

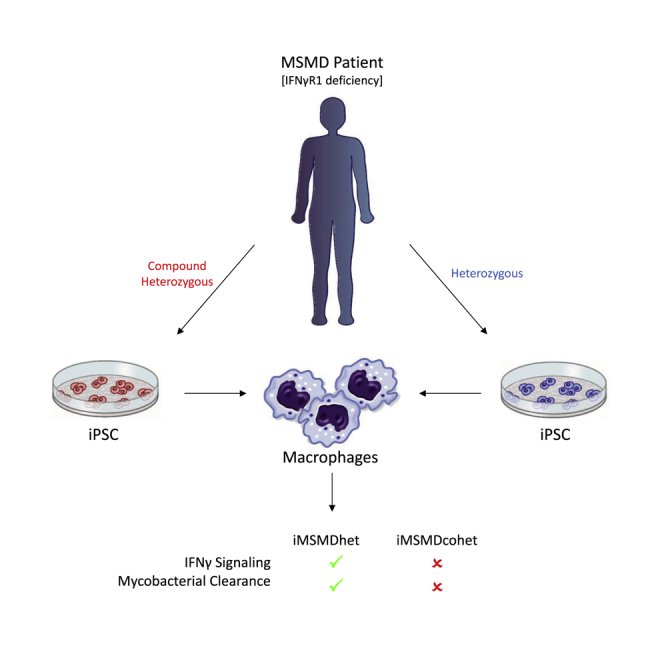
Summary
Mendelian susceptibility to mycobacterial disease (MSMD) is caused by inborn errors of interferon gamma (IFNγ) immunity and is characterized by severe infections by weakly virulent mycobacteria. Although IFNγ is the macrophage-activating factor, macrophages from these patients have never been studied. We demonstrate the generation of heterozygous and compound heterozygous (iMSMD-cohet) induced pluripotent stem cells (iPSCs) from a single chimeric patient, who suffered from complete autosomal recessive IFNγR1 deficiency and received bone-marrow transplantation. Loss of IFNγR1 expression had no influence on the macrophage differentiation potential of patient-specific iPSCs. In contrast, lack of IFNγR1 in iMSMD-cohet macrophages abolished IFNγ-dependent phosphorylation of STAT1 and induction of IFNγ-downstream targets such as IRF-1, SOCS-3, and IDO. As a consequence, iMSMD-cohet macrophages show impaired upregulation of HLA-DR and reduced intracellular killing of Bacillus Calmette-Guérin. We provide a disease-modeling platform that might be suited to investigate novel treatment options for MSMD and to gain insights into IFNγ signaling in macrophages.
Keywords: hematopoiesis, iPSC, macrophages, MSMD, IFNγR1, mycobacteria, BCG
Graphical Abstract
Highlights
-
•
Generation of patient-specific iPSCs from small volume (7 mL) peripheral blood
-
•
Heterozygous or compound heterozygous iPSCs from one chimeric MSMD patient
-
•
Patient-specific iPSC macrophages reveal impaired IFNγ downstream signaling
-
•
Compound heterozygous macrophages show decreased killing of mycobacteria
Neehus and colleagues describe the simultaneous generation of genetically diverse patient-specific iPSC lines from one chimeric patient with Mendelian susceptibility to mycobacterial disease (MSMD) due to IFNγR1 deficiency. Hematopoietic differentiation toward macrophages of heterozygous and compound heterozygous MSMD iPSC lines laid the foundation to study IFNγ signaling and susceptibility to mycobacterial infections of patient-derived macrophages.
Introduction
Since their discovery in 2006, induced pluripotent stem cells (iPSCs) have been considered an invaluable tool for drug testing, disease modeling, and regenerative therapies (Yamanaka and Takahashi, 2006). Nowadays, iPSCs can be generated from different sources of patient material and subsequently be differentiated into the affected cell types to study the underlying disease pathophysiology and to develop new forms of treatment. The feasibility of this approach has been highlighted for different cell types of the three germ layers. Similarly, mesodermal differentiation of iPSCs toward different cells of the hematopoietic lineage has been successfully proven (Ackermann et al., 2015). Generation of macrophages from patient-derived iPSCs is of particular interest (Lachmann et al., 2014, Mucci et al., 2016), as these cells play an important role in tissue homeostasis and innate immunity (Ginhoux et al., 2016). In this context, impairment of macrophage functionality due to genetic predisposition or environmental factors can lead to severe and life-threatening infections, as seen in Mendelian susceptibility to mycobacterial disease (MSMD).
MSMD is a rare primary immunodeficiency characterized by clinical infections caused by weakly virulent mycobacteria in otherwise healthy individuals. Most MSMD patients suffer from severe infections triggered by Mycobacterium bovis Bacillus Calmette-Guérin (BCG), which has been used as an attenuated vaccine for tuberculosis (Bustamante et al., 2014). Up to now, genetic defects in eight autosomal (IFNGR1, IFNGR2, STAT1, IRF8, ISG15, TYK2, IL12B, and IL12RB1) and two X-linked (CYBB and NEMO) genes, all controlling interferon gamma (IFNγ) immunity, have been identified in MSMD patients (Al-Muhsen and Casanova, 2008). Mutations in the different genes mainly affect the IFNγ-signaling pathway, resulting in impaired activation of macrophages by T or natural killer (NK) cells (Bustamante et al., 2014, Chapman and Hill, 2012). The current form of treatment for MSMD patients consists of antibiotics in combination with IFNγ therapy to restore the function of macrophages. However, patients suffering from complete autosomal recessive IFNγR1 (OMIM no.209950) or IFNγR2 (OMIM no. 614889) deficiency receive hematopoietic stem cell transplantation (HSCT) as the only curative treatment (Chantrain et al., 2006, Horwitz et al., 2003, Olbrich et al., 2015, Reuter et al., 2002, Roesler et al., 2004). Although this therapy has been proven effective in some cases, further complications caused by the impaired functionality of macrophages, such as recurrent infections and high serum levels of IFNγ, hamper the overall success of this approach. Especially the latter is of high clinical relevance, as a high serum level of IFNγ directly interferes with donor hematopoietic stem cell (HSC) proliferation and consequently leads to graft rejection (Rottman et al., 2008).
Given the limited amount of patient material available and the unsatisfactory long-term treatment alternatives for IFNγR1 or IFNγR2 deficiency, we established a disease-modeling platform for IFNγR1 deficiency using iPSC-derived macrophages. In our study, we generated patient-specific iPSCs from a small volume of peripheral blood from one patient suffering from complete autosomal recessive IFNγR1 deficiency caused by a compound heterozygous mutation in IFNγR1 who had previously received HSCT from a sibling harboring a heterozygous mutation in IFNγR1. Patient-specific macrophages, which are derived from compound heterozygous iPSCs, showed impaired IFNγ-mediated signaling and functionality, thus underlining their suitability to evaluate new therapeutic interventions or to study mycobacterial infections.
Results
Heterozygous and Compound Heterozygous IFNγR1-Deficient iPSC Lines Derived from a Single Chimeric MSMD Patient
In order to establish a disease-modeling platform for MSMD, we first isolated peripheral blood from a patient with MSMD due to complete autosomal recessive IFNγR1 deficiency (Roesler et al., 1999) (Figure 1A). The patient suffered mainly from disseminated infections caused by several mycobacteria such as M. avium, M. kansasii, and BCG affecting several organs such as lymph nodes, lungs, and bones. These infections led to episodes of fever and enhanced serum parameters indicating chronic inflammation. Liver cirrhosis and meningitis caused by Listeria monocytogenes were also observed. The patient received an HSCT from her older sister with whom she shared a mutated allele (c.373+1G > T) (Figures 1A and 1B). This mutation is based on a G to T transition at position 1 in the 5′ end of intron III and results in the skipping of the entire exon III, as described previously (Roesler et al., 1999). After an initial 100% donor chimerism, the donor cells declined to roughly 50%, which was prompted by donor lymphocyte transfusions. These transfusions stabilized the chimerism at 50%–60% and the patient substantially improved clinically (Reuter et al., 2002).
Figure 1.

Generation of Heterozygous and Compound Heterozygous IFNγR1-Deficient iPSCs
(A) Schematic view of the generation, characterization, and hematopoietic differentiation of MSMD patient-derived iPSCs.
(B) Illustrative scheme of the IFNγR1 genomic DNA and transcribed mRNA in healthy wild-type individuals, the heterozygous donor, and the compound heterozygous MSMD patient. The heterozygous donor harbors a G > T transition at nucleotide position 373, which leads to the skipping of the entire exon III. The compound heterozygous patient carries the same mutation and has a deletion of 4 base pairs (bp) in the second IFNγR1 allele, which results in a frameshift mutation and a premature stop codon 42 bp downstream of the deletion at position c.561.
(C) Sequencing and electropherogram to screen for the patient-specific 4 bp deletion in exon V in different iPSCs. Sequencing was performed on healthy wild-type iPSCs (hCD34-iPSC16) and heterozygous and compound heterozygous patient-specific iPSCs.
See also Figure S1.
After isolation of CD34+ cells from 7 mL of peripheral blood (EDTA), a “4 in 1” all-in-one lentiviral vector harboring the classical Yamanaka factors was used for reprogramming (Figure 1A). Within 10–20 days post transduction, 23 iPSC clones could be successfully established and have been subjected to further genotypic analysis. To discriminate compound heterozygous patient-specific iPSC clones from heterozygous donor cells, we performed sequencing of exon V in the IFNγR1 gene locus. The 4 bp deletion between nucleotides 561 and 564 in exon V leads to a frameshift mutation creating a premature stop codon 42 bp downstream of amino acid 187 (Roesler et al., 1999) (Figure 1B). Analysis of exon V revealed nine clones positive for the patient-specific 4 bp deletion at c561 (c.561del4), whereas the remaining 14 iPSC clones showed a wild-type sequence around c.561, proving their donor-specific origin (Figure 1C). Further genotypic analysis by semi-qPCR revealed the presence of the heterozygous mutation in the splice consensus sequence at position c.373 in exon III in all 23 iPSC clones analyzed (Figure S1A). After identification of donor- and patient-specific iPSCs, we established two heterozygous (iMSMD-het.4 or .9) and three compound heterozygous iPSC clones (iMSMD-cohet.7, .5, or .17) for further analysis. Irrespective of their origin, the iMSMD-het.4 and iMSMD-cohet.7 clones showed typical iPSC morphology in bright-field images and stained positive for alkaline phosphatase activity and TRA1-60 expression, which was similar to the human embryonic stem cell line H9 (Figure 2A). Moreover, both MSMD lines had similar surface marker expression of SSEA4 as well as TRA1-60 and showed reactivation of endogenous OCT4, NANOG, and SOX2 expression (Figures 2B–2D). Pluripotency was also proven by their ability to differentiate toward cells of the three germ layers, which was analyzed by immunofluorescence staining of β3-tubulin (ectoderm), α-fetoprotein (AFP) (endoderm), and desmin (mesoderm) (Figure 2E). Three-lineage differentiation potential was also proven in teratoma assays. Here, injection of iMSMD-het.4 and iMSMD-cohet.7 clones into immunocompromised NSGS mice led to the formation of cells derived from either endoderm (epithelium lining a luminal surface), mesoderm (muscle fibers or cartilage), or ectoderm (immature neuroepithelium) lineages (Figure 2F). Similarly, the iMSMD-het.9, iMSMD-cohet.5, and iMSMD-cohet.17 iPSC lines also fulfilled the aforementioned criteria for pluripotency (Figures S2A–S2E).
Figure 2.

Generation and Characterization of Different Patient-Specific MSMD iPSC Lines
(A) Microscopic analysis of iPSC lines derived either from heterozygous donor (iMSMD-het.4) or compound heterozygous donor (iMSMD-cohet.7) cells. The first row shows bright-field images of a human embryonic stem cell line (H9), iMSMD-het.4, and iMSMD-cohet.7. Alkaline phosphatase (AP) staining or immunohistochemistry staining for TRA1-60 is shown in the second and third row, respectively (scale bar, 500 μm).
(B) Flow-cytometric analysis of TRA1-60 (first row) and SSEA4 (second row) expression on iMSMD-het.4 and iMSMD-cohet.7 (blue line, isotype; red filled line, surface marker).
(C) qRT-PCR to evaluate endogenous expression of OCT4, NANOG, and SOX2 in MSMD iPSCs relative to H9 control cells (n = 4 independent experiments; mean ± SEM).
(D) Immunofluorescent staining of pluripotency-associated transcription factors for NANOG (green) and OCT4 (red) (scale bar, 200 μm).
(E) Immunofluorescent staining of iMSMD-het.4 and iMSD-cohet.7 iPSC derivatives on day 20 of undirected differentiation (first row represents staining performed for β3-tubulin, α-fetoprotein (AFP), and desmin (red); second row represents DAPI staining; and third row represents overlay images; scale bar, 100 μm).
(F) Representative H&E staining of teratoma formation after transplantation of iMSMD-het.4 (endoderm, epithelium lining a luminal surface; mesoderm, muscle fibers; or ectoderm, immature neuroepithelium) or iMSMD-cohet.7 (endoderm, epithelium lining a luminal surface; mesoderm, cartilage; or ectoderm, immature neuroepithelium) cells into immunocompromised NSGS mice (scale bar, 100 μm).
See also Figure S2.
Hematopoietic Differentiation of IFNγR1-Deficient iPSCs toward Macrophages
In order to study IFNγ-mediated signaling effects in patient- or donor-specific iPSC-derived macrophages, we subjected the established iPSC lines to a previously published hematopoietic differentiation protocol (Lachmann et al., 2015). Early hematopoietic specification of iMSMD-het.4 and iMSMD-cohet.7 iPSC lines resulted in the formation of myeloid-cell-forming complexes (MCFCs) within 10 days of differentiation. From day 10–15 onward, MCFCs derived from iMSMD-het.4 and iMSMD-cohet.7 iPSCs continuously produced macrophages, which could be harvested weekly in comparable quantities. After terminal differentiation, macrophages from both sources showed typical morphology in bright-field and cytospin images compared with healthy iPSC-derived macrophages (Figure 3A). Of note, throughout our studies we used the previously generated and characterized iPSC control line hCD34-iPSC16 (Ackermann et al., 2014, Lachmann et al., 2014, Lachmann et al., 2015). Detailed characterization of macrophages revealed a surface marker expression profile of CD11b+CD14+CD45+CD163+ similar to that of healthy iPSC-derived cells (Figure 3B). Moreover, macrophages from compound heterozygous or heterozygous iPSC clones displayed normal functional behavior with respect to phagocytosis of latex beads (Figure 3C). Similar observations were made when iMSMD-het.9 as well as iMSMD-cohet.5 or .17 were subjected to the macrophage generation protocol. iMSMD-derived macrophages showed typical morphology, surface marker expression profile, and the ability to phagocyte latex beads, indicating that the genetic background of our patient-specific iPSCs had no influence on the generation and differentiation potential toward macrophages (Figures S3A–S3C).
Figure 3.

Hematopoietic Differentiation of iMSMD Lines
Heterozygous (iMSMD-het.4) and compound heterozygous (iMSMD-cohet.7) iPSC lines have been differentiated toward macrophages and compared with healthy iPSC-derived macrophages (hCD34-iPSC16).
(A) Microscopic analysis of macrophages in bright-field images (left; scale bar, 100 μm) or cytospin images (right; scale bar, 20 μm) generated from iMSMD-het.4, iMSMD-cohet.7, or the control line hCD34-iPSC16.
(B) Surface marker expression evaluated by flow cytometry of CD11b, CD14, CD45, and CD163 macrophages derived from iMSMD-het.4, iMSMD-cohet.7, or the control line hCD34-iPSC16 (blue line, isotype; red filled line, surface marker).
(C) Ability of iPSC-derived macrophages to phagocytose latex beads. Left images show representative flow-cytometric analysis of macrophages (blue filled line, cells without beads; red line, cells beads). Right image shows macrophages that are positive for fluorescent latex beads (n = 4 independent experiments; mean ± SEM).
See also Figure S3.
IFNγR1-Deficient Macrophages Show Impaired IFNγ Signaling and Functionality
As a next step, we investigated the phenotypic consequence of IFNγR1 deficiency in terminally differentiated macrophages derived from healthy, iMSMD-het, or iMSMD-cohet iPSCs. Flow-cytometric analysis of IFNγR1 (CD119) revealed the absence of the receptor on macrophages derived from all iMSMD-cohet clones, whereas expression could be observed in healthy cells or macrophages harboring the heterozygous c.373+1G > T frameshift mutation only (Figures 4A and S4A). Of note, no change in delta mean fluorescent intensity of CD119 could be observed between macrophages derived from heterozygous iMSMD or healthy iPSCs (Figure 4A). In order to study the loss of IFNγR1, we next stimulated iPSC-derived macrophages with IFNγ and analyzed the downstream signaling pathway. Due to the absence of IFNγR1, macrophages derived from iMSMD-cohet.7, iMSMD-cohet.5, or iMSMD-cohet.17 iPSCs were not able to phosphorylate STAT1 in the presence of IFNγ. In contrast, macrophages derived from healthy, iMSMD-het.4, or iMSMD-het.9 iPSCs showed significant phosphorylation of STAT1 after stimulation with IFNγ (Figures 4B and S4B). Of note, background phosphorylation of STAT1 could be observed in some of the experiments, which could be explained by the residual presence of and activation by M-CSF (Novak et al., 1995) (Figure S4B). As a consequence of defective IFNγ-mediated downstream signaling, macrophages from iMSMD-cohet.7 and iMSMD-cohet.5 showed impaired upregulation of known IFNγ targets such as interferon regulatory factor 1 (IRF-1), suppressor of cytokine signaling 3 (SOCS-3), or indolamin-2,3-dioxygenase (IDO). In contrast, macrophages derived from healthy or iMSMD-het.4 iPSCs were able to induce expression of IRF-1, SOCS-3, and IDO, after stimulation with IFNγ (Figures 4C and S4C). A similar effect could be observed for the surface marker expression of MHC-class II (HLA-DR). Here, stimulation of cells with IFNγ led to the upregulation of HLA-DR on healthy, iMSMD-het.4, and iMSMD-het.9 macrophages, whereas macrophages derived from either iMSMD-cohet.7, iMSMD-cohet.5, or iMSMD-cohet.17 iPSCs showed no upregulation of HLA-DR after stimulation with IFNγ (Figures 4D and S4D). As a consequence of impaired IFNγ signaling, patients suffering from MSMD frequently show disseminated infections by weakly virulent mycobacteria such as non-tuberculous environmental mycobacteria or BCG vaccine. Moreover, MSMD patients are also prone to infections triggered by more virulent Mycobacterium tuberculosis, as well as non-typhoidal Salmonella spp. Thus, as a final assessment of IFNγ-mediated functionality, we studied the ability of iPSC-derived macrophages to intracellularly clear BCG. To this end, we challenged our iPSC-derived macrophages with BCG in the presence of IFNγ and compared the intracellular killing of iMSMD-het and iMSMD-cohet macrophages with macrophages derived from healthy hCD34-iPSC16 or macrophages derived from peripheral blood mononuclear cells (PBMCs). As expected, macrophages derived from iMSMD-cohet iPSCs showed only 20% intracellular killing activity of BCG, whereas healthy macrophages derived either from PBMC, hCD34-iPSC16, or iMSMD-het could clear around 85%, 80%, and 70% of intracellular BCG, respectively (Figure 4E).
Figure 4.

Impaired IFNγ Signaling and Functionality in iMSMD-Derived Macrophages
(A) Left images show representative flow-cytometric analysis of IFNγR1 (CD119) surface marker expression by flow cytometry on iMSMD- and hCD34-iPSC16-derived macrophages (gray filled line, unstained; red line, CD119). Right images show delta mean fluorescent intensity (MFI) of CD119 expression (delta MFI was calculated by subtracting unstained [negative] MFI from CD119 [positive] MFI) relative to unstained control (n = 3 independent experiments; mean ± SEM; ∗∗p < 0.01 by one-way ANOVA).
(B) Analysis of STAT1 phosphorylation (pSTAT1) in macrophage subsets stimulated with IFNγ. Left image shows western blot analysis of pSTAT1 (approx. 90 kDa) in iMSMD- and hCD34-iPSC16-derived macrophages (tubulin [50 kDa] served as a control). Right image shows densitometry of pSTAT1 relative to unstimulated control (n = 5 independent experiments; mean ± SEM; ∗∗p < 0.01 by one-way ANOVA).
(C) qRT-PCR of IRF-1 (first row), SOCS-3 (second row), and IDO (third row) in iMSMD- and hCD34-iPSC16-derived macrophages after stimulation with IFNγ. Values are normalized to GAPDH as a housekeeping gene (n = 3 independent experiments; mean ± SEM; ∗p < 0.05 by unpaired, two-tailed Students’ t test; n.s., not significant).
(D) Flow-cytometric analysis of MHC-II (HLA-DR) surface marker expression by flow cytometry on iMSMD- and hCD34-iPSC16-derived macrophages before (−IFNγ) and after stimulation (+IFNγ) with IFNγ (gray filled line, isotype; green line, non-stimulated control; blue line, IFNγ stimulated).
(E) Intracellular killing of Bacillus Calmette-Guérin (BCG) in iMSMD- and hCD34-iPSC16-derived macrophages. PBMC-derived macrophages were used as additional control. Intracellular killing of BCG is given as a percentage, calculated to the initial time point analyzed (1–24 hr time point). The ability of mycobacterial killing was evaluated by plating a macrophage cell suspension on Middlebrook 7H10 agar plates (n = 4 independent experiments, iMSMD-het (.4 and .9); iMSMD-cohet (.7 and .17); mean ± SEM; ∗∗p < 0.01, ∗∗∗p < 0.001 by one-way ANOVA).
See also Figure S4.
Discussion
In the present study, we established an iPSC-based disease-modeling platform for MSMD, which is based on an autosomal recessive, complete IFNγR1 deficiency. This is of high clinical relevance as current forms of treatment for patients suffering from either IFNγR1 or IFNγR2 deficiency are limited (Chantrain et al., 2006, Horwitz et al., 2003, Olbrich et al., 2015). Moreover, this study has also profound biological relevance, as the impact of IFNγR1 deficiency had never been studied in the main target cell of IFNγ, namely macrophages. Indeed, IFNγ is first and foremost a macrophage-activating-factor. Thus, we established patient-specific iPSCs from a single MSMD patient, who previously received HSCT from a healthy family member carrying a heterozygous mutation in IFNγR1 (Reuter et al., 2002, Roesler et al., 1999). Lentiviral vector-based reprogramming resulted in iPSC colonies within 15–20 days post transduction. Genotypic screening of iPSC clones revealed a colony subset of 40%, which were derived from compound heterozygous patient cells. This observation is similar to the patient's blood chimerism of approx. 50% at the time of reprogramming, demonstrating that loss of IFNγR1 had no detrimental effects on the reprogramming process. Of note, IFNγ has been proven to directly interfere with long-term repopulating HSCs and to induce proliferation in these cells (Baldridge et al., 2010, Rottman et al., 2008). While we enriched CD34+ cells for reprogramming, it remains to be elucidated whether the discrepancy of 10% in the patient blood chimera is of biological relevance in our system.
After establishing donor- and patient-specific iPSCs, we subjected the different clones to a hematopoietic differentiation protocol to generate macrophages. Here, irrespective of their genetic background, generation and IFNγ independent functionalities of iPSC-derived macrophages revealed no abnormalities. In contrast, stimulation of iMSMD-cohet macrophages with IFNγ revealed deregulated STAT1 downstream signaling, which was associated with impaired breakdown of mycobacteria. Especially the latter is of high interest, as MSMD patients suffer from severe and/or recurrent infections. In our study, we used BCG, a live attenuated strain of M. bovis, which is used as a vaccine against M. tuberculosis (Colditz et al., 1994). While BCG and other environmental mycobacteria are the most common pathogens in the context of IFNγR1 deficiency, MSMD patients are also vulnerable to other pathogens such as M. tuberculosis, Salmonella spp., or Listeria monocytogenes (Al-Muhsen and Casanova, 2008, Bustamante et al., 2014). Whether or not these co-infections are directly linked to the IFNγR1 defect is still questionable and could be addressed using our established disease-modeling platform. However, here more advanced differentiation systems would be favorable in order to study the complex role of IFNγ in both innate and adaptive immunity. iPSC-based differentiation systems that are able to generate B, T, or NK cells (Ackermann et al., 2015, French et al., 2015, Sturgeon et al., 2014) may be employed and could be combined with iPSC-derived macrophages to study the crosstalk between different immune cells in an autologous setting. Using this approach, other MSMD-causing mutations could also be investigated. Thus, iPSC-derived macrophages could be used to gain new insights into deficiencies due to mutations in IFNγR2, STAT1, NEMO, IRF8, or CYBB, whereas iPSC-derived lymphocytes could be used to study functional defects due to IL12RB1 deficiency. In addition, genome editing tools such as CRISPR/Cas9 could be used in order to correct the disease-causing mutation or to perform further loss-of-function studies on an isogenic background (Casanova et al., 2014). This being said, targeted disruption of IRF8 by CRISPR/Cas9 genome editing in iPSCs has been used to study the development of dendritic cells (Sontag et al., 2017). These genetically modified iPSCs and their isogenic controls could also be used to study the effect of IFNγ signaling in the context of MSMD.
Taken together, we provide a disease-modeling platform for MSMD due to complete IFNγR1 deficiency. Considering the importance of IFNγ signaling throughout the hematopoietic system, the established iPSC lines could be used as a screening platform to investigate new forms of treatment for MSMD. Moreover, our iPSC disease-modeling platform might also have broader applicability and may also be used to study the biology and treatment options for other mycobacterial infections, such as tuberculosis.
Experimental Procedures
Human peripheral blood samples were collected after written informed consent of the donor at Universitätsklinikum Dresden (Germany). Ethical vote no. 2127-2014 was approved by the Institutional Ethical Committee at Hannover Medical School (Hannover, Germany).
Work related to the human ESC-line H9 was approved by the Robert Koch Institut (No: 1710-79-1-4-81).
Hematopoietic Differentiation of iPSCs toward Monocytes and Macrophages
iPSC-derived macrophages were generated using a modified version of the previously established embryoid body (EB)-based hematopoietic protocol (Lachmann et al., 2014, Lachmann et al., 2015). iPSCs were disrupted by collagenase V and transferred into six-well suspension plates for EB formation in embryonic stem cell medium without basic fibroblast growth factor and 10 μM Rock inhibitor (Y-27632; Tocris, Bristol, UK) for 5 days on a rotator shaker (85 rpm). Thereafter, myeloid-cell-forming complex formation was performed in the presence of X-Vivo15 medium (Lonza, Hessisch Oldendorf, Germany) supplemented with 2 mM L-glutamine, 1% penicillin/streptomycin (PS), 50 ng/mL human macrophage colony stimulating factor (hM-CSF), and 25 ng/mL human interleukin 3 (Preprotech). The macrophages generated were harvested once a week and filtered through a 100 μM mesh. Harvested cells were cultured in RPMI1640 supplemented with 10% fetal calf serum, 2 mM L-glutamine, 1% PS, and 50 ng/mL hM-CSF.
Additional experimental procedures are provided in Supplemental Experimental Procedures.
Author Contributions
A.-L.N., J.L., and N.L. designed the study and performed experiments. K.H., S.M., N.S., A.M., M.A., M.S., C.H., M.P.K., R.G., P.B., F.P., and D.J. performed experiments and analyzed data. J.R. provided patient material and discussed the results. U.M., U.B., U.K., and A.S. provided conceptual advice and discussed the results. All authors contributed to manuscript preparation and approved the final version of the manuscript.
Acknowledgments
The authors thank Doreen Lüttge and Theresa Buchegger (both Hannover Medical School) and Kristin Laarmann (Stiftung Tierärztliche Hochschule Hannover) for excellent technical assistance. We would like to thank Juliane Wilhelmine Shott (Hannover Medical School) for preparing conceptual schemes. The authors would also like to thank Jean-Laurent Casanova (Howard Hughes Medical Institute, The Rockefeller University, and Necker Hospital for Sick Children, Paris) and Jacinta Bustamante (Necker Hospital for Sick Children, Paris) for critical comments and proofreading of the manuscript. This work was supported by grants from the Deutsche Forschungsgemeinschaft (Cluster of Excellence REBIRTH; Exc 62/1 to N.L., U.M., and A.S. as well as DFG LA 3680/2-1, Sonderforschungsbereich SFB738 to A.S.), the Else Kröner-Fresenius-Stiftung (EKFS; 2015_A92 to N.L.), the German Center for Lung Research (DZL; 82DZL00201), the Joachim Herz Stiftung (N.L.), and MH-Hannover (“Young Academy” program to N.L.).
Published: December 14, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and four figures and can be found with this article online at https://doi.org/10.1016/j.stemcr.2017.11.011.
Supplemental Information
References
- Ackermann M., Lachmann N., Hartung S., Eggenschwiler R., Pfaff N., Happle C., Mucci A., Gohring G., Niemann H., Hansen G. Promoter and lineage independent anti-silencing activity of the A2 ubiquitous chromatin opening element for optimized human pluripotent stem cell-based gene therapy. Biomaterials. 2014;35:1531–1542. doi: 10.1016/j.biomaterials.2013.11.024. [DOI] [PubMed] [Google Scholar]
- Ackermann M., Liebhaber S., Klusmann J.H., Lachmann N. Lost in translation: pluripotent stem cell-derived hematopoiesis. EMBO Mol. Med. 2015;7:1388–1402. doi: 10.15252/emmm.201505301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Muhsen S., Casanova J.L. The genetic heterogeneity of mendelian susceptibility to mycobacterial diseases. J. Allergy Clin. Immunol. 2008;122:1043–1051. doi: 10.1016/j.jaci.2008.10.037. quiz 1052–1043. [DOI] [PubMed] [Google Scholar]
- Baldridge M.T., King K.Y., Boles N.C., Weksberg D.C., Goodell M.A. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465:793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante J., Boisson-Dupuis S., Abel L., Casanova J.L. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin. Immunol. 2014;26:454–470. doi: 10.1016/j.smim.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova J.L., Conley M.E., Seligman S.J., Abel L., Notarangelo L.D. Guidelines for genetic studies in single patients: lessons from primary immunodeficiencies. J. Exp. Med. 2014;211:2137–2149. doi: 10.1084/jem.20140520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantrain C.F., Bruwier A., Brichard B., Largent V., Chapgier A., Feinberg J., Casanova J.L., Stalens J.P., Vermylen C. Successful hematopoietic stem cell transplantation in a child with active disseminated Mycobacterium fortuitum infection and interferon-gamma receptor 1 deficiency. Bone Marrow Transplant. 2006;38:75–76. doi: 10.1038/sj.bmt.1705399. [DOI] [PubMed] [Google Scholar]
- Chapman S.J., Hill A.V. Human genetic susceptibility to infectious disease. Nat. Rev. Genet. 2012;13:175–188. doi: 10.1038/nrg3114. [DOI] [PubMed] [Google Scholar]
- Colditz G.A., Brewer T.F., Berkey C.S., Wilson M.E., Burdick E., Fineberg H.V., Mosteller F. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. JAMA. 1994;271:698–702. [PubMed] [Google Scholar]
- French A., Yang C.T., Taylor S., Watt S.M., Carpenter L. Human induced pluripotent stem cell-derived B lymphocytes express sIgM and can be generated via a hemogenic endothelium intermediate. Stem Cells Dev. 2015;24:1082–1095. doi: 10.1089/scd.2014.0318. [DOI] [PubMed] [Google Scholar]
- Ginhoux F., Schultze J.L., Murray P.J., Ochando J., Biswas S.K. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat. Immunol. 2016;17:34–40. doi: 10.1038/ni.3324. [DOI] [PubMed] [Google Scholar]
- Horwitz M.E., Uzel G., Linton G.F., Miller J.A., Brown M.R., Malech H.L., Holland S.M. Persistent Mycobacterium avium infection following nonmyeloablative allogeneic peripheral blood stem cell transplantation for interferon-gamma receptor-1 deficiency. Blood. 2003;102:2692–2694. doi: 10.1182/blood-2003-04-1268. [DOI] [PubMed] [Google Scholar]
- Lachmann N., Ackermann M., Frenzel E., Liebhaber S., Brennig S., Happle C., Hoffmann D., Klimenkova O., Luttge D., Buchegger T. Large-scale hematopoietic differentiation of human induced pluripotent stem cells provides granulocytes or macrophages for cell replacement therapies. Stem Cell Reports. 2015;4:282–296. doi: 10.1016/j.stemcr.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann N., Happle C., Ackermann M., Luttge D., Wetzke M., Merkert S., Hetzel M., Kensah G., Jara-Avaca M., Mucci A. Gene correction of human induced pluripotent stem cells repairs the cellular phenotype in pulmonary alveolar proteinosis. Am. J. Respir. Crit. Care Med. 2014;189:167–182. doi: 10.1164/rccm.201306-1012OC. [DOI] [PubMed] [Google Scholar]
- Mucci A., Kunkiel J., Suzuki T., Brennig S., Glage S., Kuhnel M.P., Ackermann M., Happle C., Kuhn A., Schambach A. Murine iPSC-derived macrophages as a tool for disease modeling of hereditary pulmonary alveolar proteinosis due to Csf2rb deficiency. Stem Cell Reports. 2016;7:292–305. doi: 10.1016/j.stemcr.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak U., Harpur A.G., Paradiso L., Kanagasundaram V., Jaworowski A., Wilks A.F., Hamilton J.A. Colony-stimulating factor 1-induced STAT1 and STAT3 activation is accompanied by phosphorylation of Tyk2 in macrophages and Tyk2 and JAK1 in fibroblasts. Blood. 1995;86:2948–2956. [PubMed] [Google Scholar]
- Olbrich P., Martinez-Saavedra M.T., Perez-Hurtado J.M., Sanchez C., Sanchez B., Deswarte C., Obando I., Casanova J.L., Speckmann C., Bustamante J. Diagnostic and therapeutic challenges in a child with complete interferon-gamma receptor 1 deficiency. Pediatr. Blood Cancer. 2015;62:2036–2039. doi: 10.1002/pbc.25625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter U., Roesler J., Thiede C., Schulz A., Classen C.F., Oelschlagel U., Debatin K.M., Friedrich W. Correction of complete interferon-gamma receptor 1 deficiency by bone marrow transplantation. Blood. 2002;100:4234–4235. doi: 10.1182/blood-2002-02-0433. [DOI] [PubMed] [Google Scholar]
- Roesler J., Horwitz M.E., Picard C., Bordigoni P., Davies G., Koscielniak E., Levin M., Veys P., Reuter U., Schulz A. Hematopoietic stem cell transplantation for complete IFN-gamma receptor 1 deficiency: a multi-institutional survey. J. Pediatr. 2004;145:806–812. doi: 10.1016/j.jpeds.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Roesler J., Kofink B., Wendisch J., Heyden S., Paul D., Friedrich W., Casanova J.L., Leupold W., Gahr M., Rosen-Wolff A. Listeria monocytogenes and recurrent mycobacterial infections in a child with complete interferon-gamma-receptor (IFNgammaR1) deficiency: mutational analysis and evaluation of therapeutic options. Exp. Hematol. 1999;27:1368–1374. doi: 10.1016/s0301-472x(99)00077-6. [DOI] [PubMed] [Google Scholar]
- Rottman M., Soudais C., Vogt G., Renia L., Emile J.F., Decaluwe H., Gaillard J.L., Casanova J.L. IFN-gamma mediates the rejection of haematopoietic stem cells in IFN-gammaR1-deficient hosts. PLoS Med. 2008;5:e26. doi: 10.1371/journal.pmed.0050026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag S., Forster M., Qin J., Wanek P., Mitzka S., Schuler H.M., Koschmieder S., Rose-John S., Sere K., Zenke M. Modelling IRF8 deficient human hematopoiesis and dendritic cell development with engineered iPS cells. Stem Cells. 2017;35:898–908. doi: 10.1002/stem.2565. [DOI] [PubMed] [Google Scholar]
- Sturgeon C.M., Ditadi A., Awong G., Kennedy M., Keller G. Wnt signaling controls the specification of definitive and primitive hematopoiesis from human pluripotent stem cells. Nat. Biotechnol. 2014;32:554–561. doi: 10.1038/nbt.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka S., Takahashi K. Induction of pluripotent stem cells from mouse fibroblast cultures. Tanpakushitsu Kakusan Koso. 2006;51:2346–2351. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.