

Abstract
Single biomarker detection is common in clinical laboratories due to the currently available method spectrum. For various diseases, however, no specific single biomarker could be identified. A strategy to overcome this diagnostic void is to shift from single analyte detection to multiplexed biomarker profiling. Mass spectrometric methods were employed for biomarker discovery in body fluids. The enormous complexity of biofluidic proteome compartments implies upstream fractionation. For this reason, mass spectrometry (MS) was coupled to two-dimensional gel electrophoresis, liquid chromatography, surface-enhanced laser desorption/ionization, or capillary electrophoresis (CE). Differences in performance and operating characteristics make them differentially suited for routine laboratory applications. Progress in the field of clinical proteomics relies not only on the use of an adequate technological platform, but also on a fast and efficient proteomic workflow including standardized sample preparation, proteomic data processing, statistical validation of biomarker selection, and sample classification. Based on CE-MS analysis, we describe how proteomic technology can be implemented in a clinical laboratory environment. In the last part of this review, we give an overview of CE-MS-based clinical studies and present information on identity and biological significance of the identified peptide biomarkers providing evidence of disease-induced changes in proteolytic processing and posttranslational modification.
Keywords: Biomarker pattern, bladder cancer, capillary electrophoresis, chronic kidney disease, coronary artery disease, graft-versus-host disease, mass spectrometry, multivariate statistical analysis, prostate cancer, proteome database, proteomics/peptidomics, urine
Introduction
Biomarkers are molecules being used as indicators of pathological changes, disease states, or drug responses. Many of these are proteins and peptides, which are the exclusive focus of this review. For clinical laboratory diagnosis, the molecular structure and biochemical characteristics of a biomarker are only of secondary interest. The collection of clinical data is of greater importance, allowing exact specification of diagnostic and prognostic capabilities of biomarkers. Specificity for a particular disease state, time of occurrence, abundance, accessibility, resistance to metabolic decay, and protease degradation are only some factors that must be taken into account when evaluating a biomarker. This evaluation process inevitably leads to the imposition of restrictions on the use of a biomarker, e.g. indication of the disease only at an advanced state.
Most of the analytes currently used in the clinical laboratory for screening and diagnostic purposes have been discovered after extensive physiological and biochemical characterization of diseases. This tedious and laborious procedure resulted in the identification of single markers with often moderate diagnostic value, mostly due to low specificity. For example, prostate-specific antigen (PSA) is currently widely used as a marker for prostate cancer. Its prognostic relevance, however, is the subject of ongoing debate due to a lack of specificity when PSA levels are only moderately increased (4–10 ng/mL). This kind of uncertainty not only results in unnecessary biopsies, but also in higher rates of false-positive prostate cancer diagnosis [for review see1]. As another example, early detection of renal impairment, which is of vast importance for the initiation of renoprotective intervention, mainly relies on the detection of microalbuminuria. The significance of this marker is underlined by the observation of Mogensen2 that glomerular filtration rate (GFR) and serum creatinine levels as alternative estimates of kidney function did not change within the microalbuminuria range. However, microalbuminuria is also found in apparently healthy individuals and cannot be utilized as a predictive marker of renal disease.3 These two examples underline the need for other more accurate biomarkers in order to overcome the limitations of today’s available diagnostic strategies.
To address this diagnostic challenge, the question arises of whether screening for a single marker can fulfill the requirements of reliably detecting a disease as early as possible, unambiguously distinguishing it from other pathological conditions, and monitoring the efficacy of therapy. An alternative strategy, instead of trying to find this “ideal diagnostic marker,” is identification of several markers, which may not be optimal for use as single, stand-alone markers, but work in concert, and subsequently combining these to a disease-specific pattern.4 Over the past few years, a number of different proteomic technologies have been introduced in clinical investigation in order to establish disease-specific marker patterns for clinical diagnosis and therapeutic monitoring as an alternative to single-biomarker-based approaches. It appears now that the multi-marker approach is generally being accepted, but it also comes with several challenges that were either not known or not fully appreciated and therefore disregarded in the beginning of the proteomics era. As outlined in the recently published suggestions for guidelines for clinical proteome analysis, the general criteria that are applied to a biomarker to be used for clinical assessment (e.g. known identity, reproducible detection, known deviation) also apply to the single biomarkers in a multi-marker panel.5 The initial enthusiasm and subsequent failure to deliver valid results were mostly a consequence of the over-interpretation of data, the use of inappropriate technologies and statistics, and a lack of knowledge, which was simply not available a few years ago. It is now evident that an ill-defined “pattern” does not constitute a multi-marker panel and cannot serve as a clinical diagnostic tool.
Selection of sample source
In principle, proteomic profiling can be carried out using tissue extracts, cell lysates, or body fluids (such as blood, urine, cerebrospinal fluid, and saliva) as biological materials.
Due to close proximity to the primary site of the pathological occurrence, affected cells and tissues should contain the highest amounts of biomarkers. This property makes them an ideal source for biomarker definition. Most of the tissues, however, are not easily accessible, and their sampling depends on invasive methods. The same issue has to be considered when collecting samples from cerebrospinal, synovial, and body cavity fluids. The poor accessibility of tissue sites is reflected by the fact that, to date, proteomic studies using tissue samples are rare.6,7 However, as outlined recently by Lescuyer et al.8 such approaches may nevertheless be more successful than serum- (or plasma-) based approaches, which, as the authors point out, have not resulted in measurable diagnostic success to date.
Malignant transformation and increased death of cells in disease-affected tissues and organs is most commonly associated with diffusion of tissue- and organ-specific proteins into the extracellular space and hence into the blood circulation. The role of blood as a transport medium for molecules to and from tissues, together with the ease of sample collection, makes blood an attractive source for biomarker discovery at first glance. Unfortunately, there are a number of problems to overcome before blood can be effectively used for clinical proteomic research.9–11 Complete identification of the human plasma proteome is currently impossible due to the high dynamic range between low-level and high-abundance proteins. With the removal of highly abundant proteins, most notably albumin, this dynamic range can be reduced, but this removal creates new challenges, such as the loss of low-abundance proteins by their binding to albumin or to the resin of the subtraction column. As reported by Kolch et al.12 persistent proteolytic activity in the blood sample is another source of experimental inaccuracy, resulting in an increase in polypeptide fragments of the serum during clotting time, making meaningful comparison of serum proteome or peptidome profiles between individual samples even more challenging. These reports and considerations suggest that, while blood is certainly the richest source for biomarkers, it is highly unlikely that these can actually be identified using current technologies.
Via proteolytic degradation, protease-resistant, and therefore relatively stable, peptide fragments are generated that traverse epithelial barriers and pass into the urine by glomerular filtration. Although peptides and small proteins of the blood that enter the lumen of the renal tubule are, for the most part, reabsorbed by proximal tubular cells, a small quantity escapes this process and is excreted into urine. As a consequence, urine can be used as a biomarker source for various diseases if the detection method is sensitive enough and if the interpretation of experimental data is not confounded by the proteolytic processing of the target markers.
In contrast to serum or plasma, urine as sample matrix provides several advantages. First, it is non-invasively accessible so that it can be obtained in large quantities whenever needed. Second, urine is relatively stable in its composition if handled properly. As reported by Schaub et al.13 this is also the case after long storage times. The higher stability of urinary peptides may be in part explained by the completion of endogenous proteolysis at the time of urine voidance. Third, since urine originates as the ultrafiltrate of plasma, with ~1000-fold lower protein concentrations,14 the composition of the urinary peptidome is highly susceptible to changes caused not only by renal, but also by a wide range of non-renal diseases including cardiovascular, autoimmune, and infectious diseases as well as certain types of cancer.15 Finally, urine is enriched in low-molecular-weight proteins and peptides, which can be transferred without an initial protease digestion step directly to protein mass spectrometry (top-down MS).
At this point, it is worth mentioning several sources of difficulties encountered when dealing with urine. As demonstrated by Kolch et al.12 the peptide composition of urine changes rapidly during the day and is affected by many factors, including variations in fluid intake, exercise, diet, circadian rhythms, response to hormones, to name just a few. Hence, to ensure the reliability of proteomic profiling, this biological variability should be kept to a minimum by the inclusion of statistically relevant numbers of samples, the selection of adequately matched case and control groups, and the use of standardized sample collection and preparation procedures.
Proteome analysis is best carried out using midstream samples of second morning urine. Nevertheless, exceptions exist. In a clinical trial pertaining capillary electrophoresis (CE)-MS-based prostate cancer diagnosis, Theodorescu et al.16 identified first void rather than midstream urine as a rich source for prostate cancer-specific urinary biomarker peptides.
Another factor with an impact on protein and peptide composition of urine is aging. Zürbig et al.17 found 325 of 5,000 CE-MS-identified peptides to be age-dependently regulated. Whereas the majority underwent changes in conjugation with pre- and peri-pubertal renal development, 49 peptides were related to aging in adults. Most of the latter are fragments of different types of collagens. Their down-regulation can be interpreted as morphological evidence for the previously described decrease in kidney function, i.e. decline of GFR and reduced capacity of urine concentration.18 In consequence, where pathophysiological conditions are restricted to a particular age group, i.e. chronic kidney disease (CKD), age-matched controls must be used for unbiased disease marker discovery.
Besides keeping non-disease-related variability of urine samples in the initial biomarker discovery phase minimal, it is of equal importance to vigorously evaluate application-specific characteristics such as biomarker stability or insensitivity to interferents in a later validation phase. Only biomarkers that pass this strict validation will enable appropriate classification accuracy over a broad range of samples in a clinical laboratory setting.
Separation technologies coupled to MS
Two-dimensional separation of proteins according to their isoelectric point and molecular weight in sodium dodecylsulfate polyacrylamide gels (SDS-PAGE), first described by O’Farrell,19 provided the experimental basis for the first proteomic analyses. It soon became apparent that, besides high-resolution protein separation, the unambiguous identification of the proteins in the gel spots is a critical task. Initially, proteins excised from the gel were examined by Edman degradation, but this was often unsuccessful due to a blocked N-terminus.20,21 Therefore, whenever possible, detection was performed by nitrocellulose transfer and staining with specific antibodies according to the method used by Burnette.22
Subsequently, several technologies emerged that improved the efficacy of proteome analysis. Electrophoretic and chromatographic separation technologies, such as two-dimensional electrophoresis (2DE), liquid chromatography (LC), surface-enhanced laser desorption/ionization (SELDI), and CE, were developed and were coupled to mass spectrometers with different ionization sources, i.e. matrix-assisted laser desorption/ionization (MALDI) and electrospray ionization (ESI), and analyzing systems, i.e. quadrupole, time-of-flight (TOF) and fourier transform ion cyclotron resonance (FT-ICR). As an alternative to these MS-based methods, protein-detecting microarrays have emerged due to advances in recombinant antibody engineering and automated spotting techniques for biomolecule immobilization onto solid supports.23,24
We will briefly outline the main characteristics of the different technological platforms and then focus on CE-MS. For a more detailed review on the technologies, we refer the reader to Reference 4.
Two-dimensional gel electrophoresis followed by mass spectrometry
In 2DE fractionation, proteins are first resolved by electrofocusing according to their isoelectric point followed by the orthogonal separation afforded by SDS-PAGE. MS detection of proteolytic digests has become the method of choice for protein identification from the 2D gels: tryptic digestion of excised gel spots, extraction of the proteolytic fragments, and, as soon as exact mass information of at least three fragment ions is available, comparison with public database records provided, for example, by the National Center for Biotechnology Information. Matches can subsequently be verified by tandem mass spectrometric (MS/MS) sequencing or immunoblotting.
Gel-to-gel variability compromising comparison between samples can be prevented to some extent by two-dimensional differential in gel electrophoresis (2D-DIGE),25 which enables simultaneous resolution of two differentially labelled samples within the same 2D gel. Nevertheless, comparison of several different experiments remains a challenge. Furthermore, the approach is too time-consuming to be applied in a clinical laboratory setting. Hence, potential biomarkers defined by 2DE-MS have to be transferred to an application platform, where they need to be validated. Despite these limitations, 2DE-MS remains a commonly used technique for comparative analysis of large proteins and definition of potential biomarkers above 10 kDa molecular mass.15
Liquid chromatography coupled to mass spectrometry
LC represents a high resolution separation method for proteins and peptides prior to MS detection. A great variety of LC columns suitable for e.g. reversed phase, ion exchange, or size exclusion chromatography, are available that enable separation of large amounts of analytes, and these can be combined with virtually any mass spectrometer. In recent years, LC-MS was further extended to multi-dimensional protein identification by sequential separation with different separation media. Most important in this field is multidimensional protein identification technology, which uses cation exchange prefractionation followed by reversed phase separation and MS/MS detection.26 A major disadvantage of this method is that it generally takes a considerably long period of time to analyze one sample, making its use for high-throughput screening of hundreds of samples rather difficult. Furthermore, comparative examination of datasets obtained from multi-dimensionally separated samples also still represents a substantial technical challenge.27 LC-MS has proven its usefulness as a research platform for the identification of potential biomarkers in an initial discovery phase.28,29 As is the case for 2DE, the identified biomarkers must then be transferred to other assay systems in order to allow their rapid laboratory detection. A more detailed description on the opportunities and limitations of LC-MS is provided by Nagele et al.30 and Qian et al.31
Surface-enhanced laser desorption/ionization coupled to mass spectrometry
SELDI uses selective adsorption of proteins to different active surfaces, e.g. hydrophilic matrix, reversed phase material, or affinity reagents, such as lectins or antibodies, to reduce the complexity of a biological sample. Matrix material absorbs energy and allows laser ionization for MS detection. SELDI has been used for the tentative identification of potential biomarkers in a variety of diseases.32–34 The advantages of SELDI are that it represents an easy-to-use system with a high automation grade and high-throughput capabilities and low sample volume requirements. Limitations include that proteome profiles generated by SELDI are influenced by a variety of factors such as the type of surface coating, pH and salt condition, and protein concentration of the sample, which makes standardization difficult to perform.13,35–37 For a recent description of SELDI technology the interested reader is referred to Poon.38
Capillary electrophoresis coupled to mass spectrometry
CE separates analytes from a complex mixture with high resolution based on differential migration through a liquid-filled capillary column in a strong electric field. By on-line coupling of CE to an electrospray, (ESI-TOF-MS), analysis of several thousand polypeptides within a time range of 45 to 60 min can be performed. CE and LC are similar with respect to resolution and compatibility with mass spectrometers. The advantages of CE over LC are that CE can operate at lower flow rates, which improves sensitivity, that no buffer gradients are required for separation, and that migration of analyte can be controlled by varying the electric field strength.39 Moreover, the capillary can be reconditioned with NaOH, enabling efficient cleaning after each run.
CE-MS is limited in the separation of larger proteins (above 20 kDa), which tend to precipitate at low pH. However, sensitivity to analyte precipitation is generally less an issue in CE compared to LC. Another limitation is the small sample volume that can be applied onto the capillary, which hampers CE-MS/MS applications, but due to the high sensitivity of mass spectrometers in the low fmol range, is of very little concern in CE-MS. With these restrictions kept in mind, CE-MS has developed into a stable and versatile platform for peptidomic profiling.
Adaptation of CE-MS for clinical diagnosis
In order to ensure reliability of multivariate analyses, robustness over a wide range of sample sizes, in addition to high analytical sensitivity, was an important factor that first required intense optimization before clinical studies could be initiated. In this early development phase, on-line coupling of CE to MS necessitated substantial modifications to the established CE and MS protocols, which are summarized in Figure 1 and will now be discussed in brief. With respect to clinical application, CE-MS was optimized to generate patient-specific proteome profiles, such as the one presented in Figure 2, in conveniently short measurement times for subsequent automated comparison with diseased and control reference patterns.40,41
Figure 1.

Schematic representation of on-line coupling of CE for peptide m/z separation and MS for mass detection, which is accomplished by a coaxial sheath-flow system. In the established setting, the nebulizer gas can be turned off during acquisition without disturbing ion spray stability.
Figure 2.
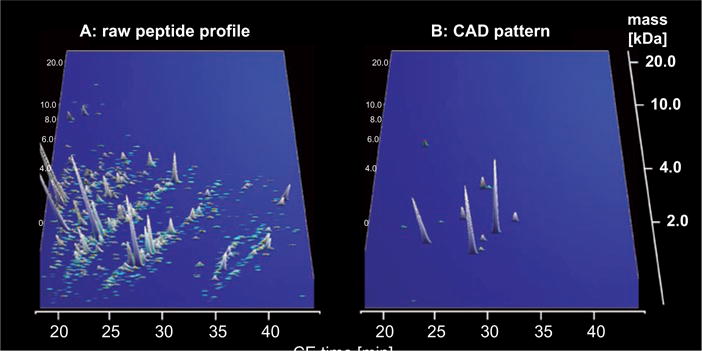
CE-MS data from an individual urine sample. In A, all relevant peptides in the sample are shown. In B, peptides are presented that form a coronary artery disease (CAD)-specific marker pattern. Based on the CE-MS analysis, the patient presented here scored positive for CAD. X axis: CE migration time [min], y axis: log molecular mass [kDa], and z axis: relative ion signal.
Due to interference with CE separation and MS detection, salts and detergents should be removed to a large extent from the sample prior to injection into the CE capillary.42 Typically, the mobile phase is an aqueous solution supplemented with methanol and formic acid. Methanol in a final concentration of 20–50% increases ionization efficiency and stabilizes the electrospray due to its low vaporization flux. The addition of 0.5% formic acid reduces the pH of the solution to ~2.2, enabling efficient generation of the positively charged peptides required for migration and subsequent ionization. Another beneficial impact of the low pH is that bare fused silica columns become fully protonated during electrophoresis, resulting in effective suppression of electroosmotic flow disturbances, normally caused by the bare silica capillary wall. This is important as it eliminates the need for special capillary coatings, which were found to be susceptible to obstruction by continuous exposure to biological fluids.12
As the entire mass-to-charge range of the CE-separated analytes should be continuously monitored during sample analysis, the ability to perform fast and sensitive scanning in full-scan mode represents an important criterion for the selection of an appropriate mass spectrometer. Due to lower scan speeds of currently available ion trap and FT-ICR-MS instruments, ESI-TOF-MS is considered to best fulfill the requirements of combining reasonable resolution and mass accuracy with fast scanning speed and adequately high sensitivity.43
High electric field densities are required to simultaneously achieve high resolution and fast separation of analytes in CE. In order to prevent interference with the downstream electrospray ionization process, the electrical circuits of CE and ESI must be separated from each other. This is ideally achieved by grounding of the ESI-needle and the introduction of an MS internal anode for the setting up of a separate low-current MS electrical circuit.12 In the context of CE-MS interfacing, it is worth mentioning that sheath flow coupling44 better fulfills stability requirements needed in a clinical setting than sheathless systems. The latter uses the outer coating of the capillary as one of the two electrical poles.45 In sheath flow systems, a sheath liquid circumflows the capillary end and closes the electrical circuit. The stability improvement of the sheath flow method outweighs a generally lower sensitivity of analyte detection, the severity of which can be reduced by keeping the sheath flow rate to an absolute minimum.12
In a typical run, 100 nL of sample are injected into the CE system. Separation is performed with 30 kV on the injection site. Due to the pH gradient, the ionized polypeptides and small proteins in the sample are initially focused before electroosmotic flow driven migration starts. To achieve highest signal intensities, the sheath flow rate is set to an absolute minimum of 100–1000 nL/min, while the nebulizer gas is turned off during acquisition. From first attempts to calculate peptide masses in complex mixtures, it became apparent that high signal intensities are more essential for convenient signal deconvolution than the perfect resolution of analytes. In this respect, it is worth mentioning that the detection limit of the CE-MS system was determined for 3 kDa peptides to be ~1 fmol.46 Under the applied operating conditions, 7 fmol of each of a set of different standard proteins and peptides results in signals with signal-to-noise ratios between 50 and 500.40
After completion of CE-MS assembly and optimization, a series of experiments was initiated with human urine as sample material to validate the analytical performance of the CE-MS platform. In these proof-of-principle experiments, mass accuracy of the CE-TOF-MS method was determined to be <25 ppm for monoisotopic resolution and <100 ppm for unresolved peaks (z>6). Moreover, a resolution above 8,000 ensured the detection of monoisotopic mass signals for z≤6.
Proteomic data mining
Due to the large amount of information provided by a single CE-MS run, which consists of ~1,200 individual spectra, adequate software solutions are required to correctly interpret and process proteomic data. Some features that must be implemented in the software include the ability to determine the charge of a particular peak, to identify and combine peaks of the same mass, but different charge states, and to perform efficient normalization of migration times and amplitudes to compensate for differences between individual measurements.
MosaiquesVisu software (http://www.proteomiques.com) was used for data evaluation.47 MosaiquesVisu employs a probabilistic clustering algorithm and uses both isotopic distribution and conjugated masses for peptide charge state determinations. The total number of distinct peptides resolved from a urine spectrogram by this software ranges from 900 to ~4,000 in healthy individuals. Subsequently, all peptides identified in the CE-MS spectrum of a particular urine sample were deposited in a Microsoft SQL database. In this database, peptides are defined by molecular mass, CE migration time and signal intensity providing a unique identification mark.
Comparative analysis of marker expression is only possible if peptides can be identified with a high level of confidence in consecutive samples. In this respect, efforts were undertaken to develop a reliable normalization and calibration procedure for CE retention times and MS signal amplitudes on the basis of 200 peptides found in more than 1,000 samples with frequencies above 90%.46,48 After normalization of the CE parameters, a clustering algorithm is applied to combine identical peptide entities to a proteomic pattern. In this latter process, the maximum allowed mass deviation is set relative to the molecular mass of the peptide, reasonably due to peak broadening at higher migration times. In the clustering algorithm, mass deviation limits are set from ±50 ppm for 800 Da peptides to ±75 ppm for 15 kDa peptides. The CE migration time deviation is linearly increased over the entire electropherogram from 2 to 5%. Finally, to compensate for mass inaccuracies caused by the deconvolution, normalization, and clustering processes, the TOF-MS-derived masses are re-calibrated by linear regression fitting to FT-ICR-MS precisely determined reference masses derived for 80 of the 5,000 in the SQL database annotated peptides with mass deviation <0.5 ppm.
After initial processing of peak spectra to identify protein or peptide targets, the next step is to use the compiled datasets for the conduction of comparative studies on the basis of multivariate statistical analyses. The classification methods may be based on logistic regression analysis49 or support vector machine (SVM).50 As with any classification procedure, these methods have their own advantages and drawbacks. It should be noted, however, that neither of these supervised learning methods include a variable selection procedure per se. In this context, statistical evaluation of the different peptides may be seen as the best-suited tool at the present time for reducing the high dimensionality of the problem. Nevertheless, a given biomarker showing statistical significance does not automatically perform well as a class-discriminating item. The statistical analysis must correct for multiple testing artefacts inherent to such an analysis.
In this context, Bonferroni corrections, and their relatives such as the Holm procedure, are the most widespread approach to control experiment-wide false positive rates.51 Distribution-free resampling methods, like these from Westfall and Young,52 are also popular approaches. A major drawback of these procedures is that they may lack sufficient statistical power. This has led Benjamini and Hochberg to introduce a false discovery rate that conserves sufficient statistical power of looking for biomarkers that are differentially expressed between two samples when subjected to two different treatments (e.g. disease/no disease).53
Finally, the importance of using proper statistics and validation of the generated classifiers in a blinded study group must be emphasized. If this practice is not strictly followed, the data obtained will likely hold no value and will be proven invalid in the next set of experiments, as was the case in a study of SELDI-TOF-MS whole-serum proteome profiling for prostate cancer detection by McLerran et al.54
Clinical validation
The performance of the biomarker(s) defined to correctly identify samples of healthy and diseased subjects may best be expressed as a receiver operating characteristic (ROC) plot, as this will indicate the degree of overlap between the two groups by plotting the sensitivity against 1-specificity at each level.55 ROC plot analysis is the method of choice since it has the remarkable property to be independent of the prevalence of the disease in the sample cohort.
After identification of candidate biomarkers in a training set of well-defined samples, it is imperative to validate their discriminating and prognostic value by applying them to a second blinded sample collective, which ideally should include diseased patients, healthy individuals, and, if necessary, patients with other related diseases. In addition, the validation process should also answer the question of how adequately the biomarker is able to discriminate between the disease of interest and all other diseases and health conditions. For example, heat shock proteins (HSPs), in particular HSP70, described by some authors as biomarkers for certain types of cancer,56–58 are frequently released by affected cells after the onset of other, non-cancer-associated stimuli such as oxidative stress, heavy metals, hypoxia, acidosis, tobacco smoke, and metabolic poisons.59 In most of the cases, it is therefore difficult to specify the cause of HSP up-regulation. On the other hand, a differentially excreted albumin-derived peptide may serve as a useful biomarker due to its uncommon occurrence as a degradation product of a disease-specific protease.
Finally, the identified biomarkers should be further characterized, e.g. by subjection to MS-based sequencing methods, in order to identify their protein provenance and gain new insights into the underlying disease mechanisms.
Biomarker identification
Separation technologies coupled to MS can make use of top-down or bottom-up approaches for protein or peptide identification. In a typical top-down experiment, intact proteins and peptides are subjected to analysis by MS/MS. In bottom-up analyses, proteins are digested by proteases, and the generated peptides are analyzed by MS/MS. Whereas the latter method is best suited for the identification of large proteins, the former is more applicable for peptides and small proteins less than 10–20 kDa and gives a more accurate definition of the potential biomarkers. As outlined recently, any biomarker should be defined by its accurate molecular composition.60
The identification of a theoretical protein based on a few tryptic fragments may be quite misleading, as it generally does not account for post-translational modifications (PTMs). However, these may in fact confer “biomarker quality” to the protein; glycated albumin may serve as a biomarker for diabetes, while albumin precursor, which would be defined as a biomarker based on several tryptic peptides, certainly does not.61 If albumin precursor was defined as a biomarker for diabetes based on a top-down 2DE- or LC-MS/MS approach, and an antibody was raised against it, then the subsequent validation of the results using an alternative technology for clinical application would have failed, due to the inaccurate definition of the original biomarker.
With the exploitation of top-down strategies for CE-MS, LC-MS, and SELDI-MS, the low-molecular-mass range of the proteome, also sometimes termed peptidome, came into focus as the source of information. The peptidome consists of all naturally occurring peptides, many of which are the products of proteolytic degradation. The rationale behind performing peptidomic analysis is provided by the finding that many disease states are reflected by changes in the peptide composition of biological fluids. As suggested by Haubitz et al.62 Villanueva et al.63 or Rossing et al.64 some of these diagnostically relevant peptides are produced by (disease−) specific proteases. These proteases, and in particular their increased activity, may be more readily assessed by analysis of their reaction products, the specific proteolytic fragments generated, than by direct identification of the proteases themselves.
It appears that the top-down approaches are currently better suited for clinical applications than the bottom-up ones. This is in part due to the more accurate definition of biomarkers, including PTMs, and the higher resolution obtained in the top-down approach (while distinguishing between e.g. 4,700 and 4,701 Da can easily be accomplished, it is almost impossible to distinguish between 60,000 and 60,001 Da), as well as the higher throughput, producing larger numbers of independent samples for statistical analysis, a prerequisite in a multiple parameter setting. Hence, while the high-molecular-weight proteome, currently analyzed generally via the bottom-up approach, may hold more information than the low-molecular-weight proteome/peptidome, its information appears to be generally not accessible in a statistically sound approach. In contrast, the low-molecular-weight proteome/peptidome can be assessed in a statistically meaningful way, owing mainly to its lesser complexity. From a practical, application-driven view, the deciphering of the low-molecular-weight proteome/peptidome using top-down approaches should be the centre of interest for clinical proteomics studies.
Since CE-MS can be used both as discovery as well as application platform, the amino acid sequence of a peptide biomarker is not absolutely required for CE-MS-based clinical diagnosis. Nevertheless, in order to gain more insights into disease processes, top-down peptide sequencing strategies were applied to obtain the amino acid sequence of peptides of interest. For verification of sequence authenticity, the number of basic and neutral polar amino acids of the database-obtained sequence candidates was correlated with their expected position in the CE-MS spectrogram.65 This assignment is possible, since the peptides are arranged in the latter in distinct charge-specific lines. This is indicated in the contour plot of Figure 3. The line marked with z=1 contains peptides with no arginine, histidine, and lysine residue in their sequence, and only the N-terminal ammonium group is positively charged. The other lines represent peptides with one, two, or more basic amino acids (z=2; z=3; z>3). This charge-specific arrangement of peptides in the CE-MS spectrogram serves as a second independent identification mark in addition to the ESI-MS-determined molecular mass. This facilitates identification of target peptides after subjection of the sample to LC-MS/MS for sequence analysis. For further, more detailed, discussion, e.g. on the advantages and limitations of the different peptide sequencing platforms, the reader is referred to Zürbig et al.65
Figure 3.
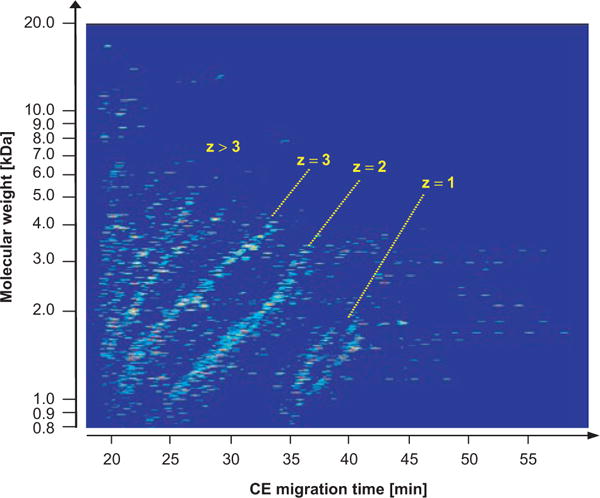
Contour plot showing the organization of peptides in the CE-MS spectrogram in distinct charge-specific lines. The membership to a certain charge line allows reliable prediction of the number of basic amino acids and therefore has considerable predictive value for peptide identification in other MS systems.
Biomarker quantification
In the past, one major limitation of all proteomic methods with respect to their clinical applicability was the inability to specify the amount of a particular protein or peptide present in a given sample. To solve this problem, several MS-based quantification strategies have been developed in the last few years, many of which are based on the use of isotope-coded affinity tags.66 These techniques, however, are time consuming, error prone, and cost expensive. As a consequence, further efforts have been made to develop simple and more straightforward quantification strategies based on signal intensity/ion counting.67 While these measures, in contrast to stable isotope-labelled internal standards,68 do not permit absolute analyte quantification, they were specially adapted to perform relative quantification with acceptable deviation characteristics (±10%,69 and own observation). In recent experiments, we were able to show that both absolute and relative quantification give highly similar results, with a slightly better performance of the ion-counting approach.70 These findings provide the rationale for us to establish a universally applicable relative quantification procedure by the selection of a set of naturally occurring, highly abundant collagen fragments as internal standards in near future.
Establishment of a low-molecular-weight proteome database
On the basis of CE-MS measurements, a database was established specifying more than 5,000 different polypeptides that were found in more than 20% of the currently analyzed 3,900 human urine samples. In this database, peptides are unambiguously characterized by their molecular mass, CE migration time, and average MS signal intensity. The storage of CE-MS-specific peptide parameters was driven by the need to efficiently and reliably retrieve all required information for the development of disease-specific peptide marker patterns and to quickly compare individual patient profiles against the same. For patient classification, no additional information other than the CE-MS parameters is required.
Since the identified peptides represent end products of specific metabolic pathways, they are biologically relevant. The composition of the peptide termini can potentially provide information on the identity of e.g. disease-associated proteases,63 whereas PTMs may offer clues to the translational capacity and function of cells.71–73 Therefore, it can be stated that the data stored in the database represent a comprehensive description of the urinary low-molecular-weight proteome. In order to make this, important from a (patho)physiological point of view, information available, the amino acid sequence of all relevant peptides has to be reliably determined by MS/MS sequence analysis.65 As a result of these efforts, 443 naturally occurring urinary peptides have been identified to date by their amino acid sequence.74 As presented in Figure 4, most of them are fragments from different types of collagens, predominantly type I; common blood proteins, like alpha-1-antitrypsin; hemoglobin, serum albumin, fibrinogen; and the major urinary protein uromodulin. Additional, less common, protein fragments include osteopontin, clusterin, epithelial cadherin, proSAAS, and gelsolin. Although these native peptides are not accessible to bottom-up sequencing, many of the precursor proteins were identified in urine by other research groups using this strategy.75,76
Figure 4.

List of all currently sequence-identified native human urinary peptides defined by their protein precursor. Given are the numbers of sequenced peptides for a particular protein, the SwissProt protein name for Homo sapiens, and the gene symbol. Additional information, like all peptides amino acid sequence, is accessible from the Mosaiques homepage, located at http://mosaiques-diagnostics.de/diapatpcms/mosaiquescms/front_content.php?idcat=257/.
The fact that the database in its current state contains sequences from only 9% of the 5,000 urinary peptides detected by CE-MS together with the restricted types of PTMs that have been identied so far, namely proline and lysine hydroxylation and methionine oxidation, reveals that improved MS-based methods for better identification and characterization of peptides carrying modifications are required to extend the informational content of the peptide database.
The organization of proteomic and clinical data in the low-molecular-weight urinary proteome database is shown in Figure 5. A major advantage of the database system is that, with the help of appropriate software, patient groups can be selected retrospectively for inclusion in a comparative study on the basis of the recorded clinical data and by a direct linkage to the patient’s proteomic profile.
Figure 5.
Establishment of a database as a core information system of patient proteome and clinical data. Storage and retrieval of peptide profiles, peptide sequences, and patient clinical records allowing sample selection and differential proteomic profiling for the purpose of biomarker discovery and patient classification.
Clinical application of CE-MS
In 2003, CE-MS reached a level of maturity that allowed its use in clinical studies. First efforts in this direction were mainly focused on methodological aspects such as the development of appropriate study designs; the establishment of advanced methods for signal detection, processing, and normalization; or the selection of adequate statistical methods resulting in the formulation of guidelines for the conduct of clinical proteomic studies.5 After several pilot studies resulting in the generation of discriminative pattern for a variety of chronic renal diseases, the focus moved to large-scale clinical trials including large cohorts of patients with various related diseases separated into training sets for biomarker identification and reference or blinded test sets for subsequent validation. An overview of all these studies is given in Table 1. Some of these studies will now be described in more detail, with particular emphasis on the identity and significance of the selected urinary peptide biomarkers.
Table 1.
Overview of clinical proteomic studies using CE-MS
| Study groups
|
Origin of sequenced biomarkers** | |||||
|---|---|---|---|---|---|---|
| Disease | Aim | Discovery phase | Validation phase | Performance***: | Reference | |
| Diabetes type 2, Diabetic nephropathy (DN*) | Renal damage evaluation | 66 diabetics type 2 w/albuminuria 46 diabetics type 2 w/o albuminuria 39 HC | – |
DN makers:
|
Renal damage pattern conspicuity 35% diabetics w/albuminuria 4% diabetics w/o albuminuria | 95 |
| Diabetes type 1, DN | Diabetes staging and early diagnosis of DN | 44 diabetics type 1 (>5 yrs diabetes duration) 9HC | – | – | Early detection of diabetic renal alterations | 96 |
| Membranous glomerulonephritis (MGN) | SELDI and CE-MS comparison for MGN-specific urinary peptide biomarker search | 8 MGN, 8 HC | – | – | Numbers of potential MGN-specific urinary peptide biomarkers identified by SELDI: 3 and by CE-MS: 200 | 35 |
| Focal segmental glomerulosclerosis (FSGS) Minimal change disease (MCD) MGN |
Establisment of normal and disease-specific urinary peptide profiles | 16 MCD, 18 MGN, 10 FSGS, 57 HC | – | – | HC vs. MCD/FSGS vs. MGN 84% true classification after cross-validation: 94% HC, 71% MCD/FSGS, 93% MGN | 41,62 |
| IgA nephropathy (IgA-N) | IgA-N diagnosis | 45 IgA-N, 13 MGN, 57 HC | – |
IgA-N markers:
|
HC vs. IgA-N 100% sensitivity, 90% specificity MGN vs. IgA-N 77% sensitivity, 100% specificity FSGS, MCD, DN vs. IgA-N 100% sensitivity/specificity | 77, 80 |
| Chronic renal diseases | IgA-N differentiation | 45 IgA-N, 25 vasculitis, 30 FSGS, 106 DN, 24 lupus nephritis, 7 hypertension, 4 nephrosclerosis, 3 amyloidosis | 49 blinded samples including IgA-N, HSP w/ and w/o nephritis, HCV-induced GN, non-IgA-N GN and HC |
Renal damage markers:
|
IgA-N vs. others 90% sensitivity, 82% specificity | 79 |
| Diabetes type 2, DN | DN diagnosis and candesartan therapy monitoring | 20 normo −DR, 20 normo +DR, 20 micro +DR, 18 macro +DR | – |
Candesartan influenced markers:
|
Profit of candesartan treatment 86% to 54% decrease of diabetic proteome pattern recognition | 81 |
| Diabetes, DN, nondiabetic proteinuric renal diseases | Differentiation of proteinuric renal diseases | 30 normo, 29 micro, 30 macro/DN, 30 HC | 211 blinded samples including normo/micro/macro diabetics (Diabetes), other macro diabetics (Macro), IgA-N/FSGS/MGN/MCD (Nondiabetic renal diseases), HC |
Diabetes and DN markers:
|
HC vs. Diabetes 89% sensitivity, 91% specificity HC vs. Macro 97% sensitivity/specificity Diabetic vs. nondiabetic renal diseases 81% sensitivity, 91% specificity | 82 |
| Coronary artery disease (CAD) | CAD diagnosis | 30 patients w/angiographically confirmed CAD, 20 controls w/o CAD history, 233 healthy university recruits (prevention of center specific bias), 17 paired samples of hypertension/type II diabetes patients before and after 12 wkrampiril treatment (exclusion of medication effects) | 47 blinded samples of patients w/angiographically confirmed CAD and of age-matched controls w/o CAD history |
CAD markers:
|
CAD vs. no CAD 98% sensitivity, 83% specificity | 116,119 |
| CAD and DN | CAD risk prediction for diabetes type 1 | 15 diabetes type 1 w/CAD, 4 nondiabetes w/CAD, 19 type 1 diabetes w/o CAD, all from the CACTI study group [97] | Angiographic follow-up of patients |
CAD risk markers:
|
Prevalence of proteomic CAD score and CAD events: 2.2 (1.3-5.2) odds ratio (95% CI) in Prediction of CAD events 1.4 ± 1.3 yrs in advance | 98 |
| Acute tubulointerstitial rejection (AIR) | Detection of AIR | 29 w/o AIR/UTI, 19 w/subclinical or clinical AIR, 10 w/UTI | 26 blinded samples w/o AIR/UTI, w/subclinical or clinical AIR and w/UTI |
Renal transplant markers:
|
Correct classification: 8/10 w/o AIR/UTI, 6/9 w/subclinical or clinical AIR, 3/7 w/UTI | 99 |
| ANCA associated vasculitis (AAV) | AAV diagnosis and therapy response | AAV diagnosis: 18 active AAV, 200 HC, 225 other glomerular diseases AAV therapy response: 18 active AAV, 19 AAV in stable remission (>18mo) | 40 blinded samples including active AAV, MGN, IgA-N, FSGS, MCD, lupus nephritis and HC |
AAV markers:
|
sensitivity: 94%, specificity: 93% for AAV positive classification of only 3 IgA-N (n=18) | Haubitz et al., submitted |
| Ureteropelvic junction obstruction (UPJ) | UPJ prognosis | 19 severe UPJ obstruction, 19 low-grade UPJ, 13 HC | Prospective blinded study on 36 newborns |
UPJ markers:
|
Prediction of clinical outcome 94% precision (9 mo in advance, n=36) | 101,102 |
| Urothelial carcinoma | Urothelial carcinoma prediction | 46 urothelial carcinoma, 11 benign prostate hyperplasia, 22 HC | Masked specificity assessment 366 including non-malignant genitourinary disease, renal cancer, prostate cancer and HC Prospective masked assessment nephrolithiasis and HC |
Urothelial carcinoma markers:
|
HC vs. urothelial carcinoma: 100% sensitivity/specificity non-malignant genitourinary disease vs. Urothelial Carcinoma: specificity range: 86–100% | 46 |
| Prostate cancer (PCa) | PCa diagnosis | 51 biopsy-proven PCa, 35 NDE | 264 blinded samples of biopsy-proven PCa and NDE |
PCa markers:
|
PCa vs. NDE89% sensitivity, 51% specificity combined with age score and free PSA91% sensitivity, 69% specificity | 16 |
| Urothelial bladder cancer (BCa) | BCa staging | 127 BCa (71 Tis-T1, 56 T2-T4), 11 genitourinary disease, 34 nephrolithiasis, 81 smokers, 171 HC | 130 blinded samples of BCa-stages Tis-T1 and T2–T4 |
BCa stage markers:
|
Muscle invasive (Tis-T1) vs. muscle non-invasive (T2-T4) tumors 81% sensitivity, 57% specificity combined with cytology 92% sensitivity, 68% specificity | Schiffer et al., submitted |
| Acute graft-versus-host disease (aGvHD) | aGvHD grade>I diagnosis after allogeneic HSCT | 13 HSCTw/aGvHD grade > I, 50 HSCT w/o aGvHD, for nonspecific marker exclusion: 69 renal disease controls including IgA-N, DN, FSGS, MCD, MGN, vasculitis, SLE and 20 HC | 599 blinded HSCT samples w/ and w/o aGvHD grade I–IV |
aGvHD grade>I markers:
|
HSCT w/aGvHD grade > I vs. HSCT w/o aGvHD or w/aGvHD grade I 83% sensitivity, 76% specificity Correct classification of aGvH D>grade I even before clinical diagnosis | 115 |
abbreviations used in the table: DR, diabetic retinopathy; GN, glomerulonephritis; HSP, Henoch-Schoenlein purpura; HSCT, hematopoietic stem cell transplantation; macro, macroalbuminuria; micro, microalbuminuria; NDE, no disease evidence; normo, normoalbuminuria; RTR, renal trasplant recipients; UTI, urinary tract infection.
peptides from the specified protiens,
if not otherwise indicated in blinded fashion.
Urinary biomarkers for renal diseases
In an attempt to define urinary polypeptide markers specific for membranous glomerulonephritis (MGN), Neuhof et al. used SELDI and CE-MS as proteomic platforms.35 Analysis of identical urine samples resulted in the definition of three potential biomarkers by SELDI and 200 potential biomarkers by CE-MS analysis, demonstrating the greater potential of the latter platform.
CE-MS analysis of samples from patients with various types of CKD, indicated that marker panels consisting of 20 to 50 urinary polypeptides allow diagnosis and discrimination of IgA nephropathy (IgA-N), focal-segmental glomerulosclerosis (FSGS), MGN, minimal-change disease (MCD), and diabetic nephropathy (DN).41,62,77–80 For better illustration, the established biomarker patterns for the diagnosis of CKD in general and DN and IgA-N in particular are presented in Figure 6.
Figure 6.

Performance characteristics of urinary peptide patterns for the diagnosis of chronic kidney disease (CKD), diabetic (DN), and IgA nephropathy (IgA-N). The compiled peptide patterns of healthy controls (NC) and patients consist of 20 to 100 single measurements. Besides biomarkers indicative for CKD present in both DN and IgA-N patients, other markers could be identified that are either specific for DN or IgA-N, allowing their differentiation. X axis: CE migration time [min], y axis: log molecular mass [kDa], and z axis: relative ion signal.
In these studies, several of the disease-associated peptide biomarkers could be identified by sequence analysis. Biomarkers for the above-mentioned renal diseases are mainly fragments from different types of collagens, serum albumin, alpha1-antitrypsin, and uromodulin.79,81,82
Increased detection of albumin-derived peptides indicated that albumin fragments serve as earlier markers of renal impairment than the parental protein itself.83,84 Whereas leakage of the latter across the glomerular wall is a rather late event, an inflammation-induced increase in proteolytic activity and an enhanced escape of albumin fragments from proximal tubular reabsorbtion resemble preceding stages of the pathological process.85,86
Another intriguing observation is a gradual decrease in specific collagen fragments in the course of diabetes to DN. This is in good correlation with a decrease in elastase activity leading to increased renal extracellular matrix (ECM) deposition87,88 and the observation that thickening of the ECM is one of the earliest detectable morphological changes in diabetes.89
Identification of various alpha1-antitrypsin fragments specifically in IgA-N-affected patients indicates that differentiation of CKD into various subforms can be conveniently performed by analysis of the urinary proteome. The crucial role of alpha1-antitrypsin peptides in IgA-N is also supported by recently published findings.90 Using 2-DE, the authors identified lower isoforms of albumin and alpha1-antitrypsin in the renal tissues of patients with IgA-N that were absent in normal controls, which they suggested are cleavage products of proteinase attack. Thus, it can be hypothesized that urinary detection of alpha1-antitrypsin in IgA-N patients is the consequence of a disease-specific imbalance between serine proteases and their tissue inhibitors.91–94
Rossing et al.81 demonstrated in a randomized double-blinded study that treatment of macroalbuminuric patients with the angiotensin II receptor blocker candesartan had significant impact on the expression of 15 of the 113 urinary peptides indicative for DN. Eleven of these candesartan-sensitive DN-specific biomarkers could be identified as fragments from serum albumin, uromodulin, and types of collagens.
In clinical studies from Mischak et al.95 and Meier et al.96 CE-MS spectra from patients with type 1 or 2 diabetes with/without macroalbuminuria and healthy volunteers were analyzed to create stage-specific polypeptide patterns. In patients with type 2 diabetes and unchanged albumin excretion rate, the detected peptide pattern differed significantly from that in patients with high-grade albuminuria. Comparable results were obtained for patients with type 1 diabetes and renal involvement, suggesting that the urinary proteome contains a much greater variety of polypeptides than previously demonstrated. These results were recently confirmed by a study from Rossing et al. involving 500 case and control samples.82 In this study, the authors demonstrate the efficiency of urinary peptidome profiling by CE-MS to detect both diabetes and DN and to predict the development of DN in a blinded, prospective manner.
By CE-MS analysis of urine samples of type 1 diabetic patients enrolled in the Coronary Artery Calcification in Type 1 Diabetes (CACTI) study97 who later developed a cardiovascular event, Snell-Bergeon et al.98 prospectively investigated arteriosclerosis risk factors and renal dysfunction in this well-characterized patient cohort. Therefore, this study provides a good example of how different biomarker models can be combined to gain a detailed picture of the patient’s current and future health status.
Wittke et al.99 used CE-MS to analyze urine samples from patients with different grades of subclinical or clinical acute allograft rejection and urinary tract infection and without evidence of rejection or inflammation. Substantial differences were found between patients with transplanted kidney and patients with native kidney, most likely due to treatment with the immunosuppressive calcineurin-inhibitor cyclosporin A. Additional biomarkers were identified that allow differentiation between infection and acute rejection. Most important, the results were not confounded by acute tubular lesions, tubular atrophy, tubulointerstitial fibrosis, calcineurin inhibitor toxicity, proteinuria, hematuria, allograft function, or different immunosuppressive regimens.
As the number of individual proteome profiles stored in the low-molecular-weight urinary database increased rapidly over the years, proteomic analysis became more and more refined to detect even slightest changes between different, presumably related, disease conditions. This point is best illustrated by a recent study of Haubitz et al. (manuscript submitted) in which biomarkers for the renal manifestation of anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) were identified in urine samples from 18 patients with active renal AAV. In the first step of the evaluation, urinary peptide profiles of the AAV patients were compared with those of 200 healthy controls and those of 225 patients with other glomerular diseases, such as FSGS, DN, IgA-N, MCD, and MGN. From a total of 113 potential vasculitis biomarkers, 18 were selected by take-one-out cross-validation and combined to an AAV-specific discriminatory biomarker model that allowed classification of a 40-sample blinded test set with 94% sensitivity and 93% specificity in both SVM- and linear combination-based regression models. Most notably, only three of the 18 IgA-N patients included in the study scored positive, providing evidence for the ability of the AAV-specific model to exclude other forms of vasculitis.
To further examine specificity, more patient groups like those with cytomegalovirus infection after renal transplantation, nephrolithiasis-induced microhematuria, bladder or renal cancer, hypertension, and type 2 diabetes were investigated and reported specificity values greater than 90%. In a second step, a search for biomarkers indicative for therapy was initiated by the comparison of the 18 active AAV patient profiles with 19 profiles from those AAV patients who had undergone disease treatment and had been under stable remission for more than 18 months. From the 266 peptides found to be affected by immunosuppressive treatment, 122 were also observed to be significant in the AAV-indicative model. All 122 biomarkers were combined to a marker set for disease activity. Some of the relevant markers could be sequenced. Besides up-regulation of alpha1-antitrypsin and albumin fragments and down-regulation of collagen fragments, the most intriguing finding is the appearance of several C- and N-terminal haemoglobin fragments. In this context, it was speculated that the latter emerge as consequence of increased protease activity after activation of neutrophiles by ANCA.100
Besides diagnosis and therapy monitoring, the studies of Decramer et al.101,102 provide a striking case for the potential of urinary proteomics for accurate estimation of prognosis. The authors used CE-MS-based urinary proteome analysis for the definition of specific biomarker patterns indicative of different grades of ureteropelvic junction (UPJ) obstruction, a frequently encountered pathology in newborns. In this study, newborns with different stages of UPJ obstruction were divided into non-operated and operated groups based on the grade of hydronephrosis and the thresholds of renal pelvic diameter and together with age-matched non-diseased controls used as training sets for UPJ obstruction-specific biomarker definition. A UPJ obstruction-specific hierarchic disease model was established and subsequently used for the classification of a clinically non-classifiable group of newborns. From the clinical outcome of the latter group, it could be deduced that prediction of the clinical outcome was possible nine months in advance with 94% precision.
A peptide fragment of the proprotein convertase subtilisin/kexin type 1 inhibitor (ProSAAS), which is known as a 26 kDa neuroendocrine protein with prohormone convertase 1 inhibition activity,103 was found by amino acid sequence analysis to be differentially down-regulated in newborns with severe UPJ obstruction.102 This finding is in line with recent data from animal studies indicating ureteral obstruction being linked to activation of the renin-angiotensin system.104
Urinary biomarkers for urological disorders
Theodorescu et al.46 described the detection and validation of biomarkers of urothelial carcinoma using CE-MS. A bladder cancer-specific biomarker pattern was established by initial definition in a training set composed of 46 patients with urothelial carcinoma and 33 healthy subjects and further refinement using CE-MS spectra of 366 urine samples from healthy volunteers and patients with malignant and nonmalignant genitourinary diseases. By this two-step biomarker discovery approach, the authors established a prediction model composed of 22 urinary peptides, which, when applied to a blinded test set containing 31 urothelial carcinoma patients, 11 healthy individuals, and 138 non-malignant genitourinary disease patients, correctly classified all urothelial carcinoma patients and all healthy controls. Differentiation between bladder cancer and other malignant and non-malignant diseases (such as renal nephrolithiasis) was accomplished with 86 to 100% specificity.
In an effort to identify stage-specific urinary peptide biomarkers for bladder cancer, Schiffer et al. (manuscript submitted) established a biomarker set composed of four peptides. Given an estimate that 30% of tumors at initial presentation have invaded the bladder muscle,105 the staging model allowed prognosis of the progressive muscle invasive form with a negative predictive value of 77% and a positive predictive value of 90% in a blinded test set composed of 130 biopsy-proven carcinoma patients.
A MS-based strategy was developed for the definition of urinary peptide biomarkers that are indicative for prostate cancer (PCa).48 Urine samples from 47 patients who underwent prostate biopsy were analyzed. On the basis of prostate biopsy, 26 patients in this group were diagnosed as suffering from PCa and 21 as afflicted with benign prostatic hyperplasia (BPH). In this set of well-characterized patient samples, the data indicated several polypeptides, allowing prediction of PCa with sensitivity and specificity values >90%. To confirm these findings, a blinded prospective study was subsequently initiated. In this follow-up study, proteomic profiling with the defined biomarker showed increased numbers of false-negative diagnoses when discrimination of malignant and benign forms of PCa in a blinded manner was attempted. Efforts were undertaken to discover the cause of the inconsistency of the results. Due to several reports describing the occurrence of polypeptides in urine that originate from prostatic fluid,106–108 it was concluded that the absence of prostatic peptides in some of the collected midstream urine samples is responsible for the increased false-negative rates. Consequently, first voided urine was tested as sample material and found to be a better source for the definition of PCa-specific biomarkers than midstream urine.
After refinement of the PCa-specific biomarker pattern using urine samples from 54 PCa and 62 BPH patients, a model with ten potential biomarkers in combination with age and percentage of free PSA resulted in the accurate prediction of 89% (32/36) of the PCa and 67% (16/24) of the BPH patients included in a blinded set of patient samples.16 In the latter study it was found that the majority of identified biomarkers were down-regulated in PCa patients. This observation was attributed to the activity of as yet unknown proteases as a consequence of malignant transformation of the prostate. MS sequencing led to the identification of Na+,K+-ATPase gamma chain, collagen alpha-1 type III, collagen alpha-1 type I, and psoriasis susceptibility 1 candidate gene 2 protein-derived peptides as being significantly reduced in their excretion levels. Na+,K+-ATPase was described by Duran et al.109 as required by the prostate for the production of citrate, the latter being an important constituent of the seminal fluid. More interestingly, reduced amounts of the targeted Na+,K+-ATPase gamma chain-derived peptide in the urine of PCa patients is in accordance with a previously reported down-regulation of the Na+,K+-ATPase complex in the membrane of androgen-dependent human prostate cancer cells when these were cultured in the presence of androgens.109,110
Collagen alpha-1 type III and collagen alpha-1 type I are substrates of matrix metalloproteinases (MMP), a group of zinc finger endopeptidases described by Brew et al.111 as differentially regulated in a variety of cancers. For the psoriasis susceptibility 1 candidate gene 2 protein, also designated as small proline-rich protein 1 (SPR1), in vitro data exist for its specific down-regulation in lung cancer. Using reverse transcriptase polymerase chain reaction (RT-PCR), DeMuth et al.112 reported a complete loss of SPR1 gene expression in malignant human bronchial epithelial cell cultures, which is normally required by this cell type for terminal differentiation. Lau et al.113 observed a gradual decline of SPR1 expression during the progression of carcinogenesis in cells of a simian virus-transformed epithelial and a lung carcinoma cell line. In conclusion, it can be deduced from the results of the proteome studies that prostate cancer can be detected by CE-MS urinary peptide profiling due to the disappearance of several prominent peptide signals, which could, in part, be attributed to a down-regulation of certain prostate proteins.
Application of urinary proteome analysis to non-renal diseases
Clinically important examples of urinary proteome analysis in non-renal diseases include the clinical follow up of patients after allogeneic hematopoietic stem cell transplantation (HSCT)114,115 and non-invasive diagnosis of coronary artery disease (CAD).116,117
For the diagnosis of graft-versus-host-disease (GVHD), urine samples from 40 patients after HSCT (35 allogeneic, five autologous) and five patients with sepsis were collected during a period of 100 days with a maximum of ten samples per patient. A pattern consisting of 16 differentially excreted polypeptides indicated early GVHD. The pattern of markers discriminated patients with early GVHD from patients without complications with 82% specificity and 100% sensitivity.114 A subsequent blinded multicenter validation study of 100 patients with more than 600 samples collected prospectively confirmed the results.115 Three polypeptides of the GVHD pattern could be successfully sequenced. The sequence information obtained revealed reduced levels of a peptide derived from collagen-1 type I chain and increased levels of two peptides from collagen-1 type III. In accordance with these findings, Pihusch et al.118 reported elevated serum levels of procollagen type III peptide, being previously described as a sensitive marker of inflammation and fibrinogenesis in hepatic GVHD.119
For non-invasive CAD diagnosis, Zimmerli et al.116 and von zur Muhlen119 examined patients undergoing coronary artery bypass grafting or patients after acute coronary infarction. In the former study, urine samples from 88 patients and 282 controls were analyzed using CE-MS to identify CAD specific biomarkers. Fifteen peptides could be defined in the training set and were combined to a characteristic CAD signature panel. Five peptide biomarkers could be identified as collagen type I and type III fragments. Whereas the collagen-derived peptide biomarkers for CKD (see above) are generally down-regulated, all sequenced collagen fragments were found significantly up-regulated in CAD samples. On the basis of recently published results on increased circulating levels of collagenases, such as MMP-9, in patients with stable angiographic coronary atherosclerosis120,121 or intermittent claudication,122 CAD-specific up-regulation of collagen fragments was interpreted to be a consequence of elevated collagen degradation. A finding that further supports this hypothesis is that elevated MMP-9 activity has been found in unstable plaques, suggesting a crucial role in plaque rupture.123,124
In a blinded assessment of more than 200 samples, specific urinary biomarkers identified CAD patients with 98% sensitivity and 83% specificity. Some of these peptides significantly changed towards “normal” in correlation with the level of physical activity after therapeutic intervention. Furthermore, these data provide evidence for association between arteriosclerosis risk factors and renal dysfunction.125
The prognostic value of the CAD model was recently demonstrated by Snell-Bergeon et al.98 In their longitudinal study carried out on retrospective samples of 19 type 1 diabetic patients derived from the CACTI study population, who were clinically asymptomatic for CAD at the date of enrollment but later developed CAD, the cardiovascular event could be predicted 1.4 ± 1.3 years in advance (range 0.2–4.5 years). These data demonstrate, for the first time, that biomarkers can predict the risk for future CAD events.
Adaptation of CE-MS for clinical routine
CE-MS, like no other proteomic platform, combines advances in CE separation and MS detection useful for both biomarker discovery and classification of patient samples with established biomarker sets. The clinical studies described in this review can be seen as proof of the principal applicability of this technology in a clinical laboratory setting. A prominent example for large-scale use of CE-MS beyond the stage of clinical trails is a GVHD screening of HSCT patients, which provides the basis for preemptive steroid therapy of patients before the onset of clinical signs.115
Of great importance for clinical application of CE-MS is the ability to deduce the amount of a protein or peptide from the CE-MS spectrum of a clinical sample. Recently, a strategy was developed by Jantos-Siwy et al.70 that allows relative quantification of a urinary biomarker by referencing a set of 29 endogenous peptide standards. The latter were selected due to their high abundance and low signal amplitude variability in more than 1,000 urine samples. This method was compared to signal normalization by the addition of isotope-labelled calibrants and found to result in highly comparable quantification results as indicated by slopes and R2 values of 1 in linear regression analyses. One advantage of this method is that biomarker excretion can be quantified even in cases where no sequence information is available, as essentially required for synthesis of an isotope-labelled biomarker in absolute quantification. Moreover, the established single-step procedure for calculation of a biomarker’s relative abundance allows both correction of analytical variances during proteomic profiling and correction for different dilution levels of individual urine samples.
In order to bring CE-MS a step closer to clinical use, the CE-MS platform was validated with respect to a system’s performance and diagnostic accuracy. In addition, the influence of temperature, time course, storage, and preparation conditions on the analytical performance of the biomarker were extensively investigated. Analytical validation was performed in the context of CE-MS-based diagnosis of muscle-invasive bladder cancer (Schiffer et al., manuscript submitted). Statistical analyses using the Mann-Whitney U test for median comparison and the F-test for comparison of deviations were performed on the classification score of the corresponding peptide biomarker panel. The latter parameter decisively reflects overall variability since it is influenced by all data recording and processing steps, including CE separation, MS detection, mass deconvolution, clustering, peak normalization, and sample classification.
In conformity with guidelines EP7-A2 and #135 of the FDA (www.fda.gov), intra- and inter-sample variations were assessed by processing three individual urine samples in 15 replicates, whereas intermediate precision was addressed by preparation of one urine sample by two operators independent from each other at 15 different days and measurement on two different CE-MS devices. Stability of urine samples was assessed at room temperature and 4°C over a time range of 24 h and after three frost/defrost cycles. Post-preparative stability was investigated by repeated measurements of a prepared sample over 24 h at 4°C in an autosampler. Reliability of classification was determined by monitoring the classification results of two male and two female healthy probands over a time course of one month. The duration of this observational period takes into account variations in work-life balance or, for the females, in menstrual cycles. In all of these control experiments, the interquartile ranges were observed to lie in the FDA-approved range, which indicates appropriateness for routine clinical application in the case of bladder cancer staging.
Despite these improvements, further optimization can be done to better meet laboratory requirements. Challenging tasks include the development of new capillary coating protocols for higher signal resolution, faster migration, and more sensitive detection; or the optimization of coupling geometry to achieve constant sheath flow soaking and to avoid the destabilizing effects of inconsistent sheath flow pumping, to name just a few.
Pathophysiological role of biomarkers
Albeit the majority of potential urinary biomarkers described to date have not been sequenced yet, informative sequences are available for more than 400 different urinary peptides.74 Not unexpectedly, most of these peptides are derived from the most abundant proteins in the blood and urine: albumin, beta 2-macroglobulin, uromodulin, and collagen, mainly types I, II, and III. Consequently, a valid question is whether peptidomics is not just another way to measure glomerular injury, which could probably be assessed with similar precision, but less effort, by measuring albuminuria.126 However, the fact, that differential diagnosis based on urinary proteome analysis is possible4,62,77,82 and that patients in complete remission without albuminuria still exhibit apparently disease-specific changes in urinary polypeptides41 strongly suggests that these peptides contain clues to the underlying pathogenesis and are not merely degradation products. It is tempting to speculate that the disease-specific peptides may be indirect indicators of the activity of disease-specific proteases, as recently suggested by Haubitz.77 This hypothesis is further strengthened by work published by Nemirovskiy et al.127 in which the presence of specific collagen fragments correlated with the disease-specific activity of MMPs.
While the evidence is still scarce, it is an attractive hypothesis that urinary peptides of diagnostic value are not merely degradation products of abundant larger proteins, but a result of distinct, disease-specific processes, in many cases due to significant changes in the activity of proteases. This assumption is supported by sometimes apparently unrelated findings; for example, the increase of collagen and ECM in patients with diabetes and DN has been established by a variety of methods. Our recent findings that collagen fragments are significantly reduced in diabetic urine82 fits into this scenario and further supports the hypothesis that both reduced activity of proteases and protection of the ECM from proteolysis by advanced glycosylation end products may be key pathological changes in diabetes mellitus.64
A similar scenario may be applicable to albuminuria. Consequently, an albumin-derived biomarker is not simply “an albumin fragment,” but rather a specific fragment, defined by its specific C- and N-terminus. The presence of specific urinary fragments of albumin and alpha-1-antitrypsin associated with nephrotic syndrome in CKD has recently also been described by Candiano et al.128 Unfortunately, such essential detailed information is frequently absent (e.g. see the recently published database of urinary proteins76). A thoroughly performed examination of the sequences of the urinary peptides and a comparison with protease specificities may strengthen the above hypothesis and lead to a better insight into regulation and the pathophysiological role of specific proteases in various diseases.
Conclusion
To date, separation technologies coupled to MS appear to be the preferred option for a generic approach to identification and evaluation of biomarker profiles in individuals. The currently available separation techniques 2DE, LC, SELDI, and CE differ, however, with respect to high throughput, robustness, accuracy, and reproducibility. It appears that CE-MS fulfills the requirements for broad application in routine clinical practice, as indicated by the validation of GVHD, renal disease, and prostate- and bladder-cancer specific marker patterns in hundreds of patient samples under identical conditions using the same CE and MS platforms.16,46,74,82,101 It must be stated, however, that future implementation of proteome profiling in laboratory diagnosis relies on more than just technological advancements. Of equal importance are concerted efforts to develop global standardization procedures for the planning, conduction, and reporting of clinical proteomic studies. By the adoption of standardized methods in the identification of disease-specific biomarker patterns, the information provided by proteomic platforms will bring clinical chemists a step closer to the ultimate goal of capturing all critical pieces of information of a particular disease in one single diagnostic step.
To enable widespread use of proteomics in a clinical diagnostic laboratory, complete CE-MS systems, together with software that enables proper evaluation of samples, must be available. Unfortunately, to date, no vendor is able to offer such a system, which effectively prevents routine application of the technology. However, it is anticipated that increased demand will result in the development and subsequent availability of a “turn-key” CE-MS system for clinical application.
The foreseeable, above-mentioned developments hopefully will result in the integration of MS-based proteomic methods into the armamentarium of clinical laboratories.
Acknowledgments
HM was supported in part by EUROTRANS-BIO grant ETB-2006-016 and EU Funding through InGenious HyperCare (LSHM-C7-2006-037093) and PREDICTIONS (1272568). DMG gratefully acknowledges support from an NIH pre-doctoral fellowship—the Biotechnology Training Program (NIH 5T32GM08349).
Abbreviations
- 2D-DIGE
two-dimensional differential in gel electrophoresis
- 2DE
two-dimensional electrophoresis
- AAV
ANCA-associated vasculitis
- ANCA
anti-neutrophil cytoplasmic antibody
- BPH
benign prostatic hyperplasia
- CACTI
coronary artery calcification in type 1 diabetes
- CAD
coronary artery disease
- CE
capillary electrophoresis
- CKD
chronic kidney disease
- DN
diabetic nephropathy
- ECM
extracellular matrix
- ESI
electrospray ionization
- FSGS
focal-segmental glomerulosclerosis
- FT-ICR
fourier transform ion cyclotron resonance
- GFR
glomerular filtration rate
- GVHD
graft-versus-host disease
- HSCT
allogeneic hematopoietic stem cell transplantation
- HSP
heat shock protein
- IgA-N
immunoglobulin A nephropathy
- LC
liquid chromatography
- MALDI
matrix-assisted laser desorption/ionization
- MCD
minimal change disease
- MGN
membranous glomerulonephritis
- MMP
matrix metalloprotease
- (MS/)MS
(tandem) mass spectrometry
- PCa
prostate cancer
- PSA
prostate-specific antigen
- PTM
posttranslational modification
- ProSAAS
proprotein convertase subtilisin/kexin type 1 inhibitor, a member of the subtilisin-like proprotein convertase family
- ROC
receiver operating characteristic
- SDS-PAGE
sodium dodecylsulfate polyacrylamide gel electrophoresis
- SELDI
surface-enhanced laser desorption/ionization
- SPR1
small proline-rich protein 1, a squamous differentiation marker
- SVM
support vector machine
- RT-PCR
reverse transcriptase polymerase chain reaction
- TOF
time-of-flight
- UPJ
ureteropelvic junction
Footnotes
Referee: Dr. Antonia Vlahou, Biomedical Research Foundation, Academy of Athens, Athens, Greece.
Statement of competing financial interests: HM is the founder and co-owner of Mosaiques Diagnostics, which developed the CE-MS technology for clinical application.
References
- 1.Hernandez J, Thompson IM. Prostate-specific antigen: a review of the validation of the most commonly used cancer biomarker. Cancer. 2004;101:894–904. doi: 10.1002/cncr.20480. [DOI] [PubMed] [Google Scholar]
- 2.Mogensen CE. Systemic blood pressure and glomerular leakage with particular reference to diabetes and hypertension. J Intern Med. 1994;235:297–316. doi: 10.1111/j.1365-2796.1994.tb01080.x. [DOI] [PubMed] [Google Scholar]
- 3.Rossing K. Progression and remission of nephropathy in type 2 diabetes: new strategies of treatment and monitoring. Dan Med Bull. 2007;54:79–98. [PubMed] [Google Scholar]
- 4.Fliser D, Novak J, Thongboonkerd V, Argiles A, Jankowski V, Girolami M, Jankowski J, Mischak H. Advances in urinary proteome analysis and biomarker discovery. J Am Soc Nephrol. 2007;18:1057–1071. doi: 10.1681/ASN.2006090956. [DOI] [PubMed] [Google Scholar]
- 5.Mischak H, Apweiler R, Banks RE, Conaway M, Coon JJ, Dominizak A, Ehrich JH, Fliser D, Girolami M, Hermjakob H, Hochstrasser DF, Jankowski V, Julian BA, Kolch W, Massy Z, Neususs C, Novak J, Peter K, Rossing K, Schanstra JP, Semmes OJ, Theodorescu D, Thongboonkerd V, Weissinger EM, Van Eyk JE, Yamamoto T. Clinical proteomics: a need to define the field and to begin to set adequate standards. Proteomics Clin Appl. 2007;1:148–156. doi: 10.1002/prca.200600771. [DOI] [PubMed] [Google Scholar]
- 6.Castronovo V, Kischel P, Guillonneau F, de LL, Defechereux T, De PE, Neri D, Waltregny D. Identification of specific reachable molecular targets in human breast cancer using a versatile ex vivo proteomic method. Proteomics. 2007;7:1188–1196. doi: 10.1002/pmic.200600888. [DOI] [PubMed] [Google Scholar]
- 7.Roessler M, Rollinger W, Mantovani-Endl L, Hagmann ML, Palme S, Berndt P, Engel AM, Pfeffer M, Karl J, Bodenmueller H, Ruschoff J, Henkel T, Rohr G, Rossol S, Rosch W, Langen H, Zolg W, Tacke M. Identification of PSME3 as a novel serum tumor marker for colorectal cancer by combining two-dimensional polyacrylamide gel electrophoresis with a strictly mass spectrometry-based approach for data analysis. Mol Cell Proteomics. 2006;5:2092–2101. doi: 10.1074/mcp.M600118-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Lescuyer P, Hochstrasser D, Rabilloud T. How shall we use the proteomics toolbox for biomarker discovery? J Proteome Res. 2007;6:3371–3376. doi: 10.1021/pr0702060. [DOI] [PubMed] [Google Scholar]
- 9.Omenn GS, States DJ, Adamski M, Blackwell TW, Menon R, Hermjakob H, Apweiler R, Haab BB, Simpson RJ, Eddes JS, Kapp EA, Moritz RL, Chan DW, Rai AJ, Admon A, Aebersold R, Eng J, Hancock WS, Hefta SA, Meyer H, Paik YK, Yoo JS, Ping P, Pounds J, Adkins J, Qian X, Wang R, Wasinger V, Wu CY, Zhao X, Zeng R, Archakov A, Tsugita A, Beer I, Pandey A, Pisano M, Andrews P, Tammen H, Speicher DW, Hanash SM. Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics. 2005;5:3226–3245. doi: 10.1002/pmic.200500358. [DOI] [PubMed] [Google Scholar]
- 10.Rai AJ, Gelfand CA, Haywood BC, Warunek DJ, Yi J, Schuchard MD, Mehigh RJ, Cockrill SL, Scott GB, Tammen H, Schulz-Knappe P, Speicher DW, Vitzthum F, Haab BB, Siest G, Chan DW. HUPO Plasma Proteome Project specimen collection and handling: towards the standardization of parameters for plasma proteome samples. Proteomics. 2005;5:3262–3277. doi: 10.1002/pmic.200401245. [DOI] [PubMed] [Google Scholar]
- 11.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 12.Kolch W, Neususs C, Pelzing M, Mischak H. Capillary electrophoresis-mass spectrometry as a powerful tool in clinical diagnosis and biomarker discovery. Mass Spectrom Rev. 2005;24:959–977. doi: 10.1002/mas.20051. [DOI] [PubMed] [Google Scholar]
- 13.Schaub S, Wilkins J, Weiler T, Sangster K, Rush D, Nickerson P. Urine protein profiling with surface-enhanced laser-desorption/ionization time-of-flight mass spectrometry. Kidney Int. 2004;65:323–332. doi: 10.1111/j.1523-1755.2004.00352.x. [DOI] [PubMed] [Google Scholar]
- 14.Hortin GL, Sviridov D. Diagnostic potential for urinary proteomics. Pharmacogenomics. 2007;8:237–255. doi: 10.2217/14622416.8.3.237. [DOI] [PubMed] [Google Scholar]
- 15.Thongboonkerd V. Recent progress in urinary proteomics. Proteomics Clin Appl. 2007;1:780–791. doi: 10.1002/prca.200700035. [DOI] [PubMed] [Google Scholar]
- 16.Theodorescu D, Schiffer E, Bauer HW, Douwes F, Eichhorn F, Polley R, Schmidt T, Schofer W, Zuerbig P, Good DM, Coon JJ, Mischak H. Discovery and validation of urinary biomarkers for prostate cancer. Proteomics Clin Appl. 2008;2:556–570. doi: 10.1002/prca.200780082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zurbig P, Decramer S, Dakna M, Jantos J, Good DM, Coon JJ, Bandin F, Mischak H, Bascands JL, Schanstra JP. A low molecular weight urinary proteome profile of human kidney aging. Proteomics Clin Appl. 2009 doi: 10.1002/pmic.200800560. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoang K, Tan JC, Derby G, Blouch KL, Masek M, Ma I, Lemley KV, Myers BD. Determinants of glomerular hypofiltration in aging humans. Kidney Int. 2003;64:1417–1424. doi: 10.1046/j.1523-1755.2003.00207.x. [DOI] [PubMed] [Google Scholar]
- 19.O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]
- 20.Rose K, Kocher HP, Blumberg BM, Kolakofsky D. An improved procedure, involving mass spectrometry, for N-terminal amino acid sequence determination of proteins which are N alpha-blocked. Biochem J. 1984;217:253–257. doi: 10.1042/bj2170253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suckau D, Resemann A. T3-sequencing: targeted characterization of the N- and C-termini of undigested proteins by mass spectrometry. Anal Chem. 2003;75:5817–5824. doi: 10.1021/ac034362b. [DOI] [PubMed] [Google Scholar]
- 22.Burnette WN. “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem. 1981;112:195–203. doi: 10.1016/0003-2697(81)90281-5. [DOI] [PubMed] [Google Scholar]
- 23.Zhu H, Snyder M. Protein chip technology. Curr Opin Chem Biol. 2003;7:55–63. doi: 10.1016/s1367-5931(02)00005-4. [DOI] [PubMed] [Google Scholar]
- 24.Spisak S, Tulassay Z, Molnar B, Guttman A. Protein microchips in biomedicine and biomarker discovery. Electrophoresis. 2007;28:4261–4273. doi: 10.1002/elps.200700539. [DOI] [PubMed] [Google Scholar]
- 25.Wu TL. Two-dimensional difference gel electrophoresis. Methods Mol Biol. 2006;328:71–95. doi: 10.1385/1-59745-026-X:71. [DOI] [PubMed] [Google Scholar]
- 26.Delahunty CM, Yates JR., III MudPIT: multidimensional protein identification technology. Biotechniques. 2007;43:563, 565, 567. [PubMed] [Google Scholar]
- 27.Gaspari M, Verhoeckx KC, Verheij ER, van der GJ. Integration of two-dimensional LC-MS with multivariate statistics for comparative analysis of proteomic samples. Anal Chem. 2006;78:2286–2296. doi: 10.1021/ac052000t. [DOI] [PubMed] [Google Scholar]
- 28.Cutillas PR, Norden AG, Cramer R, Burlingame AL, Unwin RJ. Detection and analysis of urinary peptides by on-line liquid chromatography and mass spectrometry: application to patients with renal Fanconi syndrome. Clin Sci (Lond) 2003;104:483–490. doi: 10.1042/CS20020342. [DOI] [PubMed] [Google Scholar]
- 29.Kreunin P, Zhao J, Rosser C, Urquidi V, Lubman DM, Goodison S. Bladder cancer associated glycoprotein signatures revealed by urinary proteomic profiling. J Proteome Res. 2007;6:2631–2639. doi: 10.1021/pr0700807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagele E, Vollmer M, Horth P, Vad C. 2D-LC/MS techniques for the identification of proteins in highly complex mixtures. Expert Rev Proteomics. 2004;1:37–46. doi: 10.1586/14789450.1.1.37. [DOI] [PubMed] [Google Scholar]
- 31.Qian WJ, Jacobs JM, Liu T, Camp DG, Smith RD. Advances and challenges in liquid chromatography-mass spectrometry-based proteomics profiling for clinical applications. Mol Cell Proteomics. 2006;5:1727–1744. doi: 10.1074/mcp.M600162-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiwa M, Nishimura Y, Wakatabe R, Fukawa A, Arikuni H, Ota H, Kato Y, Yamori T. Rapid discovery and identification of a tissue-specific tumor biomarker from 39 human cancer cell lines using the SELDI ProteinChip platform. Biochem Biophys Res Commun. 2003;309:18–25. doi: 10.1016/s0006-291x(03)01520-1. [DOI] [PubMed] [Google Scholar]
- 33.Woroniecki, RP, Orlova TN, Mendelev N, Shatat IF, Hailpern SM, Kaskel FJ, Goligorsky MS, O’Riordan E. Urinary proteome of steroid-sensitive and steroid-resistant idiopathic nephrotic syndrome of childhood. Am J Nephrol. 2006;26:258–267. doi: 10.1159/000093814. [DOI] [PubMed] [Google Scholar]
- 34.Jahnukainen T, Malehorn D, Sun M, Lyons-Weiler J, Bigbee W, Gupta G, Shapiro R, Randhawa PS, Pelikan R, Hauskrecht M, Vats A. Proteomic analysis of urine in kidney transplant patients with BK virus nephropathy. J Am Soc Nephrol. 2006;17:3248–3256. doi: 10.1681/ASN.2006050437. [DOI] [PubMed] [Google Scholar]
- 35.Neuhoff N, Kaiser T, Wittke S, Krebs R, Pitt A, Burchard A, Sundmacher A, Schlegelberger B, Kolch W, Mischak H. Mass spectrometry for the detection of differentially expressed proteins: a comparison of surface-enhanced laser desorption/ionization and capillary electrophoresis/mass spectrometry. Rapid Commun Mass Spectrom. 2004;18:149–156. doi: 10.1002/rcm.1294. [DOI] [PubMed] [Google Scholar]
- 36.Rogers MA, Clarke P, Noble J, Munro NP, Paul A, Selby PJ, Banks RE. Proteomic profiling of urinary proteins in renal cancer by surface enhanced laser desorption ionization and neural-network analysis: identification of key issues affecting potential clinical utility. Cancer Res. 2003;63:6971–6983. [PubMed] [Google Scholar]
- 37.Albrethsen J, Bogebo R, Olsen J, Raskov H, Gammeltoft S. Preanalytical and analytical variation of surface-enhanced laser desorption-ionization time-of-flight mass spectrometry of human serum. Clin Chem Lab Med. 2006;44:1243–1252. doi: 10.1515/CCLM.2006.228. [DOI] [PubMed] [Google Scholar]
- 38.Poon TC. Opportunities and limitations of SELDI-TOF-MS in biomedical research: practical advices. Expert Rev Proteomics. 2007;4:51–65. doi: 10.1586/14789450.4.1.51. [DOI] [PubMed] [Google Scholar]
- 39.Gaspar A, Englmann M, Fekete A, Harir M, Schmitt-Kopplin P. Trends in CE-MS 2005–2006. Electrophoresis. 2008;29:66–79. doi: 10.1002/elps.200700721. [DOI] [PubMed] [Google Scholar]
- 40.Kaiser T, Wittke S, Just I, Krebs R, Bartel S, Fliser D, Mischak H, Weissinger EM. Capillary electrophoresis coupled to mass spectrometer for automated and robust polypeptide determination in body fluids for clinical use. Electrophoresis. 2004;25:2044–2055. doi: 10.1002/elps.200305788. [DOI] [PubMed] [Google Scholar]
- 41.Weissinger EM, Wittke S, Kaiser T, Haller H, Bartel S, Krebs R, Golovko I, Rupprecht HD, Haubitz M, Hecker H, Mischak H, Fliser D. Proteomic patterns established with capillary electrophoresis and mass spectrometry for diagnostic purposes. Kidney Int. 2004;65:2426–2434. doi: 10.1111/j.1523-1755.2004.00659.x. [DOI] [PubMed] [Google Scholar]
- 42.Kaiser T, Hermann A, Kielstein JT, Wittke S, Bartel S, Krebs R, Hausadel F, Hillmann M, Golovko I, Koester P, Haller H, Weissinger EM, Fliser D, Mischak H. Capillary electrophoresis coupled to mass spectrometry to establish polypeptide patterns in dialysis fluids. J Chromatogr A. 2003;1013:157–171. doi: 10.1016/s0021-9673(03)00712-x. [DOI] [PubMed] [Google Scholar]
- 43.Neususs C, Pelzing M, Macht M. A robust approach for the analysis of peptides in the low femtomole range by capillary electrophoresis-tandem mass spectrometry. Electrophoresis. 2002;23:3149–3159. doi: 10.1002/1522-2683(200209)23:18<3149::AID-ELPS3149>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 44.Smith RD, Olivares JA, Nguyen NT, Udseth HR. Capillary zone electrophoresis-mass spectrometry using an electrospray ionization interface. Anal Chem. 1988;60:436–441. [Google Scholar]
- 45.Wahl JH, Gale DC, Smith RD. Sheathless capillary electrophoresis-electrospray ionization mass. J Chromatogr A. 1994;659:217–222. doi: 10.1016/0021-9673(94)85026-7. [DOI] [PubMed] [Google Scholar]
- 46.Theodorescu D, Wittke S, Ross MM, Walden M, Conaway M, Just I, Mischak H, Frierson HF. Discovery and validation of new protein biomarkers for urothelial cancer: a prospective analysis. Lancet Oncol. 2006;7:230–240. doi: 10.1016/S1470-2045(06)70584-8. [DOI] [PubMed] [Google Scholar]
- 47.Wittke S, Fliser D, Haubitz M, Bartel S, Krebs R, Hausadel F, Hillmann M, Golovko I, Koester P, Haller H, Kaiser T, Mischak H, Weissinger EM. Determination of peptides and proteins in human urine with capillary electrophoresis— mass spectrometry, a suitable tool for the establishment of new diagnostic markers. J Chromatogr A. 2003;1013:173–181. doi: 10.1016/s0021-9673(03)00713-1. [DOI] [PubMed] [Google Scholar]
- 48.Theodorescu D, Fliser D, Wittke S, Mischak H, Krebs R, Walden M, Ross M, Eltze E, Bettendorf O, Wulfing C, Semjonow A. Pilot study of capillary electrophoresis coupled to mass spectrometry as a tool to define potential prostate cancer biomarkers in urine. Electrophoresis. 2005;26:2797–2808. doi: 10.1002/elps.200400208. [DOI] [PubMed] [Google Scholar]
- 49.Shannon W, Culverhouse R, Duncan J. Analyzing microarray data using cluster analysis. Pharmacogenomics. 2003;4:41–52. doi: 10.1517/phgs.4.1.41.22581. [DOI] [PubMed] [Google Scholar]
- 50.Smola AJ, Scholkopf B. A tutorial on support vector regression. Statistics and Computing. 2004;14:199–222. [Google Scholar]
- 51.Abdi H. Bonferroni and Sidak corrections for multiple comparisons. In: Salkind NJ, editor. Encyclopedia of Measurement and Statistics. Sage; Thousand Oaks CA: 2007. pp. 103–107. [Google Scholar]
- 52.Westfall PH, Young SS. Resampling-Based Multiple Testing: Examples and Methods for P-Value Adjustment. Wiley; New York: 1993. [Google Scholar]
- 53.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc B (Methodological) 1995;57:125–133. [Google Scholar]
- 54.McLerran D, Grizzle WE, Feng Z, Thompson IM, Bigbee WL, Cazares LH, Chan DW, Dahlgren J, Diaz J, Kagan J, Lin DW, Malik G, Oelschlager D, Partin A, Randolph TW, Sokoll L, Srivastava S, Srivastava S, Thornquist M, Troyer D, Wright GL, Zhang Z, Zhu L, Semmes OJ. SELDI-TOF MS whole serum proteomic profiling with IMAC surface does not reliably detect prostate cancer. Clin Chem. 2008;54:53–60. doi: 10.1373/clinchem.2007.091496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zweig MH, Campbell G. Receiver-operating characteristic (ROC) plots: a fundamental evaluation tool in clinical medicine. Clin Chem. 1993:B 561–577. [PubMed] [Google Scholar]
- 56.Jaattela M. Escaping cell death: survival proteins in cancer. I. 1999:B 30–43. doi: 10.1006/excr.1999.4455. [DOI] [PubMed] [Google Scholar]
- 57.Helmbrecht K, Zeise E, Rensing L. Chaperones in cell cycle regulation and mitogenic signal transduction: a review. I. 2000:B 341–365. doi: 10.1046/j.1365-2184.2000.00189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jolly C, Morimoto RI. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J Natl Cancer Inst. 2000:B 1564–1572. doi: 10.1093/jnci/92.19.1564. [DOI] [PubMed] [Google Scholar]
- 59.Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988:B 631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 60.Good DM, Thongboonkerd V, Novak J, Bascands JL, Schanstra JP, Coon JJ, Dominiczak A, Mischak H. Body fluid proteomics for biomarker discovery: lessons from the past hold the key to success in the future. J Proteome Res. 2007;6:4549–4555. doi: 10.1021/pr070529w. [DOI] [PubMed] [Google Scholar]
- 61.Wa C, Cerny RL, Clarke WA, Hage DS. Characterization of glycation adducts on human serum albumin by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin Chim Acta. 2007;385:48–60. doi: 10.1016/j.cca.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haubitz M, Fliser D, Rupprecht H, Floege J, Haller H, Rossing K, Walden M, Wittke S, Mischak H. Defining renal diseases based on proteom analysis. Nephrol Dial Transplant. 2005;20:V20. [Google Scholar]
- 63.Villanueva J, Shaffer DR, Philip J, Chaparro CA, Erdjument-Bromage H, Olshen AB, Fleisher M, Lilja H, Brogi E, Boyd J, Sanchez-Carbayo M, Holland EC, Cordon-Cardo C, Scher HI, Tempst P. Differential exoprotease activities confer tumor-specific serum peptidome patterns. J Clin Invest. 2006;116:271–284. doi: 10.1172/JCI26022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rossing K, Mischak H, Rossing P, Schanstra JP, Wiseman A, Maahs DM. The urinary proteome in diabetes and diabetes-associated complications: new ways to assess disease progression and evaluate therapy. Proteomics Clin Appl. 2008;2:997–1007. doi: 10.1002/prca.200780166. [DOI] [PubMed] [Google Scholar]
- 65.Zurbig P, Renfrow MB, Schiffer E, Novak J, Walden M, Wittke S, Just I, Pelzing M, Neususs C, Theodorescu D, Root C, Ross M, Mischak H. Biomarker discovery by CE-MS enables sequence analysis via MS/MS with platform-independent separation. Electrophoresis. 2006;27:2111–2125. doi: 10.1002/elps.200500827. [DOI] [PubMed] [Google Scholar]
- 66.Righetti PG, Campostrini N, Pascali J, Hamdan M, Astner H. Quantitative proteomics: a review of different methodologies. Eur J Mass Spectrom (Chichester, Eng) 2004;10:335–348. doi: 10.1255/ejms.600. [DOI] [PubMed] [Google Scholar]
- 67.Zybailov B, Coleman MK, Florens L, Washburn MP. Correlation of relative abundance ratios derived from peptide ion chromatograms and spectrum counting for quantitative proteomic analysis using stable isotope labeling. Anal Chem. 2005;77:6218–6224. doi: 10.1021/ac050846r. [DOI] [PubMed] [Google Scholar]
- 68.Barnidge DR, Dratz EA, Martin T, Bonilla LE, Moran LB, Lindall A. Absolute quantification of the G protein-coupled receptor rhodopsin by LC/MS/MS using proteolysis product peptides and synthetic peptide standards. Anal Chem. 2003;75:445–451. doi: 10.1021/ac026154+. [DOI] [PubMed] [Google Scholar]
- 69.DeKeyser SS, Li L. Matrix-assisted laser desorption/ionization Fourier transform mass spectrometry quantitation via in cell combination. Analyst. 2006;131:281–290. doi: 10.1039/b510831d. [DOI] [PubMed] [Google Scholar]
- 70.Jantos-Siwy J, Schiffer E, Brand K, Schumann G, Rossing K, Delles C, Mischak H, Metzger J. Quantitative urinary proteome analysis for biomarker evaluation in chronic kidney disease. J Proteome Res. 2009;8:268–281. doi: 10.1021/pr800401m. [DOI] [PubMed] [Google Scholar]
- 71.Holland M, Yagi H, Takahashi N, Kato K, Savage CO, Goodall DM, Jefferis R. Differential glycosylation of polyclonal IgG, IgG-Fc and IgG-Fab isolated from the sera of patients with ANCA-associated systemic vasculitis. Biochim Biophys Acta. 2006;1760:669–677. doi: 10.1016/j.bbagen.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 72.Soucek K, Kamaid A, Phung AD, Kubala L, Bulinski JC, Harper RW, Eiserich JP. Normal and prostate cancer cells display distinct molecular profiles of alpha-tubulin post-translational modifications. Prostate. 2006;66:954–965. doi: 10.1002/pros.20416. [DOI] [PubMed] [Google Scholar]
- 73.Uzawa K, Marshall MK, Katz EP, Tanzawa H, Yeowell HN, Yamauchi M. Altered posttranslational modifications of collagen in keloid. Biochem Biophys Res Commun. 1998;249:652–655. doi: 10.1006/bbrc.1998.8955. [DOI] [PubMed] [Google Scholar]
- 74.Coon JJ, Zurbig P, Dakna M, Dominiczak AF, Decramer S, Fliser D, Frommberger M, Golovko I, Good DM, Herget-Rosenthal S, Jankowski J, Julian BA, Kellmann M, Kolch W, Massy Z, Novak J, Rossing K, Schanstra JP, Schiffer E, Theodorescu D, Vanholder R, Weissinger EM, Mischak H, Schmitt-Kopplin P. CE-MS analysis of the human urinary proteome for biomarker discovery and disease diagnostics. Proteomics Clin Appl. 2008;2:964–973. doi: 10.1002/prca.200800024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Castagna A, Cecconi D, Sennels L, Rappsilber J, Guerrier L, Fortis F, Boschetti E, Lomas L, Righetti PG. Exploring the hidden human urinary proteome via ligand library beads. J Proteome Res. 2005;4:1917–1930. doi: 10.1021/pr050153r. [DOI] [PubMed] [Google Scholar]
- 76.Adachi J, Kumar C, Zhang Y, Olsen JV, Mann M. The human urinary proteome contains more than 1500 proteins including a large proportion of membranes proteins. Genome Biol. 2006;7:R80. doi: 10.1186/gb-2006-7-9-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Haubitz M, Wittke S, Weissinger EM, Walden M, Rupprecht HD, Floege J, Haller H, Mischak H. Urine protein patterns can serve as diagnostic tools in patients with IgA nephropathy. Kidney Int. 2005;67:2313–2320. doi: 10.1111/j.1523-1755.2005.00335.x. [DOI] [PubMed] [Google Scholar]
- 78.Schiffer E, Mischak H, Novak J. High resolution proteome/peptidome analysis of body fluids by capillary electrophoresis coupled with MS. Proteomics. 2006;6:5615–5627. doi: 10.1002/pmic.200600230. [DOI] [PubMed] [Google Scholar]
- 79.Julian BA, Wittke S, Novak J, Good DM, Coon JJ, Kellmann M, Zurbig P, Schiffer E, Haubitz M, Moldoveanu Z, Calcatera SM, Wyatt RJ, Sykora J, Sladkova E, Hes O, Mischak H, McGuire BM. Electrophoretic methods for analysis of urinary polypeptides in IgA-associated renal diseases. Electrophoresis. 2007;28:4469–4483. doi: 10.1002/elps.200700237. [DOI] [PubMed] [Google Scholar]
- 80.Julian BA, Wittke S, Haubitz M, Zurbig P, Schiffer E, McGuire BM, Wyatt RJ, Novak J. Urinary biomarkers of IgA nephropathy and other IgA-associated renal diseases. World J Urol. 2007;25:467–476. doi: 10.1007/s00345-007-0192-5. [DOI] [PubMed] [Google Scholar]
- 81.Rossing K, Mischak H, Parving HH, Christensen PK, Walden M, Hillmann M, Kaiser T. Impact of diabetic nephropathy and angiotensin II receptor blockade on urinary polypeptide patterns. Kidney Int. 2005;68:193–205. doi: 10.1111/j.1523-1755.2005.00394.x. [DOI] [PubMed] [Google Scholar]
- 82.Rossing K, Mischak H, Dakna M, Zurbig P, Novak J, Julian BA, Good DM, Coon JJ, Tarnow L, Rossing P. Urinary proteomics in diabetes and CKD. J Am Soc Nephrol. 2008;19:1283–1290. doi: 10.1681/ASN.2007091025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chalmers MJ, Mackay CL, Hendrickson CL, Wittke S, Walden M, Mischak H, Fliser D, Just I, Marshall AG. Combined top-down and bottom-up mass spectrometric approach to characterization of biomarkers for renal disease. Anal Chem. 2005;77:7163–7171. doi: 10.1021/ac050983o. [DOI] [PubMed] [Google Scholar]
- 84.Comper WD, Hilliard LM, Nikolic-Paterson DJ, Russo LM. Disease-dependent mechanisms of albuminuria. an invited review. Am J Physiol Renal Physiol. 2008;295:F1589–F1600. doi: 10.1152/ajprenal.00142.2008. [DOI] [PubMed] [Google Scholar]
- 85.Birn H, Christensen EI. Renal albumin absorption in physiology and pathology. Kidney Int. 2006;69:440–449. doi: 10.1038/sj.ki.5000141. [DOI] [PubMed] [Google Scholar]
- 86.Sever S, Altintas MM, Nankoe SR, Moller CC, Ko D, Wei C, Henderson J, del Re EC, Hsing L, Erickson A, Cohen CD, Kretzler M, Kerjaschki D, Rudensky A, Nikolic B, Reiser J. Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J Clin Invest. 2007;117:2095–2104. doi: 10.1172/JCI32022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kafienah W, Buttle DJ, Burnett D, Hollander AP. Cleavage of native type I collagen by human neutrophil elastase. Biochem J. 1998;330(Pt 2):897–902. doi: 10.1042/bj3300897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Thongboonkerd V, Barati MT, McLeish KR, Benarafa C, Remold-O’Donnell E, Zheng S, Rovin BH, Pierce WM, Epstein PN, Klein JB. Alterations in the renal elastin-elastase system in type 1 diabetic nephropathy identified by proteomic analysis. J Am Soc Nephrol. 2004;15:650–662. doi: 10.1097/01.asn.0000115334.65095.9b. [DOI] [PubMed] [Google Scholar]
- 89.Nerlich A, Schleicher E. Immunohistochemical localization of extracellular matrix components in human diabetic glomerular lesions. Am J Pathol. 1991;139:889–899. [PMC free article] [PubMed] [Google Scholar]
- 90.Kwak NJ, Heo Y, Jin DC, Cha JH, Lee KH, Yang CW, Kang CS, Choi YJ, Wang EH. Proteomic analysis of alpha-1-antitrypsin in immunoglobulin A nephropathy. Proteomics Clin Appl. 2007;1:420–428. doi: 10.1002/prca.200600288. [DOI] [PubMed] [Google Scholar]
- 91.Barratt J, Feehally J, Smith AC. Pathogenesis of IgA nephropathy. Semin Nephrol. 2004;24:197–217. doi: 10.1016/j.semnephrol.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 92.Couchman JR, Beavan LA, McCarthy KJ. Glomerular matrix: synthesis, turnover and role in mesangial expansion. Kidney Int. 1994;45:328–335. doi: 10.1038/ki.1994.42. [DOI] [PubMed] [Google Scholar]
- 93.Gauer S, Yao J, Schoecklmann HO, Sterzel RB. Adhesion molecules in the glomerular mesangium. Kidney Int. 1997;51:1447–1453. doi: 10.1038/ki.1997.198. [DOI] [PubMed] [Google Scholar]
- 94.Rupprecht HD, Schocklmann HO, Sterzel RB. Cell-matrix interactions in the glomerular mesangium. Kidney Int. 1996;49:1575–1582. doi: 10.1038/ki.1996.228. [DOI] [PubMed] [Google Scholar]
- 95.Mischak H, Kaiser T, Walden M, Hillmann M, Wittke S, Herrmann A, Knueppel S, Haller H, Fliser D. Proteomic analysis for the assessment of diabetic renal damage in humans. Clin Sci (Lond) 2004;107:485–495. doi: 10.1042/CS20040103. [DOI] [PubMed] [Google Scholar]
- 96.Meier M, Kaiser T, Herrmann A, Knueppel S, Hillmann M, Koester P, Danne T, Haller H, Fliser D, Mischak H. Identification of urinary protein pattern in type 1 diabetic adolescents with early diabetic nephropathy by a novel combined proteome analysis. J Diabetes Complications. 2005;19:223–232. doi: 10.1016/j.jdiacomp.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 97.Dabelea D, Kinney G, Snell-Bergeon JK, Hokanson JE, Eckel RH, Ehrlich J, Garg S, Hamman RF, Rewers M. Effect of type 1 diabetes on the gender difference in coronary artery calcification: a role for insulin resistance? The Coronary Artery Calcification in Type 1 Diabetes (CACTI) Study. Diabetes. 2003;52:2833–2839. doi: 10.2337/diabetes.52.11.2833. [DOI] [PubMed] [Google Scholar]
- 98.Snell-Bergeon JK, Maahs DM, Ogden LG, Kinney G, Hokanson JE, Schiffer E, Rewers M, Mischak H. Evaluation of urinary biomarkers for coronary artery disease, diabetes, and diabetic kidney disease. Diabetes Technol Ther. 2009;11:1–9. doi: 10.1089/dia.2008.0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wittke S, Haubitz M, Walden M, Rohde F, Schwarz A, Mengel M, Mischak H, Haller H, Gwinner W. Detection of acute tubulointerstitial rejection by proteomic analysis of urinary samples in renal transplant recipients. Am J Transplant. 2005;5:2479–2488. doi: 10.1111/j.1600-6143.2005.01053.x. [DOI] [PubMed] [Google Scholar]
- 100.Jennette JC, Falk RJ. Pathogenic potential of anti-neutrophil cytoplasmic autoantibodies. Adv Exp Med Biol. 1993;336:7–15. doi: 10.1007/978-1-4757-9182-2_2. [DOI] [PubMed] [Google Scholar]
- 101.Decramer S, Wittke S, Mischak H, Zurbig P, Walden M, Bouissou F, Bascands JL, Schanstra JP. Predicting the clinical outcome of congenital unilateral ureteropelvic junction obstruction in newborn by urinary proteome analysis. Nat Med. 2006;12:398–400. doi: 10.1038/nm1384. [DOI] [PubMed] [Google Scholar]
- 102.Decramer S, Zurbig P, Wittke S, Mischak H, Bascands JL, Schanstra JP. Identification of urinary biomarkers by proteomics in newborns: use in obstructive nephropathy. Contrib Nephrol. 2008;160:127–141. doi: 10.1159/000125956. [DOI] [PubMed] [Google Scholar]
- 103.Fricker LD, McKinzie AA, Sun J, Curran E, Qian Y, Yan L, Patterson SD, Courchesne PL, Richards B, Levin N, Mzhavia N, Devi LA, Douglass J. Identification and characterization of proSAAS, a granin-like neuroendocrine peptide precursor that inhibits prohormone processing. J Neurosci. 2000;20:639–648. doi: 10.1523/JNEUROSCI.20-02-00639.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chevalier RL. Obstructive nephropathy: towards biomarker discovery and gene therapy. Nat Clin Pract Nephrol. 2006;2:157–168. doi: 10.1038/ncpneph0098. [DOI] [PubMed] [Google Scholar]
- 105.Dinney CP, McConkey DJ, Millikan RE, Wu X, Bar-Eli M, Adam L, Kamat AM, Siefker-Radtke AO, Tuziak T, Sabichi AL, Grossman HB, Benedict WF, Czerniak B. Focus on bladder cancer. Cancer Cell. 2004;6:111–116. doi: 10.1016/j.ccr.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 106.Lilja H. Structure and function of prostatic- and seminal vesicle-secreted proteins involved in the gelation and lique-faction of human semen. Scand J Clin Lab Invest Suppl. 1988;191:13–20. [PubMed] [Google Scholar]
- 107.Lilja H. Cell biology of semenogelin. Andrologia. 1990;22(Suppl 1):S132–S141. doi: 10.1111/j.1439-0272.1990.tb02079.x. [DOI] [PubMed] [Google Scholar]
- 108.Lodding P, Aus G, Bergdahl S, Frosing R, Lilja H, Pihl CG, Hugosson J. Characteristics of screening detected prostate cancer in men 50 to 66 years old with 3 to 4 ng./ml. prostate specific antigen. J Urol. 1998;159:899–903. [PubMed] [Google Scholar]
- 109.Duran MJ, Parish LD, Pressley TA. Regulation of Na, K-ATPase in prostate cancer: a role for PKCbeta. FASEB J. 2007;21:976.2. [Google Scholar]
- 110.Blok LJ, Chang GT, Steenbeek-Slotboom M, van Weerden WM, Swarts HG, De Pont JJ, van Steenbrugge GJ, Brinkmann AO. Regulation of expression of Na+,K+-ATPase in androgen-dependent and androgen-independent prostate cancer. Br J Cancer. 1999;81:28–36. doi: 10.1038/sj.bjc.6690647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim Biophys Acta. 2000;1477:267–283. doi: 10.1016/s0167-4838(99)00279-4. [DOI] [PubMed] [Google Scholar]
- 112.DeMuth JP, Weaver DA, Crawford EL, Jackson CM, Willey JC. Loss of spr1 expression measurable by quantitative RT-PCR in human bronchogenic carcinoma cell lines. Am J Respir Cell Mol Biol. 1998;19:25–29. doi: 10.1165/ajrcmb.19.1.3078. [DOI] [PubMed] [Google Scholar]
- 113.Lau D, Guo L, Chan A, Wu R. SPR1. An early molecular marker for bronchial carcinogenesis. Methods Mol Med. 2003;75:397–403. doi: 10.1385/1-59259-324-0:397. [DOI] [PubMed] [Google Scholar]
- 114.Kaiser T, Kamal H, Rank A, Kolb HJ, Holler E, Ganser A, Hertenstein B, Mischak H, Weissinger EM. Proteomics applied to the clinical follow-up of patients after allogeneic hematopoietic stem cell transplantation. Blood. 2004;104:340–349. doi: 10.1182/blood-2004-02-0518. [DOI] [PubMed] [Google Scholar]
- 115.Weissinger EM, Schiffer E, Hertenstein B, Ferrara JL, Holler E, Stadler M, Kolb HJ, Zander A, Zurbig P, Kellmann M, Ganser A. Proteomic patterns predict acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Blood. 2007;109:5511–5519. doi: 10.1182/blood-2007-01-069757. [DOI] [PubMed] [Google Scholar]
- 116.Zimmerli LU, Schiffer E, Zurbig P, Good DM, Kellmann M, Mouls L, Pitt AR, Coon JJ, Schmieder RE, Peter KH, Mischak H, Kolch W, Delles C, Dominiczak AF. Urinary proteomic biomarkers in coronary artery disease. Mol Cell Proteomics. 2008;7:290–298. doi: 10.1074/mcp.M700394-MCP200. [DOI] [PubMed] [Google Scholar]
- 117.von zur Muhlen C, Schiffer E, Kellmann M, Zurbig P, Brasse M, Meert N, Vanholder R, Dominiczak AF, Chen YC, Mischak H, Bode C, Peter K. Evaluation of urine proteome pattern analysis for its potential to reflect coronary artery atherosclerosis in symptomatic patients. J Proteome Res. 2009;8:335–345. doi: 10.1021/pr800615t. [DOI] [PubMed] [Google Scholar]
- 118.Pihusch M, Wegner H, Goehring P, Salat C, Pihusch V, Andreesen R, Kolb HJ, Holler E, Pihusch R. Protein C and procollagen III peptide levels in patients with hepatic dysfunction after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2005;36:631–637. doi: 10.1038/sj.bmt.1705114. [DOI] [PubMed] [Google Scholar]
- 119.Bentsen KD. Type III procollagen peptide: studies on the circulating peptide as a marker of fibrinogenesis with special reference to the liver. Dan Med Bull. 1993;40:235–246. [PubMed] [Google Scholar]
- 120.Kalela A, Koivu TA, Sisto T, Kanervisto J, Hoyhtya M, Sillanaukee P, Lehtimaki T, Nikkari ST. Serum matrix metalloproteinase-9 concentration in angiographically assessed coronary artery disease. Scand J Clin Lab Invest. 2002;62:337–342. doi: 10.1080/00365510260296483. [DOI] [PubMed] [Google Scholar]
- 121.Noji Y, Kajinami K, Kawashiri MA, Todo Y, Horita T, Nohara A, Higashikata T, Inazu A, Koizumi J, Takegoshi T, Mabuchi H. Circulating matrix metalloproteinases and their inhibitors in premature coronary atherosclerosis. Clin Chem Lab Med. 2001;39:380–384. doi: 10.1515/CCLM.2001.060. [DOI] [PubMed] [Google Scholar]
- 122.Tayebjee MH, Tan KT, MacFadyen RJ, Lip GY. Abnormal circulating levels of metalloprotease 9 and its tissue inhibitor 1 in angiographically proven peripheral arterial disease: relationship to disease severity. J Intern Med. 2005;257:110–116. doi: 10.1111/j.1365-2796.2004.01431.x. [DOI] [PubMed] [Google Scholar]
- 123.Blankenberg S, Rupprecht HJ, Poirier O, Bickel C, Smieja M, Hafner G, Meyer J, Cambien F, Tiret L. Plasma concentrations and genetic variation of matrix metalloproteinase 9 and prognosis of patients with cardiovascular disease. Circulation. 2003;107:1579–1585. doi: 10.1161/01.CIR.0000058700.41738.12. [DOI] [PubMed] [Google Scholar]
- 124.de NR, Verkleij CJ, von der Thusen JH, Jukema JW, van der Wall EE, van Berkel TJ, Baker AH, Biessen EA. Lesional over-expression of matrix metalloproteinase-9 promotes intra-plaque hemorrhage in advanced lesions but not at earlier stages of atherogenesis. Arterioscler Thromb Vasc Biol. 2006;26:340–346. doi: 10.1161/01.ATV.0000197795.56960.64. [DOI] [PubMed] [Google Scholar]
- 125.Verhave JC, Hillege HL, Burgerhof JG, Gansevoort RT, de Zeeuw D, de Jong PE, PREVEND Study Group The association between atherosclerotic risk factors and renal function in the general population. Kidney Int. 2005;67:1967–1973. doi: 10.1111/j.1523-1755.2005.00296.x. [DOI] [PubMed] [Google Scholar]
- 126.Comper WD, Osicka TM, Clark M, MacIsaac RJ, Jerums G. Earlier detection of microalbuminuria in diabetic patients using a new urinary albumin assay. Kidney Int. 2004;65:1850–1855. doi: 10.1111/j.1523-1755.2004.00585.x. [DOI] [PubMed] [Google Scholar]
- 127.Nemirovskiy OV, Dufield DR, Sunyer T, Aggarwal P, Welsch DJ, Mathews WR. Discovery and development of a type II collagen neoepitope (TIINE) biomarker for matrix metalloproteinase activity: from in vitro to in vivo. Anal Biochem. 2006;361:93–101. doi: 10.1016/j.ab.2006.10.034. [DOI] [PubMed] [Google Scholar]
- 128.Candiano G, Musante L, Bruschi M, Petretto A, Santucci L, Del BP, Pavone B, Perfumo F, Urbani A, Scolari F, Ghiggeri GM. Repetitive fragmentation products of albumin and alpha1-antitrypsin in glomerular diseases associated with nephrotic syndrome. J Am Soc Nephrol. 2006;17:3139–3148. doi: 10.1681/ASN.2006050486. [DOI] [PubMed] [Google Scholar]