

Abstract
Background:
Alzheimer’s disease (AD) is currently incurable and a majority of investigational drugs have failed clinical trials. One explanation for this failure may be the invalidity of hypotheses focus-ing on amyloid to explain AD pathogenesis. Recently, hypotheses which are centered on synaptic and met-abolic dysfunction are increasingly implicated in AD.
Objective:
Evaluate AD hypotheses by comparing neurotransmitter and metabolite marker concentrations in normal versus AD CSF.
Methods:
Meta-analysis allows for statistical comparison of pooled, existing cerebrospinal fluid (CSF) marker data extracted from multiple publications, to obtain a more reliable estimate of concentrations. This method also provides a unique opportunity to rapidly validate AD hypotheses using the resulting CSF con-centration data. Hubmed, Pubmed and Google Scholar were comprehensively searched for published Eng-lish articles, without date restrictions, for the keywords “AD”, “CSF”, and “human” plus markers selected for synaptic and metabolic pathways. Synaptic markers were acetylcholine, gamma-aminobutyric acid (GABA), glutamine, and glycine. Metabolic markers were glutathione, glucose, lactate, pyruvate, and 8 other amino acids. Only studies that measured markers in AD and controls (Ctl), provided means, standard er-rors/deviation, and subject numbers were included. Data were extracted by six authors and reviewed by two others for accuracy. Data were pooled using ratio of means (RoM of AD/Ctl) and random effects meta-analysis using Cochrane Collaboration’s Review Manager software.
Results:
Of the 435 identified publications, after exclusion and removal of duplicates, 35 articles were in-cluded comprising a total of 605 AD patients and 585 controls. The following markers of synaptic and met-abolic pathways were significantly changed in AD/controls: acetylcholine (RoM 0.36, 95% CI 0.24-0.53, p<0.00001), GABA (0.74, 0.58-0.94, p<0.01), pyruvate (0.48, 0.24-0.94, p=0.03), glutathione (1.11, 1.01-1.21, p=0.03), alanine (1.10, 0.98-1.23, p=0.09), and lower levels of significance for lactate (1.2, 1.00-1.47, p=0.05). Of note, CSF glucose and glutamate levels in AD were not significantly different than that of the controls.
Conclusion:
This study provides proof of concept for the use of meta-analysis validation of AD hypothe-ses, specifically via robust evidence for the cholinergic hypothesis of AD. Our data disagree with the other synaptic hypotheses of glutamate excitotoxicity and GABAergic resistance to neurodegeneration, given ob-served unchanged glutamate levels and decreased GABA levels. With regards to metabolic hypotheses, the data supported upregulation of anaerobic glycolysis, pentose phosphate pathway (glutathione), and anaple-rosis of the tricarboxylic acid cycle using glutamate. Future applications of meta-analysis indicate the pos-sibility of further in silico evaluation and generation of novel hypotheses in the AD field.
Keywords: Anaplerosis, anaerobic glycolysis, glutamate excitotoxicity, CSF, GABA resistance, cholinergic hypothesis, pentose phosphate pathway, glutaminolysis
1. INTRODUCTION
Alzheimer’s disease (AD) is a slowly progressing neurodegenerative disease for which there is no cure and a high failure rate of therapeutic drugs in clinical trials (99.6% drug failure rate between 2002 and 2012 [1]). While there are some promising drugs, such as the Merck BACE inhibitor [2], one possible explanation for the above reported high drug failure rate is likely the invalidity of some AD pathogenic hypotheses targeted by these trials. There are many competing AD hypotheses proposed to explain the pathological features of AD and it is becoming clear that these hypotheses need to be validated using human populations, rather than transgenic mouse models of CNS diseases [3]. Meta-analysis allows for statistical comparison of existing cerebrospinal fluid (CSF) marker data extracted from multiple publications, providing a unique opportunity to provide evidence for or against several specific AD hypotheses. We speculated that any flaws in existing AD hypotheses related to synaptic and metabolic dysfunction could be revealed by comparing CSF levels of neurotransmitters and metabolites between AD and controls.
The synaptic hypotheses related to AD are described below. The cholinergic hypotheses of AD were formulated over 30 years ago based on the observations that acetylcholine plays an important role in cognition and that there are reduced acetylcholine levels and cholinergic neuronal loss in AD [4-6]. Thus, it has been well established that synaptic loss correlates strongly with memory loss in AD. Other AD hypotheses related to synapses include the glutamatergic hypothesis [7-9], whereby elevated glutamate levels resulting in hyperactivity which then leads to neuronal cell death [9-11], and resistance of GABAergic cells to neurodegeneration [12], with an expected decline in GABA CSF levels.
The metabolic hypotheses of AD that we focused on here are related to glycolysis, the pentose phosphate pathway (PPP), and the tricarboxylic acid (TCA) cycle dysfunctions in AD. The inadequacy of these metabolic pathways were derived in part from Hoyer’s proposed lactate acidosis, as well as increased TCA anaplerosis due to hypoglycemia [13]. It is well known that hypoxic conditions, deficits in pyruvate dehydrogenase complex activity, or increased oxidative stress all result in shifts to anaerobic glycolysis and/or the PPP in AD [14-19]. Of note, PPP activity generates NADPH which serves as a protective mechanism against oxidative stress and has been found to be increased during mild cognitive impairment [15]. With regards to the TCA cycle, the pyruvate dehydrogenase complex and enzyme alpha-ketoglutarate dehydrogenase are also known to be altered and dysfunctional in AD [20, 21]. Last, it has been demonstrated by Krebs in 1960 [22] that the TCA cycle can function via utilization of the glutamate shunt (anaplerosis) [13, 23, 24]. Indeed, this truncated TCA shunt was more recently demonstrated during hypoglycemia, albeit in an animal model [25].
While these synaptic and metabolic dysfunctions in AD have been published, some controversy exists in the literature. For example, increased use of glycolysis [14-18], is at odds with reduced glucose utilization in AD [19, 26]. There is also debate as to whether either anaerobic glycolysis or oxidative phosphorylation is increased in AD [16, 27]. Interestingly, an elevation of lactic acid may reflect either anaerobic glycolysis, or increased synaptic activity [28]. With regards to synaptic hypotheses, elevated CSF glutamate levels are not consistent across reports, and thus may not support the glutamate excitoxicity. Additionally, CSF GABA levels appear to be reduced, which contradicts GABAergic neurons resistance to cell death in AD [12]. Thus, it is important to be able to readily validate these hypotheses using CSF metabolite levels.
We addressed these controversies in existing AD hypotheses by using meta-analysis to determine the concentration of important compounds/metabolites involved in the metabolic and synaptic dysfunctions in AD. Meta-analysis is a method of combining similar studies to increase accuracy and statistical power, furthermore the heterogeneity across studies can be analyzed. This method has previously been used to evaluate biomarkers in AD [29] (AlzBiomaker Database, Version 2.0, April 2017 (www.alzforum.org/ alzbiomarker), Alzforum Date of Access: June 2017). However we also realized that the data generated using this method could be used to validate AD hypotheses. An important proof-of-concept of this hypothesis would be validation of the cholinergic hypothesis, as reflected by significant lower CSF acetylcholine levels in AD patients compared to the controls.
2. METHODS
2.1. Literature Retrieval
A literature search using Hubmed, Pubmed, and Google Scholar was performed from inception to August 5, 2016 for specific markers (e.g. metabolites, amino acids, neurotransmitters) involved in glycolysis and the TCA cycle according to PRISMA (Preferred Reporting Items for Systematic Review and Meta-Analyses) guidelines [30]. Additionally, relevant publications were identified via citations within relevant publications and reviews, as well as metabolomic databases http://www.csfmetabolome.ca/ [31], allowing for an evaluation of the completeness of the search criteria for this meta-analysis.
2.2. Inclusion Criteria
Published studies measuring any of the following synaptic [acetylcholine, glutamate, GABA] or metabolic [glutathione, glucose, lactate, pyruvate, alanine, glycine, asparagine, aspartate, glutamine, isoleucine, leucine and valine] markers in CSF in both control patients and subjects with AD.
The control patients in these studies were healthy patients, without neurological disorders, and had CSF withdrawn by lumbar puncture as part of the study (Appendix Table 1). The majority of AD patients were evaluated according to Diagnostic and Statistical Manual of Mental Disorders (DSM) III and higher or National Institute of Neurological and Communicative Disorders and Stroke- the Alzheimer's Disease and Related Disorders Association (NINCDS-ADRA) criteria, with comorbidities assessed in some publications (Appendix Table 1). Studies both with and without Mini-mental state exam (MMSE) scores for AD were included in the analysis so as to reduce bias in the selection of studies and to increase the pool of data. These studies were observational studies and included both unmatched/unadjusted and matched/adjusted publications. Details of the studies (e.g. number of enrolling centers, enrolment period, how were patients identified) are not known.
Table 1.
Demographics of AD and Ctl patients in the meta-analysis sorted by publication. Demographic data for age and mini-mental state exam (MMSE, a measure of cognitive impairment). Data was collected from 35 publications, however 2 did not report mean ± standard deviations and were not included in this table (Kuroda 1983, Tosca 1992).
| Author | Compound | Ctl Age | AD Age | Ctl MMSE | AD MMSE | |
|---|---|---|---|---|---|---|
| Bareggi 1982 | GABA | 50.2±11.9 | 62.3±10.7 | |||
| Basun 1990 | Arg, Ala, Glu, Gln, Gly, His, Ile, Leu, Lys, Val | 79.0±2 | 79.0±2 | |||
| D'Aniello 2005 | Arg, Ala, Asp, Asn, GABA, Glu, Gln, Gly, His, Ile, Leu, Lys, Val | 72.0±10 | 72.0±10 | |||
| Degrell 1989 | Arg, Ala, Glu, Gln, Gly, His, Ile, Leu, Lys, Val | 29.0±9 | 77.3±2.7 | |||
| Fisher 1998 | Arg, Ala, Asp, Asn, GABA, Glu, Gln, Gly, His, Ile, Leu, Lys, Val | 70.1±7.8 | 75.2±10.4 | |||
| Frolich 1998 | Acteylcholine | 58.8±2.7 | 69.1±8.3 | 29.7±0.7 | 16.2±5.5 | |
| Jia 2004 | Acteylcholine | 65.6±5 | 68.0±6 | 28.5±1.2 | 15.6±2.9 | |
| Jimenez 1998 | Arg, Asp, Asn, GABA, Glu, Gln, Gly | 67.9±9.2 | 70.9±8.5 | 11.6±5.5 | ||
| Kadurah-Daouk | Glutathione (GSH) | 69.5 | 69.0 | 30.0±0 | 23.0±3 | |
| Konings 1999 | Glutathione (GSH) | 65.0±10 | 65.0±8 | |||
| Kuiper 2000 | Arg, Glu | 65.8±11.9 | 64.5±7.8 | |||
| Liguiori 2016 | Lactate | 68.07±7.64 | 71.786.75 | 27.79±0.91 | 19.72±5.81 | |
| Madiera 2015 (suppl table) | Glycine | 71.6±6.65 | 72.1±8.46 | 26.3±2.14 | 12.7±6.22 | |
| Malm 1991 | Lactate | 73.8±8.2 | 71.2±4 | |||
| Martinez 1993 | Arg, Ala, Asp Asn, Glu, Gln, Gly, His, Ile, Leu, Lys, Val | 66.0±8 | 68.0±6 | 17.8±9 | 17.7±10 | |
| Mochizuki 1996 | Arg, Ala, Asp, Asn, GABA, Glu, Gln, Gly, His, Ile, Leu, Lys, Val | 62.0±10 | 66.0±10 | 29.0±1.1 | 16.1±5.7 | |
| Mohr 1986 | GABA | 57.0±1.8 | 58.0±2 | |||
| Molina 1998 | Arg, Ala, GABA, His, Ile, Leu. Lys, Val | 67.9±9.2 | 70.9±8.5 | 11.6±5.5 | ||
| Oishi 1996 | GABA | 66.7±8.9 | 66.3±9.2 | 30.0±1 | 17.0±5 | |
| Parnetti 1995 | Glucose, Pyruvate, Lactate | 69.0±5 | 71.0±7 | 29.0±1 | 15.5±5.4 | |
| Parnetti 2000 | Lactate | 69±1 | 71.0±7 | 28.0±1.5 | 15.0±6.7 | |
| Pomara 1989 | Arg, Asp, Asn, GABA, Glu, Gln, Gly, His, Ile, Lys, Val | 64.0±11.8 | 60.7±9.4 | 17.1±9.9 | ||
| Pomara 1992 | Arg, Asp,Asn, Glu, Gln, Gly, His, Ile, Lys, Val | 63.8±1.7 | 59.7±10.8 | 16.6±3.5 | ||
| Proctor 1988 | Gln | 73.3±10.2 | 72.5±11.4 | |||
| Redjems-Bennani 1998 | Glucose, Pyruvate, Lactate | 75.0±11 | 78.0±7 | 9.9±4.6 | ||
| Shuvaeva 2001 | Glucose | 27.0±19 | 79.0±8 | |||
| Smith 1985 | Asp, GABA, Glu, Gln, Gly, Ile | 62.0±6 | 61.0±5 | |||
| Tato 2016 | Glucose | 67.0±11 | 70.0±7 | 17.5±7.4 | ||
| Toghi 1992 | Arg, Ala, sp, Asn, GABA, Glu, Gly, acetylchol | 65.8±6 | 69.0±10 | 13.20±8.2 | ||
| Vitvisky 2012 | Glu, Gln | 77.0±15 | 79.0±8 | |||
| Weiner 1996 | GABA | 69.7±5 | 75.9±4.3 | 29.0±0.8 | 16.2±7.9 | |
| White 2014 | Ala, Glu, GLn, Glucose, lactate, pyruvate | 57.3±7.3 | 61.7±10.2 | |||
| Zimmer 1984 | GABA | 69.9±10.1 | 73.6±8.8 | 2.8±2.3 | ||
2.3. Exclusion Criteria
Rejection criteria for data included literature reviews, publications with no numerical data on metabolites of interest (including data only in graphical format), animal studies, pooled CSF samples, or those publications that did not include paired AD and control data acquired under similar conditions. Studies with values acquired by using brain extracts were omitted. However, studies of brain extracts were included for enzymes since intracellular metabolic enzymes are not in the CSF. Metabolome databases such as the http://www.csfmetabolome.ca [31] were omitted as they typically did not contain paired AD and Control (Ctl) CSF data
Several metabolites involved in glycolysis and the TCA cycle were not included in this publication. For example, only a single publication each was identified as meeting inclusion criteria for ATP, ammonia, and fumarate values in CSF, so these data were not included in the meta-analysis. The CSF data for the following compounds also did not meet inclusion criteria (e.g. data not containing both control and AD values): succinyl-CoA, acetyl-CoA, NADPH, NAD, alpha-ketoglutarate, malate, and oxaloacetate.
2.4. Data Extraction
Six (SS, MP, AN, MC, ST, NK) investigators independently extracted data for each metabolite and entered them into an excel database. The data were independently checked for errors by three investigators (IM, BB, RM). Data extracted from the publications included authors and publication year, NCBI Pubmed publication URL, the number of subjects (AD and controls), and marker levels in CSF with their appropriate units.
2.5. Calculations for Meta-Analysis
Data were pooled using Ratio of Means (RoM) on the logarithmic scale. RoM is calculated as the mean CSF concentration in the AD group divided by the mean CSF concentration in the Ctl group for each marker in each study. Standard error for the natural logarithm for the RoM in each study, SE(ln[RoM]) was calculated according to Friedrich [32, 33]: SE[ln(RoM)] = SQRT((1/n AD) (Standard Deviation AD/mean AD)2 + (1/n Ctl) (Standard Deviation Ctl/mean Ctl)2), where “n” is the number of patients in the AD and Ctl groups, respectively. These calculations were performed in an Excel spreadsheet prior to entering the resulting data into the meta-analysis software of the Cochrane Collaboration, Review Manager, Version 5 (Revman, Cochrane Collaboration, Oxford, England, freely available at http://ims.cochrane.org/revman/download) and analyzed using the random effects model. Revman software uses standard equations for inverse variance weighting. Random effects analysis was performed using the DerSimonian and Laird [34] method which incorporates heterogeneity leading to wider (more conservative) confidence intervals when heterogeneity is present. Statistical heterogeneity, or variability of results between studies beyond that expected by chance was quantified using the I2 statistic, with I2 >50% considered to indicate a high degree of heterogeneity. Funnel plots were created to assess for publication bias. We should note that for the enzyme data that lacked standard error data, these errors were imputed from the publications with the largest error to allow for meta-analysis. This is noted in the figure legends.
3. RESULTS
3.1. Inclusion/Exclusion of Publications
We document the inclusion/exclusion for different categories, and not individual markers below: antioxidants, amino acids, and metabolites/neurotransmitters (Fig. 1). In total thirty-five (35) publications met the selection criteria, including publications with paired control data, and the included markers and included publications are listed in Tables 2-4. Excluded publications for each group are below. For the antioxidant glutathione (GSH), 13 publications were rejected [35-47]. For the amino acids, 20 publications were rejected [41, 48-67]. Last, for the metabolites/neurotransmitters 14 publications were rejected [5, 58, 68-78].
Fig. (1).

PRISMA Flow diagram showing meta-analysis selection of publications for all biomarkers, using PubMed database search as an example. Data represents pooled publications collected for 15 different markers for both space limitations and clarity. The duplicates were removed later in the work flow diagram, because this more accurately reflects the work flow which involved several authors independently searching for publications. Furthermore, several publications were common for several compounds, especially when amino acid analyses were performed. Additional publications from other sources highlight creativity of medical students in their searches. The workflow diagram is modified from Moher et al. [34]. Details of the individual markers investigated in this study are in Tables 1-4.
Table 2.
Ratios of the means (RoM) of selected metabolites and amino acids.RoM of selected metabolites and amino acids are presented as the ratio of CSF concentrations between patients with Alzheimer’s disease and controls. For each individual publication or study (author name and year), the filled squares are the ratio value and the size of the square indicates the weight of the study. The horizontal lines represent the 95% CIs for each individual study. The average ratio for the pooled studies is indicated by a diamond, with the diamond width indicating the 95% CI. Heterogeneity (Tau2, Chi2 and I2) and effect size (Z), and effect size statistical significance (P) for the pooled data is also indicated for each subgroup.
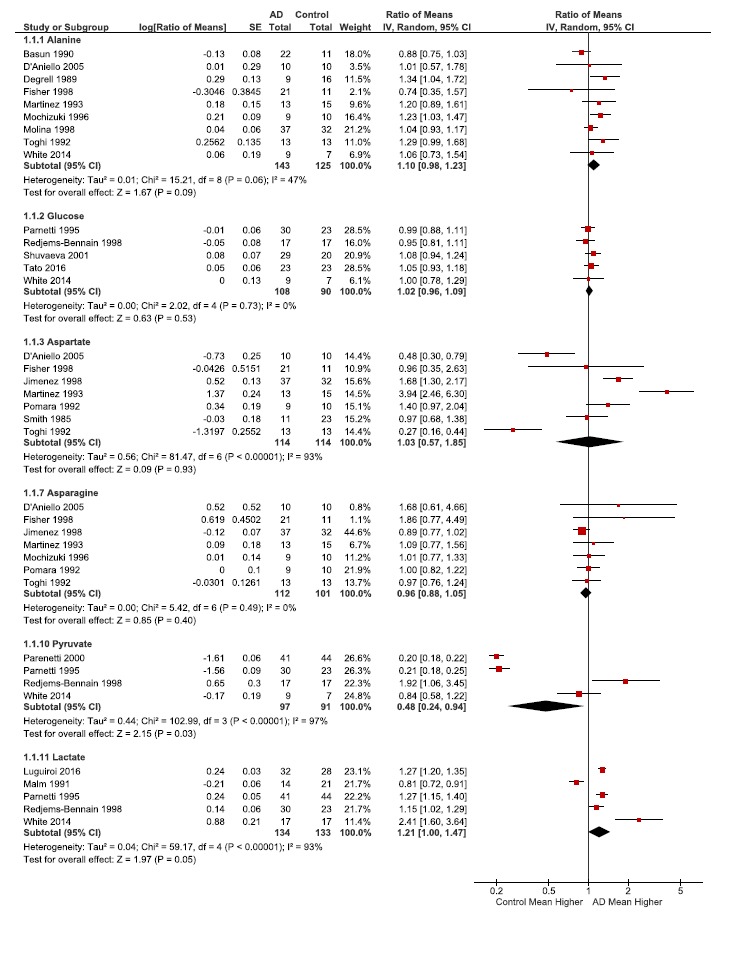
Table 4.
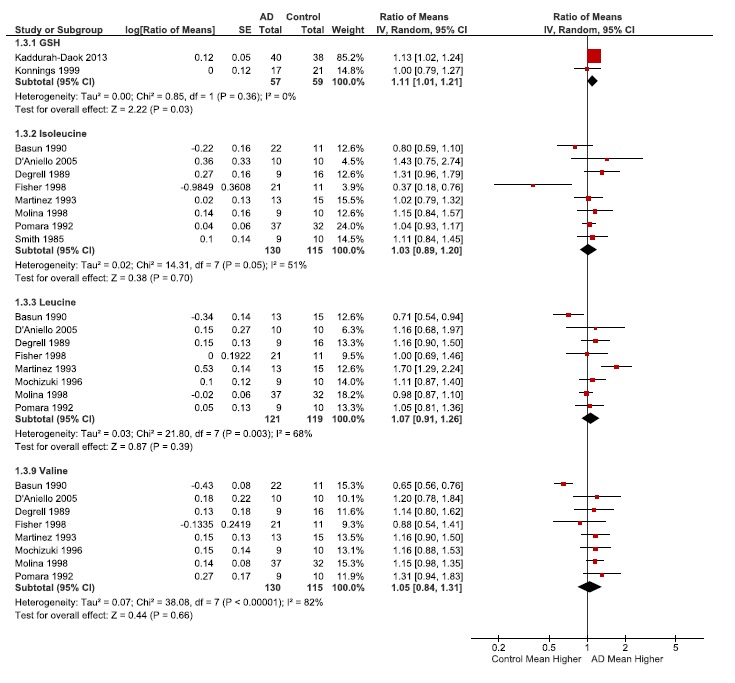
Ratios of the means (RoM) of antioxidant glutathione and selected branched chain amino acids. RoM of the antioxidant glutathione and selected branched chain amino acids (isoleucine, leucine, and valine) presented as the ratio of CSF concentrations between patients with Alzheimer’s disease and controls. For each individual publication or study (author name and year), the filled squares are the ratio value and the size of the square indicates the weight of the study. The horizontal lines represent the 95% CIs for each individual study. The average ratio for the pooled studies is indicated by a diamond, with the diamond width indicating the 95% CI. Heterogeneity (Tau2, Chi2 and I2) and effect size (Z), and effect size statistical significance (P) for the pooled data is also indicated for each subgroup.
3.2. Clinical Diversity of Subjects in Publications Used for this Study
We attempted to address clinical heterogeneity by recording age and cognitive abilities (MMSE), however, this data was not included in all studies. Table 1 provides data for metabolites, average age, and MMSE for all studies for each metabolite. It is clear that the age of the controls did not match that of AD in two publications (Degrell and Shuvaev, Ctls 29 and 27 in Table 1), while other studies had young ages relatively near or below 60 years of age (Bareggi, Mohr, Smith, Pomara 1989, and White in Table 1). However these age differences did not appear to affect the variation in the results (Tables 2-4), with the exception of Mohr which may be an outlier (lower GABA, Table 3).
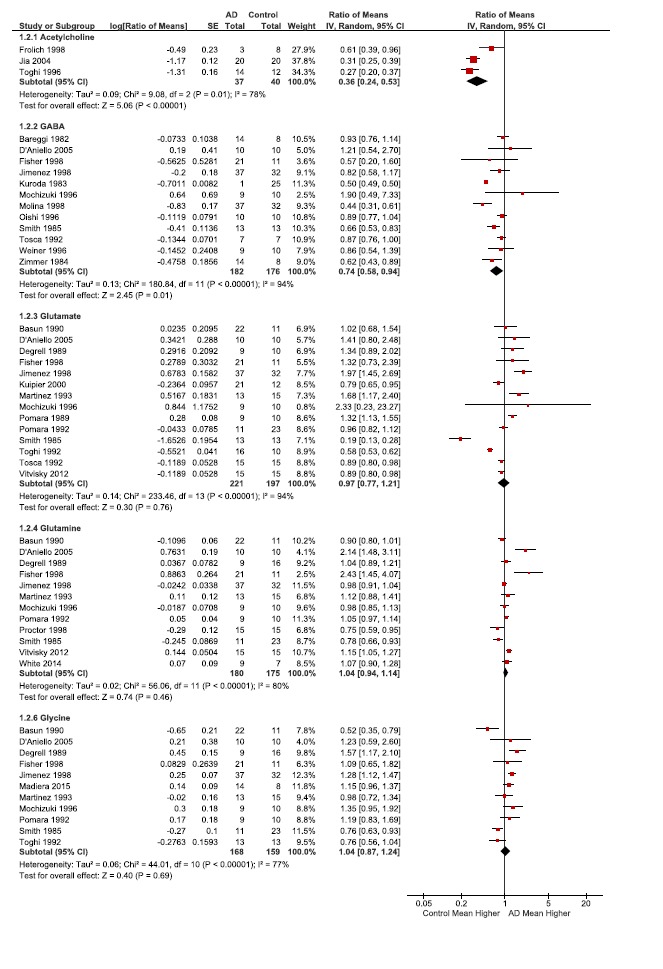
Table 3.
Ratios of the means (RoM) of selected neurotransmitters. RoM of selected metabolites and amino acids presented as the ratio of CSF concentrations between patients with Alzheimer’s disease and controls. For each individual publication or study (author name and year), the filled squares are the ratio value and the size of the square indicates the weight of the study. The horizontal lines represent the 95% CIs for each individual study. The average ratio for the pooled studies is indicated by a diamond, with the diamond width indicating the 95% CI. Heterogeneity (Tau2, Chi2 and I2) and effect size (Z), and effect size statistical significance (P) for the pooled data is also indicated for each subgroup.
With regards to cognitive scores, the mini-mental state evaluation (MMSE) is scored out of a total of 30 points, allowing AD cognitive impairment to be ranked as severe (≤9 points), moderate (10-18 points) or mild (19-23 points). Several publications had relatively mild AD (Kaddurah-Daouk, Table 1) or abnormally low MMSE for the control groups (Martinez) or the AD groups (Redjems-Bennani, Zimmer, Table 1). These cognitive differences appeared to have greater effect than age, with GSH RoMs being slightly elevated with less cognitive deficits or increased MMSE in AD patients (Kaddurah-Daouk, Table 4). Other metabolites affected include, increased aspartate with lower MMSE in controls (Martinez, Table 2), and increased pyruvate and lactate with low MMSE in AD patients (Redjems-Bennani, Table 2). The last publication with low MMSE was Zimmer, however there was no effect on the only metabolite measured (GABA, Table 3). However, MMSE is of limited value in this study as it was reported in only a subset of the publications.
3.3. Publication Bias (Funnel Plots Appendix Fig. 1)
For publication bias one is looking for asymmetry in the “funnel” of the funnel plots. However, for most of the metabolites there are less than 10 studies which are generally too few to assess funnel plots for asymmetry. In addition many of the studies have similar SE yielding little discrimination along the y-axis of the funnel plots.
3.4. Heterogeneity of Included Studies
Most variations were noted for GABA, glutamate, aspartate, asparagine, pyruvate, isoleucine, and lactate (Tables 2 and 3). For the most part this was also reflected in their I2, a statistical measure of heterogeneity. In general the largest variations were noted in Fisher and D’Aniello publications which used postmortem CSF (Appendix Table 1). The majority of compounds had an I2 below 50, such as alanine, asparagine, glucose, and glutathione. The low heterogeneity in glucose levels (I2=0), is likely because the measurement of glucose is fairly standardized and routine (Table 2). Surprisingly asparagine had an I2 of 0% even though there was variation in the data, and this reduced variation was due to low weighting of the publications with more variability (D’Aniello and Fisher, Table 2). Of note, the acetylcholine data still had high heterogeneity (78%), even though it had the fewest included publications for the metabolites (n=3) and highest effect size significance (<0.00001, Table 3).
Methodological variation may have also accounted for some variation in the results of this analysis. Use of several different methods may also have contributed to variation. For example lactate and pyruvate levels were relatively higher in the Redjems-Benneni reference where gas chromatography-mass spectrometry (GC-MS) was used, and their levels were lower in the Parnetti publication which used enzymatic assays (Tables 2 and 3, Appendix Table 1). It is also known that certain CSF collection, storage or preparation methods can alter levels of asparagine, aspartate, glutamine, glutamate. For example, acid hydrolysis-mediated deamidation of asparagine and glutamine can result in conversion to aspartic and glutamic acids [79]. Additionally, improper CSF storage can cause artificial elevations in glutamate and aspartate as well as decreases in glutamine and asparagine, while hemolysis results in increased aspartate and glutamate [79]. This deamidation and improper CSF storage are unlikely to explain the variation in aspartic and glutamic acid levels (Table 2), as there were no inverse changes in asparagine and glutamine, with the exception of glutamine and glutamate in Fisher and D’Aneillo. Of note, the latter publications used post mortem CSF. It is more likely that aspartate and glutamate elevations are due to hemolysis.
3.5. Meta-Analysis
Meta-analyses of the ratios of several amino acid and metabolites (Tables 2-4, Fig. 2) were mapped to biochemical pathways (Fig. 3), providing insight into changes relevant to metabolic and synaptic AD hypotheses. The majority of the metabolites and amino acids studied had fold changes, or effect sizes, that were not markedly different from one (unity) and thus indicating CSF concentrations similar to control values (Fig. 2).
Fig. (2).

Meta-analysis of AD-specific changes in CSF markers. Bubble plot of the meta-analysis data plotting 3 values: effect size, z-score, and a number of subjects. The effect size, or the Ration of the Means (RoM), on the y-axis is the ratio of the pooled means (AD/Ctl concentrations). A ratio greater than unity indicates that CSF concentrations in AD are greater than the control, while a lower ratio indicates decreased CSF concentrations in AD vs Ctrl. The Z-score, shown on the x-axis, is a statistic indicating the deviation of the mean from the standard error. For example, any Z-score > 1.645 relates to significant p-value < 0.10. The bubble size reflects the total number of subjects in each meta-analysis (e.g. the smallest and largest bubbles represent pooled subject sizes of 67 and 323, respectively).
Fig. (3).

Mapping of meta-analysis data onto biochemical pathways. Several metabolic and synaptic pathways are depicted. Metabolic pathways include anaplerosis, glycolysis, and the pentose phosphate pathway (PPP), all shown as boxes. The synaptic pathways include the cholinergic (boxed), glutamatergic (boxed) and GABA. The relevant metabolic enzymes and CSF metabolites and amino acids were mapped to these biochemical pathways, with increases (green arrows) or decreases (red arrows), and significant changes denoted with their p-values. Note: for simplicity, several additional sources of glutamate are not depicted e.g. from the TCA cycle. For enzyme data lacking standard deviation, these values were imputed. The GDH fold change based on 1 publication [80].
3.6. Synapse
Our data provides an important proof-of-principle which supports the validity of this meta-analysis method. Neuronal death and synaptic dysfunction are well described pathological features of AD that underlie cognitive deficits and memory loss. The cholinergic hypothesis, one of the oldest AD hypotheses, and its effects on cognition, and loss of acetylcholine-secreting neurons are well described. For support of this hypothesis, low acetylcholine levels are expected. Importantly, this was supported by our data with a highly significant decrease in CSF acetylcholine levels (Figs. 2, 3, and Table 3, 0.36-fold change, p <0.00001). Three additional neurotransmitters were analyzed in this study: the major excitatory neurotransmitter glutamate, and the inhibitory neurotransmitters GABA and glycine (Figs. 2, 3, and Table 3). Of these 3 additional neurotransmitters, only GABA CSF levels were found to be significantly decreased in AD (Table 3, GABA 0.74-fold change, p=0.01). It is likely that GABA production was reduced in order to maintain glutamate levels since glutamate is crucial for detoxification and TCA anaplerosis (Fig. 3). We were unable to find data meeting meta-analysis criteria for the enzyme that converts glutamate to GABA (glutamate decarboxylase) to corroborate the assertion of reduced GABA synthesis. The second neurotransmitter evaluated was glycine. The levels of glycine were slightly, but not significantly elevated (Table 3). Last, glutamate concentrations in AD were normal, and due to glutamates additional role in metabolism, it is addressed in more detail in that section below. Thus, our meta-analysis supports the idea that synaptic perturbations are a feature of AD, specifically cholinergic and GABAergic systems.
3.7. Metabolism
CSF glucose levels were found to be similar between AD and Ctl patients (Table 2), suggesting that there is no deficit in glucose supply to the brain for metabolism. Increased anaerobic glycolysis was found to be present in AD observed as a significant increase in lactate and a significant decrease in pyruvate (Fig. 2 and Table 2). A second glycolytic alteration suggested in AD is the increased activity of the Pentose Phosphate Pathway (PPP). Glutathione (GSH) has been used as a marker for the PPP [32], although it is an imperfect marker that is susceptible to oxidative loss. Glutathione (GSH) was found to be slightly but significantly increased in AD overall (p= 0.03, Table 4). We should note, NADPH generated by PPP, and is needed as coenzyme for glutamate dehydrogenase the latter of which was increased in AD. Our data supports both increased anaerobic glycolysis and increased PPP activity in AD.
Elevated glutamate was speculated to be central to the glutamate excitotoxicity hypothesis in AD [9]. Pooling of several (14) studies increased the subject numbers to 418, allowed for a more accurate estimate of CSF glutamate levels (Glu), which we found to be not different between AD and Ctl groups (Table 3). To further investigate this result, we examined the four (4) major metabolic pathways and enzymes that produce glutamate in the brain.
Glutamate is formed by transamination of branched chain amino acids (isoleucine, leucine and valine) taken up from the blood. In the brain, this reversible reaction favors the direction whereby there is usage of alpha-ketoglutarate and synthesis of glutamate. Branched chain amino acids exhibited only slight and non-significant elevations which indicate close to normal metabolism (Fig. 3, Table 4).
Glutamate is formed by glutamate dehydrogenase in a reaction using alpha-ketoglutarate, free ammonia and NADPH, with the NADPH for this reaction likely provided by the PPP. The reaction is reversible, however in the brain it is used to detoxify ammonia by incorporating ammonia into glutamate (Fig. 3). Independent support of
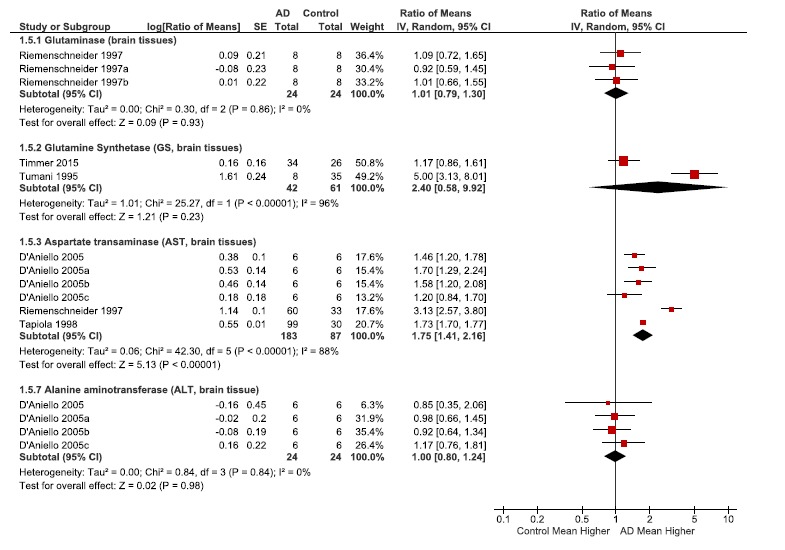
the activity of this pathway is from enzyme levels of glutamate dehydrogenase (GDH). The level of GDH was 2.46 fold increased in the AD brain as noted in one publication [80].
Glutamate can be formed by alanine aminotransferase (ALT) using α-ketoglutarate and alanine as substrates. This reaction is also reversible. While brain ALT activity was not altered (1.0 RoM, Table 5a), metabolite levels suggest that the direction of the reaction does not favor glutamate production. E.g. the significantly increased alanine levels (Fig. 2, Table 2) suggest reduced utilization of this pathway to form glutamate, instead with preferential formation of alpha-ketoglutarate. This reduced formation of glutamate via ALT is also supported by the decreased levels of pyruvate, however reduced pyruvate levels can also result from increased lactate formation (Fig. 3).
Glutamate can be formed by aspartate aminotransferase (AST). Although data for the TCA metabolites oxaloacetate and alpha-ketoglutarate were not obtained, there was a slight non-significant increase in aspartate (Table 2), and the level of AST enzyme was increased 1.75 fold (p < 0.00001) in the AD brain (Table 5a). We should note that the meta-analysis of the enzyme data relied on imputing the standard deviation for some publications.
In summary, our data indicate a possible shunt in the AD brain metabolism using glutamate to refill the TCA cycle at the level of alpha-ketoglutarate as shown in Fig. 3. This allows the bypassing of the pyruvate dehydrogenase (PDH) complex which is less functional in the AD brain due to low levels of pyruvate (Table 2). Use of this shunt in the TCA cycle can start with alpha-ketoglutarate and finishes with oxaloacetate formation. Instead of using oxaloacetate and acetyl CoA for citrate formation, oxaloacetate and glutamate may be used by AST to form aspartate and alpha-ketoglutarate to refill the modified TCA cycle.
With regards to glutamine synthesis, glutamate is also a substrate for glutamine synthetase and results in sequestering of ammonia. While glutamine levels were slightly elevated (non-significant changes, Fig. 2, Table 3), the levels of glutamine synthetase (2.4 RoM, non-significant, Table 5a) and also of glutamate dehydrogenase were increased in AD [80]. Thus, another reason why glutamate levels were likely kept within the normal range was due to the importance of glutamate in both maintaining the TCA cycle as well as detoxification of ammonia as shown in Fig. 3.
4. DISCUSSION
This meta-analysis has evaluated several published hypotheses related to metabolic and synaptic dysfunction in AD. As a proof-of-concept, this research approach validates the Cholinergic hypothesis, as evidenced by the significant lower CSF acetylcholine levels in AD patients compared to the controls. As for the synaptic hypotheses, our data also support significant reductions in CSF GABA levels in AD patients. However, the glutamate excitotoxicity hypothesis is not supported because CSF glutamate levels in AD were normal, or not significantly different from controls. With reference to the metabolic hypotheses, this meta-analysis of CSF metabolites supports a metabolic shift towards increased anaerobic glycolysis, PPP, and TCA cycle anaplerosis using glutamate in AD.
We have attempted to validate synaptic hypotheses utilizing CSF metabolite concentrations. Our meta-analysis data strongly supports the cholinergic hypothesis by demonstrating a statistically significant reduction in CSF acetylcholine levels in AD (p < 0.00001). These data should be interpreted cautiously, as it is derived from only 3 publications and more data is required to reduce confirmation bias. Regardless, the decreased levels of acetylcholine are even more remarkable given that cholinergic neurons make up about 1% and 10% of total brain cells and nerve terminal populations, respectively [81]. The highly significant findings are supported by studies which suggest that degeneration of acetylcholine releasing cholinergic neurons in the brain are one of the earliest changes in AD [82]. The decreased production of acetylcholine is likely due to a reduction in the substrate acetyl-CoA, which itself is predominantly synthesized from pyruvate via PDH [81, 83]. (Note: pyruvate levels were significantly lowered). Indeed, cholinergic marker loss has been shown to correlate with reduced energy metabolism and dementia severity prior to death [81]. Thus, reductions of CSF acetylcholine levels reflect metabolic dysfunction and contribute to cognitive deficits in AD.
GABA is the primary inhibitory neurotransmitter in the brain and is synthesized within GABAergic neurons in a single step from glutamate via glutamate decarboxylase (GAD). In AD, there are conflicting reports on the levels of GABA and the enzymes and neurons which synthesize it. For example, increases in brain GABA levels in AD have been cited [84], however, pronounced deficits were observed in our meta-analysis and in other reports [85]. There are also conflicting reports on GAD enzyme levels in AD, with cited increases [84], no significant changes, and decreases simply being attributed to postmortem changes [86]. Controversy over the loss of neurons producing GABA in AD reflects the discrepancy between the widely held beliefs that these neurons are resistant to neurodegeneration [12], whereas some reports indicate neuronal loss [87]. From a metabolic standpoint, GABA production reflects both glutamine-glutamate concentrations and synthesis by GAD. It is likely that increased glutamate flux to glutamine, in order to sequester toxic ammonia in the brain, results in this reduced GABA synthesis in AD (Fig. 3). This reduced GABA level found in our meta-analysis data likely underlies the early association of depression, insomnia, and seizures with AD [88-92].
Our meta-analysis finding of normal glutamate CSF levels has important ramifications on the glutamate excitotoxicity hypothesis or the idea that neuronal death is due to excessive glutamatergic activity [7, 8]. Notably, this hypothesis has the inherent assumption that toxicity is directly due to elevated glutamate levels in the brain. Our data indicate that glutamate levels are normal in AD with slight elevations in aspartate, which is supported by similar amino acid changes in early AD [93]. Based on our data, any apparent glutamate hyperactivity and related toxicity could be attributed to elevated aspartate. It has been suggested elevated aspartate exerts toxic effects via binding to glutaminergic receptors [13, 93]. Alternatively, this glutamatoxicity could be attributed to disinhibition of glutamatergic neurons resulting from the significantly reduced GABA levels. Reduced neuronal inhibition by GABA could explain the hippocampal hyperactivity observed in early AD [94, 95], which may itself be a compensation for neuronal loss [94-96]. This is an interesting concept which ultimately suggests that in pre-AD stages, neuronal hyperactivity may lead to increased neuronal glutamate release. Later in the progression of AD, glutamatergic neuronal death makes the elevated secretion of glutamate by remaining neurons appear “normal”. Lastly, since cancers also utilize anaplerosis [97], it is possible that competition for this common metabolic pathway may explain the inverse relationship between AD and cancer occurrence [98, 99].
We have used a similar approach to validate metabolic hypotheses in AD, as several lines of evidence implicate metabolic dysfunction in AD. First, the high energy requirement and limited energy stores of the brain [14, 96] leads to neuronal dysfunction and death in AD because of the pathological reduction of glucose utilization and hypoperfusion [17, 26, 100-102]. Even though the AD brain may utilize lactate, pyruvate, glutamate, and glutamine, it is glucose which is the major energy source for ATP production [24]. There is also a correlation between reduced glucose utilization and AD severity [26]. Indeed, this glucose dysregulation have been described as insulin resistance in the brain, or Type 3 diabetes [103-106]. Second, nutrient deficiency via chemical inhibition of metabolism or deficiency of thiamine (important for pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase activities) both mimic AD pathology [21, 103]. Third, reduced nutrient supply triggers endoplasmic reticulum (ER) stress responses such as protein aggregation and autophagy [104, 105], which are also pathological features of AD. Examples of metabolic dysregulation proposed in AD include glycolytic changes, such as upregulated anaerobic glycolysis and pentose phosphate pathway (PPP) [14-19], and tricarboxylic cycle (TCA) changes [14, 17, 23, 106].
Our data demonstrate that CSF glucose levels are normal in AD brain. This seems contradictory to the reduced glucose utilization/reduced flurodeoxyglucose (FDG) uptake in AD described above. This can be explained by several different observations. First, CSF levels of glucose are different than cellular utilization of glucose. This reduced glucose utilization may be a result of insulin resistance in the brain (type 3 diabetes) [107, 108], or alterations to vascular flow or glucose uptake by astrocytes [109]. Second and alternatively, there are two concurrent processes taking place: loss of metabolically active neurons [26] and neuronal hyperactivity [96, 110]. An overall decrease in glucose utilization with increased disease severity [26] may, therefore, reflect neuronal loss, while the elevated lactate reflects increased synaptic activity [111] and thus also glycolytic flux. Regardless of glucose levels and glucose utilization, it is clear that there are changes in glycolysis and the TCA cycles as described below.
Our meta-analysis strongly suggests altered glycolysis in the brains of AD patient. An elevation in anaerobic glycolysis is supported by pyruvate and lactate data which suggest increased conversion of pyruvate to lactate in the AD brain. Importantly, aerobic glycolysis is increased in the default mode network (DMN), an area which is affected by AD pathology [112]. There are several published explanations for the elevation of CSF lactate levels in AD. First, switching from aerobic to anaerobic glycolysis may be an adaptive response to the hypoperfusion and oxygen deficiency, the latter which is known to occur in AD [113, 114]. Second, decreased pyruvate levels, along with damaged pyruvate dehydrogenase complex and alpha-ketoglutarate dehydrogenase [115-120], result in pyruvate not being able to participate in the TCA cycle. Pyruvate dehydrogenase deficiency diseases cause marked increases in lactate [121].
The PPP branches out of glycolysis, and increased PPP metabolic activity has been described in AD [14-19] likely in response to increased oxidative stress [15]. Using glutathione as a marker for PPP activity [122], our analysis does support an elevation of PPP in AD. It is likely that such changes occur in the early stages of the disease [18, 123]. However, this finding should be interpreted with caution due to limited data being available and glutathione not being an ideal marker for PPP up-regulation. Although reduced glutathione should theoretically give an indication of PPP activity, it is also subject to loss by oxidative stress. Better markers are required to accurately evaluate the alterations in the PPP within AD. Indirectly an increase of NADPH formation in the PPP is suggested by the increased activity of glutamate dehydrogenase, which needs NADPH in its reaction.
TCA cycle dysfunction is likely due to reduced pyruvate utilization by a deficient pyruvate dehydrogenase complex as indicated previously. One response to this TCA dysfunction is the utilization of glutamate as an alternative substrate to fuel the TCA cycle [23, 124]. The inter-conversion of glutamate to aspartate was demonstrated by Krebs in 1960 [22], and importantly this TCA shunt was demonstrated to be utilized under hypoglycemic conditions [25]. Of note the latter condition may occur in AD under conditions of reduced glucose utilization. This shunt uses glutamate and aspartate to refill the TCA cycle, thus bypassing TCA enzymes known to be dysfunctional in AD.
Why are the glutamate levels normal? There are several sources of glutamate in the brain. Of note, glutamate has to be formed in the brain as it cannot cross the blood-brain barrier [125]. The brain/CSF glutamate stores originate not only from the neurotransmitter pool (including neuronal and astrocytic uptake, and astrocytic conversion from glutamine via glutaminase) [7, 125, 126], but also from metabolic sources [125]. Fig. 3 depicts four metabolic sources of glutamate formation: transamination reactions of branched-chain amino acids aminotransferase, ALT and AST, as well as synthesis from alpha-ketoglutarate. Importantly, glutamate formation from glutamate dehydrogenase scavenges free ammonia. Normal glutamate levels allow for the continuation of clearing this toxic compound from the brain by synthesis of glutamine [11, 125, 126]. Indeed, an excess of glutamate was not required for removal of ammonia, and the amount of glutamate formed was proportional to the amount or ammonia removed [127]. Thus the normal levels of CSF glutamate result from different synthetic pathways, as well as glutamate’s role as the major excitatory neurotransmitter and in several other metabolic processes in the brain.
We now come back to discuss these findings in relation to AD brain in general and to potential metabolic and synaptic therapeutic avenues to remedy the failure of AD therapeutic drugs mentioned in the introduction. First we will discuss potential metabolic interventions. Our re-evaluation of the metabolic hypotheses supports the idea that the brain is “starving” in AD [14]. While anaerobic glycolysis and the anapleurotic shunt still can generate energy for the brain, they would do so at reduced levels. Levels of ATP have previously been determined to decrease with AD progression by 7% in early stages, then by 20%, and 35% - 50% in advanced AD stages [109]. Thus increasing energy production in the brain are viable strategies. Treatment with phenylbutyrate, an FDA approved drug for treating pyruvate dehydrogenase deficiencies [128], may be useful in treating AD as well [129, 130]. Increasing glucose utilization with insulin sensitizing drugs is another strategy. Insulin and insulin-like drugs are in trials for AD treatment, likely aiming to address the underlying insulin resistance exhibited in AD by increasing glucose utilization [131]. The insulin-sensitizing FDA approved drug metformin, in population studies resulted in a reduction of dementia risk, however, the effects on AD cognition are controversial [132]. Another hypoglycemic treatment that may have therapeutic potential is the natural plant compound berberine. In addition to having anti-atherosclerotic effects [133, 134], berberine also reduces amyloid beta aggregation in an in vitro assay [135], representing a multifactorial treatment.
Are there any synaptic therapies that appear promising in light of our data? Yes, there are already several FDA-approved therapies for AD, that already target cholinergic, GABAergic, and glutamatergic systems. The majority of the FDA approved AD drugs target acetylcholinesterases, aiming to compensate for the acetylcholine deficits that have historically been presumed to underlie short-term memory loss in AD [82]. However, the acetylcholinesterase inhibitor drugs donepezil, galantamine, rivastigmine, and huperzine A only provide short-term symptomatic improvement and are not useful as a long term cure [136]. Additionally, treatments aimed at recovery of GABA levels in patients with low GABA did not have therapeutic benefit [59]. Finally, targeting glutamatergic neurons with memantine, an uncompetitive NMDA antagonist approved by the FDA in 2004 for moderate to severe AD, also only alleviates symptoms [137]. Our meta-analysis re-evaluation of synaptic hypotheses has also generated insight into existing therapeutics and their mechanisms of action. The evidently normal levels of glutamate may be one reason why memantine inhibition of glutamatergic activity only offers symptomatic effects and does not constitute a cure. Our meta-analysis also identified significantly reduced levels of acetylcholine and GABA and serves to highlight the effects of neuronal loss on neurotransmitter levels in AD. Here, synaptic therapies fail to provide a cure because therapeutic efficacy wanes as the targeted neuronal populations are lost. We must point out that there is some evidence that synaptic loss itself causes AD, as exposure to organophosphates, such as paraquat, and dieldrin, which affect the cholinergic system, are associated with increased AD risk [138]. Some studies have demonstrated a link between benzodiazepine use and AD risk, however, a recent study indicated that there was no increased risk [139]. Such varied reports likely indicate that AD etiogenesis is multifactorial, with some cases being metabolic in origin, while others have a synaptic pathology. However, the majority of synaptic drugs have only had symptomatic amelioration. This may reflect the idea that synaptic dysfunction is secondary to neuronal loss and cognitive decline later on in the progression of the disease.
It is important to note that the present work is unable to estimate sensitivities and specificities for detection of Alzheimer's disease since group averages, not individual patient values, were reported in the studies included in our meta analysis. As such we are unable to suggest specific neurotransmitter and metabolite cutoff levels for clinical guidance.
CONCLUSION
This investigation has effectively demonstrated that meta-analysis of existing publications can yield considerable insight into metabolic and synaptic hypotheses of AD. Synaptic involvement in AD is clear, and drug development based on the cholinergic hypothesis has been successful. We have invalidated the glutamatergic hypothesis, suggesting instead that excitotoxicity could be due to reduced CSF GABA levels. Of course these correlations need to be further evaluated.
The role of metabolic dysfunction in AD is less clear cut, however it is undisputed that reduced perfusion and glucose utilization occur in AD (reviewed [102]). These changes are expected to result in metabolic dysfunction. Further examples of metabolic dysfunction are oxidative damage to TCA cycle enzymes [102], and autoantibody attacks on several metabolic enzymes [140]. The connection is strengthened by the fact that metabolic diseases, obesity and type 2 diabetes, are risk factors for AD (reviewed [141]). From a prevention standpoint, epidemiological studies indicate that metabolic diseases of obesity and diabetes increase the risk of AD, while specific diets, such as MIND and Mediterranean [141], and caloric restriction [142] appear to reduce AD incidence. Indeed, metabolic intervention, albeit in a small study group, slowed cognitive decline and in some cases reversed the cognitive deficits in AD [143]. However, systematic reviews indicate that many dietary, exercise multi-interventions have a modest or inconclusive effect on cognitive abilities. These reviews were performed by both the Agency for Healthcare Research & Quality and National Academies of Science, Engineering, and Medicine, and included well known studies such as Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability FINGER), Multidomain Alzheimer Preventive Trial (MAPT), Advanced Cognitive Training for Independent and Vital Elderly (ACTIVE), and Prevention of Dementia by Intensive Vascular Care (PreDIVA) [144, 145].
This meta-analytical correlation of metabolites to cognitive dysfunction evaluated the various metabolic hypotheses of AD to allow for more precise interventions and therapeutic targets in the future. We are not proposing a catch-all theory of AD, but instead are using metabolite correlations with AD diagnoses in order to validate several of the hypotheses investigated and invalidate others. Our study demonstrates the need for incorporation of routine measurements of these metabolites and amino acids in future CSF studies. It is our hope that a continued re-evaluation of AD hypotheses will lead to more effective therapeutic options and eventually to a cure for AD. Finally, our study also demonstrates an important proof-of-concept of this meta-analysis method for rapid validation of AD hypotheses, and evaluation of therapeutics.
AUTHOR CONTRIBUTION
Dr. Jan O. Friedrich designed research/study and analyzed the meta-analysis data, in excel and the Cochrane Review Manager software, as well as expert critical review of the manuscript. We should make special note that Dr Friedrich was instrumental in training IM in the meta-analysis method. The following authors spent significant time searching the literature and collected data from the following number of publications: Sean Scarpiello (21), Anthony Nanajian (19), Stefani Thompson (12), Matthew Chang (11), Matthew Protas (10), and Neil Khoury (8). Dr. Margit Trotz analyzed the data into existing biochemical pathways and wrote the biochemical sections of the manuscript. Dr. Lucy Clunes wrote the pharmacological sections of the manuscript. Marisa Deliso, Brittany Bass, Roni Manyevitch, Rachel Gonnella, Emily Harms, Dr George Perry, and Dr. Blaine Moore all made significant contributions to writing manuscript. Angélica Ortiz spent many hours to contributing to the creation of Figure 3 used in this publication. Dr. Ian Murray conceived of the hypothesis, designed the research/study, collated data, supervised the students, performed the meta-analysis and wrote the manuscript.
CONSENT FOR PUBLICATION
Not applicable.
Table 5.
(A). Changes in enzymes related to anaplerosis. Meta-analysis data are shown for enzyme protein level changes glutaminase, glutamate dehydrogenase (GDH), and glutamine synthetase (GS) , and activity changes for aspartate aminotransferase (AST) and alanine aminotransferase (ALT). Since some publications did not report standard deviation, we imputed the largest standard deviation among the other studies reporting to allow estimation of SE. The errors for glutaminase were estimated using the of 44% from D’Aneillo. Glutamine synthase (GS) errors for Timmer were estimated using the error of 60% from Tumani. Meta-analysis was not performed on Glutamate dehydrogenase (GDH), as there was only 1 study. However the study reported a 2.05 fold increase protein concentration in AD vs Ctl [80].
B.
Biochemical reactions for the enzymes above.
| Enzyme | Biochemical Reactions |
|---|---|
| Glutaminase levels | Glutamine → glutamate + ammonia |
| Glutamate dehydrogenase (GDH) | α-Ketoglutarate + ammonia + NADPH → glutamate |
| Glutamine synthetase (GS) l | Glutamate + ammonia + ATP → glutamine |
| Aspartate aminotransferase (AST) | Aspartate + α-ketoglutarate ↔ oxaloacetate + glutamate |
| Alanine aminotransferase (ALT) | Alanine + α-ketoglutarate ↔ pyruvate + glutamate |
ACKNOWLEDGEMENTS
We wish to thank Joselle O'Brien, Danielle Wolfe, Hae-Sun La, Yoon Sean Ho, Kerry Vlaming, and Ashley Yearwood for their initial contribution to data collection. I would also like to thank Drs Marios Loukas and Ronald Chamberlain for suggesting that I utilize the meta-analysis methodology.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Cummings J.L., Morstorf T., Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res. Ther. 2014;6(4):37. doi: 10.1186/alzrt269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kennedy M.E., Stamford A.W., Chen X., Cox K., Cumming J.N., Dockendorf M.F., et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med. 2016;8(363):363ra150. doi: 10.1126/scitranslmed.aad9704. [DOI] [PubMed] [Google Scholar]
- 3.Geets H. Disconnect transgenic mice and CNS drug discovery. CNS Drugs. 2009;23:915–926. doi: 10.2165/11310890-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Bartus R.T., Dean R.L., Beer B., Lippa A.S. Cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217:408–417. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- 5.Davis B.M., Mohs R.C., Greenwald M.D., Mathe A.A., Johns C.A., Horvath T.B., et al. Clinical studies of the cholinergic deficit in Alzheimer’s disease. J. Am. Geriatr. Soc. 1985;33:741–748. doi: 10.1111/j.1532-5415.1985.tb04184.x. [DOI] [PubMed] [Google Scholar]
- 6.Perry E., Tomlinson B., Blessed G., Bergmann K., Gibson P., Perry R. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. BMJ. 1978;2:1457–1459. doi: 10.1136/bmj.2.6150.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butterfield D.A., Pocernich C.B. The glutamatergic system and Alzheimer’s disease: therapeutic implications. CNS Drugs. 2003;17:641–652. doi: 10.2165/00023210-200317090-00004. [DOI] [PubMed] [Google Scholar]
- 8.Lewerenz J., Maher P. Chronic glutamate toxicity in neurodegenerative diseases-what is the evidence? Front. Neurosci. 2015;9:1–20. doi: 10.3389/fnins.2015.00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maragos W.F., Greenamyre J.T., Penney J.B., Young A.B. Glutamate dysfunction in Alzheimer’s disease: an hypothesis. Trends Neurosci. 1987;10:65–68. [Google Scholar]
- 10.Drake J., Link C.D., Butterfield D.A. Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid beta-peptide (1-42) in a transgenic Caenorhabditis elegans model. Neurobiol. Aging. 2003;24:415–420. doi: 10.1016/s0197-4580(02)00225-7. [DOI] [PubMed] [Google Scholar]
- 11.Salminen A., Jouhten P., Sarajärvi T., Haapasalo A., Hiltunen M. Hypoxia and GABA shunt activation in the pathogenesis of Alzheimer’s disease. Neurochem. Int. 2016;92:13–24. doi: 10.1016/j.neuint.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 12.Rissman R.A., Mobley W.C. Implication for tratement: GABAa receptors in aging, down syndrome and Alzheimer’s disease. J. Neurochem. 2011;117:613–622. doi: 10.1111/j.1471-4159.2011.07237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoyer S. Glucose and related brain metabolism in normal aging. 1988;11:150–156. [Google Scholar]
- 14.Mamelak M. Sporadic Alzheimer’s disease: the starving brain. J. Alzheimers Dis. 2012;31:459–474. doi: 10.3233/JAD-2012-120370. [DOI] [PubMed] [Google Scholar]
- 15.Palmer A.M. The activity of the pentose phosphate pathway is increased in response to oxidative stress in Alzheimer’s disease. J Neural Transm Springer-Verlag. 1999;106:317–328. doi: 10.1007/s007020050161. [DOI] [PubMed] [Google Scholar]
- 16.Newington J.T., Pitts A., Chien A., Arseneault R., Schubert D., Cumming R.C. Amyloid beta resistance in nerve cell lines is mediated by the Warburg effect. PLoS One. 2011;6:e19191. doi: 10.1371/journal.pone.0019191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gavai A.K., Supandi F., Hettling H., Murrell P., Leunissen J.A., Van Beek J.H. Using bioconductor package BiGGR for metabolic flux estimation based on gene expression changes in brain. PLoS One. 2015;10:1–21. doi: 10.1371/journal.pone.0119016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orešič M., Hyötyläinen T., Herukka S-K., Sysi-Aho M., Mattila I., Seppänan-Laakso T., et al. Metabolome in progression to Alzheimer’s disease. Transl. Psychiatry. 2011;1:e57. doi: 10.1038/tp.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferreira I.L., Resende R., Ferreiro E., Rego A.C., Pereira C.F., Ferriera I.L., et al. Multiple defects in energy metabolism in Alzheimer’s disease. Curr. Drug Targets. 2010;11:1193–1206. doi: 10.2174/1389450111007011193. [DOI] [PubMed] [Google Scholar]
- 20.Bubber P., Haroutunian V., Fisch G., Blass J.P., Gibson G.E. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann. Neurol. 2005;57:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- 21.Petrofes Chapa R.D., Emery M.A., Fawver J.N., Murray I.V. Amyloids as sensors and protectors (asap) hypothesis. journal of alzheimer’s disease, this paper propounds the amyloids as sensors and protectors (asap) hypothesis. in this novel hypothesis, we provide evidence that amyloids are capable of sensing dysfunction, and after misfolding, initiate protective cellular responses. Amy Prot a. 2012;29:503–514. doi: 10.3233/JAD-2012-112015. [DOI] [PubMed] [Google Scholar]
- 22.Krebs H.A., Bellamy D. The interconversion of glutamic acid and aspartic acid in respiring tissues. Biochem. J. 1960;75:523–529. doi: 10.1042/bj0750523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Procter A.W., Palmer A.M., Francis P.T., Lowe S.L., Neary D., Murphy E., et al. Evidence of glutamatergic denervation and possible abnormal metabolism in Alzheimer’s disease. J. Neurochem. 1988;50:790–802. doi: 10.1111/j.1471-4159.1988.tb02983.x. [DOI] [PubMed] [Google Scholar]
- 24.Hoyer S. Oxidative energy metabolism in Alzheimer brain. Mol. Chem. Neuropathol. 1992;16:207–224. doi: 10.1007/BF03159971. [DOI] [PubMed] [Google Scholar]
- 25.Rink C., Gnyawali S., Stewart R., Teplitsky S., Harris H., Roy S., et al. Glutamate oxaloacetate transaminase enables anaplerotic refilling of TCA cycle intermediates in stroke-affected brain. FASEB J. 2017;31:1709–1718. doi: 10.1096/fj.201601033R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mosconi L., Berti V., Glodzik L., Pupi A., De Santi S., de Leon M.J. Pre-clinical detection of alzheimer’s disease using fdg-pet, with or without amyloid imaging. J. Alzheimers Dis. 2010;20:843–854. doi: 10.3233/JAD-2010-091504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Demetrius L.A., Simon D.K. An inverse-Warburg effect and the origin of Alzheimer’s disease. Biogerontology. 2012;13:583–594. doi: 10.1007/s10522-012-9403-6. [DOI] [PubMed] [Google Scholar]
- 28.Liguori C., Chiaravalloti A., Sancesario G., Stefani A., Sancesario G.M., Mercuri N.B., et al. Cerebrospinal fluid lactate levels and brain [18F]FDG PET hypometabolism within the default mode network in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging. 2016;43(11):2040–2049. doi: 10.1007/s00259-016-3417-2. [DOI] [PubMed] [Google Scholar]
- 29.Olsson B., Lautner R., Andreasson U., Öhrfelt A., Portelius E., Bjerke M., et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol. 2016;•••:673–684. doi: 10.1016/S1474-4422(16)00070-3. [DOI] [PubMed] [Google Scholar]
- 30.Moher D., Liberati A., Tetzlaff J., Altman D.G., Grp P. Preferred reporting items for systematic reviews and meta-analyses: the prisma statement (reprinted from annals of internal medicine). Phys. Ther. 2009;89:873–880. [PubMed] [Google Scholar]
- 31.Wishart D.S., Lewis M.J., Morrissey J.A., Flegel M.D., Jeroncic K., Xiong Y., et al. The human cerebrospinal fluid metabolome. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008;871(2):164–173. doi: 10.1016/j.jchromb.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Friedrich J.O., Adhikari N.K., Beyene J. The ratio of means method as an alternative to mean differences for analyzing continuous outcome variables in meta-analysis: a simulation study. BMC Med. Res. Methodol. 2008;8:32. doi: 10.1186/1471-2288-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedrich J.O., Adhikari N.K., Beyene J. Ratio of means for analyzing continuous outcomes in meta-analysis performed as well as mean difference methods. J. Clin. Epidemiol. 2011;64:556–564. doi: 10.1016/j.jclinepi.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 34.Dersimonian R., Laird N. Meta-analysis in clinical trials. Cont Clin Trails. 1986;7:177–188. doi: 10.1016/0197-2456(86)90046-2. [DOI] [PubMed] [Google Scholar]
- 35.Chang Y., Chang W., Tsai N., Huang C., Kung C., Su Y., et al. The roles of biomarkers of oxidative stress and antioxidant in Alzheimer ’ s Disease: a systematic review. BioMed Res. Int. 2014;2014:182303. doi: 10.1155/2014/182303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Christen Y. Oxidative stress and Alzheimer disease. Am. J. Clin. Nutr. 2000;71:621s–6629. doi: 10.1093/ajcn/71.2.621s. [DOI] [PubMed] [Google Scholar]
- 37.Cooper A.J., Kristal B.S. Multiple roles of glutathione in the central nervous system. Biol. Chem. 1997;378:793–802. [PubMed] [Google Scholar]
- 38.Schulz J.B., Lindenau J., Seyfried J., Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000;267:4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- 39.Lovell M.A., Xie C., Markesbery W.R. Decreased glutathione transferase activity in brain and ventricular fluid in Alzheimer’s disease. Neurology. 1998;51:1562–1566. doi: 10.1212/wnl.51.6.1562. [DOI] [PubMed] [Google Scholar]
- 40.Mandal P.K., Saharan S., Tripathi M., Murari G. Brain glutathione levels - a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol. Psychiatry. 2015;78:702–710. doi: 10.1016/j.biopsych.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 41.Van Gool W.A., Bolhuis P.A. Cerebrospinal fluid markers of Alzheimer’s disease. Geriatric Biosci. 1991;39:1025–1039. doi: 10.1111/j.1532-5415.1991.tb04052.x. [DOI] [PubMed] [Google Scholar]
- 42.Bergen A.A., Kaing S., Ten Brink J.B., Netherlands B.B., Gorgels T.G., Janssen S.F. Gene expression and functional annotation of human choroid plexus epithelium failure in Alzheimer’s disease. BMC Genomics. 2015;16:956. doi: 10.1186/s12864-015-2159-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Veitinger M., Oehler R., Umlauf E., Baumgartner R., Schmidt G., Gerner C., et al. A platelet protein biochip rapidly detects an Alzheimer’s disease-specific phenotype. Acta Neuropathol. 2014;128:665–677. doi: 10.1007/s00401-014-1341-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodrigues L., Dutra M.F., Ilha J., Biasibetti R., Quincozes-Santos A., Leite M.C., et al. Treadmill training restores spatial cognitive deficits and neurochemical alterations in the hippocampus of rats submitted to an intracerebroventricular administration of streptozotocin. J. Neural Transm. (Vienna) 2010;117:1295–1305. doi: 10.1007/s00702-010-0501-9. [DOI] [PubMed] [Google Scholar]
- 45.Kuhla B., Lüth H-J., Haferburg D., Boeck K., Arendt T., Münch G., et al. Methylglyoxal, glyoxal, and their detoxification in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2005;1043:211–216. doi: 10.1196/annals.1333.026. [DOI] [PubMed] [Google Scholar]
- 46.Kölsch H., Linnebank M., Lütjohann D., Jessen F., Wüllner U., Harbrecht U., et al. Polymorphisms in glutathione S-transferase omega-1 and AD, vascular dementia, and stroke. Neurology. 2004;63:2255–2260. doi: 10.1212/01.wnl.0000147294.29309.47. [DOI] [PubMed] [Google Scholar]
- 47.Volicer L., Chen J.C., Crino P.B., Vogt B.A., Fishman J., Rubins J., et al. Neurotoxic properties of a serotonin oxidation product: possible role in Alzheimer’s disease. Prog. Clin. Biol. Res. 1989;317:453–465. [PubMed] [Google Scholar]
- 48.Csernansky J.G., Bardgett M.E., Sheline Y.I., Morris J.C., Olney J.W. CSF excitatory amino acids and severity of illness in Alzheimer’s disease. Neurology. 1996;46:1715–1720. doi: 10.1212/wnl.46.6.1715. [DOI] [PubMed] [Google Scholar]
- 49.Enna S.J., Stern L.Z., Wastek G.J., Yamamura H.I. Cerebrospinal fluid gamma-aminobutyric acid variations in neurological disorders. Arch. Neurol. 1977;34:683–685. doi: 10.1001/archneur.1977.00500230053008. [DOI] [PubMed] [Google Scholar]
- 50.Ferrarese C., Aliprandi A., Tremolizzo L., Stanzani L., De Micheli A., Dolara A., et al. Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology. 2001;57:671–675. doi: 10.1212/wnl.57.4.671. [DOI] [PubMed] [Google Scholar]
- 51.Gueli M.C., Taibi G. Alzheimer’s disease: amino acid levels and brain metabolic status. Neurol. Sci. 2013;34:1575–1579. doi: 10.1007/s10072-013-1289-9. [DOI] [PubMed] [Google Scholar]
- 52.Greenamyre J.T., Young A.B. Excitatory amino acids and Alzheimer’s disease. Neurobiol. Aging. 1989;10:593–602. doi: 10.1016/0197-4580(89)90143-7. [DOI] [PubMed] [Google Scholar]
- 53.Hartikainen P., Reinikainen K.J., Soininen H., Sirvio J., Soikkeli R., Riekkinen P.J. Neurochemical markers in the cerebrospinal fluid of patients with Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis and normal controls. J. Neural Transm. (Vienna) 1992;4:53–68. doi: 10.1007/BF02257622. [DOI] [PubMed] [Google Scholar]
- 54.Hardy J., Cowburn R., Barton A., Reynolds G., Dodd P., Wester P., et al. A disorder of cortical GABAergic innervation in Alzheimer’s disease. Neurosci. Lett. 1987;73:192–196. doi: 10.1016/0304-3940(87)90016-4. [DOI] [PubMed] [Google Scholar]
- 55.Iribar M.C., Montes J., Maldonado R.G., Peinad J.M. Alanyl-aminopeptidase activity decrease in cerebrospinal fluid of Alzheimer patients. Dement. Geriatr. Cogn. Disord. 1998;9(1):44–49. doi: 10.1159/000017021. [DOI] [PubMed] [Google Scholar]
- 56.Kaiser E., Schoenknecht P., Kassner S., Hildebrandt W., Kinscherf R., Schroeder J., et al. Cerebrospinal fluid concentrations of functionally important amino acids and metabolic compounds in patients with mild cognitive impairment and Alzheimer’s disease. Neurodegener. Dis. 2010;7:251–259. doi: 10.1159/000287953. [DOI] [PubMed] [Google Scholar]
- 57.Manyam N.V., Katz L., Hare T.H., Gerber J.C., Grossman M.H. Levels of gamma-aminobutyric acid in in various neurologic disorders. Arch. Neurol. 1980;37:352–355. doi: 10.1001/archneur.1980.00500550054006. [DOI] [PubMed] [Google Scholar]
- 58.Moats R.A., Ernst T., Shonk T.K., Ross B.D. Abnormal cerebral metabolite concentrations in patients with probable Alzheimer disease. Magn. Reson. Med. 1994;32:110–115. doi: 10.1002/mrm.1910320115. [DOI] [PubMed] [Google Scholar]
- 59.Mohr E., Bruno G., Foster N., Gillespie M., Cox C., Hare T.A., et al. GABA-agonist therapy for Alzheimer’s disease. Clin. Neuropharmacol. 1986;9:257–263. doi: 10.1097/00002826-198606000-00004. [DOI] [PubMed] [Google Scholar]
- 60.Raskind M.A., Peskind E.R., Lampe T.H., Risse S.C., Taborsky G.J., Dorsa D. Cerebrospinal fluid vasopressin, oxytocin, somatostatin, and beta-endorphin in Alzheimer’s Disease. Arch. Gen. Psychiatry. 1986;43:382–388. doi: 10.1001/archpsyc.1986.01800040092013. [DOI] [PubMed] [Google Scholar]
- 61.Samakashvili S., Ibáñez C., Simó C., Gil-Bea F.J., Winblad B., Cedazo-Mínguez A., et al. Analysis of chiral amino acids in cerebrospinal fluid samples linked to different stages of Alzheimer disease. Electrophoresis. 2011;32:2757–2764. doi: 10.1002/elps.201100139. [DOI] [PubMed] [Google Scholar]
- 62.Schmand B., Huizenga H.M., van Gool W.A. Meta-analysis of CSF and MRI biomarkers for detecting preclinical Alzheimer’s disease. Psychol. Med. 2010;40:135–145. doi: 10.1017/S0033291709991516. [DOI] [PubMed] [Google Scholar]
- 63.Schmidt D., Loscher W. Plasma and cerebrospinal fluid gamma-aminobutyric acid in neurological disorders. J. Neurol. Neurosurg. Psychiatry. 1982;45:931–935. doi: 10.1136/jnnp.45.10.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seidl R., Cairns N., Singewald N., Kaehler S.T., Lubec G. Differences between GABA levels in Alzheimer’s disease and Down syndrome with Alzheimer-like neuropathology. Naunyn Schmiedebergs Arch. Pharmacol. 2001;363:139–145. doi: 10.1007/s002100000346. [DOI] [PubMed] [Google Scholar]
- 65.Zepeda A.T., Nesme O.F., Mendez-Franco J., Otero-Siliceo E., de la Mora P. Free-GABA levels in the cerebrospinal fluid of patients suffering from several neurological diseases Its potential use for the diagnosis of diseases which course with inflammation and tissular necrosis. Amino Acids. 1995;9:207–216. doi: 10.1007/BF00805952. [DOI] [PubMed] [Google Scholar]
- 66.Li Y., Sun H., Chen Z., Xu H., Bu G., Zheng H. Implications of GABAergic neurotransmission in Alzheimer’s disease. Front. Aging Neurosci. 2016;8:1–12. doi: 10.3389/fnagi.2016.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kuroda H. Gamma-aminobutyric acid (GABA) in cerebrospinal fluid. Acta Med. Okayama. 1983;37:166–177. doi: 10.18926/AMO/32437. [DOI] [PubMed] [Google Scholar]
- 68.Butterfield D.A., Hardas S.S., Lange M.L. Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer’s disease: many pathways to neurodegeneration. J. Alzheimers Dis. 2010;20:369–393. doi: 10.3233/JAD-2010-1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Elble R., Giacobini E., Higgins C. Choline levels are increased in cerebrospinal fluid of Alzheimer patients. Neurobiol. Aging. 1989;10:45–50. doi: 10.1016/s0197-4580(89)80009-0. [DOI] [PubMed] [Google Scholar]
- 70.Lleó A., Cavedo E., Parnetti L., Vanderstichele H., Herukka S.K., Andreasen N., et al. Cerebrospinal fluid biomarkers in trials for Alzheimer and Parkinson diseases. Nat. Rev. Neurol. 2014;11:41–55. doi: 10.1038/nrneurol.2014.232. [DOI] [PubMed] [Google Scholar]
- 71.Karami A., Eyjolfsdottir H., Vijayaraghavan S., Lind G., Almqvist P., Kadir A., et al. Changes in CSF cholinergic biomarkers in response to cell therapy with NGF in patients with Alzheimer’s disease. Alzheimers Dement. 2015;11:1316–1328. doi: 10.1016/j.jalz.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 72.Smith R.C., Kumar V., Khera G., Elble R., Giacobini E., Colliver J. Brain morphological measures and CSF acetylcholine in Alzheimer dementia. Psychiatry Res. 1988;23:111–114. doi: 10.1016/0165-1781(88)90039-x. [DOI] [PubMed] [Google Scholar]
- 73.Sorbi S., Tonini S., Mortilla M., Piacentini S., Amaducci L. Lactate production and glycolytic enzymes in sporadic and familial Alzheimer’s disease. J. Neural Transm. (Vienna) 1989;1:18. [Google Scholar]
- 74.Toghi H., Kimura A.M., Saheki M., Takahashi S. Cerebrospinal fluid acetylcholine and choline in vascular dementia of Binswanger and multiple small infarct types as compared with Alzheimer-type dementia. J Neu Transm. 1996;104:931–941. doi: 10.1007/BF01271206. [DOI] [PubMed] [Google Scholar]
- 75.Giacobini E. Brain acetylcholine--a view from the cerebrospinal fluid (CSF). Neurobiol. Aging. 1986;7:392–396. [Google Scholar]
- 76.Gil-Bea F.J., García-Alloza M., Domínguez J., Marcos B., Ramírez M.J. Evaluation of cholinergic markers in Alzheimer’s disease and in a model of cholinergic deficit. Neurosci. Lett. 2005;375:37–41. doi: 10.1016/j.neulet.2004.10.062. [DOI] [PubMed] [Google Scholar]
- 77.Heywood W.E., Galimberti D., Bliss E., Sirka E., Paterson R.W., Magdalinou N.K., et al. Identification of novel CSF biomarkers for neurodegeneration and their validation by a high-throughput multiplexed targeted proteomic assay. Mol. Neurodegener. 2015;10:64. doi: 10.1186/s13024-015-0059-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vijayaraghavan S., Karami A., Aeinehband S., Behbahani H., Grandien A., Nilsson B., et al. Regulated extracellular choline acetyltransferase activity- the plausible missing link of the distant action of acetylcholine in the cholinergic anti-inflammatory pathway. PLoS One. 2013;8(6):e65936. doi: 10.1371/journal.pone.0065936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haas R.H., Parikh S., Falk M.J., Russell P., Wolf N.I., Darin N., et al. The in-depth evaluation of suspected mitochondrial disease: the mitochondrial medicine society’s committee on diagnosis. Mol. Genet. Metab. 2008;94(1):16–37. doi: 10.1016/j.ymgme.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Korolainen M.A., Goldsteins G., Nyman T.A., Alafuzoff I., Koistinaho J., Pirttilä T. Oxidative modification of proteins in the frontal cortex of Alzheimer’s disease brain. Neurobiol. Aging. 2006;27:42–53. doi: 10.1016/j.neurobiolaging.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 81.Szutowicz A., Bielarczyk H., Jankowska-Kulawy A., Pawełczyk T., Ronowska A. Acetyl-CoA the key factor for survival or death of cholinergic neurons in course of neurodegenerative diseases. Neurochem. Res. 2013;38:1523–1542. doi: 10.1007/s11064-013-1060-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mufson E.J., Counts S.E., Perez S.E., Ginsberg S.D. Cholinergic system during the progression of Alzheimer’s disease: therapeutic implications. Expert Rev. Neurother. 2009;8:1703–1718. doi: 10.1586/14737175.8.11.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tohgi H., Abe T., Hashiguchi K., Saheki M., Takahashi S. Remarkable reduction in acetylcholine concentration in the cerebrospinal fluid from patients with Alzheimer type dementia. Neurosci. Lett. 1994;177:139–142. doi: 10.1016/0304-3940(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 84.Boissière F., Faucheux B., Duyckaerts C., Hauw J., Agid Y., Hirsch E.C. Striatal expression of glutamic acid decarboxylase gene in Alzheimer’ s disease. J. Neurochem. 1998;71:767–774. doi: 10.1046/j.1471-4159.1998.71020767.x. [DOI] [PubMed] [Google Scholar]
- 85.Lanctôt K.L., Herrmann N., Mazzotta P., Khan L.R., Ingber N. GABAergic function in Alzheimer’s disease: evidence for dysfunction and potential as a therapeutic target for the treatment of behavioural and psychological symptoms of dementia. Can J Psychiat Revue Canadienne de Psychiatrie. 2004;49:439–453. doi: 10.1177/070674370404900705. [DOI] [PubMed] [Google Scholar]
- 86.Reinikainen K.J., Paljgtrvi L., Huuskonen M., Soininen H., Laakso M., Riekkinen P.J. A post-mortem study of noradrenergic, serotonergic and GABAergic neurons in Alzheimer’s disease. J. Neurol. Sci. 1988;84:101–116. doi: 10.1016/0022-510x(88)90179-7. [DOI] [PubMed] [Google Scholar]
- 87.Limon A., Reyes-Ruiz J.M., Miledi R. Loss of functional GABAA receptors in the Alzheimer diseased brain. Proc. Natl. Acad. Sci. USA. 2012;109:10071–10076. doi: 10.1073/pnas.1204606109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Garcia-Alloza M., Tsang S.W., Gil-Bea F.J., Francis P.T., Lai M.K., Marcos B., et al. Involvement of the GABAergic system in depressive symptoms of Alzheimer’s disease. Neurobiol. Aging. 2006;27:1110–1117. doi: 10.1016/j.neurobiolaging.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 89.Nicastro N., Assal F., Seeck M. From here to epilepsy: the risk of seizure in patients with Alzheimer’s disease. Epileptic Disord. 2016;18:1–12. doi: 10.1684/epd.2016.0808. [DOI] [PubMed] [Google Scholar]
- 90.Dauvilliers Y. Insomnia in patients with neurodegenerative conditions. Sleep Med. 2007;8:S27–S34. doi: 10.1016/S1389-9457(08)70006-6. [DOI] [PubMed] [Google Scholar]
- 91.Möhler H. GABAA receptors in central nervous system disease: anxiety, epilepsy, and insomnia. J Recept Sig Transd. 2006;26:731–740. doi: 10.1080/10799890600920035. [DOI] [PubMed] [Google Scholar]
- 92.Ting Wong C.G., Bottiglieri T., Snead O.C. GABA, gamma-hydroxybutyric acid, and neurological disease. Ann. Neurol. 2003;54:S3–S12. doi: 10.1002/ana.10696. [DOI] [PubMed] [Google Scholar]
- 93.Hoyer S., Nitsch R. Cerebral excess release of neurotransmitter amino acids subsequent to reduced cerebral glucose metabolism in early-onset dementia of Alzheimer type Short. J. Neural Transm. (Vienna) 1989;75:227–232. doi: 10.1007/BF01258634. [DOI] [PubMed] [Google Scholar]
- 94.Celone K.A., Calhoun V.D., Dickerson B.C., Atri A., Chua E.F., Miller S.L., et al. Alterations in memory networks in mild cognitive ompairment and Alzheimer’s disease: an independent component analysis. J. Neurosci. 2006;26:10222–10231. doi: 10.1523/JNEUROSCI.2250-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stargardt A., Swaab D.F., Bossers K. The storm before the quiet: Neuronal hyperactivity and Aβ in the presymptomatic stages of Alzheimer’s disease. Neurobiol. Aging. 2015;36:1–11. doi: 10.1016/j.neurobiolaging.2014.08.014. [DOI] [PubMed] [Google Scholar]
- 96.Bass B., Upson S., Roy K., Montgomery E.L., Jalonen T.O., Murray I.V. Glycogen and amyloid-beta:key players in the shift from neuronal hyperactivity to hypoactivity observed in Alzheimer’s disease? Neural Regen. Res. 2015;10:1023–1025. doi: 10.4103/1673-5374.160059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Altman B.J., Stine Z.E., Dang C.V. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Cancer. 2016;16:619–634. doi: 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Driver J.A., Beiser A., Au R., Kreger B.E., Splansky G.L., Kurth T., et al. Inverse association between cancer and Alzheimer’s disease: results from the Framingham Heart Study. BMJ. 2012;344:e1442. doi: 10.1136/bmj.e1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tirumalasetti F., Han L., Birkett D.P. The Relationship between Cancer and Alzheimer’s Disease. J. Am. Geriatr. Soc. 1991;39:840. doi: 10.1111/j.1532-5415.1991.tb02713.x. [DOI] [PubMed] [Google Scholar]
- 100.Girouard H., Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J. Appl. Physiol. 2006;100:328–335. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- 101.de la Torre J.C. Is Alzheimer’s disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol. 2004;3:184–190. doi: 10.1016/S1474-4422(04)00683-0. [DOI] [PubMed] [Google Scholar]
- 102.Murray I.V., Proza J.F., Sohrabji F., Lawler J.M. Vascular and metabolic dysfunction in Alzheimer’s disease: a review. Exp. Biol. Med. 2011;236:772–782. doi: 10.1258/ebm.2011.010355. [DOI] [PubMed] [Google Scholar]
- 103.Gabuzda D., Busciglio J., Chen L.B., Matsudaira P., Yankner B.A. Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J. Biol. Chem. 1994;269:13623–13628. [PubMed] [Google Scholar]
- 104.Kim I., Xu W., Reed J.C. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008;7:1013. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 105.Velliquette R.A., O’Connor T., Vassar R. Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer’s disease pathogenesis. J. Neurosci. 2005;25:10874–10883. doi: 10.1523/JNEUROSCI.2350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Trushina E., Dutta T., Persson X.M., Mielke M.M., Petersen R.C. Identification of altered metabolic pathways in plasma and CSF in mild cognitive impairment and Alzheimer’s disease using metabolomics. PLoS One. 2013;8:e63644. doi: 10.1371/journal.pone.0063644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Craft S., Cholerton B., Baker L.D. Insulin and Alzheimer’s disease: untangling the web. J. Alzheimers Dis. 2013;33:S263–S275. doi: 10.3233/JAD-2012-129042. [DOI] [PubMed] [Google Scholar]
- 108.de la Monte S.M. Insulin resistance and Alzheimer’s disease. BMB Rep. 2009;42:475–481. doi: 10.5483/bmbrep.2009.42.8.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Belanger M., Allaman I., Magistretti P.J. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 110.Putcha D., Brickhouse M., O’Keefe K., Sullivan C., Rentz D., Marshall G., et al. Hippocampal hyperactivation associated with cortical thinning in Alzheimer’s disease signature regions in non-demented elderly adults. J. Neurosci. 2011;31:17680–17688. doi: 10.1523/JNEUROSCI.4740-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bero A.W., Yan P., Roh J.H., Cirrito J.R., Stewart F.R., Raichle M.E., et al. Neuronal activity regulates the regional vulnerability to amyloid-[beta] deposition. Nat. Neurosci. 2011;14:750–756. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vlassenko A.G., Vaishnavi S.N., Couture L., Sacco D., Shannon B.J., Mach R.H., et al. Spatial correlation between brain aerobic glycolysis and amyloid-beta deposition. Proc. Natl. Acad. Sci. USA. 2010;107(41):17763–17767. doi: 10.1073/pnas.1010461107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.de la Torre J.C. Alzheimer’s disease is a vasocognopathy: a new term to describe its nature. Neurol. Res. 2004;26:517–524. doi: 10.1179/016164104225016254. [DOI] [PubMed] [Google Scholar]
- 114.de la Torre J.C. Critically attained threshold of cerebral hypoperfusion: can it cause Alzheimer’s disease? Neurobiol. Aging. 2000;903:424–436. doi: 10.1111/j.1749-6632.2000.tb06394.x. [DOI] [PubMed] [Google Scholar]
- 115.Sultana R., Butterfield D. Oxidatively modified, mitochondria-relevant brain proteins in subjects with Alzheimer disease and mild cognitive impairment. J. Bioenerg. Biomembr. 2009;41:441–446. doi: 10.1007/s10863-009-9241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Butterfield D.A., Sultana R. Redox proteomics identification of oxidatively modified brain proteins in Alzheimer’s disease and mild cognitive impairment: insights into the progression of this dementing disorder. J. Alzheimers Dis. 2007;12:61–72. doi: 10.3233/jad-2007-12107. [DOI] [PubMed] [Google Scholar]
- 117.Casadesus G., Moreira P.I., Nunomura A., Siedlak S.L., Bligh-Glover W., Balraj E., et al. Indices of metabolic dysfunction and oxidative stress. Neurochem. Res. 2007;32:717–722. doi: 10.1007/s11064-007-9296-y. [DOI] [PubMed] [Google Scholar]
- 118.Blass J.P., Gibson G.E., Hoyer S. The role of the metabolic lesion in Alzheimer’s disease. J. Alzheimers Dis. 2002;4:225–232. doi: 10.3233/jad-2002-4312. [DOI] [PubMed] [Google Scholar]
- 119.Gibson G.E., Karuppagounder S.S., Shi Q. Oxidant-induced changes in mitochondria and calcium dynamics in the pathophysiology of Alzheimer’s disease. Ann NY Acad. 2008;1147:221–232. doi: 10.1196/annals.1427.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rustin P, Bourgeron T, Parfait B, Chretien D, Munnich A, Rötig A. Inborn errors of the Krebs cycle: a group of unusual mitochondrial diseases in human. 1997. [DOI] [PubMed]
- 121.Robinson B.H. Lactic acidemia and mitochondrial disease. Mol. Genet. Metab. 2006;89:3–13. doi: 10.1016/j.ymgme.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 122.Riganti C., Gazzano E., Polimeni M., Aldieri E., Ghigo D. The pentose phosphate pathway: an antioxidant defense and a crossroad in tumor cell fate. Free Radic. Biol. Med. 2012;53:421–436. doi: 10.1016/j.freeradbiomed.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 123.Russell R.L., Siedlak S.L., Raina A.K., Bautista J.M., Smith M.A., Perry G. Increased neuronal glucose-6-phosphate dehydrogenase and sulfhydryl levels indicate reductive compensation to oxidative stress in Alzheimer disease. Arch. Biochem. Biophys. 1999;370:236–239. doi: 10.1006/abbi.1999.1404. [DOI] [PubMed] [Google Scholar]
- 124.Hoyer S. Oxidative energy metabolism in Alzheimer brain - Studies in early-onset and late-onset cases. Mol. Chem. Neuropathol. 1992;16:207–224. doi: 10.1007/BF03159971. [DOI] [PubMed] [Google Scholar]
- 125.Hawkins R.A. The blood-brain barrier and glutamate 1–4. Am. J. Clin. Nutr. 2009;90:867S–874S. doi: 10.3945/ajcn.2009.27462BB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bak L.K., Schousboe A., Waagepetersen H.S. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006;98:641–653. doi: 10.1111/j.1471-4159.2006.03913.x. [DOI] [PubMed] [Google Scholar]
- 127.Krebs H.A. Metabolism of amino-acids. Biochem. J. 1935;29:1951–1969. doi: 10.1042/bj0291951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ferriero R., Manco G., Lamantea E., Nusco E., Mariella I.F., Sordino P., et al. Phenylbutyrate therapy for pyruvate dehydrogenase complex deficiency and lactic acidosis. Sci. Transl. Med. 2013;5(175):175ra31. doi: 10.1126/scitranslmed.3004986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cuadrado-Tejedor M., Ricobaraza A.L., Torrijo R., Franco R., Garcia-Osta A. Phenylbutyrate is a multifaceted drug that exerts neuroprotective effects and reverses the Alzheimer disease-like phenotype of a commonly used mouse model. Curr. Pharm. Des. 2013;19(28):5076–5084. doi: 10.2174/1381612811319280006. [internet]. [DOI] [PubMed] [Google Scholar]
- 130.Corbett G.T., Roy A., Pahan K. Sodium phenylbutyrate enhances astrocytic neurotrophin synthesis via protein kinase C (PKC)-mediated activation of cAMP-response element-binding protein (CREB): Implications for alzheimer disease therapy. J. Biol. Chem. 2013;288:8299–8312. doi: 10.1074/jbc.M112.426536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Watson G.S., Craft S. Modulation of memory by insulin and glucose: neuropsychological observations in Alzheimer’s disease. Eur. J. Pharmacol. 2004;490:97–113. doi: 10.1016/j.ejphar.2004.02.048. [DOI] [PubMed] [Google Scholar]
- 132.Moreira P.I. Metformin in the diabetic brain: friend or foe? Ann. Transl. Med. 2014;2:54. doi: 10.3978/j.issn.2305-5839.2014.06.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Xia X., Yan J., Shen Y., Tang K., Yin J., Zhang Y., et al. Berberine improves glucose metabolism in diabetic rats by inhibition of hepatic gluconeogenesis. PLoS One. 2011;6(2):e16556. doi: 10.1371/journal.pone.0016556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cai Z., Chuanling W., Yang W. Role of berberine in Alzheimer ’ s disease. Neuropsychiatr. Dis. Treat. 2016;2016(12):2509–2520. doi: 10.2147/NDT.S114846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fawver J.N., Duong K.T., Wise-Scira O., Schall H.E., Coskuner O., Zhu X., et al. Probing and trapping a sensitive conformation: Amyloid beta fibrils, oligomers and dimers. J. Alzheimers Dis. 2012;32:197–215. doi: 10.3233/JAD-2012-120880. [DOI] [PubMed] [Google Scholar]
- 136.Craig L.A., Hong N.S., McDonald R.J. Revisiting the cholinergic hypothesis in the development of Alzheimer’s disease. Neurosci. Biobehav. Rev. 2011;35:1397–1409. doi: 10.1016/j.neubiorev.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 137.Lleó A. Current therapeutic options for Alzheimer’s disease. Curr. Genomics. 2007;8:550–558. doi: 10.2174/138920207783769549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yan D., Zhang Y., Liu L., Yan H. Pesticide exposure and risk of Alzheimer’s disease: a systematic review and meta-analysis. Sci. Rep. 2016;6:32222. doi: 10.1038/srep32222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gray S.L., Dublin S., Yu O., Walker R., Anderson M., Hubbard R.A., et al. Benzodiazepine use and risk of incident dementia or cognitive decline: prospective population based study. BMJ. 2016;352:190. doi: 10.1136/bmj.i90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Di Domenico F., Pupo G., Giraldo E., Lloret A., Badìa M., Schininà M.E., et al. Autoantibodies profile in matching CSF and serum from AD and aMCI patients: potential pathogenic role and link to oxidative damage. Curr. Alzheimer Res. 2016;13(2):112–122. doi: 10.2174/1567205013666151218131424. [DOI] [PubMed] [Google Scholar]
- 141.Morris M.C., Tangney C.C., Wang Y., Sacks F.M., Bennett D.A., Agrawal N.T. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. 2015;11(9):1007–1014. doi: 10.1016/j.jalz.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Maalouf M.A., Rho J.M., Mattson M.P. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res. Brain Res. Rev. 2009;59:293–315. doi: 10.1016/j.brainresrev.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bresden D.E., Bredesen D.E. Reversal of cognitive decline: A novel therapeutic program. Aging (Albany NY) 2014;6:707–717. doi: 10.18632/aging.100690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Blazer D.G., Yaffe K., Liverman C.T. Cognitive Aging: Progress in Understanding and Opportunities for Action. Washington, DC: The National Academies Press; 2015. Internet. [PubMed] [Google Scholar]
- 145.Kane R.L., Butler M., Fink H.A., Brasure M., Davila H., Desai P., Jutkowitz E., McCreedy E., Nelson V.A., McCarten J.R., Calvert C., Ratner E., Hemmy L.S. B.T. Interventions To Prevent Age-Related Cognitive Decline , Mild Cognitive Impairment , and Clinical Alzheimer’s- Type Dementia [Internet]. Comp. Eff. Rev. No 188. AHRQ Publ. No. 17-EHC008-EF. Rockville, MD (2017). 2017. [PubMed] [Google Scholar]