

Abstract
Hyperglycemia (HG) induces genome-wide cytosine demethylation. Our previous work recognized miR-200b as a critical angiomiR, which must be transiently downregulated to initiate wound angiogenesis. Under HG, miR-200b downregulation is not responsive to injury. Here, we demonstrate that HG may drive vasculopathy by epigenetic modification of a miR promoter. In human microvascular endothelial cells (HMECs), HG also lowered DNA methyltransferases (DNMT-1 and DNMT-3A) and compromised endothelial function as manifested by diminished endothelial nitric oxide (eNOS), lowered LDL uptake, impaired Matrigel tube formation, lower NO production, and compromised VE-cadherin expression. Bisulfite-sequencing documented HG-induced miR-200b promoter hypomethylation in HMECs and diabetic wound-site endothelial cells. In HMECs, HG compromised endothelial function. Methyl donor S-adenosyl-L-methionine (SAM) corrected miR-200b promoter hypomethylaton and rescued endothelial function. In vivo, wound-site administration of SAM to diabetic mice improved wound perfusion by limiting the pathogenic rise of miR-200b. Quantitative stable isotope labeling by amino acids in cell culture (SILAC) proteomics and ingenuity pathway analysis identified HG-induced proteins and principal clusters in HMECs sensitive to the genetic inhibition of miR-200b. This work presents the first evidence of the miR-200b promoter methylation as a critical determinant of diabetic wound angiogenesis.
Keywords: miR-200b, DNA methylation, wound, diabetic vasculopathy
Graphical Abstract
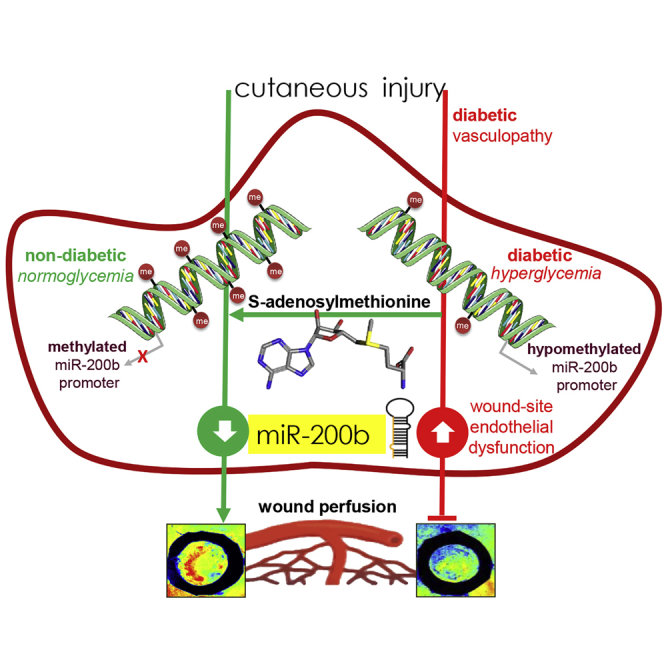
Sen and colleagues demonstrate the critical role of hyperglycemia-induced hypomethylation of the miR-200b gene promoter in causing endothelial dysfunction and impairing diabetic wound angiogenesis. Interestingly, local wound-site administration of the methyl donor S-adenosylmethionine, a common dietary supplement, may correct such hypomethylation, restoring wound responsiveness to wound-edge miR-200b and subsequent angiogenesis in diabetics.
Introduction
Hyperglycemia (HG) is known to induce specific genome-wide cytosine demethylation.1 Catalyzed by the ten-eleven translocation (TET) family of enzymes, such demethylation involves poly (ADP-ribose) polymerase (PARP)-dependent hydroxylation and oxidation of 5-methylcytosine (5mc) to 5-formylcytosine (5fc).1 HG-induced gene demethylation is implicated in the development of insulin resistance and related secondary complications like vasculopathy.2 Reactive aldehydes, such as methylglyoxal, a common by-product of HG, are also capable of driving the chemistry of epigenetics by removing methyl residues from the 5mc of CpG dinucleotides.3 Such modifications may regulate gene transcription by interfering with transcription factor binding or by changing chromatin conformation.4
Although the notion of epigenetic modification as a powerful mechanism to modify gene function originated in the context of coding genes, emergent works recognize epigenetics as a major mechanism to regulate the function of non-coding small genes, such as microRNA (miRNA).5, 6 For example, DNA methyltransferase 3a (DNMT3A)-dependent DNA methylation of miR-143 promoter in vascular smooth muscle cells enhances cell proliferation and is directly implicated in atherogenesis7. Promoter methylation of miRs is being increasingly evident in the current literature.8, 9 In 2011, our work was the first to recognize miR-200b as a critical angiomiR, the expression of which must be transiently downregulated to initiate angiogenesis.10 Subsequent work from our and other laboratories support that notion upholding miR-200b as a critical regulator of inducible angiogenesis.11, 12, 13 Importantly, it has been noted that under conditions of diabetes miR-200b downregulation is not responsive to injury posing barrier to wound angiogenesis. Patient-based studies revealed elevated levels of miR-200b under conditions of diabetes.14 In this work, we present the first evidence demonstrating that HG may drive diabetic vasculopathy by epigenetic modification of a miR promoter. We test the hypothesis that HG-induced hypomethylation in miR-200b promoter causes pathologic upregulation of miR-200b in diabetic tissues such that wound angiogenesis is impaired. Remethylation of the promoter under diabetic conditions rescued wound angiogenesis.
Results
HG-Induced miRNA-200b Elevation Causes Endothelial Cell Dysfunction
Exposure of microvascular endothelial cells (HMECs) to HG (25 mM glucose) led to consistent increase in miR-200b expression (Figures 1A and S1A). The expression of other miRNA, miR-200c and miR-429, sharing the same seed sequence remain unchanged pointing toward a specific effect of HG on miR-200b expression (Figures S1B and S1C). Instability of miR-200c and miR-429, compared to that of miR-200b, may explain the contrast in finding (Figure S1D). Induction of miR-200b was also achieved following exposure to methylglyoxal, a dicarbonyl metabolite of glucose and precursor of advance glycation end (AGE) products formation (Figure 1B). HG is known to cause endothelial dysfunction.15 Consistently, exposure of HMEC to hyperglycemic insult downregulated VEGF expression, basal nitrite/nitrate levels, endothelial nitric oxide (eNOS) expression, Matrigel tube formation, uptake of acetylated low-density lipoprotein (LDL), vascular E-cadherin expression as well as Von Willebrand factor (vWF) expression (Figures 1C–1L and S1E–S1H). Taken together, these parameters represent a comprehensive assessment of endothelial dysfunction.16 We utilized this experimental setting to test the functional significance of miR-200b induction as caused by HG and related biochemical metabolites. Of outstanding interest was the observation that successful blunting of miR-200b induction by HG using miR-200b inhibitor rescued HMEC from HG-induced dysfunction as evaluated by the above-mentioned parameters (Figures 1C–1L and S1E–S1H).
Figure 1.

Hyperglycemia-Induced MicroRNA-200b Elevation Causes Endothelial Cell Dysfunction
(A) qRT-PCR analysis of miR-200b expression in HMECs exposed to high D-glucose (HG) (25 mM, 4 days); osmotic control (L-glucose, LG); or cultured in normal glucose (NG) conditions. n = 3, *p < 0.05, F = 12.04 (one-way ANOVA). (B) qRT-PCR analysis of miR-200b levels in HMECs challenged with methylglyoxal (MG, 500 μM, 4 days). n = 3, *p < 0.05 (Student’s t test). (C) qRT-PCR analysis of VEGF expression under NG/HG and co-treatment with miRIDIAN microRNA hsa-miR-200b-3p hairpin inhibitor (MI) or miRIDIAN microRNA hairpin inhibitor negative control (control inhibitor, CI) (100 nM, 48 hr). n = 3, *p < 0.001, F = 22.17 (one-way ANOVA). (D) Total nitrate/nitrite production in HMECs under NG/HG and co-treatment with control (CI) or miR-200b inhibitor (MI). n = 4, *p < 0.05, F = 9.03 (one-way ANOVA). (E and F) Immunocytochemical analysis of eNOS production (E) and its quantitation (F) in HMECs under NG/HG and co-treatment with control (CI) or miR-200b inhibitor (MI). Scale bar, 20 μm. n = 5, *p < 0.001, F = 27.22 (one-way ANOVA). (G and H) Matrigel tube formation (G) and tube length analysis (H) in HMECs in NG/HG and co-treatment with control (CI) or miR-200b inhibitor (MI). Scale bar, 100 μm. n = 4, *p < 0.05, F = 22.24 (one-way ANOVA). (I and J) Ac-LDL uptake assay in HMECs (I) and intensity quantitation (J) under NG/HG and co-treatment with control (CI) or miR-200b inhibitor (MI). Scale bar, 100 μm. n = 6, *p < 0.05, F = 5.86 (one-way ANOVA). (K and L) Immunofluorescence staining of VE-cadherin (green) (K) and intensity quantitation (L) in HMECs under NG/HG and co-treatment with control (CI) or miR-200b inhibitor (MI). Scale bar, 100 μm. n = 4, *p < 0.05, F = 12.94 (one-way ANOVA). Data are represented as the mean ± SD. HMEC, human microvascular endothelial cells; LDL, low density lipoprotein; AU, arbitrary unit.
HG-Induced Promoter Hypomethylation Elevates Endothelial miRNA-200b
The expression of miR-200 family genes is known to be regulated by the methylation status of the genome.17, 18 Treatment of endothelial cells with global DNA demethylation agent, 5-Aza-2′-deoxycytidine (d-aza), resulted in significant increase in the levels of miR-200b, miR-200c, and miR-429 during normoglycemic as well as hyperglycemic conditions (Figures S2A–S2C). In pursuit to delineate the mechanism underlying HG-induced elevation of miR-200b (Figure 1A) our attention was turned toward promoter methylation status of the miR. Two CpG islands in miR-200b promoter, consisting of total 31 CpGs, were analyzed using bisulfite sequencing (Figure 2A). In addition, as reference, the promoter methylation status of miR-200c promoter was analyzed. HG resulted in marked hypomethylation of the miR-200b promoter (Figures 2A and 2B). Hypomethylation of miR-200b was also evident following exposure to methylglyoxal (Figures S2D and S2E). The reported promoter hypomethylation in response to HG was specific for miR-200b because such insult did not influence the promoter methylation status of miR-200c (Figures S2F and S2G). Efforts to determine the mechanistic basis of such hypomethylation led to the observation that HG downregulated the abundance of DNA methyltransferase 1 and 3A in HMECs (Figures S2H and S2I). Interestingly, such downregulation of DNMT1 as well as DNMT3A in response to HG could be prevented by miR-200b inhibition (Figures S2J and S2K). Indeed, established computational algorithms predict DNMT3A to be reasonable target of miR-200b (Figure S2L). Supplementation of HMEC with a methyl group donor S-adenosylmethionine (SAM) during hyperglycemic exposure not only brought the levels of miR-200b down by correcting promoter methylation but also rescued from endothelial dysfunction (Figures 2D–2G). SAM treatment of hyperglycemic HMEC corrected basal nitrite/nitrate levels, restored Matrigel tube formation, increased Ac-LDL uptake, as well as increased VE-cadherin, vWF, eNOS, and VEGF expression (Figures 2E–2G and S2M–S2P).
Figure 2.

Hyperglycemia-Induced Promoter Hypomethylation Elevates Endothelial MicroRNA-200b
(A) Schematic diagram showing the regions (1, 2) of miR-200b promoter analyzed through bisulfite genomic sequencing of DNA. Methylation profile of the miR-200b promoter in HMECs cultured in normal glucose (NG), exposed to high glucose (HG) and HG supplemented with S-adenosylmethionine (SAM) (80 μM, 48 hr) conditions (methylated CpG, black; unmethylated CpG, white). Number of clones = 5. (B) Total number of methylated CpG sites obtained from bisulfite sequencing analysis. n = 5, *p < 0.05 (Student’s t test). (C) qRT-PCR analysis of miR-200b expression in HMECs treated with SAM under HG condition (80 μM, 48 hr). n = 3, *p < 0.05 (Student’s t test). (D) Total number of methylated CpG sites obtained from bisulfite sequencing analysis in HG condition with or without SAM administration. n = 5. (E) miR-200b promoter luciferase activity was measured by calculating the GLUC/SEAP ratio in NG- or HG-exposed HMECs with or without SAM. n = 4, *p < 0.05, F = 10.80 (one-way ANOVA). (F) Endothelial function analysis: matrigel tube length (upper panel), ac-LDL uptake (middle panel), and VE-cadherin expression (lower panel) after SAM treatment in HG condition. n = 5, *p < 0.05 (Student’s t test). (G) Immunofluorescence staining of vWF in SAM-treated HMECs in HG condition. n = 5, *p < 0.001 (Student’s t test). Data represented as the mean ± SD.
Promoter Hypomethylation Renders miRNA-200b Non-responsive to Injury in Diabetic Wounds
Injury-induced transient repression of miR-200b, critical for initiating wound angiogenesis, is compromised in murine diabetic wounds.13 Endothelial tissue elements laser captured from wound-edge of chronic wound patients were subjected to DNA isolation followed by bisulfite sequencing (Figures 3A and 3B). Quantification of 31 CpGs analyzed in the miR-200b promoter revealed significant hypomethylation in diabetics compared to that of patients without diabetes (Figures 3C and 3D). Hypomethylated miR-200b promoter in diabetics was associated with elevated miR-200b expression (Figure 3E). This finding is consistent with our in vitro evidence establishing that the state of promoter methylation directly regulates miR-200b expression (Figures 1A and 2B). As reference, the study of miR-200c expression identified that the state of promoter methylation remains unchanged in diabetes for this non-coding gene sharing the same seed sequence with miR-200b (Figures 3F–3H). Wound fluid from diabetics, but not from non-diabetics, elevated miR-200b expression in HMECs (Figures 3I–3K). This finding supports the notion that the trigger for promoter hypomethylation and subsequent miR-200b elevation is derived from the wound microenvironment.
Figure 3.

Promoter Hypomethylation Renders MicroRNA-200b Non-responsive to Injury in Diabetic Wounds
(A) Schematic diagram showing experimental design of miR-200b promoter methylation analysis in human chronic wounds. (B) Representative figure shows the selection of CD31+ tissue elements (red) and their collection before and after the laser capture microdissection (LCM). Scale bar, 150 μm. (C and D) Methylation profile (C) and quantitation of methylated CpG islands in miR-200b promoter (D) in diabetic wounds compared to normoglycemic wounds. N = 5, *p < 0.001 (Student’s t test). (E) qRT-PCR analysis of miR-200b levels in LCM captured endothelial elements from diabetic and non-diabetic wounds. n = 3, *p < 0.05 (Student’s t test). (F) Schematic diagram showing the region of miR-200c promoter analyzed by bisulfite genomic sequencing. Methylation profile of the miR-200c gene promoter in LCM captured endothelial elements from diabetic and non-diabetic wounds with methylated CpGs shown in black. (G) Quantitation of methylated CpG islands in miR-200c promoter in diabetic wounds compared to normoglycemic wounds. n = 5, p = NS (Student’s t test). (H) qRT-PCR analysis of miR-200c expression in diabetic wounds compared to normoglycemic wounds. n = 4, p = NS (Student’s t test). (I) qRT-PCR analysis of miR-200b levels in HMECs after exposure to wound fluid (10%, 4 days). n = 3, *p < 0.05 (Student’s t test). (J and K) qRT-PCR analysis of VEGFR-2 (J) and vWF (K) in HMECs after exposure to wound fluid. n = 3, p = NS (Student’s t test). Data represented as the mean ± SD.
SAM Reversed Diabetes-Associated Impairment in Wound Vascularization
SAM, an established methyl donor, was tested for its ability to rescue HG-induced hypomethylation of miR-200b promoter.19 In HMECs challenged with HG, post-treatment with SAM after 48 hr of HG rescued miR-200b from elevated expression (Figure 2C). In vivo intradermal administration of SAM (Figure 4A) to the wound-edge of diabetic (db/db) mice significantly lowered abundance of miR-200b in endothelial tissue elements (Figures 4B and 4C). This finding, taken together with corresponding in vitro studies, point toward the efficacy of SAM as a productive countermeasure for HG-induced elevation of miR-200b. The favorable effect of SAM on wound-edge endothelial miR-200b was functionally manifested as improved wound perfusion (Figure 4D). Wound closure causes regression of wound angiogenesis.20 Such physiological regression was intact following SAM treatment (Figure 4D). Thus, SAM improved diabetic wound perfusion and allowed physiological regression of angiogenesis in a timely manner. The density of wound-edge vasculature was quantified using angiogenic markers CD31 and eNOS (Figure 4E). Analysis of co-localization demonstrated higher prevalence of CD31+/eNOS+ vasculature following SAM treatment (Figure 4F). This observation was consistent with SAM-dependent restoration of endothelial function as marked by increased vWF and smooth muscle actin (SMA) (Figures 4G, 4H, S3A, and S3B). Vascular functionality was further tested by the quantitative assessment of pulsatile blood flow using color Doppler (Figures S3C and S3D). Taken together, the therapeutic methyl donor SAM was effective in restoration of vascular function in diabetic wounds. Compromised wound angiogenesis is recognized as a key limiting factor in diabetic wound closure.21 SAM improved diabetic wound re-epithelialization and closure (Figures S3E–S3H).
Figure 4.

SAM Reversed Diabetes-Associated Impairment in Wound Vascularization
(A) Schematic diagram showing intradermal delivery of S-adenosylmethionine (SAM) or vehicle to the dorsal skin of db/db mice. Two 8-mm diameter full thickness stent wounds were created on dorsal skin on day 0. (B) Representative figure shows the selection of CD31+ tissue elements (red), and their collection before and after the LCM from the wound edge tissues. Scale bar, 150 μm. (C) qRT-PCR analysis of miR-200b levels in endothelial elements of SAM-treated diabetic wounds compared to placebo. n = 3, *p < 0.05 (Student’s t test). (D) Perimed Laser speckle-assisted wound perfusion analysis of diabetic wounds administered with SAM or placebo. n = 5, *p < 0.05 (Student’s t test). (E and F) Immunohistochemical analysis of eNOS+/CD31+ co-expression (E) and its co-localization analysis (F) in SAM- or PBS-administered diabetic wounds. (G and H) Immunohistochemical analysis of vWF+/CD31+ co-expression (G) and its co-localization analysis (H) in SAM- or PBS-administered diabetic wounds. n = 5, *p < 0.05 (Student’s t test). Data represented as the mean ± SD.
Quantitative Proteomics Identifying Novel Targets of miRNA-200b during HG
Epigenetic dysregulation of miR-200b under conditions of HG emerged as a significant contributor to endothelial dysfunction. We therefore sought to elucidate the significance of miR-200b inhibition on the hyperglycemic endothelial cell proteome. The objective was to identify candidate pathways that are miR-200b sensitive in the hyperglycaemic HMECs. Stable isotope labeling by amino acids in cell culture (SILAC) was employed to detect proteome-wide changes following miR-200b inhibition in hyperglycemic endothelial cells.22 HMEC were grown in light (12C6,14N4 L-Arginine + 12C6,14N2 L-lysine) and heavy media (13C6, 15N4 L-Arginine + 13C6, 15N2 L-Lysine) for six passages until there was 100% incorporation of heavy isotope labeling in heavy cells as detected by mass spectrometry (Figures 5A and S4A). Heavy isotope-labeled cells were grown in hyperglycemic media alone or in hyperglycemic media supplemented with miR-200b inhibitor. Appropriate normoglycemic controls cultured in light media were used for profiling basal protein levels (Figure 5A). Differential protein ratio was calculated using unique peptide intensity between heavy and light isotope labeled peptides using 2D LC-MS/MS. A total of 3,818 proteins were detected using SILAC, and, from this total, 319 were upregulated and 30 were downregulated in response to miR-200b inhibition during HG (Figure 5B; Tables S1, S2, and S3). Majority of the proteins detected were unchanged being expressed within the ± 1 range of log ratios for both the conditions (Figure S4B). Gene ontology analysis of upregulated proteins using PANTHER identified specific set of proteins involved in nucleic acid binding (Figure S4C). Chromatin binding and histone family proteins were over-represented in endothelial cells subjected to miR-200b inhibition (Figure S4C). A representative mass spectra of peptides FEDEELQQILDDIQTK (m/z = 982.47622+) and FEDEELQQILDDIQTK (13C(6)15N(2)) (m/z = 986.48262+) is shown in Figure 5C. This corresponds to DNA-directed RNA polymerase II subunit RPB4 (accession: O15514), a protein involved in guiding DNA methylation. RPB4 showed higher absolute ratio in HG plus miR-200b inhibitor compared to HG alone.23 Ingenuity pathway analysis (IPA) (Ingenuity Systems, http://www.ingenuity.com) was conducted to identify specific biological pathways altered by miR-200b inhibition during hyperglycemic insult (Figures 5D and 5E). Therapeutic miR-200b inhibition targeted proteins involved in rescue of mitochondrial dysfunction (p = 1.65 × 10−5) and in fatty acid metabolism (p = 1.43 × 10−5; Table S4). Cluster of genes upregulated in response to miR-200b inhibition under conditions of HG are shown in Figure 5D. Dominant pathways in the resultant network were represented by RAB proteins, histones, ATP generation and heat shock proteins (Tables S5 and S6). Cluster of genes downregulated in response to miR-200b inhibition under conditions of HG are shown in Figure S4D and Table S7. Dominant downregulated clusters were represented by interleukin, interferon and STAT1 pathways. In addition to this, SILAC analysis for target identification demonstrated that the inhibition of miR-200b rescue HG-induced endothelial dysfunction. This was enacted by a number of functionally clustered proteins directed at improving wound tissue vascularization. Ras-related small guanosine triphosphate (GTP)-binding Rab proteins regulate VEGFR2 trafficking and signaling linked to endothelial cell migration.24 Induction of Rab3A and Rab7A following miR-200b inhibition during HG marks the rescue of endothelial function. Elevated levels of hnRNPs and HSPs also supported the notion that miR-200b inhibition during HG enables the vascular functionality (Figure S4H).
Figure 5.

Quantitative Proteomics Identify Novel Targets of MicroRNA-200b during Hyperglycemia
(A) Schematic diagram showing SILAC quantitative proteomics workflow. (B) Total number of identified (up) and differentially expressed (down) unique proteins by SILAC analysis. (C) Respective mass spectra of peptides FEDEELQQILDDIQTK (m/z = 982.47622+) and FEDEELQQILDDIQTK (13C(6)15N(2)) (m/z = 986.48262+); the ratio between heavy isotopic-labeled peptide and light isotopic-labeled peptide represents the differential expression of the peptide (protein) in two different conditions. (D and E) Ingenuity pathway analysis (IPA) showing the inhibition of miR-200b during HG-exposed HMEC target proteins like core histones and heat shock protein (D) and in mitochondrial dysfunction and fatty acid metabolism pathways (E). (F) Oxygen consumption rate measurement in HMECs under NG/HG and co-treatment with control (CI) or miR-200b inhibitor (MI). n = 5, *p < 0.05, F = 9.30 (one-way ANOVA). (G) Normalized ATP production in HMECs under NG/HG and co-treatment with control (CI) or miR-200b inhibitor (MI). n = 3, *p < 0.001, F = 260.90 (one-way ANOVA). Data represented as the mean ± SD.
TargetScan (http://www.targetscan.org) and miRnalyze (http://www.mirnalyze.in) were utilized to predict potential targets of miR-200b. Such predicted outcomes were compared with findings of proteomic data. ATP synthase, heat shock protein 40 (HSP40), HSP70, Talin-2, and others listed in Table S8 emerged as novel candidate proteins that were backed by SILAC quantitative proteomics and supported by established predictive computational algorithms. Furthermore, STRING (search tool for the retrieval of interacting genes/proteins; http://string-db.org/) was used to predict interaction between detected candidate proteins. In the network of upregulated proteins, based on PFAM domains, proteins of the following clusters significantly interacted with each other: RAS signaling (FDR = 1.06 × 10−15), core histones (FDR = 6.57 × 10−15), RNA recognition motifs (FDR = 1.67 × 10−5), intermediate filament proteins (FDR = 6.29 × 10−5), and HSP family (FDR = 0.000429) (Figure S4H; Table S4).
Rescue of mitochondrial function under conditions of HG emerged as a top candidate pathway that was brought into effect by miR-200b inhibition as detected by SILAC (Table S5). Thus, HMEC cultured under hyperglycemic condition with or without miR-200b inhibition were subjected to mitochondrial function analyses (Seahorse Biosciences, Santa Clara, CA, USA). Hyperglycemic insult caused endothelial mitochondrial dysfunction. Inhibition of miR-200b under conditions of HG rescued mitochondrial function as indicated by increased oxygen consumption rate (OCR) and restored ATP production (Figures 5F and 5G). Consistently, miR-200b inhibition lowered cytochrome C release (Figure S4E). Independent studies using JC-1 to detect mitochondrial transmembrane potential revealed loss of potential caused by hyperglycemic insult. Inhibition of miR-200b rescued mitochondrial transmembrane potential (Figures S4F and S4G). Taken together, findings of this study are consistent with the literature in demonstrating that HG compromises mitochondrial function. Inhibition of miR-200b under hyperglycemic conditions rescued mitochondrial function lending credence to our SILAC approach aimed at detecting novel targets of miR-200b during HG.
Discussion
Persistent HG caused by metabolic dysregulation is a critical factor underlying the pathogenesis of micro- and macrovascular complications in type 2 diabetes mellitus (T2DM). Chronic HG influences the epigenome through aberrant DNA methylation, covalent chromatin modifications and editing of noncoding regulatory RNA.2 miRNAs have been known to control vascular function.25 Under the direction of specific signals, these miRNAs control endothelial specific gene expression and thus fine-tune endothelial cell physiology.26 Classical examples of such regulation include, but are not limited to, miR-126 controlling expression of VEGF,27 the let-7 family promoting endothelial cell proliferation,28 and miR-210-mediated control of endothelial cell tube formation.29 Our work, supported by independent validation, recognize the role of miR-200b as a switch for induction of angiogenesis in adults.10, 11, 12, 13The miR-200 family of non-coding genes is organized into two groups based on the differences in their seed sequence. Group A consists of miR-141 and -200a, whereas group B includes miR-200b, -200c, and -429. Our previous work recognized miR-200b as an angiomiR.10 Transient repression of miR-200b, such as that induced by injury, is necessary to initiate inducible adult wound angiogenesis.30 Such repression drives endothelial cell migration and induces endothelial growth factors.10 Diabetic conditions upregulate miR-200b such that it is more abundant than that in non-diabetic tissue. Additionally, injury inducible repression of miR-200b is lost under hyperglycemic conditions.13 Reduction of wound site miR-200b in diabetic wounds rescue the endothelial cell phenotype marked by increased endothelial cell abundance and increased blood flow.13 In trying to explain the anomalous behavior of miR-200b under diabetic conditions, it is important to recognize that the expression of miR-200b is subject to epigenetic alterations of its promoter.18, 31 For example, promoter methylation status of miR-200b is known to determine the tumor outcome in humans.17 The hypo-methylated promoter elevated miR-200b expression, thereby limiting its mesenchymal properties in that neoplastic context.32 Findings of this work present that first evidence demonstrating direct control of adult angiogenesis by epigenetic modification of a miR promoter. That HG-induced hypomethylation may induce miR-200b causing endothelial dysfunction thereby posing a barrier toward diabetic wound angiogenesis establishes a new paradigm in the pathogenesis of diabetic vasculopathy.
S-adenosylmethionine (SAM, also known as AdoMet and SAMe in the pharmacy) is a common cofactor for methyltransferase.33 During the process of transmethylation, the methyl group of SAM is transferred to an array of acceptor molecules like CpG sites of DNA, phospholipids, and proteins.33 Apart from its principal role as biological methyl donor, SAM also acts as precursor of glutathione (GSH) through its conversion to cysteine via the trans-sulfuration pathway in the liver.34 In addition to methylating DNA non-enzymatically, SAM is also known to inhibit active DNA demethylation thereby protecting against global hypomethylation.19, 35 In cancer therapeutics, SAM is commonly used to influence genes subject to regulation by DNA methylation.36, 37 Recent reports underscore the significance of SAM as intervention to prevent endothelial dysfunction. In rats fed high-fat diet, SAM administration not only corrected the impaired endothelium-dependent vascular relaxation but also lowered serum triglycerides and free fatty acids.38 In human aortic endothelial cells, SAM prevented endothelial dysfunction by inducing heme oxygenease.38 In diabetic patients, high plasma SAM levels was associated with healthy endothelial functioning.39 Findings of the current study is the first to demonstrate that HG-dependent hypomethylation of miR promoter may be reversed by SAM. Such reversal rescues diabetic wound angiogenesis improving wound perfusion.
Post-transcriptional gene silencing caused by any individual miR typically focuses on the limited number of coding genes that any given miR silences in any given cell type. This approach employs predictive computational algorithms, e.g., TargetScan and miRanda, followed by systematic validation of those targets. While this approach is powerful in identifying the immediate miR-dependent mechanism, it is limited in its ability to assess the global functional significance of that miR. Such assessment must consider the impact on all target coding genes as well as the ripple effect of such gene silencing throughout the cell biology system. Quantitative SILAC proteomics is an emergent powerful tool that has been productively employed to assess the system-wide effects of miR inhibition in several cell lines. Among the 319 proteins upregulated in response to miR-200b inhibition during HG, primary clusters were represented by Rab proteins (Ras family), core histones (involved in the regulation and organization of nucleosome), heterogeneous nuclear ribonucleoproteins (hnRNP) (RNA recognition motif family), and the heat shock protein (HSP) family. In response to HG-associated miR-200b inhibition, upregulation of ATP synthases (mitochondrial pathway) and long-chain acyl-CoA synthetases (fatty acid metabolism pathway) was recognized by Ingenuity pathway analyses.
The rescue of HG-induced endothelial dysfunction, as enabled by miR-200b inhibition, was enacted by a number of functionally clustered proteins directed at improving wound tissue vascularization. Ras-related small GTP-binding Rab proteins regulate VEGFR2 trafficking and signaling linked to endothelial cell migration.24 Rab also control the trafficking of glucose transporters (GLUT) to the plasma membrane for insulin mediated glucose uptake.40 Rab3A deletion during HG results in insufficient insulin secretion in vivo.41 Induction of histones drives cell proliferation. Such rescue is necessary under conditions of HG when endothelial cell proliferation is blunted.42 It is plausible that the observed change in histone levels during HG is caused by correction of CpG hypomethylation. Whether miR-200b inhibition may expedite nucleosome assembly and facilitate gene packaging in chromatin remain to be tested. Upregulated hnRNPs are likely to favor angiogenesis by active stabilization of eNOS and elevated VEGF.43, 44 Indeed, forced expression of hnRNPK during experimental HG is known to rescues diabetic vasculopathy.45 Elevated HSP, as observed, is also likely to support wound angiogenesis by promoting endothelial cells migration capillary-like tube formation.46 Hsp90 associates with eNOS in vascular endothelial cells to generate NO.46 HG diminishes Hsp90-eNOS interaction, compromising bioavailability of NO to vascular endothelial cells.46 High Hsp70, as observed, is expected to rescue HG-induced vasculopathy by increasing insulin sensitivity and glucose tolerance.47 HG is known to dampen cellular respiration and bioenergetics.48, 49 Compromised levels of ATP synthases in the hyperglycemic tissue is a significant cause of such dysregulation.50 Elevated ATP5B is therapeutic in decreasing the rate of AGE-induced secondary complications of diabetes.51 In this work, selective inhibition of miR-200b during HG improved mitochondrial ATP5B level. Such effect was associated with increased ATP production and oxygen consumption rate in hyperglycemic endothelial cells reflecting improved cellular bioenergetics.
HG induces proinflammatory cytokines and chemokines, which, in turn, compromise glucose metabolism via limiting insulin sensitivity. Cytokines, such as interferon gamma (IFNγ), interleukin 6 (IL-6), and tumor necrosis factor-α (TNF-α), are directly implicated in the pathogenesis of obesity-linked insulin resistance.52 These cytokines induce pro-inflammatory STAT1 pathways.53 Selective inhibition of STAT1 prevented HG-induced of endothelial dysfunction.54 In this work, inhibition of miR-200b during HG favorably blunted IFN, IL, and LDL signaling as well as lowered STAT1 levels.
In conclusion, our study introduces a new paradigm recognizing HG-induced promoter hypomethylation of miR-200b as a critical event leading to endothelial dysfunction. Wound-site local administration of the DNA methylation agent SAM improved diabetic wound healing by reducing endothelial miR-200b levels and restoring tissue perfusion in a murine model of diabetes.
Materials and Methods
Reagents and Antibodies
All tissue culture materials were either obtained from GIBCO-BRL/Life Technologies (Gaithersburg, MA, USA) or Lonza (Allendale, NJ, USA). miRIDIAN miRNA hairpin inhibitor negative control (cat. no. IN-001005-01-05) and miRIDIAN miRNA hsa-miR-200b-3p hairpin inhibitor (cat. no. IH-300582-08-0005) were purchased from GE Dharmacon (Lafayette, CO, USA). Antibodies against eNOS (cat. no. ab66127), VE-cadherin (cat. no. ab33168), vWF (cat. no. ab6994), and SMA (cat. no. ab5694) were obtained from Abcam (Cambridge, MA, USA). Purified Rat Anti-Mouse CD31 (also known as PECAM-1) (cat. no. 550274) was obtained from BD PharMingen (San Jose, CA, USA). We purchased allophycocyanin (APC) conjugated anti-human CD31 antibody (clone: WM59, cat. no. 303115), BioLegend (San Diego, CA, USA). Anti-mouse β-3-tubulin (cat. no. 4466S) was purchased from Cell Signaling Technology (Danvers, MA, USA). Horseradish peroxidase-conjugated anti-rabbit-IgG (cat. no. NA934V), anti-mouse-IgG (cat. no. NA931V), and Amersham ECL Prime Western Blotting Detection Reagent were procured from GE Healthcare Bio-Sciences (Pittsburgh, PA, USA). Calcein AM (cat. no. C3099) was purchased from Molecular Probes, Thermo Fisher Scientific (Waltham, MA, USA). We procured Corning Matrigel Matrix (cat. no. 354234) from Corning (New York, NY, USA). S-adenosylmethionine (SAM) (cat. no. B9003S) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). U6 small nuclear RNA (snRNA) primer (cat. no. 4427975; ID: 001973), hsa-miR-200b primer (cat. no. 4427975; ID: 002251), hsa-miR-200c primer (cat. no. 4427975; ID: 002300), and hsa-miR-429 primer (cat. no. 4427975; ID: 001024) were obtained from Applied Biosystems (Foster City, CA, USA). EpiQuik Nuclear Extraction Kit II (Nucleic Acid-Free) (cat. no. OP-0022), EpiQuik DNMT1 Assay Kit (cat. no. P-3011-2), and EpiQuik DNMT3A Assay Kit (cat. no. P-3012-2) were obtained from Epigentek Group (Farmingdale, NY, USA). All other chemicals were procured from Sigma-Aldrich.
Cells and Cell Culture
Human dermal HMECs were cultured in MCDB-131 medium (GIBCOTM/Life Technologies, cat. no. 10372-019) as described.13 To induce HG in vitro, the cells were maintained in MCDB-131 medium that was supplemented with 25 mM D-glucose.55
miRNA-200b Inhibitor Transfection
HMECs were seeded (0.1 × 106 cells/well) in a 12-well plate in antibiotic-free medium for 24 hr prior to transfection to achieve ∼70% confluency at the time of transfection. Transfection was performed by liposome-mediated delivery of miRIDIAN miRNA hsa-miR-200b-3p hairpin inhibitor (cat. no. IH-300582-08-0005) or miRIDIAN miRNA hairpin inhibitor negative control (cat. no. IN-001005-01-05) (100 nM) using DharmaFECT transfection reagent (GE Dharmacon) and OptiMEM serum-free medium (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA). Samples for quantification of miRNA, mRNA, or functional assessment were collected after 48 hr of transfection with control and miR-200b inhibitor.56, 57
Human Samples
Wound fluid and biopsy samples were collected from chronic wound patients at the OSU Comprehensive Wound Center (CWC) (Table S9). All human studies were approved by The Ohio State University’s (OSU) Institutional Review Board (IRB). The Declaration of Helsinki protocols were followed, and patients gave their written informed consent.
Animal Studies
Mice homozygous (BKS.Cg-m+/+Leprdb/J or db/db; stock no. 000642) for spontaneous mutation of the leptin receptor (Leprdb) (aged 10–12 weeks) were obtained from Jackson Laboratory (Bar Harbor, ME, USA). All animal studies were performed in accordance with the protocols approved by the Laboratory Animal Care and Use Committee of The Ohio State University. The animals were tagged and randomly assigned to specific experimental groups.
Wound Models
Two 8-mm biopsy punch excisional wounds were created on the dorsal skin, equidistant from the midline and adjacent to the 4 limbs and splinted with a silicon sheet to prevent contraction, thereby allowing wounds to heal through granulation and re-epithelialization.58 During the wounding procedure, mice were anesthetized by low-dose isoflurane inhalation as per standard recommendations. S-adenosyl methionine (SAM) dissolved in PBS (12.5 μg/wound) was intradermally injected near the wound edge 2 days before surgery, on the day of surgery, 3 days post-surgery, and 5 days post-surgery. Intradermal injection was preferred over gavazing based on the previous report that SAM has low bioavailability following oral administration.59 Furthermore, SAM metabolism is disturbed in the case of diabetes mellitus and is associated with hyperhomocysteinemia.60 The dosage of SAM was chosen based on prior reports in which SAM is used for methylation experiments in vitro.61, 62 Wounds from sex- and age-matched animals receiving only a vehicle (PBS) served as controls. Each wound was digitally photographed, and perfusion was checked by laser speckle (PeriCam PSI HR System, PeriMed, Sweden) at the different time points mentioned.63 The wound area was analyzed by the ImageJ software. The animals were euthanized at the indicated time, and wound edges were collected for analyses. The tissues were collected either in 4% paraformaldehyde or in optimal cutting temperature (OCT) compound.
RNA Extraction and Real-Time qPCR
RNA was isolated from cells or the wound edge tissue sample using an miRVana miRNA Isolation Kit (Ambion, Thermo Fisher Scientific, cat. no. AM1560) and according to the manufacturer’s instructions. The RNA quantity was measured using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). For miRNA expression analysis, specific TaqMan assays for miRNAs were performed using a TaqMan miRNA RT Kit (Applied Biosystems, ThermoFisher Scientific, Foster City, CA, USA; cat. no. 4366596), followed by real-time PCR using the Taqman Universal PCR Master Mix (Applied Biosystems, ThermoFisher Scientific, cat. no. 4324018). U6 snRNA was simultaneously amplified in separate reactions and used as a housekeeping control (Applied Biosystems, ThermoFisher Scientific, cat. no. 4324018, cat. no. 4427975). 100 ng of total RNA was initially used for cDNA preparation for each miRNA or housekeeping control. For mRNA expression, cDNA was prepared from 500 ng of total RNA using qScript cDNA SuperMix (Quanta Biosciences, cat. no. 101414) as per the manufacturer’s instructions. SYBR green-based real-time quantitative PCR (real-time qPCR) reactions (Applied Biosystems) and gene-specific primers were used. vWF, forward primer: 5′-CTGGCAGCTGTTCTTATGTCCTATT-3′, reverse primer: 5′-CTCATGCATGATGGCACCATAA-3′; VEGF: forward primer: 5′-TGCCCACTGAGGAG TCCAACAT-3′, reverse primer: 5′-CACGTCTGCGGATCTTGTACAAACA-3′; VEGFR2, forward primer: 5′-AGCGCTGTGAACGCTTGCCT-3′, reverse primer: 5′-CATGAGAGGCCCTCCCG GCT-3′. The specificity of the product was ensured through melt curve analysis. 18S rRNA was simultaneously amplified in separate reactions and used as a housekeeping control. 18S, forward primer: 5′- GTAACCCGTTGAACCCCATT-3′, reverse primer: 5′- CCATCCAATCGGTAGTAGCG-3′. Gene expression was quantified using the delta-delta Ct relative quantization method using 18S as a normalization control and was represented as fold change.
Nitric Oxide Level Determination
Extracellular nitric oxide (NO) release was determined by a Nitric Oxide Calorimetric Assay Kit (BioVision Nitric Oxide Colorimetric Assay Kit [Cat; K262-200]) as recommended. Briefly, culture media collected after treatment was added to each assay well and added with nitrate reductase, enzyme cofactor, and Griess reagents as per the manufacturer’s protocol. Absorbance at 540 nm was recorded as a function of nitrate and/or nitrite concentration.64
LDL Uptake Assay
HMECs were transfected with either control or miR-200b inhibitor in normoglycemic and hyperglycemic media. On day 4 post-HG, cells were incubated with DyLight 550-labeled human LDL (10 μg/ml) at 37°C for 4 hr. HMECs maintained in normoglycemic and hyperglycemic media for 4 days were used as an appropriate control. At the end of incubation, cells were washed in PBS and fixed with 4% paraformaldehyde for 30 min. The uptake of Ac-LDL was analyzed by fluorescence microscopy and quantified using AxioVision Rel (v.4.8) software (Zeiss).
In Vitro Angiogenesis Assay
In vitro angiogenesis was assessed by the tube formation ability on Matrigel as described previously.10, 13 Briefly, HMECs were transfected with control or miR-200b inhibitor in normoglycemic and hyperglycemic media. Following 4 days of hyperglycemic exposure, the cells were seeded on a Matrigel pre-coated 4-well plates at 5 × 104 cells/well. Cells were labeled with 3 μM of calcein AM 15 min at 37°C in 5% CO2. The angiogenic property was assessed by measuring the tube length after 6 hr of cell seeding using fluorescence microscopy and AxioVision Rel (v.4.8) software (Zeiss).65
Immunocytochemistry, Immunohistochemistry, and Confocal Microscopy
For immunocytochemistry (ICC), HMECs (3 × 104 cells/well) were seeded on Nunc Lab-Tek II Chamber Slides (Thermo Fisher Scientific), fixed with ICC fixation buffer (BD Biosciences, San Jose, CA, USA; cat. no. 550010), blocked with 10% normal goat serum, and incubated overnight with primary antibody against eNOS (1:200), VE-cadherin (1:400), VEGF (1:250), and vWF (1:200). The signal was visualized by subsequent incubation with fluorescence-tagged Alexa 488-tagged α-rabbit antibody (1:1,000 dilution) and counterstained with DAPI. Images were captured by an inverted fluorescence microscope (Carl Zeiss Microscopy, Germany) or a confocal microscope (Olympus Spectral FV1000), and image analysis was performed using Axiovision Rel (v.4.8) software (Axiovert 200M; Carl Zeiss Microscopy, Germany) or FV10-ASW 3.0 (Olympus Europa SE & Co. KG, Medical Systems & Micro-Imaging Solutions Group).66 The image intensity was quantified using ImageJ software (NIH). Immunohistochemistry (IHC) was performed on cryosections or formalin-fixed paraffin-embedded sections of wound sample using specific antibodies as described previously.66 Briefly, OCT-embedded tissue were cryosectioned (10 μm), fixed with cold acetone, blocked with 10% normal goat serum (NGS), and incubated with specific primary antibodies. Paraffin tissue sections (10 μm) were deparaffinized using xylene and rehydrated using decreasing gradient of ethanol. Antigen retrieval was done using citrate buffer, blocked with 10% NGS, and incubated with primary antibodies against CD31 (1:250), smooth muscle actin (SMA) (1:400), and keratin 14 (1: 1,000), followed by appropriate fluorescence-conjugated secondary antibodies (Alexa 568-tagged α-rat, 1:1,000 dilution; Alexa 488-tagged α-488 rabbit, 1:1,000 dilution). Images were collected using a Zeiss Axio Scan Z1 and Zeiss Axiovert 200 inverted fluorescence microscope supported by an AxioCam digital camera and a motorized stage and guided by Axiovision software (Zeiss).65
DNA Methyltransferase Assay
Nuclear protein was extracted from HMECs with the EpiQuik Nuclear Extraction Kit (Epigentek, OP-0022). Protein concentration was determined using BCA assay. The activity of DNMT1 and DNMT3A in the nuclear extracts was determined using the Epiquick DNMT1 and DNMT3A Assay Kit, which measures total DNMT levels (Epigentek, P-3012-2 and P-3011-2). Protein (20 μg) was added to a 96-well plate that was pre-coated with substrate specific for DNMT1 and DNMT3A and incubated for 90 min at 37°C, followed 30 min incubation at 37°C in 150 μl of blocking buffer. It was then incubated with the capture (1:1,200) and detection (1:1,200) antibody for 60 and 30 min, respectively. Addition of 100 μL of the developing solution resulted in development of blue color, which after adding stop solution turned yellow. The absorbance was immediately read at 450 nm.
Western Blot
The protein concentration of tissue extract or cell lysates was determined by the BCA method, and protein samples were resolved on SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA; cat no. 548 IPVH00010). The membranes were first blocked in 10% skim milk and then incubated with primary antibody, followed by specific secondary antibody conjugated with horseradish peroxidase at 1:2,000 dilutions. The signal was visualized using ECL Prime Western Blotting Detection Reagent (Amersham).56, 66 Pixel densitometry analysis was performed for individual band using ImageJ software. Anti-mouse β-3-tubulin served as loading control.
Laser Capture Microdissection of Wound Endothelial Cells
Laser capture microdissection was performed using the laser microdissection system from PALM Technologies (Zeiss, Germany) as described previously by our group.67 For endothelial cell-rich region capture, 10-μm tissue sections were taken on membrane slides, fixed with acetone, stained with α-CD31-APC for 60 s, subsequently washed with DEPC-H2O, and dehydrated in ethanol. Endothelial elements were identified based on the red fluorescence. Tissue sections were typically cut and captured under a 10× ocular lens. The samples were catapulted into 25 μL of cell direct lysis extraction buffer (Invitrogen). Approximately 10,00,000 μm2 of tissue area was captured into each cap, and the lysate was then stored at −80°C for further processing.
Bisulfite Conversion of DNA
Bisulfite conversion of DNA from HMECs or wound endothelial elements was performed using the Cells-to-CpG Bisulfite Conversion Kit (Thermo Fisher Scientific, part number: 4445555), as per the manufacturer’s protocol. Treating DNA with bisulfite converts unmethylated cytosines to uracil but does not change methylated cytosines. Briefly, cells were lysed, and DNA was denatured using the denaturation reagent in a PCR tube at 50°C for 10 min. Unmethylated cytosines were converted to uracil in the denatured DNA samples via treatment with conversion buffer containing bisulfite in a PCR reaction under following conditions: (1) 65°C for 30 min, (2) 95°C for 1.5 min, (3) 65°C for 30 min, (4) 95°C for 1.5 min, (5) 65°C for 30 min, and (6) 4°C up to 4 hr. This was followed by the removal of salts and desulfonation of the converted DNA in a binding column, a series of washing steps, and then elution of DNA. The converted DNA was stored at −20°C until further used.
Bisulfite Sequencing for miR-200 Promoters
The primers for sequencing region of each promoter were as follow: miR-200b/200a/429, promoter sequence 1, forward primer, 5′- TATAGAAGTTTTTTTATTTTGGTTTTTGTTT-3′ reverse primer, 5′- ATATATCCCCTA AACTCCCATAA-3′, promoter sequence 2, forward primer, 5′- TGGGAGTTTAGGGGATATATTTG-3′, and reverse primer, 5′- TCTACCTCAACCAAAA TCAAACC-3′.18 Primers for sequencing of miR-200c/141 promoter were as follows: forward primer, 5′- GGTAGTTTATGGTAGGAGGATA-3′ and reverse primer, 5′ -AAAACAAAAAAACTTTAAAACCCCAA-3′. The PCR products were gel extracted (GenElute Gel Extraction Kit, Sigma, cat. no. NA1111-1KT) and sequenced using ABI PRISM 3700 Genetic Analyzer. Results were confirmed by cloning the purified DNA into pGEM-T Easy Vector System II (Promega, cat. no. A1380), followed by sequencing.
miRNA-200b Promoter Luciferase Reporter Assay
HMECs were transfected with 100 ng of a human miR-200b promoter vector for 48 hr using Lipofectamine LTX/Plus Reagent. The promoter reporter construct containing the CpG sites within miR-200b promoter (hg38_dna range = chromosome 1:1161254-1163054) (cat. no. CS-HPRM344L-pGL3) was obtained from GeneCopoeia. Cells were lysed, and luciferase activity was determined using the dual-luciferase reporter assay system (Promega, Madison, WI, USA), according to manufacturer’s protocol. Data normalization was achieved by co-transfecting cell with Renilla plasmid (10 ng). Data are presented as a ratio of firefly to Renilla luciferase activity.
Ultrasound Data Acquisition and Analysis
Mice were anesthetized using 1.5% isoflurane and carbogen (95% O2 and 5% CO2) mixture and placed on a mouse bed setup of vevo 2100 system (Vevo 2100 System, FUJIFILM VisualSonics, Toronto, ON, Canada). Using B-Mode protocol, video clips of the axial view of the wound area were recorded using a solid-state transducer (probe #MS550D). A 40-MHz ultrasound frequency was used for all the measurements described in this work. Normal skin images were used to measure baseline skin thickness. The skin-adipose border being the brightest was used as an anatomical landmark to measure the wound depth, diameter, and re-epithelialization area. The ultrasound Doppler color flow imaging (color Doppler mode) feature of the system was used to acquire color-coded videos representing blood flow. This method enabled measuring blood flow in the vessels of the skin and wound regions. The pulse wave color Doppler (PW mode) feature of the system generated the blood flow velocity profiles from the color coded flow videos. Real-time velocity profiles were recorded and used to measure velocity at end systole and velocity at end diastole. Three repeated-measurements were performed on three different peaks. From the velocity data, pulse pressure was computed using modified Bernoulli Equation as explained in our previous report.68 Mean and standard deviation were calculated.
Extracellular Flux Assays
Bioenergetics of HMECs under HG with or without miR-200b inhibition were determined using the XF96e Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA, USA). Cells were seeded in specialized tissue culture plates in an optimized concentration of 100,000 cells per well. 1 hr before measurement, cells were incubated at 37°C in a CO2-free atmosphere. The oxygen consumption rate (OCR) was detected under basal conditions. It was followed by the sequential addition of oligomycin, FCCP, rotenone, and antimycin A.
ATP Level Measurement
The ATP content of HMECs under different culture conditions were measured using the commercial Enzylight Assay Kit (Bioassay Systems, Hayward, CA, USA) based on the luciferin-luciferase reaction. An assay was performed as per the manufacturer’s instructions. Briefly, 10 μL of cell suspension was mixed with 90 μL of ATP reagent and incubated at room temperature for 1 min. Luminescence was subsequently measured in Berthold Luminometer (Berthold Technologies). Normalized ratio of luminescence intensities (RLU, relative light unit) of ATP was calculated for each sample as the ratio of RLU to the cell number.
Mitochondrial Potential Assay
Cells were stained with JC-1 dye (2 μM, MitoProbe JC-1 Assay Kit, Life Technologies) and incubated at 37°C, 5% CO2, for 30 min. There is potential-dependent accumulation of JC-1 in mitochondria. JC-1 forms J aggregates and produces a red fluorescence at high membrane potentials. At low membrane potentials, JC-1 exists as a monomer and produces a green fluorescence. Images were collected using a Zeiss Axiovert 200 inverted fluorescence microscope.
SILAC Labeling
DMEM deficient in L-lysine and L-arginine for the SILAC assay was obtained from Thermo Scientific (catalog number: 88420). For labeling experiments, L-lysine-2HCl (50 mg/L, Thermo Scientific, catalog number: 88429) and L-arginine-HCl (50 mg/L, Thermo Scientific, catalog number: 88427) were added to form the medium referred to as light medium. L-lysine-2HCl, 13C6, 15N2 (50 mg/L, Thermo Scientific, catalog number: 88209) and L-arginine-HCl, 13C6, 15N4 (50 mg/L, Thermo Scientific, catalog number: 89990) were added to form the medium referred to as heavy medium. HMECs were grown in light and heavy SILAC media supplemented with 10% dialyzed fetal bovine serum (Thermo Scientific, catalog number: 88212) and 1% antibiotics in a humidified atmosphere with 5% CO2 in air for six passages. The 100% incorporation of heavy isotope was assured by mass spectrometry on the basis of mass shift compared to light isotope. After confirmed labeling, two different sets of experiments were performed. In the first set, the heavy-labeled cells were exposed to HG (HG) for 4 days and then compared with normoglycemic light cells (NG). In the second set, the heavy-labeled cells were exposed to HG for 2 days and then transfected with miR-200b inhibitor for 2 days in HG (HG + MI) and were compared with light-labeled normoglycemic cells transfected with control inhibitor for 2 days (NG + CI). Cells were harvested and mixed at a 1:1 ratio (set 1: NG and HG cells; set 2: NG + CI and HG + MI) and subjected to protein extraction.
Sample Preparation for Proteomics Analysis
2 × 106 cells were lysed in lysis buffer (30 mM Tris [pH 8.5], 7 M Urea, 2 M Thiourea, and 4% CHAPS), followed by 2 × 10 s probe sonication and supernatants were collected after centrifugation at 13,000 rpm for 10 min. Protein (125 μg) was precipitated using a 2D cleanup kit as per the manufacturer’s instructions (GE, 80-6484-51). Peptide concentration was determined by nanodrop (A280nm).
2D Liquid Chromatography-Tandem Mass Spectrometry
2D liquid chromatography-tandem mass spectrometry (LC-MS/MS) separations were performed on Acquity UPLC M-Class (Water’s Corp., Milford, MA, USA) equipment. The first dimension chromatographic separations were performed on a BEH C18 column (part #186007471, Water’s Corp.) (5 mm × 300 μm; particle size: 3 μm; pore size: 130 Å). A 2D Symmetry C18 Column (part # 186007497, Water’s Corp.) (20 mm × 180 μm; particle size: 5 μm; pore size: 100 Å) was used for desalting. Peptides were eluted from the first dimension fraction column in 12 successive fractions using 8.0, 10.7, 12.4, 13.7, 14.9, 16.0, 17.2, 18.4, 19.7, 21.4, and 24.1 and 50% acetonitrile. Each eluted fraction was then trapped, desalted, and separated using pepmap C18 column (part # ES800, Thermo Scientific) (15 cm × 75 μm; particle size: 3 μm; pore size: 100 Å). MS/MS data were acquired with a spray voltage of 1.7 KV and a capillary temperature of 275°C. The scan sequence of the mass spectrometer was based on the preview mode data-dependent TopSpeed method. The full scan was performed at Fourier transform (FT) mode and at a resolution of 120,000 to achieve high mass accuracy. The AGC Target ion number for the FT full scan was set at 2 × 105 ions; the maximum ion injection time was set at 50 ms; and the micro scan number (MSn) was set at 1. MSn was performed using ion trap mode to ensure the highest signal intensity of MSn spectra using both CID (for 2+ and 3+ charges) and ETD (for 4+–6+ charges) methods. The AGC Target ion number for ion trap MSn scan was set at 1,000 ions; maximum ion injection time was set at 100 ms; and micro scan number was set at 1. The CID fragmentation energy was set to 35%. Dynamic exclusion was enabled with a repeat count of 1 within 60 s and within a mass width of 10 ppm.
Computational Analysis
Data were searched on Proteome Discoverer using Sequest against Human Uniprot database (Thermo Scientific).The mass accuracy of the precursor ions were set to 10 ppm, and accidental pick of 1 13C peaks was also included into the search. The fragment mass tolerance was set to 0.5 Da. Considered variable modifications were oxidation (Met), deamidation (N and Q), and carbamidomethylation (Cys), as well as the heavy isotope labeling. Four missed cleavages for the enzyme were permitted. A decoy database was also searched to determine the false discovery rate (FDR), and peptides were filtered according to the FDR. The significance threshold was set at p < 0.05, and bold red peptides were required for valid peptide identification. Proteins with less than 1% FDR as well as a minimal of 2 significant peptides detected were considered as valid proteins. The ratio was calculated using unique peptide intensity between heavy and light isotope-labeled peptides. The protein ratios were normalized with beta-3 tubulin and significantly different proteins having a p value less than 0.05 and percent change greater than 10% were included for further bioinformatics analysis. For the analysis of protein-protein interactions of significantly different proteins, STRING (search tool for the retrieval of interacting genes/proteins) (v.10.0) (http://string-db.org/) was used. The parameters used for the analysis included interaction sources based on neighborhood, databases, gene fusion, co-expression, text mining, and an experiment using high confidence (0.9). In addition, the IPA tool (Ingenuity Systems, http://www.ingenuity.com) and PANTHER (http://pantherdb.org/) were used to identify novel biological pathways affected because of HG.
Statistical Analysis
The data analysis was performed in a blinded fashion. Student’s t test (two-tailed) was used to determine significant differences. Comparisons among multiple groups were tested using ANOVA. p < 0.05 was considered statistically significant.
Author Contributions
Conceptualization, C.K.S. and K.S.; Methodology, C.K.S., K.S., and D.P.; Investigation and Validation, K.S., D.P., M.S., S.G., and S.C.G., Formal Analysis, K.S., D.P., M.S., S.K., and S.R.; Writing – Original Draft, K.S., D.P., M.S., S.G., S.C.G., S.K., S.R., and C.K.S.; Writing – Review & Editing, K.S., D.P., S.K., S.R., and C.K.S.; Visualization, K.S. and C.K.S.; Funding Acquisition, C.K.S., S.K., and S.R.; Resources, S.K., S.R., and C.K.S.; Supervision, S.K., S.R., and C.K.S.
Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgments
We thank Laser Capture Molecular Core, CMIF core, and Genomics Shared Resource core, OSU. This work was supported, in part, by grants from the NIH (GM108014, GM077185, NR013898, NR015676, and NS42617 to C.K.S.; NR015676 to S.R.; and NS085272 to S.K; and DK076169 subaward and R56DK076566 to S.R.). We thank Liwen Zhang (Proteomic Shared Resources, The Ohio State University) for her help in MS. The Fusion Orbitrap instrument was supported by NIH grant S10 OD018056.
Footnotes
Supplemental Information includes four figures and nine tables and can be found with this article online at https://doi.org/10.1016/j.ymthe.2017.09.009.
Supplemental Information
References
- 1.Dhliwayo N., Sarras M.P., Jr., Luczkowski E., Mason S.M., Intine R.V. Parp inhibition prevents ten-eleven translocase enzyme activation and hyperglycemia-induced DNA demethylation. Diabetes. 2014;63:3069–3076. doi: 10.2337/db13-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reddy M.A., Zhang E., Natarajan R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia. 2015;58:443–455. doi: 10.1007/s00125-014-3462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palsamy P., Bidasee K.R., Ayaki M., Augusteyn R.C., Chan J.Y., Shinohara T. Methylglyoxal induces endoplasmic reticulum stress and DNA demethylation in the Keap1 promoter of human lens epithelial cells and age-related cataracts. Free Radic. Biol. Med. 2014;72:134–148. doi: 10.1016/j.freeradbiomed.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Portela A., Esteller M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 5.Saito Y., Liang G., Egger G., Friedman J.M., Chuang J.C., Coetzee G.A., Jones P.A. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–443. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 6.Saito Y., Saito H., Liang G., Friedman J.M. Epigenetic alterations and microRNA misexpression in cancer and autoimmune diseases: a critical review. Clin. Rev. Allergy Immunol. 2014;47:128–135. doi: 10.1007/s12016-013-8401-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang H.P., Wang Y.H., Cao C.J., Yang X.M., Ma S.C., Han X.B., Yang X.L., Yang A.N., Tian J., Xu H. A regulatory circuit involving miR-143 and DNMT3a mediates vascular smooth muscle cell proliferation induced by homocysteine. Mol. Med. Rep. 2016;13:483–490. doi: 10.3892/mmr.2015.4558. [DOI] [PubMed] [Google Scholar]
- 8.Lujambio A., Ropero S., Ballestar E., Fraga M.F., Cerrato C., Setién F., Casado S., Suarez-Gauthier A., Sanchez-Cespedes M., Git A. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67:1424–1429. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- 9.Kozaki K., Imoto I., Mogi S., Omura K., Inazawa J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008;68:2094–2105. doi: 10.1158/0008-5472.CAN-07-5194. [DOI] [PubMed] [Google Scholar]
- 10.Chan Y.C., Khanna S., Roy S., Sen C.K. miR-200b targets Ets-1 and is down-regulated by hypoxia to induce angiogenic response of endothelial cells. J. Biol. Chem. 2011;286:2047–2056. doi: 10.1074/jbc.M110.158790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi Y.C., Yoon S., Jeong Y., Yoon J., Baek K. Regulation of vascular endothelial growth factor signaling by miR-200b. Mol. Cells. 2011;32:77–82. doi: 10.1007/s10059-011-1042-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McArthur K., Feng B., Wu Y., Chen S., Chakrabarti S. MicroRNA-200b regulates vascular endothelial growth factor-mediated alterations in diabetic retinopathy. Diabetes. 2011;60:1314–1323. doi: 10.2337/db10-1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan Y.C., Roy S., Khanna S., Sen C.K. Downregulation of endothelial microRNA-200b supports cutaneous wound angiogenesis by desilencing GATA binding protein 2 and vascular endothelial growth factor receptor 2. Arterioscler. Thromb. Vasc. Biol. 2012;32:1372–1382. doi: 10.1161/ATVBAHA.112.248583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dangwal S., Stratmann B., Bang C., Lorenzen J.M., Kumarswamy R., Fiedler J., Falk C.S., Scholz C.J., Thum T., Tschoepe D. Impairment of wound healing in patients with type 2 diabetes mellitus influences circulating microRNA patterns via inflammatory cytokines. Arterioscler. Thromb. Vasc. Biol. 2015;35:1480–1488. doi: 10.1161/ATVBAHA.114.305048. [DOI] [PubMed] [Google Scholar]
- 15.Avogaro A., Albiero M., Menegazzo L., de Kreutzenberg S., Fadini G.P. Endothelial dysfunction in diabetes: the role of reparatory mechanisms. Diabetes Care. 2011;34(Suppl 2):S285–S290. doi: 10.2337/dc11-s239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gifford S.M., Grummer M.A., Pierre S.A., Austin J.L., Zheng J., Bird I.M. Functional characterization of HUVEC-CS: Ca2+ signaling, ERK 1/2 activation, mitogenesis and vasodilator production. J. Endocrinol. 2004;182:485–499. doi: 10.1677/joe.0.1820485. [DOI] [PubMed] [Google Scholar]
- 17.Kurashige J., Mima K., Sawada G., Takahashi Y., Eguchi H., Sugimachi K., Mori M., Yanagihara K., Yashiro M., Hirakawa K. Epigenetic modulation and repression of miR-200b by cancer-associated fibroblasts contribute to cancer invasion and peritoneal dissemination in gastric cancer. Carcinogenesis. 2015;36:133–141. doi: 10.1093/carcin/bgu232. [DOI] [PubMed] [Google Scholar]
- 18.Lim Y.Y., Wright J.A., Attema J.L., Gregory P.A., Bert A.G., Smith E., Thomas D., Lopez A.F., Drew P.A., Khew-Goodall Y., Goodall G.J. Epigenetic modulation of the miR-200 family is associated with transition to a breast cancer stem-cell-like state. J. Cell Sci. 2013;126:2256–2266. doi: 10.1242/jcs.122275. [DOI] [PubMed] [Google Scholar]
- 19.Detich N., Hamm S., Just G., Knox J.D., Szyf M. The methyl donor S-adenosylmethionine inhibits active demethylation of DNA: a candidate novel mechanism for the pharmacological effects of S-adenosylmethionine. J. Biol. Chem. 2003;278:20812–20820. doi: 10.1074/jbc.M211813200. [DOI] [PubMed] [Google Scholar]
- 20.Koh T.J., DiPietro L.A. Inflammation and wound healing: the role of the macrophage. Expert Rev. Mol. Med. 2011;13:e23. doi: 10.1017/S1462399411001943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rendell M.S., Milliken B.K., Finnegan M.F., Finney D.A., Healy J.C. The skin blood flow response in wound healing. Microvasc. Res. 1997;53:222–234. doi: 10.1006/mvre.1997.2008. [DOI] [PubMed] [Google Scholar]
- 22.Ong S.E., Mann M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC) Nat. Protoc. 2006;1:2650–2660. doi: 10.1038/nprot.2006.427. [DOI] [PubMed] [Google Scholar]
- 23.He X.J., Hsu Y.F., Pontes O., Zhu J., Lu J., Bressan R.A., Pikaard C., Wang C.S., Zhu J.K. NRPD4, a protein related to the RPB4 subunit of RNA polymerase II, is a component of RNA polymerases IV and V and is required for RNA-directed DNA methylation. Genes Dev. 2009;23:318–330. doi: 10.1101/gad.1765209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jopling H.M., Odell A.F., Hooper N.M., Zachary I.C., Walker J.H., Ponnambalam S. Rab GTPase regulation of VEGFR2 trafficking and signaling in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2009;29:1119–1124. doi: 10.1161/ATVBAHA.109.186239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poliseno L., Tuccoli A., Mariani L., Evangelista M., Citti L., Woods K., Mercatanti A., Hammond S., Rainaldi G. MicroRNAs modulate the angiogenic properties of HUVECs. Blood. 2006;108:3068–3071. doi: 10.1182/blood-2006-01-012369. [DOI] [PubMed] [Google Scholar]
- 26.Landskroner-Eiger S., Moneke I., Sessa W.C. miRNAs as modulators of angiogenesis. Cold Spring Harb. Perspect. Med. 2013;3:a006643. doi: 10.1101/cshperspect.a006643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuhnert F., Mancuso M.R., Hampton J., Stankunas K., Asano T., Chen C.Z., Kuo C.J. Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Development. 2008;135:3989–3993. doi: 10.1242/dev.029736. [DOI] [PubMed] [Google Scholar]
- 28.Otsuka M., Zheng M., Hayashi M., Lee J.D., Yoshino O., Lin S., Han J. Impaired microRNA processing causes corpus luteum insufficiency and infertility in mice. J. Clin. Invest. 2008;118:1944–1954. doi: 10.1172/JCI33680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fasanaro P., Greco S., Lorenzi M., Pescatori M., Brioschi M., Kulshreshtha R., Banfi C., Stubbs A., Calin G.A., Ivan M. An integrated approach for experimental target identification of hypoxia-induced miR-210. J. Biol. Chem. 2009;284:35134–35143. doi: 10.1074/jbc.M109.052779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sinha M., Ghatak S., Roy S., Sen C.K. microRNA-200b as a Switch for Inducible Adult Angiogenesis. Antioxid. Redox Signal. 2015;22:1257–1272. doi: 10.1089/ars.2014.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davalos V., Moutinho C., Villanueva A., Boque R., Silva P., Carneiro F., Esteller M. Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene. 2012;31:2062–2074. doi: 10.1038/onc.2011.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li A., Omura N., Hong S.M., Vincent A., Walter K., Griffith M., Borges M., Goggins M. Pancreatic cancers epigenetically silence SIP1 and hypomethylate and overexpress miR-200a/200b in association with elevated circulating miR-200a and miR-200b levels. Cancer Res. 2010;70:5226–5237. doi: 10.1158/0008-5472.CAN-09-4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu S.C. S-adenosylmethionine. Int. J. Biochem. Cell Biol. 2000;32:391–395. doi: 10.1016/s1357-2725(99)00139-9. [DOI] [PubMed] [Google Scholar]
- 34.Mato J.M., Alvarez L., Ortiz P., Pajares M.A. S-adenosylmethionine synthesis: molecular mechanisms and clinical implications. Pharmacol. Ther. 1997;73:265–280. doi: 10.1016/s0163-7258(96)00197-0. [DOI] [PubMed] [Google Scholar]
- 35.Barrows L.R., Magee P.N. Nonenzymatic methylation of DNA by S-adenosylmethionine in vitro. Carcinogenesis. 1982;3:349–351. doi: 10.1093/carcin/3.3.349. [DOI] [PubMed] [Google Scholar]
- 36.Zhao Y., Li J.S., Guo M.Z., Feng B.S., Zhang J.P. Inhibitory effect of S-adenosylmethionine on the growth of human gastric cancer cells in vivo and in vitro. Chin. J. Cancer. 2010;29:752–760. doi: 10.5732/cjc.010.10046. [DOI] [PubMed] [Google Scholar]
- 37.Luo J., Li Y.N., Wang F., Zhang W.M., Geng X. S-adenosylmethionine inhibits the growth of cancer cells by reversing the hypomethylation status of c-myc and H-ras in human gastric cancer and colon cancer. Int. J. Biol. Sci. 2010;6:784–795. doi: 10.7150/ijbs.6.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim S.Y., Hong S.W., Kim M.O., Kim H.S., Jang J.E., Leem J., Park I.S., Lee K.U., Koh E.H. S-adenosyl methionine prevents endothelial dysfunction by inducing heme oxygenase-1 in vascular endothelial cells. Mol. Cells. 2013;36:376–384. doi: 10.1007/s10059-013-0210-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spijkerman A.M., Smulders Y.M., Kostense P.J., Henry R.M., Becker A., Teerlink T., Jakobs C., Dekker J.M., Nijpels G., Heine R.J. S-adenosylmethionine and 5-methyltetrahydrofolate are associated with endothelial function after controlling for confounding by homocysteine: the Hoorn study. Arterioscler. Thromb. Vasc. Biol. 2005;25:778–784. doi: 10.1161/01.ATV.0000157981.57694.d2. [DOI] [PubMed] [Google Scholar]
- 40.Kaddai V., Le Marchand-Brustel Y., Cormont M. Rab proteins in endocytosis and Glut4 trafficking. Acta Physiol. (Oxf.) 2008;192:75–88. doi: 10.1111/j.1748-1716.2007.01787.x. [DOI] [PubMed] [Google Scholar]
- 41.Yaekura K., Julyan R., Wicksteed B.L., Hays L.B., Alarcon C., Sommers S., Poitout V., Baskin D.G., Wang Y., Philipson L.H., Rhodes C.J. Insulin secretory deficiency and glucose intolerance in Rab3A null mice. J. Biol. Chem. 2003;278:9715–9721. doi: 10.1074/jbc.M211352200. [DOI] [PubMed] [Google Scholar]
- 42.Santilli S.M., Fiegel V.D., Aldridge D.E., Knighton D.R. The effect of diabetes on the proliferation of aortic endothelial cells. Ann. Vasc. Surg. 1992;6:503–510. doi: 10.1007/BF02000821. [DOI] [PubMed] [Google Scholar]
- 43.Ho J.J., Robb G.B., Tai S.C., Turgeon P.J., Mawji I.A., Man H.S., Marsden P.A. Active stabilization of human endothelial nitric oxide synthase mRNA by hnRNP E1 protects against antisense RNA and microRNAs. Mol. Cell. Biol. 2013;33:2029–2046. doi: 10.1128/MCB.01257-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao R., Yu Y., Inoue A., Widodo N., Kaul S.C., Wadhwa R. Heterogeneous nuclear ribonucleoprotein K (hnRNP-K) promotes tumor metastasis by induction of genes involved in extracellular matrix, cell movement, and angiogenesis. J. Biol. Chem. 2013;288:15046–15056. doi: 10.1074/jbc.M113.466136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lo C.S., Shi Y., Chang S.Y., Abdo S., Chenier I., Filep J.G., Ingelfinger J.R., Zhang S.L., Chan J.S. Overexpression of heterogeneous nuclear ribonucleoprotein F stimulates renal Ace-2 gene expression and prevents TGF-β1-induced kidney injury in a mouse model of diabetes. Diabetologia. 2015;58:2443–2454. doi: 10.1007/s00125-015-3700-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun J., Liao J.K. Induction of angiogenesis by heat shock protein 90 mediated by protein kinase Akt and endothelial nitric oxide synthase. Arterioscler. Thromb. Vasc. Biol. 2004;24:2238–2244. doi: 10.1161/01.ATV.0000147894.22300.4c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kavanagh K., Flynn D.M., Jenkins K.A., Zhang L., Wagner J.D. Restoring HSP70 deficiencies improves glucose tolerance in diabetic monkeys. Am. J. Physiol. Endocrinol. Metab. 2011;300:E894–E901. doi: 10.1152/ajpendo.00699.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trudeau K., Molina A.J., Guo W., Roy S. High glucose disrupts mitochondrial morphology in retinal endothelial cells: implications for diabetic retinopathy. Am. J. Pathol. 2010;177:447–455. doi: 10.2353/ajpath.2010.091029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Czajka A., Malik A.N. Hyperglycemia induced damage to mitochondrial respiration in renal mesangial and tubular cells: Implications for diabetic nephropathy. Redox Biol. 2016;10:100–107. doi: 10.1016/j.redox.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sivitz W.I., Yorek M.A. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 2010;12:537–577. doi: 10.1089/ars.2009.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guan S.S., Sheu M.L., Wu C.T., Chiang C.K., Liu S.H. ATP synthase subunit-β down-regulation aggravates diabetic nephropathy. Sci. Rep. 2015;5:14561. doi: 10.1038/srep14561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stentz F.B., Umpierrez G.E., Cuervo R., Kitabchi A.E. Proinflammatory cytokines, markers of cardiovascular risks, oxidative stress, and lipid peroxidation in patients with hyperglycemic crises. Diabetes. 2004;53:2079–2086. doi: 10.2337/diabetes.53.8.2079. [DOI] [PubMed] [Google Scholar]
- 53.Nareika A., Sundararaj K.P., Im Y.B., Game B.A., Lopes-Virella M.F., Huang Y. High glucose and interferon gamma synergistically stimulate MMP-1 expression in U937 macrophages by increasing transcription factor STAT1 activity. Atherosclerosis. 2009;202:363–371. doi: 10.1016/j.atherosclerosis.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manea S.A., Manea A., Heltianu C. Inhibition of JAK/STAT signaling pathway prevents high-glucose-induced increase in endothelin-1 synthesis in human endothelial cells. Cell Tissue Res. 2010;340:71–79. doi: 10.1007/s00441-010-0936-1. [DOI] [PubMed] [Google Scholar]
- 55.Caporali A., Meloni M., Nailor A., Mitić T., Shantikumar S., Riu F., Sala-Newby G.B., Rose L., Besnier M., Katare R. p75(NTR)-dependent activation of NF-κB regulates microRNA-503 transcription and pericyte-endothelial crosstalk in diabetes after limb ischaemia. Nat. Commun. 2015;6:8024. doi: 10.1038/ncomms9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Das A., Ganesh K., Khanna S., Sen C.K., Roy S. Engulfment of apoptotic cells by macrophages: a role of microRNA-21 in the resolution of wound inflammation. J. Immunol. 2014;192:1120–1129. doi: 10.4049/jimmunol.1300613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roy S., Elgharably H., Sinha M., Ganesh K., Chaney S., Mann E., Miller C., Khanna S., Bergdall V.K., Powell H.M. Mixed-species biofilm compromises wound healing by disrupting epidermal barrier function. J. Pathol. 2014;233:331–343. doi: 10.1002/path.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghatak S., Chan Y.C., Khanna S., Banerjee J., Weist J., Roy S., Sen C.K. Barrier function of the repaired skin is disrupted following arrest of dicer in keratinocytes. Mol. Ther. 2015;23:1201–1210. doi: 10.1038/mt.2015.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stramentinoli G., Gualano M., Galli-Kienle M. Intestinal absorption of S-adenosyl-L-methionine. J. Pharmacol. Exp. Ther. 1979;209:323–326. [PubMed] [Google Scholar]
- 60.Jin C.J., Park H.K., Cho Y.M., Pak Y.K., Lee K.U., Kim M.S., Friso S., Choi S.W., Park K.S., Lee H.K. S-adenosyl-L-methionine increases skeletal muscle mitochondrial DNA density and whole body insulin sensitivity in OLETF rats. J. Nutr. 2007;137:339–344. doi: 10.1093/jn/137.2.339. [DOI] [PubMed] [Google Scholar]
- 61.Zhen Z., Tang L.J., Long H., Jiang J.H. Enzymatic immuno-assembly of gold nanoparticles for visualized activity screening of histone-modifying enzymes. Anal. Chem. 2012;84:3614–3620. doi: 10.1021/ac203385v. [DOI] [PubMed] [Google Scholar]
- 62.Galm O., Rountree M.R., Bachman K.E., Jair K.W., Baylin S.B., Herman J.G. Enzymatic regional methylation assay: a novel method to quantify regional CpG methylation density. Genome Res. 2002;12:153–157. doi: 10.1101/gr.202501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gallego-Perez D., Pal D., Ghatak S., Malkoc V., Higuita-Castro N., Gnyawali S., Chang L., Liao W.C., Shi J., Sinha M. Topical tissue nano-transfection mediates non-viral stroma reprogramming and rescue. Nat. Nanotechnol. 2017 doi: 10.1038/nnano.2017.134. Published online August 7, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fu H., Khan A., Coe D., Zaher S., Chai J.G., Kropf P., Müller I., Larkin D.F., George A.J. Arginine depletion as a mechanism for the immune privilege of corneal allografts. Eur. J. Immunol. 2011;41:2997–3005. doi: 10.1002/eji.201141683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Banerjee J., Das Ghatak P., Roy S., Khanna S., Sequin E.K., Bellman K., Dickinson B.C., Suri P., Subramaniam V.V., Chang C.J., Sen C.K. Improvement of human keratinocyte migration by a redox active bioelectric dressing. PLoS ONE. 2014;9:e89239. doi: 10.1371/journal.pone.0089239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Das A., Ghatak S., Sinha M., Chaffee S., Ahmed N.S., Parinandi N.L., Wohleb E.S., Sheridan J.F., Sen C.K., Roy S. Correction of MFG-E8 resolves inflammation and promotes cutaneous wound healing in diabetes. J. Immunol. 2016;196:5089–5100. doi: 10.4049/jimmunol.1502270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roy S., Patel D., Khanna S., Gordillo G.M., Biswas S., Friedman A., Sen C.K. Transcriptome-wide analysis of blood vessels laser captured from human skin and chronic wound-edge tissue. Proc. Natl. Acad. Sci. USA. 2007;104:14472–14477. doi: 10.1073/pnas.0706793104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gnyawali S.C., Barki K.G., Mathew-Steiner S.S., Dixith S., Vanzant D., Kim J., Dickerson J.L., Datta S., Powell H., Roy S. High-resolution harmonics ultrasound imaging for non-invasive characterization of wound healing in a pre-clinical swine model. PLoS ONE. 2015;10:e0122327. doi: 10.1371/journal.pone.0122327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.