

Abstract
Cases
Testicular adrenal rest tumor (TART) is one of the possible causes of male infertility, accompanied by congenital adrenal hyperplasia (CAH). Here are reported two cases of TARTs that were referred to Kobe City Medical Center West Hospital for the treatment of infertility and testicular tumors.
Outcome
In one case, the semen analysis was improved from oligoasthenozoospermia to normozoospermia after taking oral glucocorticoid supplementation. The other case of original azoospermia showed that sperm had ejaculated into the semen after taking oral glucocorticoid supplementation.
Conclusion
Although the prevalence of TARTs in male infertility is very rare, it is important to know how to approach this disease, considering the curable pathology of spermatogenesis and tumors resembling an appearance to germ cell tumors.
Keywords: azoospermia, congenital adrenal hyperplasia, germ cell tumor, infertility, testicular adrenal rest tumor
1. INTRODUCTION
Congenital adrenal hyperplasia (CAH) is one of the most commonly found autosomal recessive diseases (~1:16 000 births).1 The defective conversion of 17‐hydroxyprogesterone (17‐OHP) to 11‐deoxycortisol, which is mediated by 21‐hydroxylase, accounts for >90% of the cases of CAH,2, 3, 4 (Figure 1), resulting in increased adrenocorticotropic hormone (ACTH) production and an associated hyperplasia of the adrenal glands, with the subsequent overproduction of adrenal androgens.5 Recently, CAH has been included in newborn screening in many countries. Therefore, the number of patients with CAH, but without any medication history, has notably decreased. In general, the chief goal of CAH treatment tends to be the adjustment of adrenal disorders.
Figure 1.
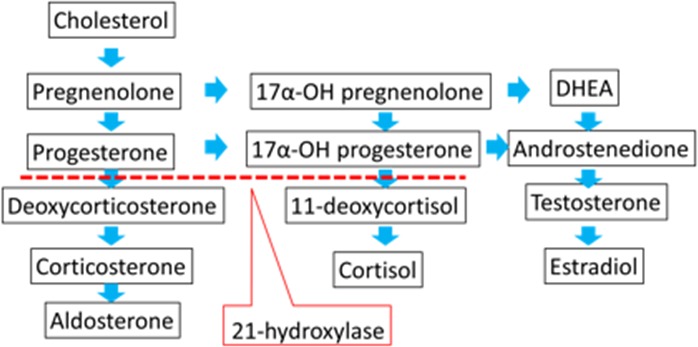
Simplified scheme of adrenal steroidogenesis. In most patients with congenital adrenal hyperplasia, the defective conversion of 17α‐hydroxy(OH)progesterone to 11‐deoxycortisol leads to impaired cortisol production. DHEA, dehydroepiandrosterone
Here are reported two cases of testicular adrenal rest tumor (TART) in patients with oligozoospermia and azoospermia. It is quite rare to highlight both the appearance of testicular tumors and the result of a semen analysis before and after the first glucocorticoid induction at middle age.
2. CASE REPORTS
2.1. Case 1
A 41 year old man was referred to Kobe City Medical Center West Hospital, Kobe City, Japan, because of 2 years of infertility. At the physical examination, the patient's physical constitution was as follows (height: 1.58 m; weight: 65 kg; body mass index: 23.9 kg/m²). Both testes had become atrophied (9 mL) (performed with a Prader orchidmeter) and pea‐sized indurations were felt around the epididymal head in both testes. A scrotal ultrasound examination confirmed the presence of bilateral heteroechogenic, hypovascularized masses near the testes (Figure 2A). The semen analysis revealed oligoasthenozoospermia (volume: 2.6 mL; concentration: 7 × 106/mL; motility: 14%). The hormonal examination revealed a normal hormonal status of serum follicle‐stimulating hormone (FSH) of 2.7 mIU/mL, luteinizing hormone (LH) of 1.3 mIU/mL, testosterone of 4.3 ng/mL, and estradiol of 13 pg/mL. All the serum markers for testicular tumors, including human chorionic gonadotropin, alpha‐fetoprotein, and lactate dehydrogenase, were within the normal range. The pelvic magnetic resonance imaging (MRI) scan showed irregular, margin, heterogeneous low‐signal‐intensity tumors that were surrounded by high‐signal normal testicular tissue in the T2‐weighted image (Figure 3). An enhanced computed tomography (CT) scan revealed diffuse, irregular enlargement of both adrenal glands, with no finding in the testes (Figure 4A,B). Bilateral testicular atrophy and bilateral enlargement of the adrenal glands without any other metastases did not seem to be typical for malignant testicular tumors. At this point, adrenal gland disease was suspected and an endocrinologist was consulted for further examination. In the endocrinological examination, both the serum ACTH and 17‐OHP levels were extremely high (69.3 pg/mL and 112 ng/mL, respectively). Genetic diagnosis by polymerase chain reaction (PCR)‐direct sequencing confirmed the 21‐hydroxylase deficiency due to CYP21A2 gene mutation (I‐172N mutation) and a low‐dose, oral glucocorticoid of 0.5 mg/d was started under the diagnosis of CAH.
Figure 2.
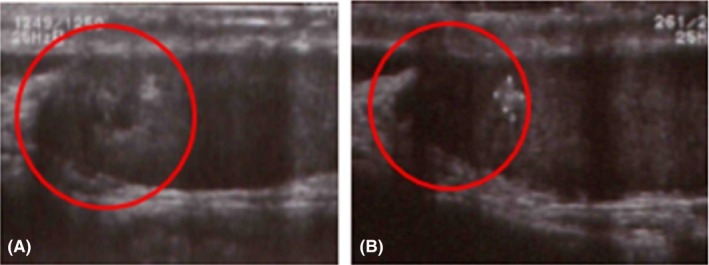
A, Testicular ultrasound examination on the patient's first visit confirmed the presence of a heteroechogenic, hypervascularized mosaic area of 1.5 cm×1.5 cm at the projection of the testis network. B, The testicular tumor had become almost undetectable after 1 month of glucocorticoid therapy
Figure 3.
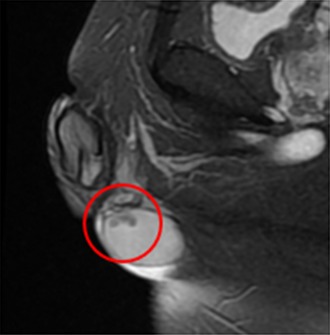
Magnetic resonance imaging‐realized T2‐weighted, low‐sign, bilateral, solid enlarged serpiginous lesions in the testicular parenchyma
Figure 4.
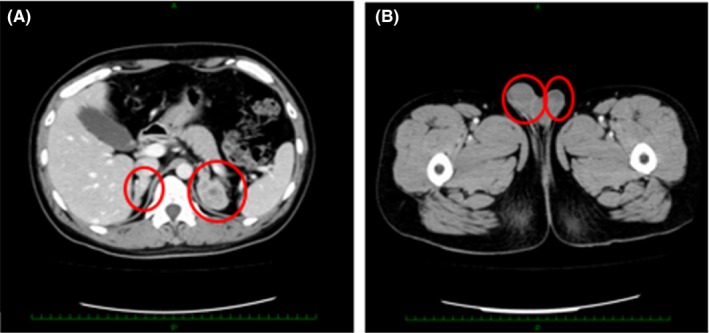
Enhanced computed tomography scan revealed diffuse, irregular enlargement of both adrenal glands (A), with no findings in the testes (B)
After 1 month of glucocorticoid therapy of 0.5 mg/d, the semen analysis revealed a significant change (concentration: 34 × 106/mL; motility: 59%), along with normalization of the testicular size (right: 14 mL; left: 16 mL). The size reduction of the TART was identified by ultrasound (Figure 2B). The hormonal status had not changed significantly from the pretreatment status (serum FSH: 2.8 mIU/mL; LH: 1.6 mIU/mL; testosterone: 3.2 ng/mL; estradiol: 18 pg/mL). After 2 years of glucocorticoid administration, the clinical pregnancy of his wife was established by using in vitro fertilization (IVF), resulting in the delivery of a female offspring (3650 g) who was confirmed as negative for CAH by serum 17‐OHP and 21‐deoxycortisol measurement.
2.2. Case 2
A 41 year old man was followed at the local clinic for polycythemia (hemoglobin [Hb]: 18.0 g/dL; red blood count: 627 × 104; hematocrit: 53.1%) and hypertension. An abdominal CT scan for the examination of these conditions revealed the enlargement of the bilateral adrenal glands (Figure 5). The patient was referred to endocrinologists in the hospital for further investigation.
Figure 5.
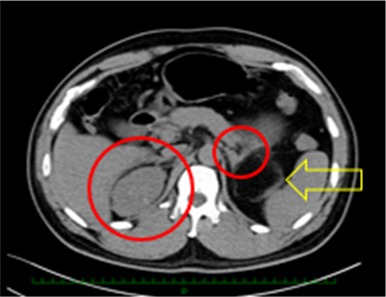
Computed tomography scan revealed diffuse, irregular enlargement of both adrenal glands (red circles) and a myelolipoma close to the left adrenal gland (yellow arrow), which was removed by laparoscopic operation afterwards
The patient was seen to have obesity (height: 165 cm; weight: 79 kg; Body Mass Index: 29.0 kg/m²). The laboratory tests revealed an extremely high level of ACTH (316.4 pg/mL) and a relatively low level of cortisol (6.8 μg/dL). Although the patient did not have any family history of CAH, the CT findings regarding the adrenal glands, unbalanced pituitary–adrenal hormones, and high level of serum 17‐OHP (62.7 ng/mL) indicated the possibility of CAH. The difference from case 1 is that the first examination was made by endocrinologists, not urologists. The differential diagnosis was made before a testicular examination. The abnormality in the pituitary–gonadal hormonal status (FSH: <0.1 IU/mL; LH: <0.1 IU/mL; testosterone: 10.9 ng/mL; estradiol: 29 pg/mL) indicated the excessive secretion of adrenal androgens. Bilateral testicular tumors were identified in a scrotal ultrasound echo scan and then a urological consultation and several additional examinations were offered in order to rule out a germ cell tumor. At the physical examination, both testes had become slightly atrophied (right: 14 mL; left: 12 mL) (performed with a Prader orchidmeter) and no induration was palpated. All the serum markers for testicular tumors were within the normal range and the testicular MRI showed the same sign as case 1 (Figure 6A). The semen analysis showed azoospermia. A genetic diagnosis by PCR‐direct sequencing proved CAH due to 21‐hydroxylase deficiency due to CYP21A2 gene mutation.
Figure 6.
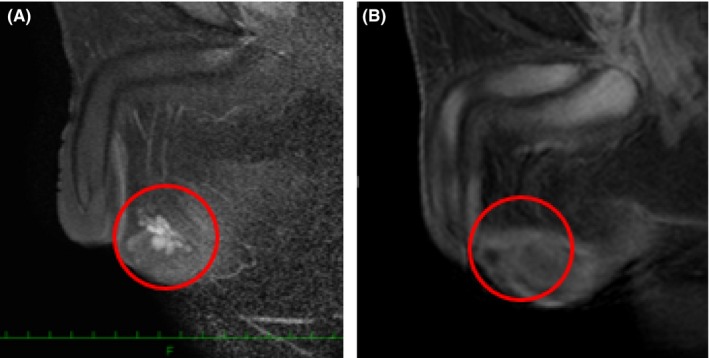
A, Before glucocorticoid therapy, the testicular tumors demonstrated uniform enhancement, with discrete margins that were readily visualized on the T1‐weighted images following i.v. gadolinium administration. B, Three years later, the testicular tumors became undetectable on the enhanced T1‐weighted images
After 6 months of low‐dose, daily oral glucocorticoid therapy (0.5 mg/d), the serum FSH, LH, testosterone, and estradiol levels returned to normal levels (9.2 IU/mL, 1.8 IU/mL, 0.8 ng/mL, and <10 pg/mL, respectively). The polycythemia also improved to a Hb level of 15.2 g/dL. Sperm emerged (concentration: 3.4 × 106/mL; motility: 26%), along with a size reduction in the adrenal hyperplasia and TART (Figure 6B).
3. DISCUSSION
The testicular adrenal rest tumor is characterized by a serpiginous tumor in a focal area. Adrenals and gonads derive from a common adrenogonadal primordium during development. It is believed that remnants of the adrenogonadal primordium can persist and proliferate to TARTs, with the preservation of adrenal‐like, hormone‐producing properties.5 The reported prevalence of TARTs in patients with CAH ranges from 27% to 47%.1
In cases in which bilateral mosaic enlarged serpiginous lesions in the testicular parenchyma are found in the scrotal ultrasound examination, an additional abdominal echo scan around the adrenal glands and serum markers for testicular tumors would be helpful to rule out a germ cell tumor in the clinic.
It is also reasonable to add a testicular MRI scan in the urologic department in general hospitals. On the T1‐weighted images, TARTs are usually isointense, while they are usually hypointense on the T2‐weighted images.6 Contrast‐enhanced, T1‐weighted images demonstrate testicular nodules in a focal area with diffuse enhancement.7 Other previous reports also showed the same ultrasound and MRI features of TARTs as in cases 1 and 2.8, 9
The prevalence of oligospermia in patients with a TART was 66%, reported in the largest series with 219 male participants.10 Congenital adrenal hyperplasia causes male infertility by several different mechanisms, regardless of the presence or absence of a TART. The first mechanism is due to the abnormal hormonal status due to CAH. Hypogonadotropism is caused by both elevated adrenal androgens and estrogens aromatized from androgens and leads to impaired Leydig cell functions.11 The second mechanism is mechanical obstruction that is caused by a TART, which often appears in the testes and therefore it can cause mechanical obstruction of the seminiferous tubules in the testes or epididymis. The third mechanism is due to the disturbance of the local hormonal status. Thus, TARTs can inhibit spermatogenesis by local steroid production, resulting in impaired function of testicular androgen.5, 11
When applying these mechanisms to the current cases, in case 1, normal levels of LH, FSH, and testosterone and partially disordered spermatogenesis support the hypothesis that local steroid production by the TART was the main cause of oligozoospermia. In case 2, hypogonadotropic–hypergonadism and completely disordered spermatogenesis indicate that hypothalamus–pituitary gonadal (HPG) dysfunction was the main cause of azoospermia.
The main difference in the treatment strategy of patients with CAH to the other male patients with infertility is the use of corticosteroids. Corticosteroid therapy, used to adjust the HPG axis and consequent shrinkage of the TART, should be tried first in order to avoid irreversible damage to the testes. In the literature, ~40% of patients with CAH had spontaneous fertility after corticosteroid therapy, having required no alteration in their routine treatment and experiencing no delay in conception.12 If hypogonadotropism still persists after the normalization of the ACTH and 17‐OHP levels, treatment with human chorionic gonadotropin and FSH should be considered in order to recover spermatogenesis.
The goal of corticosteroid therapy is not only in reducing the size of the TART, but also in optimizing the control of excess hormones, replacing deficient hormones, and avoiding potential Cushingoid side‐effects.13 It seems that corticosteroid therapy should be kept going for as long as possible, regardless of the change in the size of the TART. The suppression of excess ACTH secretion by corticosteroid treatment is not always successful in reducing the size of the TART. Usually, 75% of these lesions regress after sufficient steroid therapy.14 In the case of no possible reason for infertility, except for seminiferous tubal obstruction, an operation for the removal of the TART is a reasonable option.15 Corticosteroid therapy itself can induce HPG dysfunction, especially if patients are overdosed.1, 11 When all the treatments fail to recover spermatogenesis, testicular sperm retrieval for IVF/intracytoplasmic sperm injection (ICSI) may be proposed for the goal of conception. One study reported a new treatment approach for sperm retrieval. Two azoospermic men with CAH, orchialgia, and bilateral TARTs, who failed to respond to corticosteroid therapy, had adequate sperm retrieved for IVF/ICSI simultaneously with a TART resection. One man proceeded with the use of his sperm for IVF/ICSI and his wife achieved a successful pregnancy, resulting in a live birth. The other man had his sperm cryopreserved for future use. Both men had a complete resolution of orchialgia from the TART.1
In conclusion, the pathophysiology and background vary in each patient with CAH. The best treatment option for each patient should be adopted.
DISCLOSURES
Conflict of interest: The authors declare no conflict of interest. Human and Animal Rights: All the procedures were followed in accordance with the ethical standards of the responsible committees on human experimentation (institutional and national) and with the Helsinki Declaration of 1964 and its later amendments. This study is a case report, not clinical research. Kobe University Hospital ethics committee does not include case reports in its judgment of ethical problems. Informed consent was obtained from all the patients to be included in the study. This article does not contain any study with animal participants that was performed by any of the authors.
Tanaka M, Enatsu N, Chiba K, Fujisawa M. Two cases of reversible male infertility due to congenital adrenal hyperplasia combined with testicular adrenal rest tumor. Reprod Med Biol. 2018;17:93‐97. https://doi.org/10.1002/rmb2.12068
REFERENCES
- 1. Kavoussi PK, Summers‐Colquitt RB, Odenwald KC, et al. Sperm retrieval and concomitant tumor resection in azoospermic men with congenital adrenal hyperplasia and bilateral testicular adrenal rest tumors: a case report. J Assist Reprod Genet. 2016;33:545‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Witchel SF, Nayak S, Suda‐Hartman M, Lee PA. Newborn screening for 21‐hydroxylase deficiency: results of CYP21 molecular genetic analysis. J Pediatr. 1997;131:328‐331. [DOI] [PubMed] [Google Scholar]
- 3. Allen DB, Hoffman GL, Fitzpatrick P, Laessig R, Maby S, Slyper A. Improved precision of newborn screening for congenital adrenal hyperplasia using weight‐adjusted criteria for 17‐hydroxyprogesterone levels. J Pediatr. 1997;130:128‐133. [DOI] [PubMed] [Google Scholar]
- 4. Gruneiro‐Papendieck L, Prieto L, Chiesa A, Bengolea S, Bossi G, Bergada C. Neonatal screening program for congenital adrenal hyperplasia: adjustments to the recall protocol. Horm Res. 2001;55:271‐277. [DOI] [PubMed] [Google Scholar]
- 5. Marchini GS, Cocuzza M, Pagani R, Torricelli FC, Hallak J, Srougi M. Testicular adrenal rest tumor in infertile man with congenital adrenal hyperplasia: case report and literature review. San Paulo Med J. 2011;129:346‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nagamine WH, Metha SV, Vade A. Testicular adrenal rest tumors in a patient with congenital adrenal hyperplasia: sonographic and magnetic resonance imaging findings. J Ultrasound Med. 2005;24:1717‐1720. [DOI] [PubMed] [Google Scholar]
- 7. Ozisik H, Yurekli BS, Simsir IY, et al. Testicular adrenal rest tumor (TART) in congenital adrenal hyperplasia. Eur J Med Genet. 2017;60:489‐493. [DOI] [PubMed] [Google Scholar]
- 8. Proto G, Di‐Donna A, Grimaldi F, Mazzolini A, Purinan A, Bertolissi F. Bilateral testicular adrenal rest tissue in congenital adrenal hyperplasia: US and MR features. J Endocrinol Invest. 2001;24:529‐531. [DOI] [PubMed] [Google Scholar]
- 9. Fitoz S, Atasoy C, Adiyaman P, Berberoglu M, Erden I, Ocal G. Testicular adrenal rests in a patient with congenital adrenal hyperplasia: US and MRI features. Comput Med Imaging Graph. 2006;30:465‐468. [DOI] [PubMed] [Google Scholar]
- 10. Bouvattier C, Esterle L, Renoult‐Pierre P, et al. Clinical outcome, hormonal status, gonadotrope axis, and testicular function in 219 adult men born with classic 21‐hydroxylase deficiency. A French national survey. J Clin Endocrinol Metab. 2015;100:2303‐2313. [DOI] [PubMed] [Google Scholar]
- 11. Reisch N, Flade L, Scherr M, et al. High prevalence of reduced fecundity in men with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2009;94:1665‐1670. [DOI] [PubMed] [Google Scholar]
- 12. King TF, Lee MC, Williamson EE, Conway GS. Experience in optimizing fertility outcomes in men with congenital adrenal hyperplasia due to 21 hydroxylase deficiency. Clin Endocrinol. 2016;84:830‐836. [DOI] [PubMed] [Google Scholar]
- 13. El‐Maouche D, Arlt W, Merke DP. Congenital adrenal hyperplasia. Lancet. https://doi.org/10.1016/s0140-6736(17)31431-9. Accessed May 30, 2017. [DOI] [PubMed] [Google Scholar]
- 14. Naouar S, Braiek S, El Kamel R. Testicular tumors of adrenogenital syndrome: from physiopathology to therapy. Presse Med. 2017;46:572‐578. [DOI] [PubMed] [Google Scholar]
- 15. Stikkelbroeck NM, Otten BJ, Pasic A, et al. High prevalence of testicular adrenal rest tumors, impaired spermatogenesis, and Leydig cell failure in adolescent and adult males with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2001;86:5721‐5728. [DOI] [PubMed] [Google Scholar]