

Abstract
Tumor Necrosis Factor α (TNFα) induces both the apoptotic pathway and anti-apoptotic factors. Incubation of human dermal fibroblasts with TAPF (TNF Apoptosis Protection Fraction) protects them from apoptosis induced by the subsequent addition of TNF and cycloheximide (CHX). TAPF does not protect against apoptosis induced by CHX in combination with either TRAIL (TNF related apoptosis inducing ligand) or an agonistic Fas antibody, or against apoptosis induced by the chemotherapeutic agent doxorubicin. Incubation with TAPF does not affect the quantity of TNF that binds to the cell. TAPF prevents TNF-induced cleavage of caspases 8, 9, 3 and 7 and the apoptotic substrate PARP (poly-ADP ribose polymerase), but has no effect when these molecules are induced by an agonistic Fas antibody. TAPF induces rapid phosphorylation of the NF-κB/p65 (nuclear factor – κB) transcription factor at serine 536 which is indicative of its activation. TAPF increases the expression of cFLIP (cellular FLICE-inhibitory protein) which is a potent inhibitor of apoptosis that acts by preventing the cleavage of caspase 8. This increase in cFLIP is coincident with protection from TNF-induced apoptosis. Decreasing cFLIP levels using shRNA (short hairpin RNA) decreases protection by TAPF. TAPF also induced the anti-apoptotic A20 protein. These data indicate that TAPF protects human dermal fibroblasts from TNF-induced apoptosis by induction of cFLIP and subsequent inhibition of caspase 8 cleavage.
Keywords: TNF, apoptosis, TRAIL, Fas, NF-ĸB, cFLIP
1. INTRODUCTION
Tumor necrosis factor (TNF) has multiple functions in controlling immunity and inflammation [1]. It is also involved in many disparate pathological conditions, including cancer [2], and immune-mediated inflammatory diseases [e.g. rheumatoid arthritis, inflammatory bowel disease, and ankylosing spondylitis [3,4,5,6,7]. Many of these effects are thought to be mediated through the apoptotic cell death pathway which can be induced by TNF [8]. Apoptosis is critical for tissue homeostasis and is important for many physiological processes, especially immunity and embryological development. Because of the importance of apoptosis in many pathological processes, therapies intended to stimulate or prevent apoptosis are desirable and their rational design requires knowledge of the mechanisms by which apoptosis is induced, the pathways that confer resistance and how these differ between different cell types and agents that induce apoptosis.
Physiologic inducers of apoptosis also include other members of the TNF superfamily, including FasL (CD95L) and TRAIL. All three induce apoptosis by a virtually identical mechanism in which ligand binding stimulates recruitment of adaptor proteins to the cytosolic portion of the receptor. This in turn activates the initiator caspase, caspase 8, by cleavage. Activated caspase 8 stimulates both activation of the executioner caspase, caspase 3, and cleavage of Bid. Truncated Bid induces changes in the Bcl2 family proteins in the mitochondria which lead to breakdown of the mitochondrial membrane potential and release of cytochrome c into the cytoplasm. Cytosolic cytochrome c forms a complex with APAF-1 and caspase 9 and this leads to activation of caspase 9. Activated caspase 9 is also able to cleave and activate caspase 3. Activated caspase 3 proteolyzes multiple substrates, including the canonical substrate poly-ADP ribose polymerase (PARP), which leads to controlled cell disassembly and death [8].
TNF is different from FasL and TRAIL because it is a strong activator of the transcription factor NF-κB which induces expression of a number of proteins that inhibit the apoptotic pathway (e.g. cFLIP, IAP, A20). The relative stimulation of this anti-apoptotic pathway versus that of the apoptotic pathway determines whether the cell lives or dies. The pathways are conserved in most cell types, but the final fate of the cell depends on a complex integration of multiple pathways and factors which is often dependent on the cell type as well as other signals affecting the cell [2]. Many cells undergo apoptosis in culture only when TNF is combined with cycloheximide (CHX). Treatment of cells, including dermal fibroblasts [9] with TNF induces protective proteins which prevent apoptosis when TNF and CHX are subsequently added. We have previously determined that there are several cellular factors with different isoelectric points present in extracts from cells treated with TNF which, when incubated exogenously with fibroblasts, protect them from apoptosis induced by TNF plus CHX [9]. The TIP-B1 (pI ≈ 4.7) protective protein has been cloned and characterized [9–11].
In this study, we investigated the mechanism whereby a fraction with a pI of ≈ 5.5 inhibits the TNF-induced apoptotic pathway. This fraction, which we have termed TNF Apoptosis Protection Fraction (TAPF), activates NF-κB/p65, induces A20 and cFLIP and inhibits TNF-induced activation of caspase 8 as well as subsequent cleavage of caspases 9, 3, 7 and PARP. Decreasing the levels of cFLIP using shRNA decreased protection by TAPF, thereby verifying the role of cFLIP in TAPF-induced protection. Surprisingly, induction of cFLIP was not sufficient to protect the cells from apoptosis induced by either an agonistic Fas antibody or TRAIL.
2. EXPERIMENTAL PROCEDURES
2.1 Cell Culture
Human dermal fibroblasts (NHDF, Clonectics Corporation, San Diego, CA) were maintained in DMEM supplemented with 25 mM HEPES and 10% Fe2+-enriched calf sera. Cell monolayers were harvested when they reached 80% confluence; detachment was by using 0.05% trypsin containing 0.53 mM EDTA. Only cells growing exponentially in the first 8 passages were used. Phoenix-AMPHO retroviral packaging cells (SD-3443), obtained from ATCC (Rockville, MD), were maintained in DMEM supplemented with 1% penicillin, 1% streptomycin, 1% glutamine and 10% fetal calf sera and were harvested similarly as described for NHDF. Cultures were tested for mycoplasma at regular intervals (by the Roswell Park Tissue Culture Facility) and found to be negative.
2.2 Reagents
TNF (recombinant human, 1 unit = 0.4 ng) was a gift from the Asahi Chemical Industry Co, Ltd. (Shizwoka-ken, Japan). Agonistic Fas antibody (clone CH11) and TRAIL were purchased from Upstate (Lake Placid, NY) and BIOMOL (Plymouth Meeting, PA), respectively. Doxorubicin (DOX, Adriamycin) was a gift from Adria Labs (Columbus, OH).
2.3 Induction of Apoptosis
NHDF were dispensed into a 96-well plate (2×104 cells/well) and allowed to adhere. Apoptotic agents were added 24h later and viable cells were measured after an additional 24h (TNF, TRAIL, agonistic Fas antibody) or 48h (doxorubicin) by cellular enzymatic reduction of MTT and spectrophotometric detection of the formazan product as described previously [9]. The final volume of the assays was 200 μl. The final concentrations were: 1.3 × 10−5M doxorubicin, 50 μg/ml CHX in combination with either 70 ng/ml TNF or 0.06 μg/ml agonistic Fas antibody or 10 μg/ml TRAIL.
2.4 Protection from Apoptosis
Fractions to be tested for protective activity were incubated at multiple concentrations (serial two-fold dilutions spanning several log orders) with adhered NHDF (final volume, 100μl) for 6-18 h prior to the addition of apoptotic agents. Protection was determined by comparison with controls that were incubated with medium instead of putative protective fractions. Percent protection was calculated based on the formula: [(A570experimental– A570TNF+CHX)/(A570CHX–A570TNF+CHX)]×100.
2.5 Preparation of Cellular Extract and Partial Purification of TAPF
Cellular extracts were generated and subjected to isoelectric focusing in solution as described previously [9]. Briefly, NHDF were treated with 4 ng/ml TNF for 18h. The cells were harvested, lysed by sonication in 10 mM sodium phosphate (pH 7.4) plus protease inhibitors, centrifuged at 12,000 × g and the supernatant was centrifuged at 100,000 × g. The resulting supernatant is the cellular extract. The cellular extract (3mg protein, determined using BCA Protein Assay, Pierce, Rockford, IL) was subjected to isoelectric focusing on a Rotofor apparatus (Bio-Rad, Hercules CA) using Bio-Lyte 3/10 ampholytes (Bio-Rad, Hercules CA). Ampholytes were dissociated from proteins by incubation with 1M NaCl. Salt and ampholytes were removed by extensive buffer exchange into 10 mM sodium phosphate using 2ml centrifugal filtration devices which retained species with a molecular mass greater than 20 kDa.
2.6 Quantification of Cell Binding of TNF
Cellular binding of biotinylated TNF compared to a non-specific biotinylated probe (soybean trypsin inhibitor) was assessed using a commercially available kit (R&D Systems, Minneapolis, MN) according to the manufacturer’s directions.
2.7 Immunoblot Analysis
Samples for immunoblot analysis were prepared as follows. Preparation of cellular extracts and TAPF-enriched fractions produced by isoelectric focusing are described above. For analysis of NHDF infected with shRNA, washed cell pellets were lysed by freezing and thawing in 500 μl of 10 mM sodium phosphate plus protease inhibitors [9], centrifuged at 16,000 × g and the protein concentration of the supernatant was determined (BCA Protein Assay, Pierce). All other samples were generated by lysing equal numbers of washed NHDF monolayers following treatment directly from multiple wells of a 96 well plate with SDS-PAGE sample buffer.
Protein samples were separated on 12.5% SDS-PAGE gels, and transferred to PVDF membranes (Millipore Co., Bedford, MA) in transfer buffer (25 mM Tris base, 192 mM glycine, 15% methanol). Membranes were blocked for 1h in 5% dry milk in Tris-buffered saline plus 0.1% Tween 20 (TBST), incubated overnight in primary antibody diluted in 5% milk/TBST, washed for 3 × 5 min in TBST, incubated with the appropriate secondary antibody (conjugated to alkaline phosphatase or horseradish peroxidase) in 5% milk/TBST, then washed for 3 × 5 min in TBST prior to detection of alkaline phosphatase activity (CDP-Star, Tropix, Bedford, MA) or horseradish peroxidase activity (ECL plus, GE Healthcare, Piscataway, NJ) and exposure to film. Reactive proteins were identified based on their published mobilities as reflected by comparison to pre-stained protein migration markers resolved on and transferred from the same gel as the experimental samples. Primary antibodies, the dilutions used and suppliers are: TNF [1:1,000 gift from the Asahi Chemical Industry Co, Ltd. (Shizwoka-ken, Japan)], β-actin (1:10,000, Calbiochem, La Jolla, CA), caspases 8, 9, 3, 7, PARP, phospho ser536 NF-κB/p65, phospho thr202 p44/tyr204 p42 MAP kinase (i.e. ERK1 and ERK2), total p44/p42 MAP kinase, phospho thr308 Akt, Akt (1:1,000, Cell Signaling Danvers, MA), total NF-κB/p65 (1:10,000, StressGen, Ann Arbor, MI), cFLIP (Dave-2, 1:1,000, Axxora, San Diego, CA), A20 (1:1,500, Calbiochem, La Jolla, CA).
2.8 Retroviral Infection of NHDF with shRNA
Plasmids encoding three different 29 mer short hairpin RNAs (shRNAs) targeting cFLIP, one encoding a non-specific shRNA (targeting GFP) and a vector control were purchased (Origene, Rockville MD). NEB5-α E. coli were transformed with the individual plasmids and plasmids were purified using commercial kits (Qiagen, Valencia CA). Phoenix-AMPHO retroviral packaging cells were transfected with plasmid, the supernatant containing the virus was harvested and was then used to infect NHDF. Briefly, 5×106 Phoenix-AMPHO cells were plated (6 × 10 cm plate) and transfected 24h later using the calcium phosphate precipitation method in the presence of 25 μM chloroquin diphosphate. Viral supernatants were harvested 48h later, filtered through a 0.45 μ filter, supplemented with polybrene (hexadimethrine bromide) at 4μg/ml and used to infect NHDF (plated 24h previously at 8×105 cells per 6 × 10 cm plate). Media was changed 24h post-infection and selection with 2.5 μg/ml puromycin was initiated 24h later.
2.9 Data Analysis
Data were analyzed using Microsoft Excel. When A570 or Percent Protection are shown, data are indicated as mean ± standard deviation. For determination of p values, SigmaStat was used.
3. RESULTS
3.1 TNF treatment induces an activity within a pI ≈ 5.5 fraction which protects fibroblasts from TNF-induced cytotoxicity
As we have reported previously for several different fibroblast cell lines [9], human dermal fibroblasts (NHDF) treated with TNF are no longer sensitive to cytotoxicity induced by the subsequent addition of TNF and CHX. To determine if resistance to apoptosis was mediated by upregulation of cellular factor(s) which were active when added exogenously to cells, NHDF were treated with 4 ng/ml TNF, lysed and a cellular extract was prepared. When this extract was incubated with NHDF, the cells were no longer killed by TNF and CHX (Fig. 1A). Visual microscopic examination prior to reaction with MTT revealed both a considerable decrease in cell number and increase in apparent cellular debris following the addition of TNF+CHX compared to cell samples which were untreated, treated with CHX alone or pretreated with extract prior to the addition of TNF+CHX (data not shown). These data indicate that decreased MTT reactivity is due to cytotoxicity, rather than decreased cellular metabolic activity, in these studies. The amount of protection increased with increasing protein concentration (Fig. 1A). When the cellular extract was prepared from NHDF without TNF treatment, no protective activity was observed at concentrations up to 4 μg (Fig. 1A). However, higher amounts of cellular extract from untreated NHDF (e.g. 12 μg) did induce significant protection (data not shown) which suggests that the level of the protective factor(s) is/are low in untreated NHDF and is/are induced upon TNF treatment. The increase in the protective activity induced by TNF was dependent on the time of TNF treatment (Fig. 1B). The fact that some protection was exerted at 0 h, namely, without TNF treatment, confirms that protective activity is also present in normal extracts and is augmented in extracts from cells treated with TNF. Increased protective activity was detected in the cellular extract as early as 30 min after initiation of TNF treatment and increased thereafter (Fig. 1B). Protective activity reached a maximum at ~6 h and remained at this level until 18 h after initiation of TNF treatment (Fig. 1B). The increase in the protective activity induced by TNF was also dependent on the concentration of TNF (Fig. 1C).
Fig 1. TNF treatment induces a protective factor in NHDF (A) and this is dependent on both the time (B) and concentration (C) of treatment with TNF.
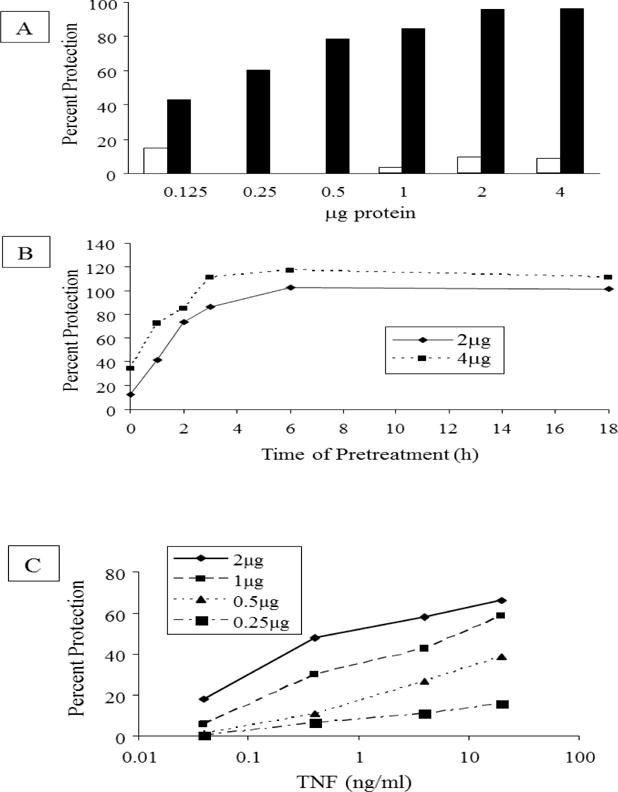
A NHDF were treated with 4 ng/ml TNF for 18h or left untreated. Cellular extracts were prepared and equal amounts of protein from extracts of treated (black bars) and untreated (white bars) cells were examined for their ability to protect NHDF from cytotoxicity induced by 70 ng/ml TNF in combination with 50μg/ml CHX. Percent protection was calculated based on the formula: [(A570experimental– A570TNF+CHX)/(A570CHX–A570TNF+CHX)]×100. The mean for each data set [n=2 for experimental fractions and n=4 for controls (CHX, CHX+TNF) where n is the number of replicates] is shown. Where white bars are not visible, the mean percent protection was zero. In different independent experiments, the A570 values for CHX+TNF varied between 2%–20% of the A570 values for CHX alone. A570 for CHX was the same as that of untreated NHDF (Fig 4). Data are representative of 3 independent experiments. B: NHDF were treated with 4 ng/ml TNF for various times as indicated. Cellular extracts were prepared and the protective activity was evaluated and compared to that present in untreated cells (0h). Percent protection was calculated as described above for Fig 1A. The protection afforded by 2 μg (◆⎯◆) and 4 μg (■---■) of protein is shown. The mean (n=2–4) for each data set is shown. Data are from one of three independent experiments. C. NHDF were treated for 6h with the indicated concentrations of TNF. Cellular extracts were prepared and the protective activity afforded by equivalent amounts of protein (as indicated in the inset legend) was evaluated and compared to that present in untreated cells. Percent protection was calculated as described in Fig. 1A. The mean (n=4) for each data set is shown. Data from one of two independent experiments are shown.
To enrich for the protective factor(s), the cellular extract from NHDF which had been treated with 4 ng/ml TNF was divided into 20 fractions by isoelectric focusing in solution and the ampholytes were removed. Unlike previous data from HEL-299 fibroblasts, which indicated multiple protective factors with differing isoelectric points [9], with NHDF there was a single peak of protective activity with a pI ≈ 5.5 (Fig. 2). The specific activity was increased ~40-fold by this single purification step, and the protective activity in the active fraction increased with increasing protein concentrations. Hereafter, this protective activity will be referred to as TAPF (TNF Apoptosis Protection Fraction). Multiple (>10) independent preparations of TAPF were used in these investigations and produced similar results. The protein profile of the active fraction was compared to that of a corresponding (same pI) inactive fraction generated identically to that described above but from cells that had not been treated with TNF. No discernible differences in the pattern or expression levels of the proteins detected following SDS-PAGE and staining with either silver or Coomassie blue were apparent. In subsequent experiments, the activity of the fraction containing TAPF was compared to that of the negative fraction (NEG) to confirm that the activity examined is due to TAPF and not to other components of the fraction.
Fig. 2. The fraction of pI ≈ 5.5 has protective activity.
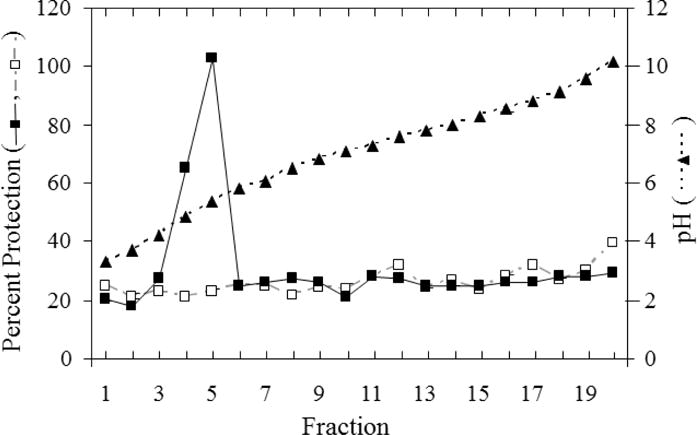
Cellular extracts (3 mg) from untreated NHDF (□, dashed gray line) and NHDF following treatment with 4 ng/ml TNF for 18h (■, solid black line) were separated by isoelectric focusing in solution. Ampholytes were removed and equivalent amounts of each fraction were assayed for protective activity. Percent protection was calculated as described for Fig. 1A. A570 for untreated NHDF was the same as that for the three controls: CHX alone (Fig 4), fraction alone (data not shown) and fraction + CHX (data not shown). The average percent protection (□,■, n=2-4) is shown. The average pH (of the same fraction number from treated and untreated cellular extracts) for each fraction is shown (▲, dashed black line). Data shown are representative of multiple (>10) experiments using multiple (>10) independently-generated cellular extracts.
3.2 TAPF is not TNF
Treatment of fibroblasts with TNF protects them from the cytolysis induced by the subsequent addition of TNF and CHX [9]. TNF treatment can induce endogenous TNF [12], and the isoelectric point of TAPF (≈ 5.5) is similar to that of TNF [5.3, [13]]. The possibility that TAPF was TNF was investigated. The levels of TNF were examined by immunoblot analysis of cellular extracts from NHDF which were either untreated or treated with TNF. Whereas TAPF activity was induced in cells which had been treated with TNF (Fig. 2), immunoreactive intracellular or extracellular TNF was not (Fig. 3). Additionally, the levels of TNF present in the fractions generated during purification were similarly examined and did not correlate with the levels of TAPF protective activity.
Fig 3. TNF treatment of NHDF does not induce endogenous TNF.
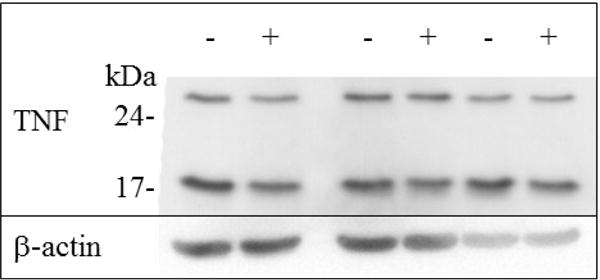
Equal amounts (15 μg) of cellular extract from cells which were untreated (−) or treated with 4 ng/ml TNF for 18h (+) were immunoblotted with anti-TNF antibody. Molecular masses of markers are indicated. Three independent matched (untreated and treated cellular extracts prepared in parallel) preparations are shown.
3.3 Although TAPF protects NHDF from TNF, it does not protect against cytotoxicity induced by agonistic Fas antibody, TRAIL, or doxorubicin
Since the apoptotic pathways induced by the TNF Superfamily members FasL and TRAIL are highly similar to that induced by TNF, whereas the chemotherapeutic drug doxorubicin induces apoptosis by a different pathway, we hypothesized that TAPF would protect against apoptosis induced by FasL and TRAIL, but not doxorubicin. Surprisingly, when NHDF were incubated with TAPF and then apoptosis was induced by agonistic Fas antibody and CHX, TRAIL and CHX, or doxorubicin alone, there was no apparent protection against any of these agents (Fig. 4). TAPF may even increase apoptosis by these agents. Protection against TNF and CHX was performed in parallel and served as a positive control (Fig. 4). Visual microscopic examination prior to reaction with MTT revealed both a considerable decrease in cell number and increase in apparent cellular debris following the addition of TNF+CHX, agonistic Fas antibody + CHX, TRAIL + CHX or doxorubicin compared to cell samples which were untreated, treated with CHX alone or pretreated with TAPF prior to the addition of lytic agents (data not shown). These data indicate that decreased MTT reactivity is due to cytotoxicity, rather than decreased cellular metabolic activity, in these studies.
Fig 4. TAPF does not protect against cytotoxicity induced by Fas, TRAIL or doxorubicin.
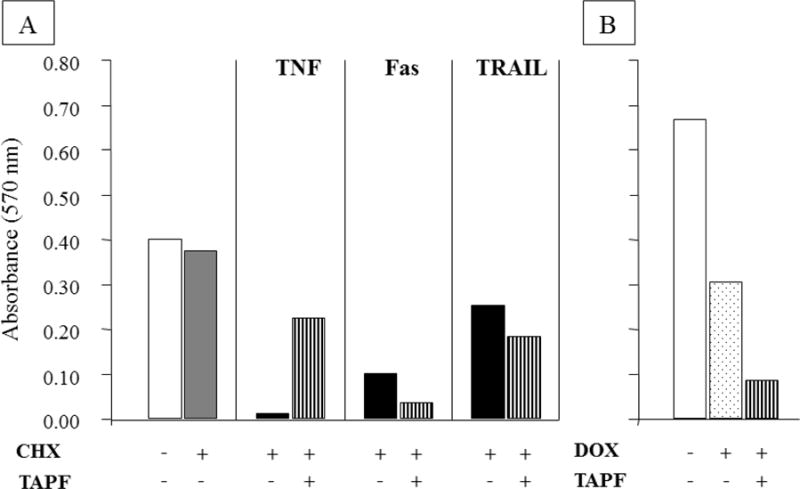
NHDF were incubated without or with TAPF prior to induction of lysis by TNF + CHX, agonistic Fas antibody + CHX, TRAIL + CHX or doxorubicin alone; lysis was measured by cellular enzymatic reduction of MTT and spectrophotometric detection of the formazan product. Induction of cytotoxicity was for 48h with doxorubicin and 24h with the three other agents. Bar height represents the mean of each data set (n=3). White bars are cells alone, the gray bar is CHX alone, black bars are the combination of lytic agents + CHX, the striped bars are TAPF incubation prior to the addition of lytic agents (± CHX as indicated) and the dotted bar is doxorubicin alone. One representative experiment out of two is shown for TRAIL and doxorubicin, one of four for agonistic Fas antibody and one of >20 for TNF.
3.4 TAPF does not affect the level of TNF:TNFR association
Since TAPF functions when added exogenously and only protects against apoptosis induced by TNF, we hypothesized that TAPF might act by blocking the binding of TNF to its receptors. To examine this, the amount of TNF bound by untreated NHDF was compared to that bound by NHDF which had been incubated with TAPF. There was significantly more binding of TNF to the cells when compared to the binding of a non-specific probe (compare bar 1 to 3 and bar 2 to 4, Fig. 5). However, there was no significant difference in the amount of TNF bound between the untreated and TAPF-treated cells (compare bar 3 to 4, Fig. 5).
Fig 5. Incubation with TAPF does not change cellular TNF binding capacity.
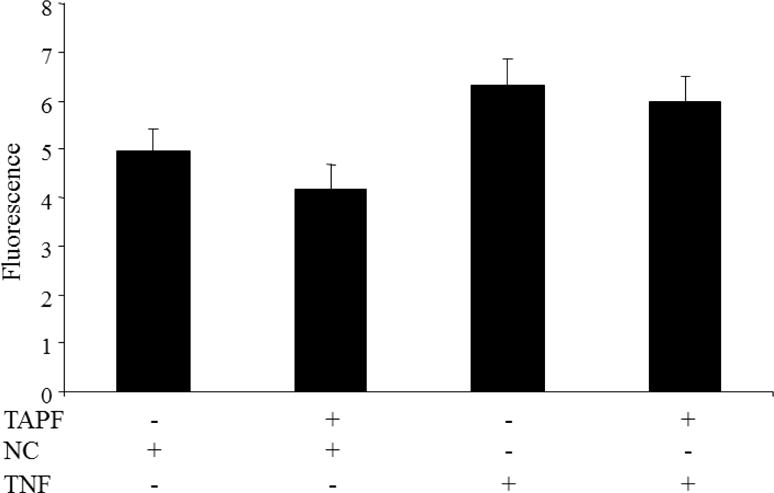
NHDF were incubated with or without TAPF prior to binding of either biotinylated soybean trypsin inhibitor (negative control, NC) or biotinylated TNF. Following washing, the bound probe was detected by fluorescence. The mean ± standard deviation for each data set is shown. The p value for comparison of the first to the third condition was 0.002, the second to the fourth condition was 0.006 and the third to the fourth condition was 0.149. The experiment was performed three times and similar results were obtained each time. The graph and statistics include the data from all three experiments combined (n=5).
3.5 TAPF prevents TNF-induced cleavage of PARP and caspases 8, 9, 3 and 7
Because TAPF protected NHDF from cytotoxicity induced by TNF, we hypothesized that TAPF inhibited a known step in the TNF-induced pathway of apoptosis and that this step and all subsequent apoptotic steps would be inhibited after incubation with TAPF. Cleavage of PARP and caspases 8, 9, 3 and 7 were examined by immunoblot analysis in samples from cells that had been untreated, treated with TAPF, or treated with the negative control fraction prior to the addition of TNF and CHX. TAPF inhibited TNF-induced cleavage of all the proteins examined, whereas the negative control fraction did not (Fig. 6, left panel). Consistent with its inability to protect against apoptosis induced by Fas, TAPF had no effect on cleavage of PARP or caspases 8, 9, 3 and 7 when they were induced by an agonistic Fas antibody and CHX (Fig. 6, right panel). These data provide additional confirmation that the decreased conversion of MTT to formazan documented in Figs 1 and 2 is indeed indicative of cytotoxicity, specifically apoptosis, rather than an effect on oxidoreductase enzyme activity.
Fig 6. TAPF prevents TNF-induced cleavage of PARP and caspases 8, 9, 3 and 7 but not cleavage induced by Fas.
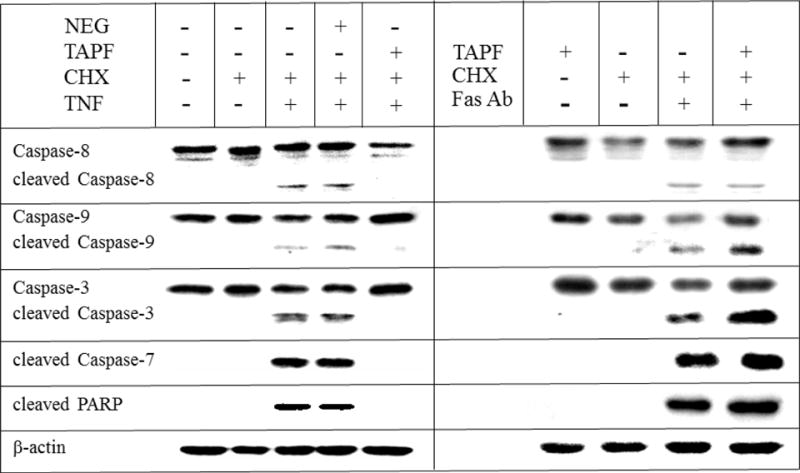
NHDF were untreated, incubated with TAPF or incubated with the negative fraction (NEG) prior to the addition of CHX + either TNF (8h) or agonistic Fas antibody (18h) as indicated. Treatment with CHX alone or no treatment whatsoever were performed in parallel as controls. Cells were washed, lysed and equal volumes were analyzed by immunoblotting for the indicated proteins. Only one representative β-actin panel is shown. Data were replicated in two to five independent experiments.
3.6 TAPF induces phosphorylation of the p65 subunit of NF-κB and IκBα
Multiple intracellular proteins which inhibit the apoptotic pathway are induced by the NF-κB transcription factor [14]. To determine if TAPF could activate NF-κB, the ability of TAPF to induce phosphorylation of the p65 subunit of NF-κB on serine 536, which is indicative of its activation, was examined by immunoblot analysis. TAPF, but not the negative control fraction, induced rapid and significant phosphorylation of NF-κB (Fig. 7, upper panel). Activation of NF-κB is typically preceded by phosphorylation and subsequent degradation of its inhibitor IκBα. Immunoblot analysis of cellular extracts generated at different times after initiation of treatment with TAPF indicated that TAPF induced phosphorylation of IκBα which led to its degradation (Fig. 7, lower panel).
Fig 7. TAPF activates NF-κB/p65 by inducing phosphorylation and subsequent degradation of its inhibitor I-κBα.

NHDF were either untreated (0 hr), incubated with TAPF or incubated with the negative fraction (NEG) for the times indicated. Cells were washed, lysed and equal volumes were analyzed by immunoblotting for the indicated proteins. For quantification, each protein was first normalized to the corresponding β-actin signal and used to calculate the ratios of phosphorylated to total NF-κB/p65 and I-κBα. One representative experiment out of two independent experiments is shown.
3.7 TAPF increases cFLIP and its induction is coincident with induction of protection
Because caspase 8 cleavage was the earliest step in the apoptotic pathway demonstrated to be inhibited by TAPF (Fig. 6) and TAPF activated NF-κB (Fig. 7), we investigated the effect of TAPF on the NF-κB-inducible caspase 8 inhibitor protein cFLIP [15] by immunoblot analysis. TAPF increased cFLIP, whereas the negative control fraction did not (Fig. 8A). When cFLIP and protection were examined in parallel, this increase in cFLIP was coincident with TAPF-induced protection against TNF and CHX (Fig 8B).
Fig 8. TAPF induces cFLIP and the levels of cFLIP increase concomitantly with protection against TNF + CHX.
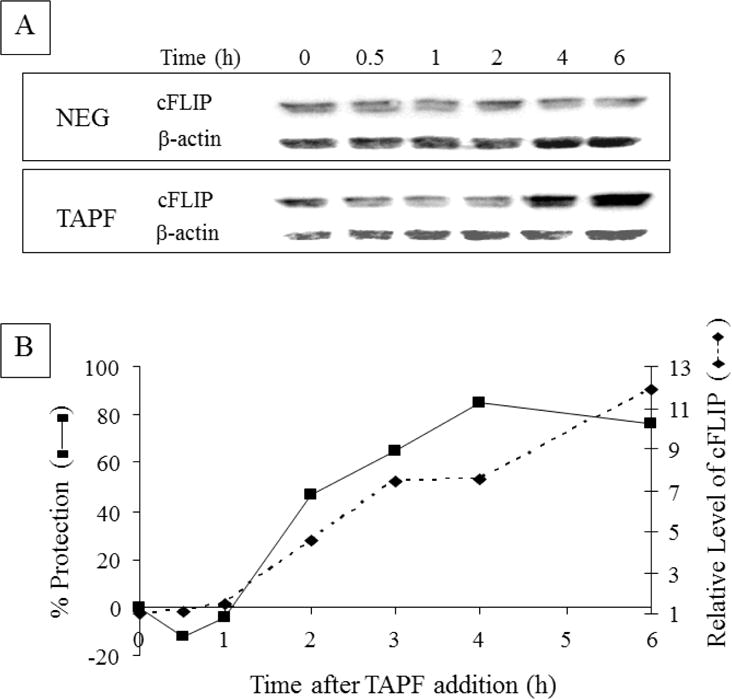
A: NHDF were either not treated (0h), incubated with TAPF or incubated with the negative fraction (NEG) for the times indicated. Cells were washed, lysed and equal volumes were analyzed by immunoblotting for cFLIP. B: Another experiment identical to A was performed. In addition, following identical TAPF treatment of samples in parallel for the times indicated on the abscissa, TNF+CHX were added and viable cells were stained 24h later. Percent protection was calculated as described for Fig. 1A. For each sample, n=3. Relative levels of cFLIP were calculated by first normalizing cFLIP to the corresponding β-actin signal; the time 0 value was set at 1 and values for all other times were reported relative to it. Data from one of three independent experiments are shown.
3.8 Decreasing cFLIP levels decreases TAPF-induced protection
To evaluate the role of cFLIP in protection against TNF and CHX mediated by TAPF, basal levels of cFLIP were lowered in NHDF using shRNA directed at cFLIP (Fig. 9A). When the ability of TAPF to protect against apoptosis induced by TNF and CHX was compared between cells with basal and lowered levels of cFLIP, those with lowered cFLIP levels were significantly less sensitive to protection by TAPF (Fig. 9B).
Fig 9. Decreasing cFLIP levels using shRNA results in decreased protection by TAPF.
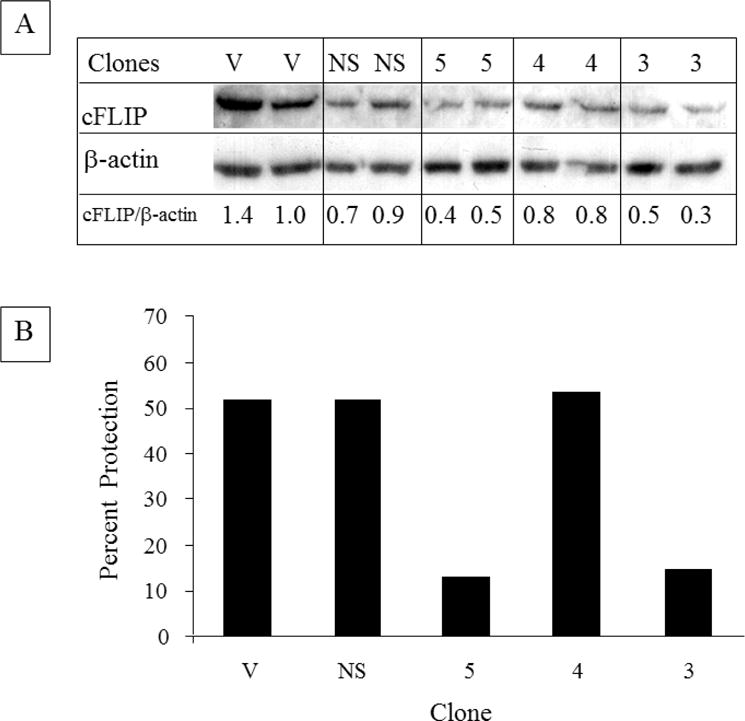
A: NHDF were infected with multiple cDNAs which encode either vector (V), an shRNA against GFP (NS), or three different shRNAs against cFLIP [5, 4, 3]. Following antibiotic selection, pools of clones from each were washed, lysed and equivalent amounts (10 μg) were analyzed by immunoblotting for cFLIP. The levels of cFLIP normalized to β-actin are indicated. Decreased cFLIP was observed in seven independent preparations of the pool of clone 3 and in five independent preparations of the pool of clone 5. B: pools of the clones described in A were treated with TAPF for 18h, TNF+CHX were added and viable cells were stained 24h later. Bar height represents the mean of each data set. For incubations with TAPF, n=2. For controls (i.e. CHX, TNF+CHX), n=4. Percent protection was calculated as described for Fig. 1A and utilized individual controls for each pool of clones. One representative experiment out of four is shown.
3.9 TAPF increases A20 but does not activate ERK or Akt
Another NF-κB-inducible protein which has been connected to resistance to apoptosis induced by TNF is A20 [17]. When the effect of TAPF on A20 levels in NHDF was examined by immunoblot analysis, A20 levels were increased by TAPF but not by the negative control fraction (Fig. 10). The kinetics of this increase in A20 paralleled the protection against TNF and CHX. (Compare Fig. 10 with Fig. 8B). The effect of TAPF on two other proteins connected to resistance to TNF, ERK and Akt [18], was also examined by immunoblot. TAPF did not increase the levels of their phosphorylated forms, thereby indicating that TAPF did not activate either of these two proteins (Fig. 10). Akt phosphorylation was examined at earlier times after TAPF treatment (5 and 15 min) with an antibody against Akt phosphorylated on ser473 and the levels were also unchanged.
Fig 10. TAPF induces A20, but does not activate ERK and Akt.
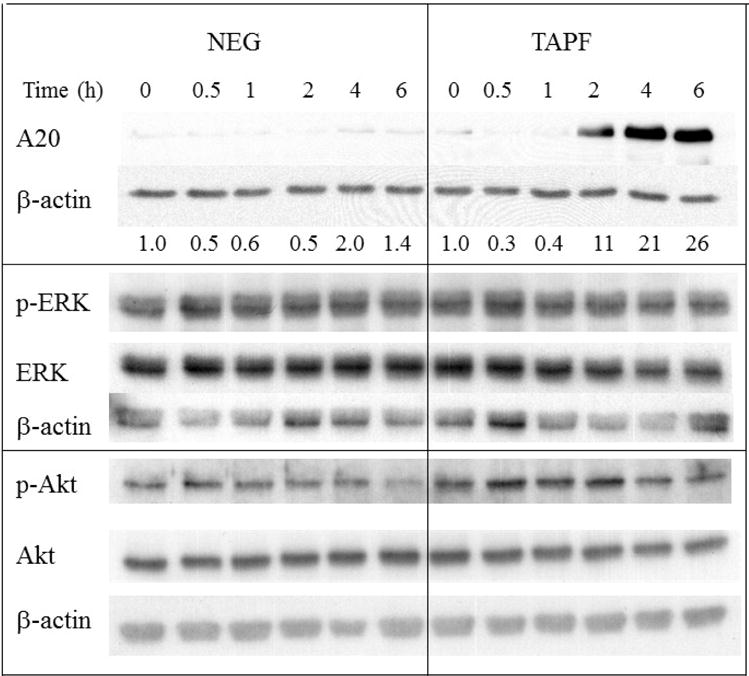
NHDF were either untreated (0 hr), incubated with TAPF or incubated with the negative fraction (NEG) for the times indicated. Cells were washed, lysed and equal volumes were analyzed by immunoblotting for the indicated proteins. Levels of A20 expression, normalized to β-actin, are shown. Normalization of p-ERK to total ERK and phospho thr308 Akt to total Akt showed no increase (highest value = 1.5). Data from one of two independent experiments are shown.
4. DISCUSSION
The data reported herein indicate that: TAPF is induced in culture by treatment with TNF, and protects fibroblasts (NHDF) from apoptosis induced by the subsequent addition of TNF and CHX. Despite their similar isoelectric points, TAPF is not TNF based on a lack of correlation between immunoreactive TNF and protective activity (Fig. 3) as well as TAPF’s inability to activate ERK and Akt (Fig. 10). Indeed, published studies indicate that TNF activates ERK [18] in fibroblast-like synoviocytes and Akt in normal human FS-4 dermal fibroblasts [19].
The spectrum of cells protected from TNF-induced apoptosis by TAPF is different from that protected by the fibroblast-derived TIP-B1 protective protein which we have studied previously. TIP-B1 protects human dermal fibroblasts [9,10], U937 human histiocytic lymphoma [9,10] and MCF7 human mammary adenocarcinoma [10,11]. TAPF, however, protects human dermal fibroblasts, but does not protect MCF7, U937, HCC1937 human mammary carcinoma, MCF10A immortalized human mammary epithelial cells and HT29 human colon adenocarcinoma. These data indicate that the cellular response to protective factors is dependent on both the cell type and the protective factor examined.
Although prepared from cell lysates, TAPF is able to protect NHDF from apoptosis induced by TNF and CHX following exogenous incubation of NHDF with TAPF. We hypothesize that TAPF is secreted by NHDF following their exposure to TNF and that secreted TAPF then acts in an autocrine fashion by binding to an unknown receptor on NHDF and inducing the signal transduction cascade documented herein.
TAPF protects NHDF from apoptosis induced by TNF and CHX by inhibiting caspase-8 cleavage and all downstream apoptotic indicators which were examined (i.e. cleavage of PARP and caspases 9, 3 and 7). The inhibition of caspase-8 activation which is induced by TAPF was mediated by an increase in cFLIP, and decreasing cFLIP levels with shRNA decreases TAPF protection. The increased cFLIP resulted from activation of the NF-κB transcription factor. Although both NF-κB and Akt can induce cFLIP expression, TAPF activated NF-κB but not Akt.
Despite its induction of cFLIP, TAPF did not inhibit apoptosis induced by CHX in combination with either an agonistic Fas (CD95) antibody, TRAIL or DOX. Additionally, TAPF did not affect the cleavage of caspase-8 (nor cleavage of PARP and caspases 9, 3 and 7) when they were induced by agonistic Fas antibody and CHX. This is surprising since a published study indicates that cFLIP is associated with resistance of normal dermal fibroblasts [20] to Fas-induced apoptosis. However, in that study, although apoptosis is induced in dermal fibroblasts only when the agonistic Fas antibody is combined with CHX, the effect of decreasing the levels of cFLIP via introduction of antisense oligonucleotides on apoptosis induced by agonistic Fas antibody in combination with CHX was not reported [20]. Instead, the data indicate that the antisense ologonucleotide combined with agonistic Fas antibody had slightly increased TUNEL staining when compared to the nonsense oligonucleotide plus agonistic Fas antibody or agonistic Fas antibody alone [5% versus 2% versus 1%, respectively]. However, the effect of the antisense oligonucleotide alone was not reported and the slight increase in apoptosis may merely result from a decrease in the cellular levels of cFLIP. As a whole, those data do not support the conclusion that cFLIP is involved in resistance to agonistic Fas antibody in dermal fibroblasts. The present study demonstrates the inability of increased cFLIP to protect against apoptosis induced by an agonistic Fas antibody and CHX.
Since both Fas and TRAIL are poor activators of NF-κB, they typically induce apoptosis more rapidly than TNF [13]. However, examination of the kinetics of caspase 8 cleavage induced by TNF and Fas in NHDF revealed that cleaved caspase 8 was detected at similar times after addition of the apoptotic agent and CHX. Thus different kinetics of caspase-8 activation did not appear to explain the inability of TAPF-induced increased cFLIP to protect against apoptosis induced by Fas.
Our hypothesis as to why increased cFLIP prevents apoptosis induced by TNF, but not that induced by Fas or TRAIL, is based on the differences in signaling between TNF versus Fas and TRAIL. Apoptosis induced by TNF involves two sequential signaling complexes. First formed is complex I which is membrane bound, contains TNFR1, TRADD, RIP1 and TRAF2 and rapidly activates NF-κB. Next formed is complex II which is cytosolic, contains TRADD, RIP1, FADD and caspase-8 and induces apoptosis [21]. In contrast, Fas and TRAIL signaling occurs through a single membrane bound complex consisting of the appropriate receptor, FADD and caspase-8 [22]. It may be that complex II formed by TNF is more efficiently inhibited by cFLIP than is the complex formed by Fas and TRAIL because of either their different subcellular locations or protein composition.
The data suggest another hypothesis for why increased cFLIP protects the cells against TNF, but not against Fas or TRAIL, which may function as either an alternative to or in addition to the one presented above. TAPF not only increased cFLIP levels, but also increased A20 levels. A20 has been implicated as a protective factor against apoptosis induced by TNF [16,17]. A20 inhibits apoptosis induced by TNF by interfering with the formation of complex I. The concomitant increase in both A20 and cFLIP which would lead to inhibition of complex I and complex II, respectively, may be required for complete protection against apoptosis induced by TNF. The increased cFLIP may be capable of binding to the complex formed by Fas and TRAIL, however, the increased cFLIP bound may be insufficient to inhibit apoptosis induced by these agents. The data utilizing shRNA against cFLIP unequivocally implicate cFLIP in the TAPF-induced resistance to TNF-induced apoptosis. However, those data cannot exclude that additional factors are also involved in the induction of resistance. Studies in which A20 protein levels were decreased using shRNA would test the hypothesis that the increase in A20 is another mediator of TAPF-induced resistance to TNF-induced apoptosis.
The data presented indicate that TAPF protects NHDF from apoptosis induced by TNF and CHX, at least in part, through an NF-κB-induced increase in cFLIP. However, this increased cFLIP does not result in protection from CHX in combination with either Fas or TRAIL. These results reinforce the importance of directly examining the pro-apoptotic and anti-apoptotic activities of each specific factor in each particular system rather than inferring the response based on the canonical apoptotic signaling pathway.
Highlights.
pI~5.5 TAPF prevents apoptosis induced by TNF but not by TRAIL, Fas or doxorubicin
TAPF prevents TNF-induced, but not Fas-induced, cleavage of caspases 8,9,3,7 & PARP
TAPF activates NF-ĸB/p65 via phosphorylation and degradation of IĸB
TAPF increases cFLIP which is required for protection; TAPF also increases A20
TAPF does not interfere with TNF:TNFR binding; TAPF does not activate ERK or Akt
Acknowledgments
We thank and acknowledge Dr. M.M. Ip, as well as members of her laboratory, for helpful discussion, gifts of reagents and critical review of the manuscript. We also acknowledge the Asahi Chemical Industry Co., Ltd. for the generous gift of TNF and the National Cancer Institute for funding support to E. Berleth (R01 CA093594) and the core resources provided through the Cancer Center Support Grant to Roswell Park Cancer Institute (P30 CA016056).
Abbrevations used
- cFLIP
cellular FLICE-inhibitory protein
- CHX
cycloheximide
- DOX
doxorubicin
- IAP
inhibitor of apoptosis protein
- NF-κB
nuclear factor - κB
- NHDF
human dermal fibroblasts
- PARP
poly-ADP ribose polymerase
- pI
isoelectric point
- shRNA
short hairpin RNA
- TAPF
TNF apoptosis protection fraction
- TNF
tumor necrosis factor α
- TRAIL
TNF related apoptosis inducing ligand
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol Therapeut. 2008;117:244–79. doi: 10.1016/j.pharmthera.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Daniel D, Wilson NS. Tumor necrosis factor: Renaissance as a cancer therapeutic? Curr Cancer Drug Tar. 2008;8:124–31. doi: 10.2174/156800908783769346. [DOI] [PubMed] [Google Scholar]
- 3.Wong M, Ziring D, Korin Y, Desai S, Kim S, Lin J, et al. TNFα blockade in human diseases: Mechanisms and future directions) Cl Immunol. 2008;126:121–36. doi: 10.1016/j.clim.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Novacek G, Dejaco C, Knoflach P, Moschen A, Petritsch W, Vogelsang H, et al. Adalimumab for the treatment of ulcerative colitis–A consensus report by the working group inflammatory bowel diseases of the Austrian Society of Gastroenterology. Gastroenterology. 2014;52:204–11. doi: 10.1055/s-0033-1355818. [DOI] [PubMed] [Google Scholar]
- 5.Song IH, Rudwalert M. Certolizumab pegol in axial spondyloarthritis. Exp Rev Clin Immunol. 2013;12:1161–72. doi: 10.1586/1744666X.2013.858859. [DOI] [PubMed] [Google Scholar]
- 6.Lowe AW, Miseley RH. Subcutaneous golimurab therapy for ulcerative colitis. Gastroenterology. 2014;146:1–2. [PubMed] [Google Scholar]
- 7.Larsen CG, Andersen PH, Lorentzen H, Zachariae C, Huldt-Nystrom T, Dottermol LK, et al. Clinical and economic impact of etanercept in real-life: A prospective, non-interventional study of etanercept in the treatment of patients with moderate to severe plaque psoriasis in private dermatological setting (ESTHER) Eur J Dermatol. 2013;23:779–81. doi: 10.1684/ejd.2013.2173. [DOI] [PubMed] [Google Scholar]
- 8.Park HH, Lo YC, Lin SC, Wang L, Yang JK, Wu H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol. 2007;25:561–86. doi: 10.1146/annurev.immunol.25.022106.141656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berleth ES, Nadadur S, Henn AD, Eppolito C, Shiojiri S, Gurtoo HL, et al. Identification, characterization, and cloning of TIP-B1, a novel protein inhibitor of tumor necrosis factor-induced lysis. Cancer Res. 1999;59:5497–506. [PubMed] [Google Scholar]
- 10.Berleth ES, Henn AD, Gurtoo HL, Wollman R, Alderfer JL, Mihich E, et al. A novel tumor necrosis factor-α inhibitory protein, TIP B1. Int J Immunopharmco. 2000;22:1137–42. doi: 10.1016/s0192-0561(00)00071-0. [DOI] [PubMed] [Google Scholar]
- 11.Berleth ES, Masso-Welch PA, Kazim LA, Ip MM, Mihich E, Ehrke MJ. Expression, tissue distribution and cellular localization of the anti-apoptotic TIP B1 protein. J Leukocyte Biol. 2001;69:995–1005. [PubMed] [Google Scholar]
- 12.Niitsu Y, Watanabe N, Neda H, Yamauchi N, Maeda M, Sone H, et al. Induction of synethsis of tumor necrosis factor in human and murine cell lines by exogenous recombinant human tumor necrosis factor. Cancer Res. 1988;48:5407–10. [PubMed] [Google Scholar]
- 13.Aggarwal BB. Signaling pathways of the TNF superfamily: A double edged sword. Nat Rev Immunol. 2003;3:745–56. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 14.Duo CC, Gong FY, He XY, Li YM, Wang J, Zhang JP, et al. Soluble calreticuline induces tumor necrosis factor α (TNFα) and interleukin (IL) 6 production by macrophages through mitogen-activated protein kinase (MAPK) and NFkB signaling pathways. Int J Mol Sci. 2014;15:2916–28. doi: 10.3390/ijms15022916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye WC, Itie A, Elia HG, Ng M, Shu HB, Wakeham A, Mirtsos C, et al. Requirement of caspes (c-FLIP) in regulation of death receptor induced apoptosis and embryonic development. Immunity. 2000;12:633–42. doi: 10.1016/s1074-7613(00)80214-9. [DOI] [PubMed] [Google Scholar]
- 16.Guo S, Messmer-Blust AF, Wu J, Song X, Philbrick MJ, Shil JL, et al. Role of A20 in CLAP2 protection against tumor necrosis factor α (TNFα)-mediated apoptosis in endothelial cells. Int J Mol Sci. 2014;15:3816–33. doi: 10.3390/ijms15033816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sass G, Shembade ND, Haimerl F, Lamoureux N, Hastemolhosseini S, Tannapfel A, et al. TNF pretreatment interferes with mitochondrial apoptosis in the mouse liver by A20-mediated down-regulation of BAX. J Immunol. 2007;179:7042–9. doi: 10.4049/jimmunol.179.10.7042. [DOI] [PubMed] [Google Scholar]
- 18.Gortz B, Hayer S, Tuerck B, Zwerina J, Smolen JS, Schett G. Tumor necrosis factor activates the mitogen-activated protein kinase p38 alpha and ERK in the synovial membrane in vivo. Arthritis Res Ther. 2005;7:R1140–7. doi: 10.1186/ar1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poppers DM, Schwenger P, Vilcek J. Persistent tumor necrosis factor signaling in normal human fibroblasts prevents the complete resynthesis of kappa B-alpha. J Biol Chem. 2000;275:29587–93. doi: 10.1074/jbc.M002806200. [DOI] [PubMed] [Google Scholar]
- 20.Santiago B, Galindo M, Palao G, Pablos JL. Intracellular regulation of Fas-induced apoptosis in human fibroblasts by extracellular factors and cycloheximide. J Immunol. 2004;172:560–6. doi: 10.4049/jimmunol.172.1.560. [DOI] [PubMed] [Google Scholar]
- 21.Micheau O, Tschopp J. Induction of TNF receptor-1 mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–90. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 22.Harper N, Hughes M, MacFarlane M, Cohen GM. Fas associated death domain protein and caspase 8 are not recruited to the tumor necrosis factor receptor 1 signaling complex during tumor necrosis factor-induced apoptosis. J Biol Chem. 2003;278:25534–41. doi: 10.1074/jbc.M303399200. [DOI] [PubMed] [Google Scholar]