

Abstract
AIM
To evaluate the pharmacodynamics of compounds in clinical development for nonalcoholic steatohepatitis (NASH) in obese mouse models of biopsy-confirmed NASH.
METHODS
Male wild-type C57BL/6J mice (DIO-NASH) and Lepob/ob (ob/ob-NASH) mice were fed a diet high in trans-fat (40%), fructose (20%) and cholesterol (2%) for 30 and 21 wk, respectively. Prior to treatment, all mice underwent liver biopsy for confirmation and stratification of liver steatosis and fibrosis, using the nonalcoholic fatty liver disease activity score (NAS) and fibrosis staging system. The mice were kept on the diet and received vehicle, liraglutide (0.2 mg/kg, SC, BID), obeticholic acid (OCA, 30 mg/kg PO, QD), or elafibranor (30 mg/kg PO, QD) for eight weeks. Within-subject comparisons were performed on changes in steatosis, inflammation, ballooning degeneration, and fibrosis scores. In addition, compound effects were evaluated by quantitative liver histology, including percent fractional area of liver fat, galectin-3, and collagen 1a1.
RESULTS
Liraglutide and elafibranor, but not OCA, reduced body weight in both models. Liraglutide improved steatosis scores in DIO-NASH mice only. Elafibranor and OCA reduced histopathological scores of hepatic steatosis and inflammation in both models, but only elafibranor reduced fibrosis severity. Liraglutide and OCA reduced total liver fat, collagen 1a1, and galectin-3 content, driven by significant reductions in liver weight. The individual drug effects on NASH histological endpoints were supported by global gene expression (RNA sequencing) and liver lipid biochemistry.
CONCLUSION
DIO-NASH and ob/ob-NASH mouse models show distinct treatment effects of liraglutide, OCA, and elafibranor, being in general agreement with corresponding findings in clinical trials for NASH. The present data therefore further supports the clinical translatability and utility of DIO-NASH and ob/ob-NASH mouse models of NASH for probing the therapeutic efficacy of compounds in preclinical drug development for NASH.
Keywords: Nonalcoholic steatohepatitis, Disease models, Pathology, Fibrosis, Liver biopsy, Transcriptomics, Pharmacodynamics, Glucagon-like peptide-1 receptor, Peroxisome proliferator-activated receptor, Farnesoid X receptor
Core tip: The pharmacodynamics of three compounds in advanced clinical development for the treatment of nonalcoholic steatohepatitis (NASH), including liraglutide, elafibranor and obeticholic acid, were evaluated in wild-type and genetically (ob/ob) obese mouse models of NASH. Prior to treatment, all mice underwent liver biopsy for confirmation and stratification of liver steatosis and fibrosis. Within-subject comparisons were performed on changes in liver histopathology. Wild-type and ob/ob-NASH obese mice showed distinct treatment effects of liraglutide, OCA, and elafibranor, being in general agreement with corresponding findings in clinical trials for NASH. In conclusion, the two obese mouse models of NASH show clinical translatability with respect to disease etiology, histopathology and drug treatment effects, which supports their utility in preclinical drug development.
INTRODUCTION
Nonalcoholic steatohepatitis (NASH) is characterized by varying degrees of hepatic steatosis, cytoskeletal damage, and lobular inflammation with or without fibrosis[1]. The pathogenesis of NASH is complex with current hypotheses involving fatty acid-mediated lipotoxicity exhausting hepatocyte adaptive and regenerative responses, whereby accumulating oxidative stress can trigger hepatocyte necroinflammation, fibrosis, and disruption of hepatic cytoarchitecture[2,3]. Immunopathological alterations likely also play a key role in NASH, presumably driven by maladaptive responses in the innate and adaptive immune system[4,5].
The primary drivers of NASH are obesity and diabetes, and the high prevalence of these major metabolic diseases is expected to make NASH one of the most common causes of advanced liver disorders in the coming decade[6]. NASH is therefore rapidly emerging as a major public health problem, however, with currently no evidence-based approved drug therapies. This has prompted substantial efforts to identify novel pharmacological concepts for correcting the underlying metabolic deficits and alleviate, or prevent, hepatic fibrosis in NASH[7,8]. To improve the understanding of NASH pathogenesis and facilitate the development of novel therapeutics for NASH, several mouse models have been employed[8]. A number of diet-induced obesity (DIO) models mimic the natural history of NASH and have demonstrated relatively good clinical translatability with respect to key metabolic and liver pathological changes with mild- to moderate-grade liver fibrosis, and these models have therefore been increasingly used in preclinical drug development[9-13]. In comparison, non-physiological diets that are low or devoid of certain essential amino acids promote more severe liver fibrosis, but also result in significant weight loss, making these NASH models more applicable for probing drug treatment efficacy on hepatic injury and regeneration[13-16].
Notably, NASH show spontaneous and unpredictable onset with varying disease severity and different rates of progression[1]. Today, liver biopsy-confirmed pathology is the mainstay for clinical trials which allows for stratification of disease severity/stage and within-subject evaluation of endpoint liver histology[17,18]. In agreement with the clinical findings, DIO mouse models of NASH show variable disease severity. Recent reports have demonstrated that all stages of NASH are represented in mice fed Western-type obesogenic diets for any period ≥ 20 wk, and a significant proportion of up to 30% of the animals fail to develop steatohepatitis and liver fibrosis[9,13,19], indicating that individual baseline disease stage is also a critical factor in the assessment of treatment effects in DIO mouse models of NASH. However, disease state heterogeneity is often overlooked in preclinical NASH studies which may result in unintentional large variability in endpoint histopathology and narrow the window for detection of therapeutic effects. Another factor to be considered in preclinical drug development for NASH is that drug treatment periods in mouse models of NASH have varied from subacute to chronic dosing settings with histological endpoints evaluated by different qualitative and quantitative methods. Comparison of preclinical treatment effects of potential anti-NASH compounds is therefore misleading in the absence of head-to-head pharmacological studies in experimental models of NASH.
We have recently reported liver biopsy-confirmed histopathology in wild-type (DIO-NASH) and genetically obese (ob/ob-NASH) mouse models of NASH[19]. Both models display hallmarks of NASH, i.e. steatosis, lobular inflammation, hepatocellular ballooning, but are distinguished histologically by the representation of mild (DIO-NASH) or moderate (ob/ob-NASH) grade hepatic fibrosis[9,19]. The present comparative study in the DIO-NASH and ob/ob-NASH mouse models of biopsy-confirmed histopathology aimed to characterize within-subject treatment responses of compounds in current advanced clinical development for NASH, including liraglutide[20], obeticholic acid (OCA, INT-747)[21], and elafibranor (GFT-505)[22]. Liraglutide is a long-acting glucagon-like peptide 1 (GLP-1) analogue with picomolar GLP-1 receptor binding potency[23], and is approved for the management of type 2 diabetes and obesity. Whereas liraglutide improves glycemia by stimulating pancreatic cell function, its anti-obesity effects are predominantly centrally mediated through suppression of hypothalamic appetite signaling[24,25]. OCA is a semi-synthetic bile acid currently approved for the treatment of primary biliary cholangitis[26]. OCA acts as a transcriptional activator through high-affinity binding to the nuclear farnesoid X receptor (FXR)[27] to regulate bile acid synthesis and transport, hepatic lipid and carbohydrate metabolism, as well as immune function[28]. Elafibranor is a selective dual agonist for peroxisome proliferator-activated α/δ receptors (PPAR-α/δ)[29], another group of ligand-activated nuclear receptors with particularly highly abundancy in the liver. As for OCA, elafibranor exerts its major effects through transcriptional regulation of key genes involved in hepatic lipid and glucose metabolism, but also modulates hepatic inflammation and collagen turnover[30]. Liraglutide, OCA and elafibranor therefore influences different disease mechanisms in NASH, making these compounds well-suited for comparing the treatment efficacy of different therapeutic concepts in the two mouse models of NASH.
MATERIALS AND METHODS
Animals
All animal experiments were conducted according to internationally accepted principles for the care and use of laboratory animals (licence no. 2013-15-2934-00784, The Animal Experiments Inspectorate, Denmark). The animal protocol was designed to minimize pain or discomfort to the animals. Male mice were obtained from Janvier Labs (Le Genest Saint Isle, France) and housed in a controlled environment (12 h light/dark cycle, light on at 3 AM, 21 ± 2 °C, humidity 50% ± 10%). Each animal was identified by an implantable microchip (PetID Microchip, E-vet, Haderslev, Denmark). Mice had ad libitum access to tap water and either regular rodent chow (Altromin 1324, Brogaarden, Hoersholm, Denmark), or a diet high in fat (40%, containing 18% trans-fat), 40% carbohydrates (20% fructose) and 2% cholesterol (AMLN diet; D09100301, Research Diets, New Brunswick, NJ)[19]. Disease progression was characterized in wild-type DIO-NASH mice, 5 wk-old at the arrival at Gubra, and fed AMLN diet for up to 50 wk (n = 8-10 animals per time point). Effects of drug treatment were evaluated in wild-type C57BL/6J and B6.V-Lepob/JRj (ob/ob) mice 5-6 wk-old at the arrival at Gubra. C57BL/6J mice were fed chow (controls) or AMLN diet (DIO-NASH mice) for 30 wk prior to treatment start. ob/ob mice were fed chow (ob/ob controls) or AMLN diet (ob/ob-NASH mice) for 21 wk prior to treatment start. Body weight was measured daily during the treatment period.
Baseline liver biopsy
All animals included in the drug treatment experiments underwent liver biopsy, as described in detail previously[9,19]. In brief, the mice were anesthetized with isoflurane (Vetflurane®, Virbac, Kolding, Denmark) in atmospheric air. A midline abdominal incision was made to expose the left lateral lobe, and a cone-shaped biopsy of 50-100 mg liver tissue was collected and fixed in 4% paraformaldehyde overnight and subsequently used for histological assessment. The cut surfaces were electrocoagulated using an electrosurgicial unit (ERBE VIO 100C, ERBE, Marietta, GA, United States), whereupon the liver was returned to the abdominal cavity, the abdominal wall was sutured and the skin was stapled. The animals received 5 mg/kg carprofen (Norodyl®, ScanVet, Fredensborg, Denmark) prior to surgery and on post-operative day 1 and 2. Animals were single-housed after the procedure and allowed to recover for 3 wk prior to treatment start. Only mice with fibrosis stage ≥ 1 and steatosis score ≥ 2, evaluated using the clinical criteria outlined by Kleiner et al[31], were included in the study.
Drug treatment
Liraglutide (Victoza™ pen) was from Novo Nordisk (Bagsvaerd, Denmark), elafibranor and OCA were from SunshineChem (Shanghai, China). Vehicles were 0.5% carboxymethyl cellulose with 0.01 % Tween-80 (PO dosing) or phosphate-buffered saline with 0.1% bovine serum albumin (SC dosing), administered in a dosing volume of 5 mL/kg. Animals were stratified (n = 9-12 per group) based on mean fibrosis and steatosis score, and treated for 8 wk with vehicle (PO, QD), liraglutide (0.2 mg/kg, SC, BID), OCA (30 mg/kg, PO, QD), or elafibranor (30 mg/kg, PO, QD). Vehicle-dosed chow-fed mice (PO, QD) served as additional controls. The compound doses used in the present study were within the dose range reported efficacious in other mouse models of diet-induced obesity and NASH[13,30,32-35]. A terminal blood sample was collected from the tail vein in non-fasted mice and used for plasma biochemistry. Animals were sacrificed by cardiac puncture under isoflurane anesthesia. Liver samples were processed as described below.
Biochemical and histological analyses
Biochemical and histological analyses were performed as reported previously[19]. Plasma analytes included alanine aminotransferase (ALT), aspartate aminotransferase (AST), triglycerides (TG) and total cholesterol (TC). Liver homogenates were analyzed for TG and TC. Paraformaldehyde-fixed liver pre- and post-biopsies were paraffin-embedded, sectioned, and stained with hematoxylin-eosin (Dako, Glostrup, Denmark), Picro-Sirius red (Sigma-Aldrich, Broendby, Denmark), anti-type I collagen (Col1a1; Southern Biotech, Birmingham, AL), or anti-galectin-3 (Biolegend, San Diego, CA, United States). The NAFLD activity score (NAS) and fibrosis staging system was applied to liver pre-biopies and terminal samples (drug treatment experiments) or only terminal samples (disease progression experiment) for scoring of steatosis, lobular inflammation, hepatocyte ballooning, and fibrosis outlined by Kleiner et al[31]. All histological assessments were performed by a pathologist blind to treatment. Because all treatment paradigms affected total liver weight, quantitative data on liver biochemistry (liver TG, TC) and histology (liver lipid, galectin-3, Col1a1) were expressed as whole-liver amounts by multiplying individual terminal liver weight with the corresponding liver lipid concentration (biochemistry data) or percent fractional area (histology data), respectively.
RNA sequencing
Hepatic transcriptome analysis was performed by RNA sequencing on RNA extracts from terminal liver samples (15 mg fresh tissue), as described in detail previously[19]. The RNA quantity was measured using Qubit® (Thermo Scientific, Eugene, OR, United States). The RNA quality was determined using a bioanalyzer with RNA 6000 Nano kit (Agilent, Waldbronn, Germany). RNA sequence libraries were prepared with NeoPrep (Illumina, San Diego, CA, United States) using Illumina TruSeq stranded mRNA Library kit for NeoPrep (Illumina, San Diego, CA, United States) and sequenced on the NextSeq 500 (Illumina, San Diego, CA, United States) with NSQ 500 hi-Output KT v2 (75 CYS, Illumina, San Diego, CA, United States). Reads were aligned to the GRCm38 v84 Ensembl Mus musculus genome using STAR v.2.5.2a with default parameters[36]. Differential gene expression analysis was performed with DEseq2[37]. Genes with a Benjamini and Hochberg adjusted P ≤ 0.05 (5% False Discovery Rate) were regarded as statistically significantly regulated. Enrichment analysis of KEGG pathways were performed using the clusterProfiler package for R[38].
Statistical analyses
Except from RNA sequencing, data were analyzed using GraphPad Prism v5.02 software (GraphPad, La Jolla, CA, United States). All results are shown as mean ± SE. A two-way ANOVA with Bonferroni’s post-hoc test was performed for body weight analysis. Fisher’s exact test was used to test for within-subject changes in histology scores before and after treatment, compared to vehicle controls. A one-way ANOVA with Dunnett’s post-hoc test was used for all other parameters. A P-value < 0.05 was considered statistically significant.
RESULTS
Disease progression in wild-type DIO-NASH mice
Terminal samples were assessed for analysis of plasma and liver biochemistry, as well as liver morphology and quantitative histology.
Metabolic data are shown in Supplemental Figure 1. Body weight in DIO-NASH mice gradually increased over the feeding period and reached a plateau (approximately 40 g) after approximately 35 wk of dieting (Supplemental Figure 1, panel A). DIO-NASH mice developed hepatomegaly and fatty liver from dietary week 10 and onwards (panel B, F). Plasma ALT and AST increased during the dieting period, with the effect on AST being transient (panel C, D). Onset of hypercholesterolemia was evident from dieting week 10 (panel E), and liver TC levels were elevated even earlier (panel G). Liver, but not plasma, TG levels were increased from dieting wk 10-50 (panel F, H).
Figure 1.
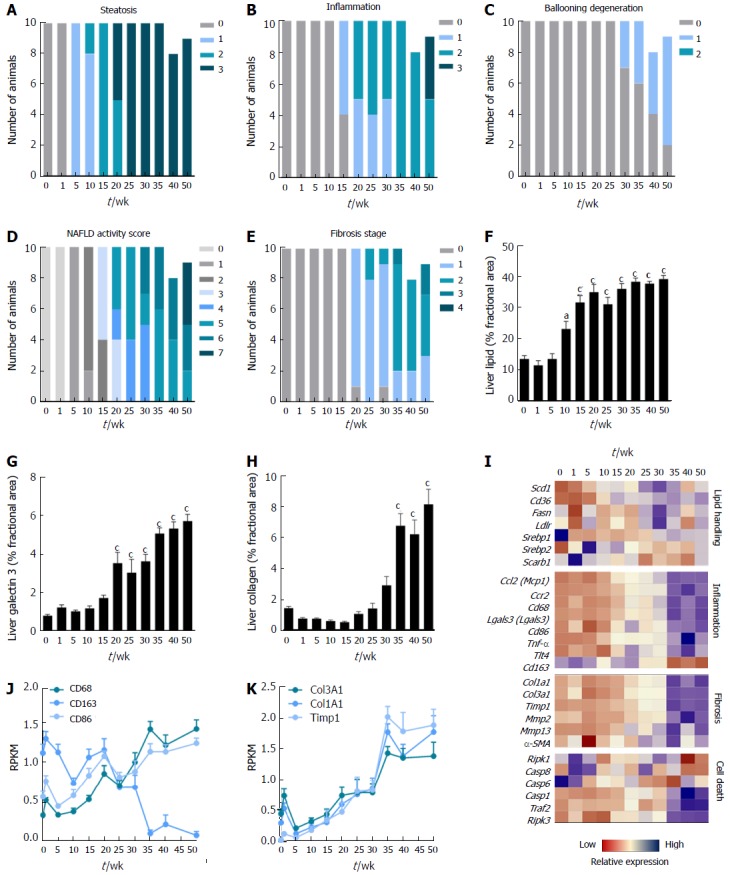
Disease progression in DIO-nonalcoholic steatohepatitis mice. A-E: Individual and composite NAFLD activity scores (NAS) and fibrosis stage; F-H: Quantitative analysis of liver lipid, galectin-3 and collagen 1a1 fractional area; I-K: Heatmap on liver transcriptome changes and expression of selected genes. aP < 0.05, cP < 0.001 vs week 0. NASH: Nonalcoholic steatohepatitis.
Histology data are presented in Figure 1. Steatosis developed gradually in DIO-NASH mice and became severe after 20-25 wk of dieting (panel A). Lobular inflammation was observed after 15 wk and progressed in severity with increasing dieting periods (panel B). Hepatocyte ballooning was detected in a few mice after 30 wk, increasing in prevalence over time, but did not progress beyond stage 1 (panel C). Manifest NASH (NAS 4-5) was consistently observed after 20-30 wk of dieting and increase in severity during the remainder of the monitoring period (panel D). Fibrosis (stage 1) was consistently detected from 20 wk of dieting. All DIO-NASH mice developed fibrosis after 35 wk (predominantly stage 2), but 20%-30% of the mice remained at stage 1 after 50 wk of dieting (panel E). Hepatic lipid amounts significantly increased from dieting week 10 and reached a maximal level from week 15 (panel F). Liver galectin-3 immunoreactivity increased after 20 wk of dieting and progressed further with increasing dieting periods (panel G). Col1a1 immunoreactivity was elevated after 35 wk of dieting and did not change further (panel H). Late-stage complications were rare in DIO-NASH mice, and occurred only after ≥ 40 wk on AMLN diet (cirrhosis and hepatic nodular formation, n = 1/19; extensive bile duct proliferation, n = 1/19, animals not included in data analysis).
A heatmap was generated for a panel of prototypical gene candidates associated with NASH pathology (Supplementary Figure 1, panel I). Signatures were markedly different during the study period, with an upregulation of genes involved in lipid handling, inflammation, fibrosis and cell death occurring over time. Expression of genes involved in lipid handling were gradually upregulated from week 20, with highest expression from week 35 wk and onwards. Gene groupings representative of monocyte/macrophage infiltration, inflammation, extracellular matrix (ECM) turnover, and apoptosis execution peaked at 35-50 wk of dieting (panel I-K).
Drug treatment in DIO-NASH mice
As expected, DIO-NASH mice gained more weight than chow-fed controls (Figure 2A). Treatment with liraglutide and elafibranor, but not OCA, resulted in weight loss (and loss of terminal whole-body fat, data not shown) which reached a maximal effect after approximately 2 wk of treatment (Figure 2A and B). Liraglutide and elafibranor, but not OCA, induced a transient reduction in daily food intake (from treatment d 1-3), whereupon food intake remained similar to vehicle-dosed DIO-NASH mice (data not shown). Whereas liraglutide and OCA reduced liver weight, elafibranor had the opposite effect (Figure 2C). Biochemical (plasma, liver) and quantitative histological (liver) analyses were applied. Liraglutide and OCA reduced total liver TG and TC content (Figure 2D and E). Only liraglutide significantly reduced plasma ALT and AST (Figure 2F and G). Liraglutide and OCA, but not elafibranor, reduced plasma TC levels (Figure 2I). None of the compounds affected plasma TG levels (Figure 2H). Liver morphometry was compared in pre- vs. post-treatment liver biopsies. All DIO-NASH mice included in the experiment showed pre-biopsy confirmed NASH (composite NAS 4-6) and liver fibrosis (stage 1-2), see Figure 3B and Supplementary Figure 2. All three compounds significantly improved NAS in DIO-NASH mice (Figure 3A and B), mainly due to reduced steatosis score (Supplementary Figure 2). In correspondence, all drug treatments also reduced total liver lipid content (Figure 4A and B). Only elafibranor improved fibrosis stage in pre- vs. post-treatment liver biopsies (P < 0.05, Figure 3D-F). In contrast, total galectin-3 and total Col1a1 levels were reduced following liraglutide and OCA, but not elafibranor, treatment (Figure 4C-F). Principal component analysis (PCA) indicated distinct uniform and stable gene transcriptional responses in the experimental groups. The major proportion of variance correlated with separation of the chow-fed and DIO-NASH controls (Figure 5A). PCA on the minor proportion of variance yielded a more mixed profile with partial separation of liraglutide and OCA effects (Figure 5B). Overall, liraglutide and OCA treatment resulted in transcriptome signatures partially resembling the lean control signature. Transcriptome changes also markedly overlapped between drug treatment groups. Notably, elafibranor induced the highest number of differentially expressed genes and evoked transcriptome modifications distinct from all other experimental groups (Figure 5A and C). Liraglutide induced conspicuous changes in transcriptional pathways associated with glucose metabolism, but had virtually no impact on gene groupings representing other liver metabolic pathways (Figure 5D). OCA modulated gene groupings linked to glucose metabolism, but also affected signaling pathways associated with NAFLD, ECM-receptor interaction, PI3 kinase signaling, and focal adhesion. Pathways perturbations promoted by elafibranor treatment mapped to NAFLD, lipid metabolism, oxidative phosphorylation, PPAR and peroxisome signaling. All drug treatments reduced expression of gene markers involved in inflammation, fibrogenesis, and apoptosis (Figure 5E and F).
Figure 2.
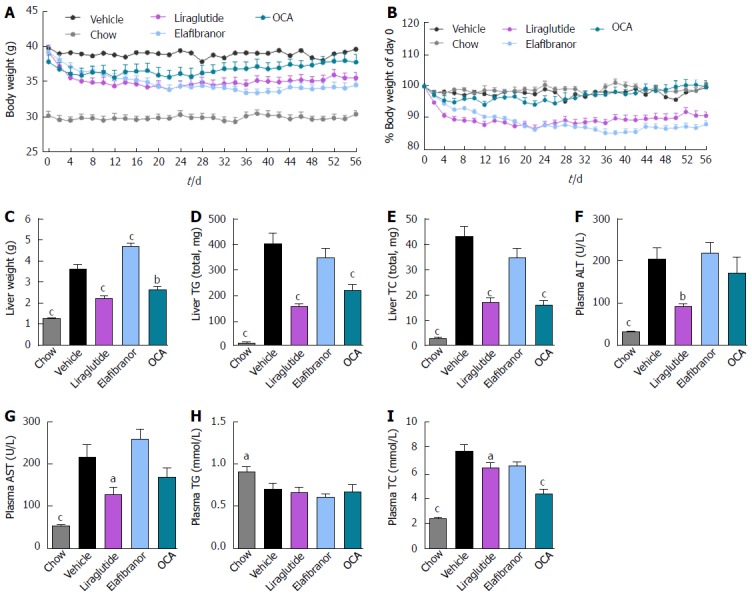
Metabolic effects of liraglutide, obeticholic acid, and elafibranor treatment in DIO-nonalcoholic steatohepatitis mice. aP < 0.05, bP < 0.01, cP < 0.001 vs vehicle controls; OCA: Obeticholic acid; NASH: Nonalcoholic steatohepatitis.
Figure 3.
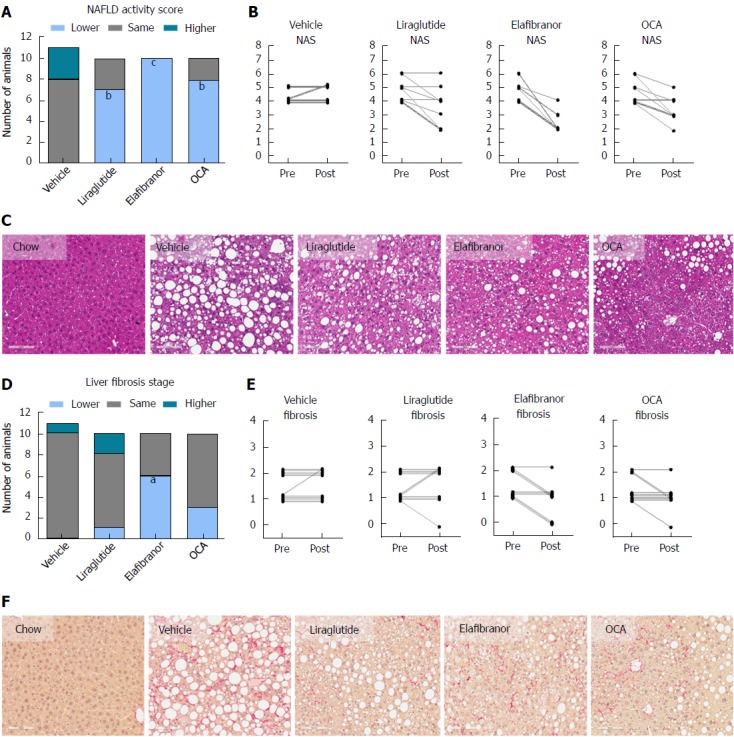
Histomorphological effects of liraglutide, obeticholic acid, and elafibranor treatment in DIO-nonalcoholic steatohepatitis mice. aP < 0.05, bP < 0.01, cP < 0.001 vs vehicle controls. OCA: Obeticholic acid; NASH: Nonalcoholic steatohepatitis.
Figure 4.
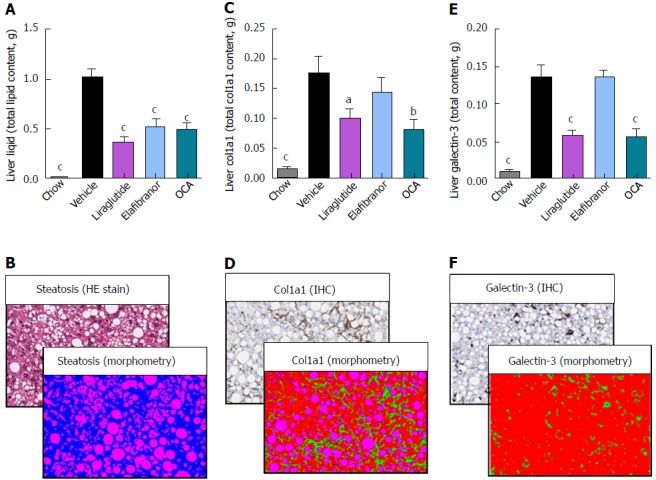
Quantitative histological assessment of liver lipids, collagen deposition and inflammation in DIO-nonalcoholic steatohepatitis mice. aP < 0.05, bP < 0.01, cP < 0.001 vs vehicle controls. OCA: Obeticholic acid; NASH: Nonalcoholic steatohepatitis.
Figure 5.
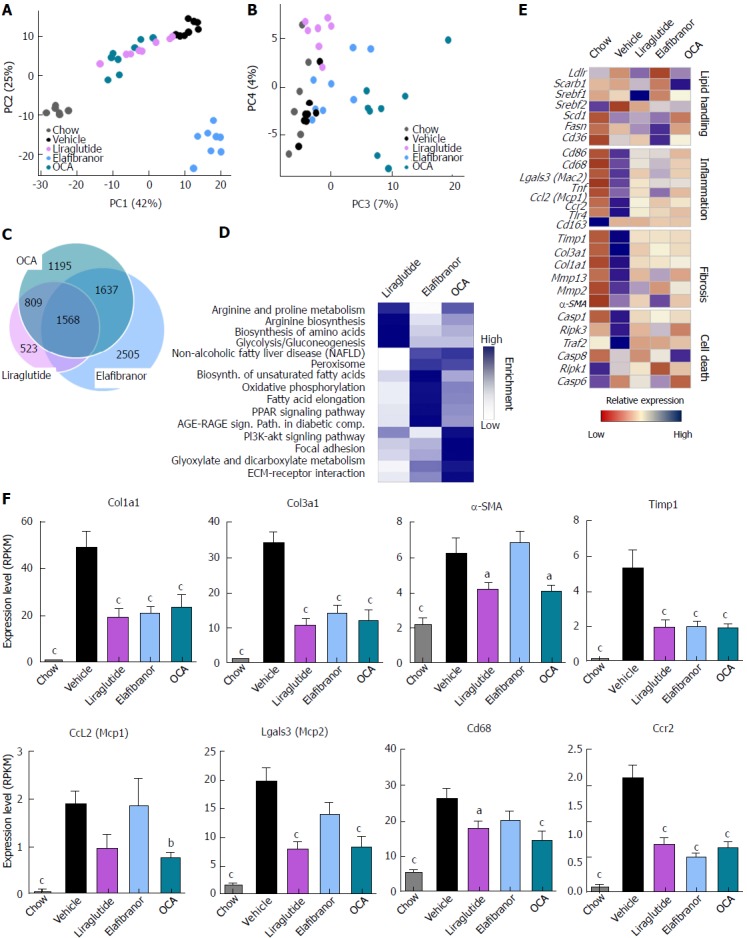
Hepatic global gene expression analysis in DIO-nonalcoholic steatohepatitis mice. A and B: Principal component analysis; C: Venn diagram of differentially expressed genes, compared to vehicle treatment; D: Overview of KEGG pathway-enriched gene groups; E: Relative expression of prototypic NASH genes; F: Relative expression of selected genes. aP < 0.05, bP < 0.01, cP < 0.001 vs vehicle controls. OCA: Obeticholic acid; NASH: Nonalcoholic steatohepatitis.
Drug treatment in ob/ob-NASH mice
ob/ob-NASH controls were obese prior to treatment (51.7 ± 1.1 g, n = 12), but showed lower body weight compared to age-matched chow-fed ob/ob controls (60.1 ± 1.3 g, n = 9) (Figure 6A). Terminal liver weight was markedly higher in ob/ob-NASH control mice compared to chow-fed controls (Figure 6A-C). Liraglutide and elafibranor treatment progressively reduced body weight with a maximal weight loss of approximately 10% compared to baseline (approximately 20% vs vehicle-dosing), see Figure 6B. OCA did not affect body weight in ob/ob-NASH mice. Liraglutide, but not OCA and elafibranor, induced a transient reduction in daily food intake (from treatment d 1-7), whereupon food intake remained similar to vehicle-dosed ob/ob-NASH mice (data not shown). Liraglutide and OCA, but not elafibranor, reduced terminal liver weight (Figure 6C). Liraglutide reduced total liver TG content (Figure 6D) as well as plasma TG levels (Figure 6H), while elafibranor and OCA reduced total liver TC content (Figure 6E). All compounds lowered plasma TC levels (Figure 6I). Liraglutide and elafibranor, but not OCA, reduced plasma ALT and AST (Figure 6F and G). All ob/ob-NASH mice included in the experiment showed pre-biopsy confirmed NASH (composite NAS 4-7) and liver fibrosis (stage 2-3), see Figure 7B and Supplementary Figure 3. Elafibranor and OCA significantly reduced composite NAS, total liver fat and galectin-3 content in ob/ob-NASH mice (Figure 7A, B, E and F). Liver morphometry was compared in pre- vs. post-treatment liver biopsies. While elafibranor reduced NAS largely by improving steatosis and inflammation scores, OCA also consistently reduced ballooning (Supplementary Figure 3). Only elafibranor significantly reduced hepatic fibrosis stage (Figure 7C and D), but did not change total levels of Col1a1 (Figure 7F). Liraglutide had no effect on any individual NAS component, as well as composite NAS, and did not alter the fibrosis stage (Figure 7A and B, Supplementary Figure 3). In contrast, liraglutide significantly reduced total liver levels of lipids, galectin-3 and Col1a1 (Figure 7E-G).
Figure 6.
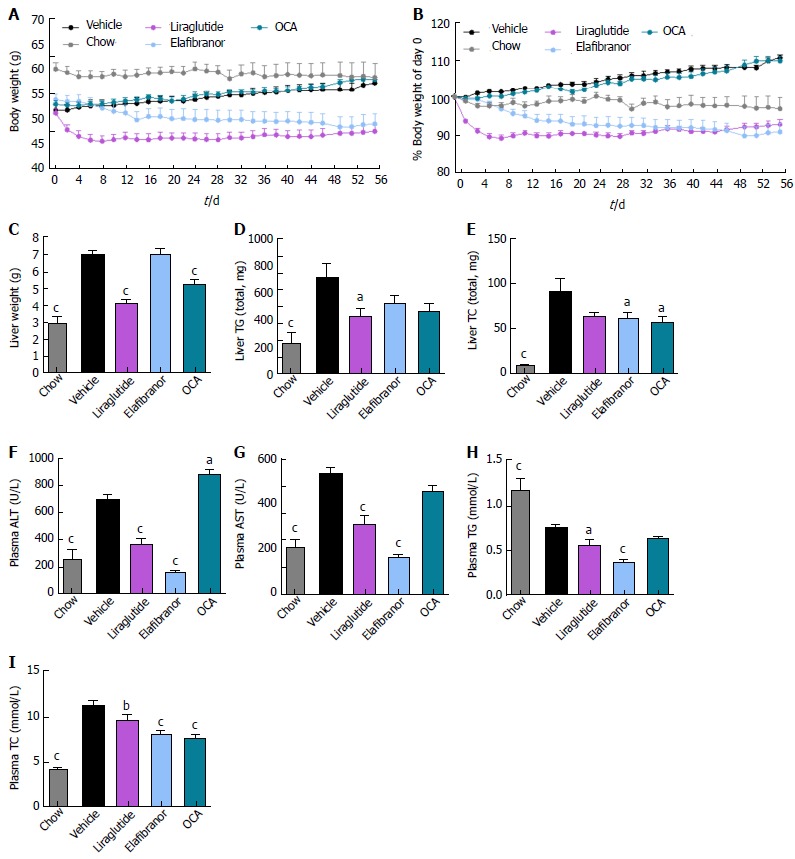
Metabolic effects of liraglutide, obeticholic acid, and elafibranor treatment in ob/ob-nonalcoholic steatohepatitis mice. aP < 0.05, bP < 0.01, cP < 0.001 vs vehicle controls. OCA: Obeticholic acid; NASH: Nonalcoholic steatohepatitis.
Figure 7.
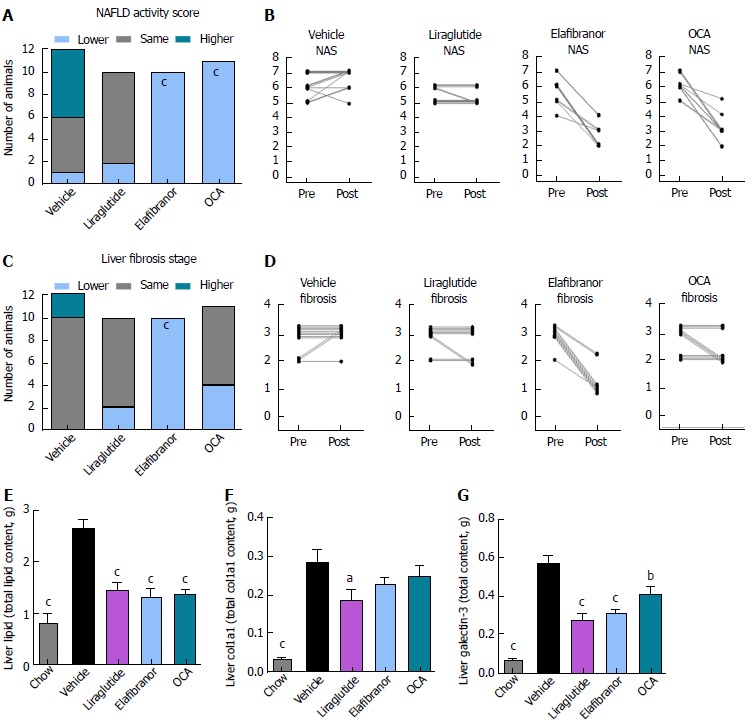
Histomorphological effects of liraglutide, obeticholic acid, and elafibranor treatment in ob/ob-nonalcoholic steatohepatitis mice. aP < 0.05, bP < 0.01, cP < 0.001 vs vehicle controls. OCA: Obeticholic acid; NASH: Nonalcoholic steatohepatitis.
DISCUSSION
We and others have previously reported that DIO-NASH mice develop low-grade liver fibrosis when maintained on a trans-fat containing obesogenic diet[9,19,39]. To obtain further information on the pathology in this translational mouse model of NASH we initially performed a detailed investigation on the disease onset and progression in DIO-NASH mice, followed by comparison of the treatment effects of liraglutide, OCA and elafibranor in both DIO-NASH and ob/ob-NASH mice.
DIO-NASH mice exhibited a relatively rapid onset of body weight gain with concurrent liver TG and TC accumulation and progressive development of hepatomegaly. DIO-NASH mice developed hypercholesterolemia, but not hypertriglyceridemia. This is a consistent finding DIO-NASH mice[9,19], as well as in ob/ob-NASH mice[10,19] and other Western diet-based mouse models of NASH[12,40]. In contrast, high-fat diets with lower cholesterol content (0.2%) are reported to induce combined hypertriglyceridemia and hypercholesterolemia in obese mice[11,41,42], suggesting that elevated dietary cholesterol influences hepatocyte TG secretion, perhaps by modifying cholesterol ester and lipoprotein synthesis[40,43]. Hepatopathology was further indicated by increased levels of plasma transaminases, most consistently observed for ALT. As persistently elevated ratios of AST:ALT has been reported to associate to advanced fibrotic NASH but not at milder disease stages[44], the moderate changes in ALT and AST levels in DIO-NASH (and ob/ob-NASH) mice presumably reflects the mild-to-moderate disease severity in these two models. Histological morphometry suggested a sequential onset of NASH pathology in DIO-NASH mice, with initial steatosis progressing to inflammation and ballooning degeneration (from week 30). Quantitative histology on steatosis (HE staining), inflammation (galectin-3) and fibrosis (Col1a1) supported a distinct temporal sequence of hepatopathological events in DIO-NASH mice. In correspondence, transcriptome pathway analyses revealed initial gene signatures of upregulated hepatic lipid synthesis and handling, over enhanced inflammation signaling to recruitment of monocytes/myofibroblasts, stimulation of fibrogenesis and extracellular matrix turnover. These findings are in general concordance with recent hypotheses that mounting lipotoxicity constitutes a triggering event for progression of NAFLD into NASH[45,46].
In agreement with previous findings[9,19], all morphological hallmarks of NASH were consistently present in DIO-NASH mice at ≥ 25 wk of dieting (NAS ≥ 4-5, corresponding to the clinical criteria for NASH[31]). NAS was largely driven by progressive changes in hepatosteatosis and inflammation as affected mice only exhibited marginal hepatocyte ballooning. The absence of conspicuous hepatocyte ballooning is in accordance with a recent comprehensive histological scoring study concluding that obese rodent models of NASH do not entirely meet the histomorphological criteria for human hepatocyte ballooning[47]. As is seen in the clinic, DIO-NASH mice represented all stages of NAFLD and fibrosis for any dieting period ≥ 20 wk, which confirms the highly individual fibrosis progression in this model[9,19]. Notably, up to 30% of the animals failed to develop steatohepatitis and fibrosis, and the severity in histopathology did not become more homogenous by extending AMLN dieting periods. Similar disease variability has been reported in other DIO mouse models of NASH[12,13,48], which therefore likely generalizes to all obese mouse models of fibrotic steatohepatitis. Therefore, the use of a prebiopsy for selection and stratification of in DIO-NASH models not only substantiates the efficacy readouts, but also allows for the employment of clinically relevant inclusion criteria and stratification procedures.
In the present pharmacological study, inherent differences in NASH pathology were therefore considered by introducing liver biopsy-based histology for evaluating NASH severity in all individual mice prior to introduction of treatment. Only mice with biopsy-confirmed fibrotic NASH were included in the study, and randomization and stratification procedures were applied to equalize mean levels of baseline NAS and fibrosis stage across treatment groups. The present study evaluated the pharmacodynamics of three individual compounds in current advanced clinical development for NASH. The selected compounds are representative of different drug classes and therapeutic concepts and included agonists for the GLP-1 receptor (liraglutide), FXR (OCA) and PPAR-α/δ (elafibranor)[20-22]. We have recently reported a comparative liver biopsy study on DIO-NASH and ob/ob-NASH mice, which confirmed that ob/ob-NASH mice generally display more marked fibrotic steatohepatitis[19]. Thus, potential differences in therapeutic efficacies as a function of overall severity in hepatopathology were addressed by applying identical treatment protocols to both DIO-NASH and ob/ob-NASH mice.
The GLP-1 analog, liraglutide, which is currently approved for the treatment of type 2 diabetes and obesity[24,25], reduced weight gain in both DIO-NASH and ob/ob-NASH mice with concurrent improvements in whole-body fat mass, hepatomegaly, liver lipid deposition, and hypercholesterolemia, which is consistent with findings in other obese mouse models of NASH[33,41]. The intrahepatic lipid-lowering effects of liraglutide are likely secondary to the progressive weight loss promoted by the GLP-1 agonist[49]. This is relevant, as weight loss per se is known to mediate benefits on NAFLD endpoints[50]. The present findings are also in agreement with a previous study assessing the effect of an exendin-4 analogue in ob/ob-NASH mice[10]. Liraglutide showed most marked effects on hepatic gene transcriptional signatures associated to the canonical effects of GLP-1 receptor agonists on hepatic glucose metabolism and production. The liver transcriptome analyses in DIO-NASH mice also suggested that liraglutide-induced improvements in lipid metabolism was linked to activation of signaling pathways involved in fatty acid degradation, oxidative phosphorylation and cholesterol handling. Perturbations in these signaling pathways could therefore potentially trigger gene expression programs that favored reduced hepatic free fatty acid uptake, as well as improving the clearance of cholesterol and fatty acids, all of which have been implicated in the pathogenesis of NASH. In contrast to OCA and elafibranor, liraglutide did not improve composite NAS in ob/ob-NASH mice. In addition, liraglutide treatment did not influence fibrosis and low-grade hepatocyte ballooning in either model, indicating that GLP-1 receptor mediated amelioration of hepatic lipid overload was perhaps not sufficient to reduce morphological indices of more advanced NASH. In contrast, total hepatic galectin-3 and Col1a1 levels were significantly lowered in both DIO-NASH and ob/ob-NASH mice after liraglutide treatment, consistent with the reduction in hepatomegaly. Overall, the pharmacodynamics of liraglutide in the DIO-NASH mice are in general agreement with a recent phase-II (LEAN) trial for NASH, where liraglutide treatment promoted a significant weight loss (approximately 5%) and improved NAS (lowered steatosis score) without worsening of fibrosis stage[20]. GLP-1 receptor agonists have not shown significant effects on liver fibrosis morphology in various mouse models of NASH[8]. Future studies must therefore aim to clarify if extended dosing periods are required for detecting antifibrotic efficacy of liraglutide, or whether the lack of a direct hepatic action could be a limiting factor for influencing liver fibrosis in obese mouse models of NASH.
OCA is a first-in-class selective synthetic FXR agonist that is approximately 100-fold more potent than its natural bile acid homologue[27]. Nuclear FXRs are abundantly expressed in the liver, intestine, and adipose tissue, serving as primary sensors for regulating enterohepatic bile acid flow. In the present study, OCA reduced hepatomegaly and NAS scores in both DIO-NASH and ob/ob-NASH mice. FXR agonists, including OCA, have shown similar effects in genetic and methinonine-choline deficient (MCD) dietary models of NASH[13,15,51]. The improvement of steatohepatitis in DIO-NASH mice corresponded to reduced expression of genes that are important transcriptional targets of FXRs and regulate hepatocyte lipid metabolism and macrophage activity[52]. Whereas several genes involved in cholesterol and lipoprotein metabolism are transcriptional targets of hepatocyte FXRs[53], it is not resolved whether FXR stimulation directly activates master transcription factors that controls the expression of genes involved in fatty acid biosynthesis[54]. As FXRs also suppress transcriptional activity of several carbohydrate responsive genes, it is assumed that FXR reduces TG levels mainly through increasing plasma lipoprotein clearance and improving hepatic insulin resistance[52-54]. The anti-inflammatory effects of FXR are well-established. Accordingly, several studies have reported that FXR agonists, including OCA, attenuate hepatic inflammation in high-fat fed obese rodents[15,51,55,56], and Fxr-null mice are highly sensitive to develop severe steatohepatitis when fed an obesogenic diet[57]. OCA treatment also reversed mild-stage hepatocyte ballooning in affected ob/ob-NASH mice. OCA has previously been reported to reverse low-grade hepatocyte ballooning in mice fed an atherogenic diet[13], and a different semisynthetic FXR agonist (INT-767) has shown a similar effect in non-fibrotic db/db mice[51]. Interestingly, enhanced hepatocyte ballooning is also reported in FXR-deficient hypercholesterolemic mice[57], thus lending further support to FXR-dependent hepatocyte protective effects. As for liraglutide, OCA treatment did not influence fibrosis morphometry, but reduced total hepatic Col1a1 expression due to reduced liver weight. A recent study in ob/ob-NASH mice (fed AMLN diet for 8 wk) and treated with a similar daily dose of OCA for 4 wk also indicated no effect on liver fibrosis[58]. In contrast, antifibrotic effects of synthetic FXR agonists, including OCA, have been demonstrated in more aggressive models of fibrotic NASH[15,59]. It should, however, be noted that OCA reduced all NAS components in ob/ob-NASH mice, suggesting that prolonged OCA treatment might lead to improved fibrosis stage in this model. The present data are in accordance with a recent phase-II (FLINT) trial on OCA treatment for NASH[21], reporting significant improvements of all NAS components without worsening of fibrosis. The FLINT trial also indicated no anti-obesity potential of OCA, analogous to the observations in the present study.
PPARs constitute another family of ligand-activated nuclear receptors. PPARs are abundantly expressed in the liver (PPAR-α, PPAR-β/δ) and adipose tissue (PPAR-δ, PPAR-γ), being prominent regulators of lipid and glucose metabolism, but are also functional in immune cells, including monocytes and macrophages (PPAR-γ)[60]. Several subtype-selective PPAR agonists have been demonstrated to reduce experimental fibrotic steatohepatitis, particularly in MCD mice[8]. In contrast, there is a sparsity in studies addressing effects of PPAR agonists in more translational mouse models of NASH. In DIO-NASH mice, elafibranor showed similar efficacy in reducing body weight and steatosis compared to liraglutide, however, the metabolic effects of elafibranor were not accompanied by improvements in liver weight. Accordingly, hepatomegaly was either accentuated (DIO-NASH mice) or unaltered (ob/ob-NASH mice) with elafibranor treatment. RNA sequencing analysis indicated stimulation of PPAR and peroxisomal signaling pathways in DIO-NASH mice, and hepatocyte peroxisome proliferation and tumorigenesis have been consistent findings in rodent studies with PPAR-α agonists (including elafibranor)[30], but not in humans[61]. Notably, elafibranor improved NAS scores with concomitant reductions in fibrosis stage, also supported by corresponding liver transcriptome changes. In contrast, total Col1a1 and galectin-3 immunoreactivity was unaffected in elafibranor-treated DIO-NASH mice due to the liver hypertrophic effect. Elafibranor treatment also promoted marked effects on hepatic signaling pathways associated with lipid metabolism, mitochondrial energy harvesting, inflammation, fibrogenesis and execution of cell death programs. Other PPAR agonists, including fibrates (bezafibrate, Wy-14,463; PPAR-α ligands), thiazolidinediones (rosiglitazone, PPAR-γ ligand) and GW501516 (PPAR-β/δ ligand), have been reported to reduce fibrotic steatohepatitis in MCD mice[14,62,63]. PPAR stimulation is suggested to ameliorate NASH primarily by enhancing β-oxidation associated hepatic fatty acid disposal, but anti-inflammatory and antifibrotic mechanisms might also contribute to improvements in liver histology independent on reduced hepatocyte lipid accumulation[63,64]. In the present study, dual PPAR-α/δ stimulation by elafibranor may thus improve fibrosis stage in DIO-NASH and ob/ob-NASH mice by resolving intrahepatic lipid deposition but also through engagement of lipid metabolism-independent signaling pathways. The pharmacokinetics of elafibranor has not been reported in detail, but a preliminary study in rats indicated elafibranor excretion in the bile suggestive of extensive enterohepatic cycling[29]. Elafibranor may therefore be considered liver-targeted, which could enhance treatment efficacy in DIO-NASH and ob/ob-NASH mice. The present data are in overall agreement with a recent clinical phase-II study (GOLDEN-505) on elafibranor treatment for NASH[22]. Interestingly, the improvements in mean NAS (approximately 2.5 points) and fibrosis score (approximately 0.5 points) in elafibranor-treated NASH patients achieving the modified primary outcome (no fibrosis worsening) was on par with the reductions in NAS and fibrosis scores determined in DIO-NASH and ob/ob-NASH mice. In contrast to the weight neutral effect of elafibranor in NASH patients, elafibranor markedly reduced body weight in both DIO-NASH and ob/ob-NASH mice. Whereas body weight loss has not been reported with PPAR agonist treatment in humans[65], stimulated PPAR-α and PPAR-δ function has been associated with weight loss, appetite suppression and reduced tissue lipid deposition in obesity-prone mice[66-68], which could imply species-dependent metabolic effects of PPAR agonists.
In conclusion, the present comparative pharmacological study in diet-induced and genetically obese mouse models of biopsy-confirmed NASH indicated both shared and distinct anti-NASH effects of liraglutide, OCA, and elafibranor. The anti-NASH effects of the compounds corresponded well to their individual modes of action, and were also in concordance to histological findings in clinical trials for NASH. The present data therefore further supports the clinical translatability and utility of DIO-NASH and ob/ob-NASH mouse models of NASH in preclinical drug development.
ARTICLE HIGHLIGHTS
Research background
Although various diet-induced obese mouse models of nonalcoholic steatohepatitis (NASH) are highly applicable in preclinical drug development, none of these models have so far been systematically evaluated with respect to comparing individual pharmacodynamics of several important compound classes in current clinical development for NASH.
Research motivation
Comparison of preclinical treatment effects of potential anti-NASH compounds is currently hampered by the absence of head-to-head pharmacological studies in experimental models of NASH. Moreover, baseline NASH disease heterogeneity is often overlooked in pharmacological studies which may introduce unintentional variability in treatment responses, thereby narrowing the window for detection of therapeutic effects of potential anti-NASH compounds.
Research objectives
The present study aimed to characterize within-subject treatment responses to liraglutide, obeticholic acid and elafibranor in two obese mouse models of biopsy-confirmed NASH.
Research methods
Comparative treatment studies were conducted in male wild-type mice (DIO-NASH) and Lepob/ob (ob/ob-NASH) mice fed a diet high in trans-fat, fructose and cholesterol. Liver biopsy was applied for stratification of liver steatosis and fibrosis, using the nonalcoholic fatty liver disease activity score (NAS) and fibrosis staging system. Individual biopsy-confirmed histopathology also allowed for evaluation of within-subject treatment responses based on differences in baseline vs. endpoint histomorphometry. DIO-NASH and ob/ob-NASH mice were treated with vehicle, liraglutide, obeticholic acid, or elafibranor for 8 weeks. Metabolic, histomorphological and quantitative histological effects of each compound were compared in the two models of biopsy-confirmed NASH.
Research results
Only liraglutide and elafibranor reduced body weight in DIO-NASH and ob/ob-NASH mice. Liraglutide improved steatosis scores in DIO-NASH mice only. Whereas elafibranor and OCA both reduced hepatic steatosis and inflammation scores in both models, only elafibranor also improved fibrosis stage. Owing to a marked reduction in liver weight, liraglutide and OCA lowered total liver fat, collagen 1a1 and galectin-3 content. Individual drug effects on NASH histological endpoints were supported by global gene expression (RNA sequencing) and liver lipid biochemistry.
Research conclusions
DIO-NASH and ob/ob-NASH mice show good clinical translatability with respect to disease etiology, histopathology and therapeutic effects of compounds in late-stage clinical development for NASH.
Research perspectives
The present data supports the utility of DIO-NASH and ob/ob-NASH mice for preclinical evaluation of the treatment efficacy of potential novel anti-NASH compounds. As also applied in clinical trials for NASH, biopsy-confirmed histopathology in the two obese mouse models of NASH allows for stratification of disease severity prior to treatment start and improves detection of treatment efficacy of test compounds by considering within-subject responses.
ACKNOWLEDGEMENTS
The authors would like to thank Louise D Fensholdt, Dan Hemmingsen, Martin Illemann, Rasmus Lind and Chen Zhang for skillful technical assistance.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Denmark
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): C, C
Grade D (Fair): D
Grade E (Poor): 0
Supported by Innovation Fund Denmark, KST; No. 5016-00168B; and MNBK, No. 5189-00040B.
Institutional animal care and use committee statement: All animal experiments conformed to the internationally accepted principles for the care and use of laboratory animals (Licence No. 2013-15-2934-00784, The Animal Experiments Inspectorate, Denmark).
Conflict-of-interest statement: The authors declare no conflict of interest.
Data sharing statement: No additional data are available.
Peer-review started: September 25, 2017
First decision: November 3, 2017
Article in press: December 5, 2017
P- Reviewer: Balaban YH, Murotomi K, Pan Q, Sutti S, Tanaka N S- Editor: Gong ZM L- Editor: A E- Editor: Ma YJ
Contributor Information
Kirstine S Tølbøl, Gubra Aps, Hørsholm DK-2970, Denmark; Department of Biomedical Sciences, Faculty of Health Sciences, University of Copenhagen, Copenhagen DK-2200, Denmark; Section for Metabolic Imaging and Liver Metabolism, The Novo Nordisk Foundation Center for Basic Metabolic Research, Faculty of Health Sciences, University of Copenhagen, Copenhagen DK-2200, Denmark.
Maria NB Kristiansen, Gubra Aps, Hørsholm DK-2970, Denmark. kst@gubra.dk; Department of Biomedical Sciences, Faculty of Health Sciences, University of Copenhagen, Copenhagen DK-2200, Denmark.
Henrik H Hansen, Gubra Aps, Hørsholm DK-2970, Denmark.
Sanne S Veidal, Gubra Aps, Hørsholm DK-2970, Denmark.
Kristoffer TG Rigbolt, Gubra Aps, Hørsholm DK-2970, Denmark..
Matthew P Gillum, Section for Metabolic Imaging and Liver Metabolism, The Novo Nordisk Foundation Center for Basic Metabolic Research, Faculty of Health Sciences, University of Copenhagen, Copenhagen DK-2200, Denmark.
Jacob Jelsing, Gubra Aps, Hørsholm DK-2970, Denmark.
Niels Vrang, Gubra Aps, Hørsholm DK-2970, Denmark; Department of Chemistry, Faculty of Science, University of Copenhagen, Copenhagen DK-2200, Denmark.
Michael Feigh, Gubra Aps, Hørsholm DK-2970, Denmark.
References
- 1.Bedossa P. Pathology of non-alcoholic fatty liver disease. Liver Int. 2017;37 Suppl 1:85–89. doi: 10.1111/liv.13301. [DOI] [PubMed] [Google Scholar]
- 2.Rosso N, Chavez-Tapia NC, Tiribelli C, Bellentani S. Translational approaches: from fatty liver to non-alcoholic steatohepatitis. World J Gastroenterol. 2014;20:9038–9049. doi: 10.3748/wjg.v20.i27.9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berlanga A, Guiu-Jurado E, Porras JA, Auguet T. Molecular pathways in non-alcoholic fatty liver disease. Clin Exp Gastroenterol. 2014;7:221–239. doi: 10.2147/CEG.S62831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sutti S, Jindal A, Bruzzì S, Locatelli I, Bozzola C, Albano E. Is there a role for adaptive immunity in nonalcoholic steatohepatitis? World J Hepatol. 2015;7:1725–1729. doi: 10.4254/wjh.v7.i13.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ganz M, Szabo G. Immune and inflammatory pathways in NASH. Hepatol Int. 2013;7 Suppl 2:771–781. doi: 10.1007/s12072-013-9468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- 7.Rotman Y, Sanyal AJ. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut. 2017;66:180–190. doi: 10.1136/gutjnl-2016-312431. [DOI] [PubMed] [Google Scholar]
- 8.Hansen HH, Feigh M, Veidal SS, Rigbolt KT, Vrang N, Fosgerau K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov Today. 2017;22:1707–1718. doi: 10.1016/j.drudis.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 9.Clapper JR, Hendricks MD, Gu G, Wittmer C, Dolman CS, Herich J, Athanacio J, Villescaz C, Ghosh SS, Heilig JS, et al. Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am J Physiol Gastrointest Liver Physiol. 2013;305:G483–G495. doi: 10.1152/ajpgi.00079.2013. [DOI] [PubMed] [Google Scholar]
- 10.Trevaskis JL, Griffin PS, Wittmer C, Neuschwander-Tetri BA, Brunt EM, Dolman CS, Erickson MR, Napora J, Parkes DG, Roth JD. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am J Physiol Gastrointest Liver Physiol. 2012;302:G762–G772. doi: 10.1152/ajpgi.00476.2011. [DOI] [PubMed] [Google Scholar]
- 11.Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, Kumar DP, Daita K, Min HK, Mirshahi F, et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J Hepatol. 2016;65:579–588. doi: 10.1016/j.jhep.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krishnan A, Abdullah TS, Mounajjed T, Hartono S, McConico A, White T, LeBrasseur N, Lanza I, Nair S, Gores G, et al. A longitudinal study of whole body, tissue, and cellular physiology in a mouse model of fibrosing NASH with high fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol. 2017;312:G666–G680. doi: 10.1152/ajpgi.00213.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haczeyni F, Poekes L, Wang H, Mridha AR, Barn V, Geoffrey Haigh W, Ioannou GN, Yeh MM, Leclercq IA, Teoh NC, et al. Obeticholic acid improves adipose morphometry and inflammation and reduces steatosis in dietary but not metabolic obesity in mice. Obesity (Silver Spring) 2017;25:155–165. doi: 10.1002/oby.21701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. 2004;39:1286–1296. doi: 10.1002/hep.20170. [DOI] [PubMed] [Google Scholar]
- 15.Zhang S, Wang J, Liu Q, Harnish DC. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol. 2009;51:380–388. doi: 10.1016/j.jhep.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 16.Lee JH, Kang YE, Chang JY, Park KC, Kim HW, Kim JT, Kim HJ, Yi HS, Shong M, Chung HK, et al. An engineered FGF21 variant, LY2405319, can prevent non-alcoholic steatohepatitis by enhancing hepatic mitochondrial function. Am J Transl Res. 2016;8:4750–4763. [PMC free article] [PubMed] [Google Scholar]
- 17.Sanyal AJ, Friedman SL, McCullough AJ, Dimick-Santos L; American Association for the Study of Liver Diseases; United States Food and Drug Administration. Challenges and opportunities in drug and biomarker development for nonalcoholic steatohepatitis: findings and recommendations from an American Association for the Study of Liver Diseases-U.S. Food and Drug Administration Joint Workshop. Hepatology. 2015;61:1392–1405. doi: 10.1002/hep.27678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ratziu V, Bellentani S, Cortez-Pinto H, Day C, Marchesini G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J Hepatol. 2010;53:372–384. doi: 10.1016/j.jhep.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Kristiansen MN, Veidal SS, Rigbolt KT, Tølbøl KS, Roth JD, Jelsing J, Vrang N, Feigh M. Obese diet-induced mouse models of nonalcoholic steatohepatitis-tracking disease by liver biopsy. World J Hepatol. 2016;8:673–684. doi: 10.4254/wjh.v8.i16.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, Hazlehurst JM, Guo K; LEAN trial team, Abouda G, Aldersley MA, Stocken D, Gough SC, Tomlinson JW, Brown RM, Hübscher SG, Newsome PN. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387:679–690. doi: 10.1016/S0140-6736(15)00803-X. [DOI] [PubMed] [Google Scholar]
- 21.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956–965. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, Romero-Gomez M, Boursier J, Abdelmalek M, Caldwell S, et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology. 2016;150:1147–1159.e5. doi: 10.1053/j.gastro.2016.01.038. [DOI] [PubMed] [Google Scholar]
- 23.Madsen K, Knudsen LB, Agersoe H, Nielsen PF, Thøgersen H, Wilken M, Johansen NL. Structure-activity and protraction relationship of long-acting glucagon-like peptide-1 derivatives: importance of fatty acid length, polarity, and bulkiness. J Med Chem. 2007;50:6126–6132. doi: 10.1021/jm070861j. [DOI] [PubMed] [Google Scholar]
- 24.Drucker DJ, Dritselis A, Kirkpatrick P. Liraglutide. Nat Rev Drug Discov. 2010;9:267–268. doi: 10.1038/nrd3148. [DOI] [PubMed] [Google Scholar]
- 25.Nuffer WA, Trujillo JM. Liraglutide: A New Option for the Treatment of Obesity. Pharmacotherapy. 2015;35:926–934. doi: 10.1002/phar.1639. [DOI] [PubMed] [Google Scholar]
- 26.Markham A, Keam SJ. Obeticholic Acid: First Global Approval. Drugs. 2016;76:1221–1226. doi: 10.1007/s40265-016-0616-x. [DOI] [PubMed] [Google Scholar]
- 27.Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, Morelli A, Parks DJ, Willson TM. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002;45:3569–3572. doi: 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- 28.Asgharpour A, Kumar D, Sanyal A. Bile acids: emerging role in management of liver diseases. Hepatol Int. 2015;9:527–533. doi: 10.1007/s12072-015-9656-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cariou B, Hanf R, Lambert-Porcheron S, Zaïr Y, Sauvinet V, Noël B, Flet L, Vidal H, Staels B, Laville M. Dual peroxisome proliferator-activated receptor α/δ agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care. 2013;36:2923–2930. doi: 10.2337/dc12-2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Staels B, Rubenstrunk A, Noel B, Rigou G, Delataille P, Millatt LJ, Baron M, Lucas A, Tailleux A, Hum DW, et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology. 2013;58:1941–1952. doi: 10.1002/hep.26461. [DOI] [PubMed] [Google Scholar]
- 31.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 32.Wang XX, Wang D, Luo Y, Myakala K, Dobrinskikh E, Rosenberg AZ, Levi J, Kopp JB, Field A, Hill A, et al. FXR/TGR5 Dual Agonist Prevents Progression of Nephropathy in Diabetes and Obesity. J Am Soc Nephrol. 2018;29:118–137. doi: 10.1681/ASN.2017020222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rahman K, Liu Y, Kumar P, Smith T, Thorn NE, Farris AB, Anania FA. C/EBP homologous protein modulates liraglutide-mediated attenuation of non-alcoholic steatohepatitis. Lab Invest. 2016;96:895–908. doi: 10.1038/labinvest.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henderson SJ, Konkar A, Hornigold DC, Trevaskis JL, Jackson R, Fritsch Fredin M, Jansson-Löfmark R, Naylor J, Rossi A, Bednarek MA, et al. Robust anti-obesity and metabolic effects of a dual GLP-1/glucagon receptor peptide agonist in rodents and non-human primates. Diabetes Obes Metab. 2016;18:1176–1190. doi: 10.1111/dom.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanf R, Millatt LJ, Cariou B, Noel B, Rigou G, Delataille P, Daix V, Hum DW, Staels B. The dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 exerts anti-diabetic effects in db/db mice without peroxisome proliferator-activated receptor gamma-associated adverse cardiac effects. Diab Vasc Dis Res. 2014;11:440–447. doi: 10.1177/1479164114548027. [DOI] [PubMed] [Google Scholar]
- 36.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Honda Y, Imajo K, Kato T, Kessoku T, Ogawa Y, Tomeno W, Kato S, Mawatari H, Fujita K, Yoneda M, et al. The Selective SGLT2 Inhibitor Ipragliflozin Has a Therapeutic Effect on Nonalcoholic Steatohepatitis in Mice. PLoS One. 2016;11:e0146337. doi: 10.1371/journal.pone.0146337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henkel J, Coleman CD, Schraplau A, Johrens, Weber D, Castro JP, Hugo M, Schulz TJ, Krämer S, Schürmann A, et al. Induction of steatohepatitis (NASH) with insulin resistance in wildtype B6 mice by a western-type diet containing soybean oil and cholesterol. Mol Med. 2017 doi: 10.2119/molmed.2016.00203. 23: Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mells JE, Fu PP, Sharma S, Olson D, Cheng L, Handy JA, Saxena NK, Sorescu D, Anania FA. Glp-1 analog, liraglutide, ameliorates hepatic steatosis and cardiac hypertrophy in C57BL/6J mice fed a Western diet. Am J Physiol Gastrointest Liver Physiol. 2012;302:G225–G235. doi: 10.1152/ajpgi.00274.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tetri LH, Basaranoglu M, Brunt EM, Yerian LM, Neuschwander-Tetri BA. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am J Physiol Gastrointest Liver Physiol. 2008;295:G987–G995. doi: 10.1152/ajpgi.90272.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma K, Malhotra P, Soni V, Hedroug O, Annaba F, Dudeja A, Shen L, Turner JR, Khramtsova EA, Saksena S, et al. Overactivation of intestinal SREBP2 in mice increases serum cholesterol. PLoS One. 2014;9:e84221. doi: 10.1371/journal.pone.0084221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313:2263–2273. doi: 10.1001/jama.2015.5370. [DOI] [PubMed] [Google Scholar]
- 45.Reccia I, Kumar J, Akladios C, Virdis F, Pai M, Habib N, Spalding D. Non-alcoholic fatty liver disease: A sign of systemic disease. Metabolism. 2017;72:94–108. doi: 10.1016/j.metabol.2017.04.011. [DOI] [PubMed] [Google Scholar]
- 46.Neuschwander-Tetri BA. Non-alcoholic fatty liver disease. BMC Med. 2017;15:45. doi: 10.1186/s12916-017-0806-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liang W, Menke AL, Driessen A, Koek GH, Lindeman JH, Stoop R, Havekes LM, Kleemann R, van den Hoek AM. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS One. 2014;9:e115922. doi: 10.1371/journal.pone.0115922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Farrell GC, Mridha AR, Yeh MM, Arsov T, Van Rooyen DM, Brooling J, Nguyen T, Heydet D, Delghingaro-Augusto V, Nolan CJ, et al. Strain dependence of diet-induced NASH and liver fibrosis in obese mice is linked to diabetes and inflammatory phenotype. Liver Int. 2014;34:1084–1093. doi: 10.1111/liv.12335. [DOI] [PubMed] [Google Scholar]
- 49.Kanoski SE, Hayes MR, Skibicka KP. GLP-1 and weight loss: unraveling the diverse neural circuitry. Am J Physiol Regul Integr Comp Physiol. 2016;310:R885–R895. doi: 10.1152/ajpregu.00520.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patel NS, Doycheva I, Peterson MR, Hooker J, Kisselva T, Schnabl B, Seki E, Sirlin CB, Loomba R. Effect of weight loss on magnetic resonance imaging estimation of liver fat and volume in patients with nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. 2015;13:561–568.e1. doi: 10.1016/j.cgh.2014.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McMahan RH, Wang XX, Cheng LL, Krisko T, Smith M, El Kasmi K, Pruzanski M, Adorini L, Golden-Mason L, Levi M, et al. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J Biol Chem. 2013;288:11761–11770. doi: 10.1074/jbc.M112.446575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuipers F, Bloks VW, Groen AK. Beyond intestinal soap--bile acids in metabolic control. Nat Rev Endocrinol. 2014;10:488–498. doi: 10.1038/nrendo.2014.60. [DOI] [PubMed] [Google Scholar]
- 53.Fiorucci S, Mencarelli A, Palladino G, Cipriani S. Bile-acid-activated receptors: targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders. Trends Pharmacol Sci. 2009;30:570–580. doi: 10.1016/j.tips.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 54.Li Y, Jadhav K, Zhang Y. Bile acid receptors in non-alcoholic fatty liver disease. Biochem Pharmacol. 2013;86:1517–1524. doi: 10.1016/j.bcp.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma Y, Huang Y, Yan L, Gao M, Liu D. Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharm Res. 2013;30:1447–1457. doi: 10.1007/s11095-013-0986-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cipriani S, Mencarelli A, Palladino G, Fiorucci S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res. 2010;51:771–784. doi: 10.1194/jlr.M001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kong B, Luyendyk JP, Tawfik O, Guo GL. Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. J Pharmacol Exp Ther. 2009;328:116–122. doi: 10.1124/jpet.108.144600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jouihan H, Will S, Guionaud S, Boland ML, Oldham S, Ravn P, Celeste A, Trevaskis JL. Superior reductions in hepatic steatosis and fibrosis with co-administration of a glucagon-like peptide-1 receptor agonist and obeticholic acid in mice. Mol Metab. 2017;6:1360–1370. doi: 10.1016/j.molmet.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang DG, Zhang C, Wang JX, Wang BW, Wang H, Zhang ZH, Chen YH, Lu Y, Tao L, Wang JQ, et al. Obeticholic acid protects against carbon tetrachloride-induced acute liver injury and inflammation. Toxicol Appl Pharmacol. 2017;314:39–47. doi: 10.1016/j.taap.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 60.Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors: new targets for the pharmacological modulation of macrophage gene expression and function. Curr Opin Lipidol. 2003;14:459–468. doi: 10.1097/00041433-200310000-00006. [DOI] [PubMed] [Google Scholar]
- 61.Gonzalez FJ, Shah YM. PPARalpha: mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators. Toxicology. 2008;246:2–8. doi: 10.1016/j.tox.2007.09.030. [DOI] [PubMed] [Google Scholar]
- 62.Nagasawa T, Inada Y, Nakano S, Tamura T, Takahashi T, Maruyama K, Yamazaki Y, Kuroda J, Shibata N. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARdelta agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur J Pharmacol. 2006;536:182–191. doi: 10.1016/j.ejphar.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 63.Pawlak M, Baugé E, Bourguet W, De Bosscher K, Lalloyer F, Tailleux A, Lebherz C, Lefebvre P, Staels B. The transrepressive activity of peroxisome proliferator-activated receptor alpha is necessary and sufficient to prevent liver fibrosis in mice. Hepatology. 2014;60:1593–1606. doi: 10.1002/hep.27297. [DOI] [PubMed] [Google Scholar]
- 64.Tanaka N, Aoyama T, Kimura S, Gonzalez FJ. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol Ther. 2017;179:142–157. doi: 10.1016/j.pharmthera.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Savkur RS, Miller AR. Investigational PPAR-gamma agonists for the treatment of Type 2 diabetes. Expert Opin Investig Drugs. 2006;15:763–778. doi: 10.1517/13543784.15.7.763. [DOI] [PubMed] [Google Scholar]
- 66.Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, Ham J, Kang H, Evans RM. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harrington WW, S Britt C, G Wilson J, O Milliken N, G Binz J, C Lobe D, R Oliver W, C Lewis M, M Ignar D. The Effect of PPARalpha, PPARdelta, PPARgamma, and PPARpan Agonists on Body Weight, Body Mass, and Serum Lipid Profiles in Diet-Induced Obese AKR/J Mice. PPAR Res. 2007;2007:97125. doi: 10.1155/2007/97125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rachid TL, Penna-de-Carvalho A, Bringhenti I, Aguila MB, Mandarim-de-Lacerda CA, Souza-Mello V. Fenofibrate (PPARalpha agonist) induces beige cell formation in subcutaneous white adipose tissue from diet-induced male obese mice. Mol Cell Endocrinol. 2015;402:86–94. doi: 10.1016/j.mce.2014.12.027. [DOI] [PubMed] [Google Scholar]