 63.Nguyen TM, Manohar N, Nicewicz DA. Angew Chem, Int Ed. 2014;53:6198–6201. doi: 10.1002/anie.201402443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wilger DJ, Grandjean JMM, Lammert TR, Nicewicz DA. Nat Chem. 2014;6:720–726. doi: 10.1038/nchem.2000. [DOI] [PubMed] [Google Scholar]
- 65.Zeller MA, Riener M, Nicewicz DA. Org Lett. 2014;16:4810–4813. doi: 10.1021/ol5022993. [DOI] [PubMed] [Google Scholar]
- 66.Galicia M, González FJ. J Electrochem Soc. 2002;149:D46–D50. [Google Scholar]
- 67.Miguel ELM, Silva PL, Pliego JR. J Phys Chem B. 2014;118:5730–5739. doi: 10.1021/jp501379p. [DOI] [PubMed] [Google Scholar]
- 68.Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Narayanam JMR, Tucker JW, Stephenson CRJ. J Am Chem Soc. 2009;131:8756–8757. doi: 10.1021/ja9033582. [DOI] [PubMed] [Google Scholar]
- 70.Romero NA, Nicewicz DA. J Am Chem Soc. 2014;136:17024–17035. doi: 10.1021/ja506228u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akerlof G. J Am Chem Soc. 1932;54:4125–4139. [Google Scholar]
- 72.Hong DP, Hoshino M, Kuboi R, Goto Y. J Am Chem Soc. 1999;121:8427–8433. [Google Scholar]
- 73.Schadt FL, Bentley TW, Schleyer PvR. J Am Chem Soc. 1976;98:7667–7675. [Google Scholar]
- 74.Suga K, Ohkubo K, Fukuzumi S. J Phys Chem A. 2006;110:3860–3867. doi: 10.1021/jp056637s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.