

Abstract
Purpose
Uveal melanoma (UM) is uniformly refractory to all available systemic chemotherapies, thus creating an urgent need for novel therapeutics. In this study, we investigated the sensitivity of UM cells to ICG-001, a small molecule reported to suppress the Wnt/β-catenin–mediated transcriptional program.
Methods
We used a panel of UM cell lines to examine the effects of ICG-001 on cellular proliferation, migration, and gene expression. In vivo efficacy of ICG-001 was evaluated in a UM xenograft model.
Results
ICG-001 exerted strong antiproliferative activity against UM cells, leading to cell cycle arrest, apoptosis, and inhibition of migration. Global gene expression profiling revealed strong suppression of genes associated with cell cycle proliferation, DNA replication, and G1/S transition. Gene set enrichment analysis revealed that ICG-001 suppressed Wnt, mTOR, and MAPK signaling. Strikingly, ICG-001 suppressed the expression of genes associated with UM aggressiveness, including CDH1, CITED1, EMP1, EMP3, SDCBP, and SPARC. Notably, the transcriptomic footprint of ICG-001, when applied to a UM patient dataset, was associated with better clinical outcome. Lastly, ICG-001 exerted anticancer activity against a UM tumor xenograft in mice.
Conclusions
Using in vitro and in vivo experiments, we demonstrate that ICG-001 has strong anticancer activity against UM cells and suppresses transcriptional programs critical for the cancer cell. Our results suggest that ICG-001 holds promise and should be examined further as a novel therapeutic agent for UM.
Keywords: uveal melanoma (UM), ICG-001, Wnt signaling, metastasis
Uveal melanoma (UM), the most common intraocular malignancy in adults,1 arises from melanin-producing melanocytes of the iris, ciliary body, or choroid. Primary UM can be treated effectively via irradiation (radiotherapy with charged particles or radioactive iodine). Unfortunately, approximately 50% of UM patients suffer metastatic disease; among those, more than 90% have involvement of the liver, with the lung (24%), bones (16%), and skin (11%) also being common metastatic sites.2–6 Metastatic UM is uniformly refractory to all available systemic chemotherapies,7,8 creating an unmet need for novel, effective, targeted therapies.
In recent years, intense efforts have been made to increase our understanding of the molecular pathophysiology of UM. Sequencing studies have shown that approximately 85% of UMs harbor an activating somatic mutation in the G-protein α subunits, Gαq or Gα11,9,10 leading to constitutive activation of the protein kinase C (PKC) and the MEK signaling pathways. Cytogenetic profiling identified frequent large-scale aberrations in chromosomes 1, 3, 6, and 8,11–15 as well as small scale changes, such as deletions (e.g., loss of the tumor suppressor gene PTEN located on chromosome 10q), and amplifications (e.g., gain of proto-oncogenes, such as MYC (43%) and BCL2 (95%).16 Moreover, gene expression profiling studies have identified at least two distinct classes of gene sets: Class 1, associated with low risk of metastasis and Class 2, associated with high risk of metastasis.17,18 Importantly, Class 2 UMs express higher levels of mRNAs related to epithelial lineage (EMP1 and EMP3) and to epithelial cell adhesion and interactions with basement membrane (such as CDH1 and SPARC) that are thought to promote UM cell plasticity, allowing them to become resistant to traditional chemotherapeutic agents.17,19 Among Class 1 UMs, further risk stratification can be performed based on the levels of preferentially expressed antigen in melanoma (PRAME) mRNA, which correlate with increased metastatic risk.20 PRAME also has been proposed as an immunotherapy target.21
Despite these advances in molecular understanding of UM, systemic therapeutic options for metastatic disease remain nonexistent. Thus far, the results from clinical trials evaluating the efficacy of PKC, MEK, c-Kit, MET/VEGF, and CTLA-4 inhibitors in UM patients have failed to improve overall survival.22 Hence, there is a dire need to identify potential compounds with therapeutic promise.
To this goal, we investigated the efficacy of ICG-00123 against UM cells in vitro and in vivo. ICG-001 exerts strong anticancer activity against colorectal cancer,23,24 pancreatic cancer,24,25 and multiple myeloma.24 It was identified based on its capacity to disrupt the interaction of β-catenin with the transcriptional coactivator CREB-binding protein (CBP) and, thus, suppress the Wnt/β-catenin–mediated transcriptional program.23,24,26,27 Our study revealed that ICG-001 induces cell death in UM cell lines. Gene expression profiling showed strong suppression of genes associated with cell cycle, DNA replication, and G1/S transition, stemness, Wnt, and mTORC1 pathways. Furthermore, we observed a strong suppression of genes that are associated with focal adhesion and aggressiveness of Class 2 UM. Application of our transcriptomic signature of ICG-001 to publicly available UM patient datasets showed that ICG-001 induced global transcriptional changes that are associated with decreased metastatic potential and improved clinical outcomes. Lastly, we found that ICG-001 is highly effective in suppressing the migration of UM cells in vitro and is a potent inhibitor of the growth of human UM cells xenografted in mice. Collectively, these results highlight that ICG-001 can be used potentially as a novel therapeutic option for the treatment of UM.
Methods
Cell Lines and Tissue Culture
The 92.1 UM cell line (carrying a GNAQ Q209L mutation) was established in the lab and was a generous gift of Martine J. Jager, Leiden University Medical Center, Leiden, The Netherlands28–30 from a primary UM. The Mel202 cell line (carrying GNAQ Q209L and GNAQ R210K mutations) was established from a previously irradiated, locally recurrent primary UM by Bruce R. Ksander (Schepens Eye Research Institute, Boston, MA, USA)31 and was generously provided by Demetrios Vavvas (Massachusetts Eye and Ear Infirmary and Schepens Eye Research Institute, Harvard Medical School, Boston, MA, USA). The Mel270 cell line (carrying a GNAQ Q209P mutation) was established from a previously irradiated, locally recurrent primary UM by Bruce R. Ksander. A year after the enucleation, the patient suffered liver metastases, from which the OMM1.3 (also known as OMM2.3) and OMM2.5 (also known as OMM1.5) lines were established.32 These lines were generously provided by Martine J. Jager and Demetrios Vavvas. Genotypes of all cell lines used in this study were authenticated by Sanger sequencing (representative sequence electropherograms have been reported previously33) and matched what has been reported previously.10,29,34 All UM lines were confirmed to be BRAF-wt. All cell lines were grown in Dulbecco's modified Eagle's medium (DMEM)/F12 (Invitrogen, Carlsbad, CA, USA) with 100 units/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum (FBS; in a 37°C incubator in a humid environment with 5% CO2) and were passaged for <6 months.
MTT Assay
Cell viability after ICG-001 was monitored using MTT assay. ICG-001 was purchased from Selleckchem (Houston, TX, USA). Briefly, cells were plated in 24-well plates and allowed to adhere to the wells for 24 hours, then treated with indicated concentrations of ICG-001 or vehicle control (dimethyl sulfoxide [DMSO], <0.1% vol/vol). After 96 hours, (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; MTT) reagent (Sigma-Aldrich Corp., St. Louis, MO, USA) was added at a final concentration of 50 μg/mL to each well and cells were incubated for 2 more hours. Media were aspirated and equal volume of DMSO:isopropanol (1:1 vol/vol) was added to each well to dissolve the precipitated crystals. The optical density was calculated by subtracting absorbance at 630 nM from absorbance at 570 nM and normalized to the respective controls. IC50 was calculated using Prism 7 v7.0b (GraphPad Software, Inc., La Jolla, CA, USA).
Transcriptomic Profiling
Global gene expression profiling was performed to examine the effects of ICG-001 in UM. Cells were treated with 3 μM ICG-001 or DMSO for 24 or 48 hours in six-well tissue culture plates. A detailed methodology is included in the Supplementary Methods.
In Vivo UM Model
All animal experiments were performed in compliance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and the study was approved by the Veterans Affairs Boston Healthcare System Institutional Animal Care and Use Committee (IACUC). Mel270 (GNAQMT) cells were injected subcutaneously (8 × 106 cells/mouse) into the flank of athymic nude mice (n = 10 mice per cohort). Mice were monitored daily and tumor measurements were acquired with digital calipers. Tumor volume was calculated with the formula (width2) × length/2. Treatment was initiated 3 weeks after subcutaneous cell injection. Mice were treated with intratumoral injection of vehicle or 50 mg/kg ICG-001 (dissolved in 20% polyethylene glycol [PEG], 5% Solutol, 3.75% dextrose, 1% dimethyl sulfoxide [DMSO], sterile PBS) 5 days/week for the duration of the experiment (the first 3 days of treatment were with an induction dose of 100 mg/kg ICG-001). Mice exhibiting any signs of distress or pain, or bearing tumors reaching diameter of 1 cm were euthanized humanely.
Additional methods for flow cytometry, apoptosis measurement, immunoblotting, and wound-healing assay are described in the Supplementary Methods.
Results
ICG-001 Inhibits Proliferation and Induces Apoptosis in UM Cells
ICG-001 treatment resulted in a potent inhibition of cellular proliferation of a panel of UM cell lines in a dose-dependent fashion (Fig. 1A; IC50 range, 0.6–2.7 μM, Supplementary Table S1). Further examination of the effects of ICG-001 on Mel202 and Mel270 cell cycle revealed a decrease in the S and G2/M phase (Fig. 1B, Supplementary Figs. S1A, S1B). An increase in the sub-G1 content also was observed, suggesting the presence of fragmented DNA from apoptotic cells. The presence of apoptotic cells was confirmed by double Annexin V/PI labeling (Supplementary Fig. S2A) and detection of cleaved caspase-3 and PARP (Supplementary Fig. S3A).
Figure 1.

ICG-001 suppresses proliferation and induces growth arrest in UM cells. (A) ICG-001 suppresses the growth of a panel of UM cells. MTT assay was performed after 96 hours of ICG-001 treatment. Results shown are average ± SEM. (B) ICG-001 induces cell cycle arrest. Mel270 and Mel202 UM cells were treated with ICG-001 for 24 and 72 hours and stained with propidium iodide. Cell cycle distribution is shown as bar graphs. Experiments were repeated three times with similar results; shown here is one representative experiment performed with at least three technical replicates. Results shown are average ± SD. Representative histograms are shown in Supplementary Figure S1.
ICG-001 Inhibits the Expression of Genes Involved in DNA Replication and Cell Cycle
To further characterize the underlying mechanism behind the inhibition of UM cell proliferation by ICG-001, we next performed global gene expression profiling after treating the Mel202 cells with 3 μM ICG-001. We found 2493 genes to be expressed differentially after 48 hours of treatment with ICG-001 (Supplementary Fig. S2B). Gene set enrichment analysis (GSEA) showed that ICG-001 suppressed a large set of genes involved in key cell cycle processes: DNA replication/synthesis, DNA repair, regulation of the mitotic cell cycle, and cell cycle checkpoints. In addition, we observed a negative enrichment for the gene targets of E2F, a transcription factor family that drives cellular proliferation (Fig. 2).
Figure 2.

ICG-001 suppresses cell cycle gene expression in UM cells. Gene expression profiling was performed with the Illumina HumanHT-12 v4 Expression BeadChip array using RNA harvested from Mel202 cells treated with 3 μM of ICG-001 for 48 hours. Gene set enrichment analyses against MSigDB pathways reveal that ICG-001 potent suppresses cell cycle proliferation, DNA replication, G1/S transition cell cycle, cell cycle checkpoints genes, and transcriptional targets of E2F.
ICG-001 Inhibits the Expression of Genes Involved in the mTOR Pathway, Wnt Pathway and Stemness
Our GSEA also demonstrated that ICG-001 suppressed the mTORC1 signaling cascade (Fig. 3A). Further evidence of mTOR pathway inhibition was provided by immunoblotting that demonstrated that ICG-001 decreases phospho-p70S6K levels (Supplementary Fig. S3).
Figure 3.

ICG-001 inhibits the expression of genes involved in the mTOR pathway, Wnt pathway, and stemness. (A, B) GSEA shows negative enrichment of transcriptional targets of mTORC1 and Wnt signaling pathways (MSigDB curated geneset). (C, D) GSEA of a WNT5A siRNA signature, derived from a prostate cancer cell line (LNCaP-abl), compared against the ICG-001 signature from Mel202 cells shows strong concordance. (E) Normalized enrichment scores from stemness gene sets (from MSigDB) compared against the ICG-001 transcriptional footprint. Strong inhibition of embryonic stem cell (ESC)-associated transcriptional programs is observed upon ICG-001 treatment of UM cells. Gene sets with a nominal P value <0.05 and FDR < 0.25 were defined as significantly enriched.
Previous studies have suggested that ICG-001 can suppress the Wnt/β-catenin signaling pathway.23,26,27,35,36 Thus, we next performed GSEA comparing the ICG-001 transcriptomic footprint against existing gene sets for the Wnt signaling pathway and stemness in MSigDB. These analyses showed that ICG-001 suppressed the Wnt signaling pathway (Fig. 3B). We also compared our ICG-001 signature against a signature that we had generated previously upon silencing WNT5A with siRNA. As shown in Figures 3C and 3D, genes suppressed after ICG-001 were enriched among genes that were suppressed after siWNT5A, while genes induced by ICG-001 were enriched among those upregulated upon silencing WNT5A. ICG-001 did not alter the levels of β-catenin protein in total cell lysates (Supplementary Fig. S3B).
The transcriptomic footprint of ICG-001 also showed a negative enrichment for several gene sets associated with stem cell-like properties (“stemness”). Specifically, we found that gene sets responsible for maintaining human embryonic stem-cell state (“WONG_EMBRYONIC_STEM_CELL_CORE” and “BHATTACHARYA_EMBRYONIC_ STEM_CELL”) were suppressed by ICG-001. Furthermore, consistent with data previously published in other models,35,37 ICG-001 suppressed expression of gene sets driven by key stemness-related transcription factors: SOX2, NANOG, OCT4, and MYC (Fig. 3E).
The ICG-001 Transcriptional Footprint Mimics the Signatures of the Transcriptional SMIs C646 and JQ1
We next compared the transcriptomic footprint of ICG-001 in UM cells with several previously published ICG-001 signatures derived in various other malignancies. The ICG-001 signature in UM showed strong concordance with previously published ICG-001 signatures from colon cancer (HCT116), pancreatic cancer (PANC1, ASPC1), and hematopoietic stem cells (HSCs; Fig. 4, Supplementary Fig. S4). Interestingly, the ICG-001 signature also mimicked the transcriptomic footprint of C646, a selective SMI of p300 histone acetyltransferase. Moreover, the ICG-001 signature from UM also had high concordance with a signature generated upon silencing EP300 (p300) via siRNA (Fig. 5). We also found that the transcriptomic signature of JQ1, an inhibitor of BET family of bromodomain proteins (BRD) that bind acetylated histones,38,39 had high concordance with our ICG-001 transcriptomic footprint. Collectively, these results suggested that ICG-001 can elicit global epigenetic changes in the UM cells that mimic the effects of p300 and BRD inhibition.
Figure 4.

Strong concordance of gene signature of ICG-001 in UM with signatures of ICG-001 and p300 inhibitor in other cancers. Gene set enrichment analysis shows high concordance between the ICG-001 signature from UM and the transcriptomic footprints of ICG-001 (A–D) as well as with the signature of C646 (E–H), a small molecule inhibitor of p300, derived from the HCT116 (colon cancer) and PANC1 (pancreatic cancer) cell lines (GSE64038).
Figure 5.
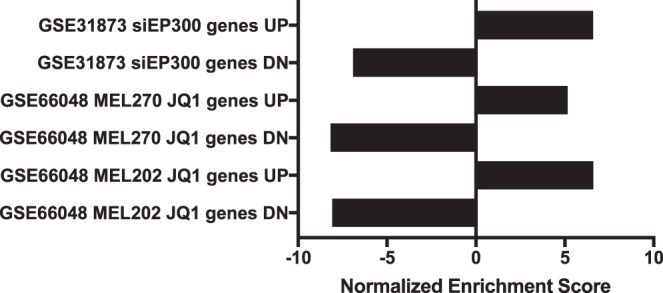
Strong concordance between the gene signatures of ICG-001, BET inhibitor JQ1, and siEP300. Comparison of publically available signatures from UM cell lines treated with JQ1, an inhibitor of BET family of bromodomain proteins, shows strong concordance with our signature of ICG-001 in UM cells. Similar concordance also is observed with the EP300 siRNA (siEP300) signature from C4-2B prostate cancer cell line. Gene sets with a nominal P value <0.05 and FDR < 0.25 were defined as significantly enriched.
Interestingly, we observed upregulation of KLF4 protein in ICG-001-treated Mel202 cells (Supplementary Fig. S3F). KLF4 was shown previously to inhibit the interaction of β-catenin with p300.40
ICG-001 Inhibits Genes Associated With UM Invasiveness and Suppresses UM Cell Invasion In Vitro
A more focused evaluation revealed that ICG-001 was very effective in suppressing genes that are critical for UM aggressiveness. Specifically, we found significant suppression of genes associated with focal adhesion (which, in turn, participates in migration and metastasis) (Fig. 6A). ICG-001 suppressed expression of the SDCBP gene (Fig. 6B), which encodes syntenin-1, a scaffolding-PDZ domain-containing protein with roles in modulating shape, and migration and invasive properties of cancer cells.41–44 SDCBP, together with CDH1, CITED1, EMP1, EMP3, and SPARC, constitute a group of genes that are linked to an epithelioid phenotype and were reported previously to be upregulated in Class 2 UMs,19,44 and also were downregulated by ICG-001 (Fig. 6B). Interestingly, ICG-001 suppressed SPP1 (Fig. 6B), which encodes the phosphoglycoprotein osteopontin. A recent study showed that osteopontin expression is increased in UM liver metastasis tissue and serum45 compared to primary and/or normal patients. We also found that GAGE cancer/testis antigen family members, that are important players in metastasis, were among the most repressed genes in our ICG-001 signature (Supplementary Fig. S5A).
Figure 6.

ICG-001 suppresses UM cell migration in vitro and the expression of genes associated in vivo with UM aggressiveness. (A) ICG-001 suppresses the expression of genes that are associated with focal adhesion in UM (left), as well as SPP1 (osteopontin) and epithelial genes that are associated with UM aggressiveness in Class 2 UM (right). (B) Normalized enrichment scores for pathways associated with kinase signaling (ERK/MAPK, MEK, and HGF signaling) when compared against the transcriptomic footprint of ICG-001 in UM. (C) In vitro migration of UM cells was assessed by measuring their ability to heal a scratch wound in a cell monolayer. ICG-001 potently suppressed UM cell migration in the Mel270 (primary) and OMM2.5 (metastatic) UM cell lines. Results shown are average ± SD. Representative images are shown in Supplementary Figure S5.
Following ICG-001 treatment, we also observed suppression of the MAPK/ERK/MEK and the hepatic growth factor (HGF)-mediated signaling pathways (Fig. 6B), which have been shown to promote growth, adhesion, migration, and invasion of UM cells. Further evidence of MAPK/ERK pathway inhibition was provided by immunoblotting, which demonstrated that ICG-001 decreases phospho-ERK1/2 levels (Supplementary Fig. S3). We also examined the levels of total and phospho-YAP1 in whole cell lysates and did not observe any significant changes.
Given our observation that ICG-001 is a potent inhibitor of various genes and signaling pathways that are important for aggressiveness and invasive behavior of UM cells, we next evaluated the effect of ICG-001 using an in vitro wound-healing assay. We found that ICG-001 potently suppressed the ability of UM cells to migrate in vitro (Fig. 6C, Supplementary Fig. S5B).
ICG-001 Inhibits the Expression of Genes Involved in UM Metastasis
Further dissection of the gene expression changes elicited by ICG-001 treatment revealed strong suppression of genes that are associated with metastasis in various UM patient datasets. First, we compared our ICG-001 transcriptomic footprint against a UM patient dataset reported by Onken et al.17,19 They established gene expression-based classification of primary UMs: Class 1, associated with low risk of metastasis and Class 2, associated with high risk of metastasis.17,18 Comparison of these respective gene sets against our ICG-001 transcriptomic footprint revealed that ICG-001 suppressed genes that are associated with high metastasis risk, while it induced genes that are associated with low metastasis risk (Figs. 7A, 7B). In subsequent studies, Class 1 UM genes were further subclassified into Class1met+ and Class1met−, with higher and lower metastatic potential, respectively.20 We also found that numerous genes that are upregulated in Class1met+ over Class1met− UMs, including PRAME, also were suppressed by ICG-001 treatment (Supplementary Fig. S6). Collectively, our data suggested ICG-001 suppresses genes that are associated with increased metastatic potential in UM.
Figure 7.

ICG-001 suppresses the expression of genes associated with metastasis in vivo and its transcriptomic footprint is associated with increased patient overall survival. (A–D) GSEA of various UM clinical datasets against the transcriptomic footprint of ICG-001 in UM demonstrates that ICG-001 potently suppresses gene expression profiles associated with UM metastasis. (E) The ICG-001 gene expression profile, applied to a UM patient dataset (TCGA) shows a statistically significant association with clinical outcomes. UM patient samples exhibiting low ICG-001 signature (lower 20% of patients) had inferior overall survival.
We also compared our ICG-001 transcriptomic footprint against two additional cutaneous melanoma patient datasets: those of Alonso et al.46 and Winnepenninckx et al.47 Genes reported to be upregulated in metastatic tumors in these studies also were enriched among those suppressed by ICG-001 in our transcriptomic signature (Figs. 7C, 7D).
The ICG-001 Transcriptomic Footprint is Associated With Better Overall Survival in UM Patients
In addition, we also performed gene expression profiling after 24 hours of treatment with ICG-001. We found 2413 genes to be expressed differentially in the 24-hour signature, compared to the 2493 genes that were expressed differentially in the 48-hour signature. These two signatures shared a large core set of genes (1510 genes) whose expression was altered concordantly in both. We next applied this core (shared) transcriptomic signature of ICG-001 to the TCGA-UM patient dataset and examined its prognostic significance. We found that the gene expression changes observed after ICG-001 treatment were associated with better overall survival (Fig. 7E).
ICG-001 Suppressed UM Xenograft Growth In Vivo
Finally, we used a Mel270 UM xenograft model to evaluate the anticancer activity of ICG-001 in vivo. For this experiment, Mel270 cells were injected subcutaneously into the flank of athymic nude mice and 50 mg/kg ICG-001 or vehicle control were administered intratumorally 5 days/week. Treatment with ICG-001 substantially reduced growth of UM xenografts compared to the vehicle controls (Fig. 8A) and prolonged animal survival (i.e., delayed the xenografts from reaching diameter of 1 cm, Fig. 8B).
Figure 8.
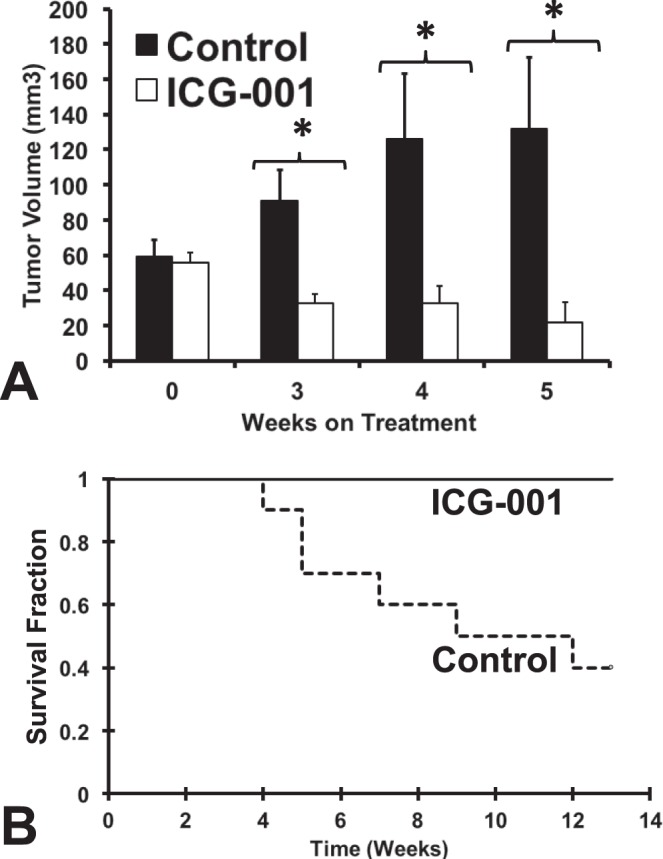
ICG-001 suppresses tumor growth in a murine model of UM. A Mel270 UM xenograft model was used to evaluate the anticancer activity of ICG-001 in vivo. Treatment with ICG-001 substantially reduced growth of UM xenografts compared to the vehicle controls. Shown in (A) are the average tumor volumes of mice treated with ICG-001 or vehicle (average±SEM; *P value <0.05). After 5 weeks of treatment, several mice (those with the largest tumors) in the vehicle cohort (but not in the ICG-001 cohort) had to be removed from the study due to tumor diameter exceeding the study threshold, thus making moot any further direct comparison of volumes between the two cohorts. ICG-001 delayed the xenografts from reaching the study threshold of 1 cm diameter, thus prolonging animal survival on the study (Kaplan-Meier survival plot is shown in [B]).
Discussion
Increased understanding and identification of numerous key driver molecules and signaling pathways in UM in the past decade have resulted in the launch of a number of clinical studies with molecularly targeted agents, such as selumetinib, sunitinib, imatinib, vorinostat, and antiangiogenic agents. However, the results of these studies have been disappointing.22,48–51 Hence, identification of compounds with therapeutic potential in UM is an area of pressing need. We identified ICG-001 as a compound with potent anticancer activity against UM cells in vitro and in vivo.
Global gene expression profiling, along with additional in vitro experiments, allowed us to illuminate several key effects of ICG-001 in UM. First, ICG-001 resulted in potent suppression of cell cycle genes (such as CDK2, MCM4, MCM7, and CCNB2) and many transcriptional targets of E2F. This suppression of cell cycle genes was accompanied by cell cycle arrest and, eventually, apoptosis. Second, we found that ICG-001 resulted in suppression of the Wnt signaling pathway and inhibited the expression of genes involved in cell “stemness.” The transcriptional footprints of the Sox2, Oct4, Myc, and Nanog transcription factors, which are essential for maintaining the pluripotent embryonic stem-like cell phenotype, were downregulated in our ICG-001 gene expression signature. Third, ICG-001 significantly downregulated a set of epithelial/focal adhesion-related markers (such as E-cadherin) that recently have been shown to be associated with increased metastatic potential in UM.19 Fourth, application of our ICG-001 signature to a UM patient cohort revealed that ICG-001 induced transcriptomic changes associated with better overall survival. Finally, ICG-001 reduced tumor growth and increased overall survival in a murine UM xenograft model.
The major site of UM metastasis is the liver. In recent years, several studies have focused on understanding the role of HGF and its corresponding receptor c-Met in UM pathophysiology. These studies have found that, similar to other cancers, in UM, these particular signaling molecules function as mediators of proliferation, survival, and cell migration.52–55 In our study, we found that the transcriptomic footprint of ICG-001 showed downregulation of genes activated by HGF/cMet. Recent studies also have highlighted the GAGE cancer/testis antigen family to be an important player in metastasis56,57; specifically, knockdown of GAGE family members abolished the migratory capacity of cutaneous melanoma cells.58 Interestingly, we found that GAGE family members were among the most repressed genes in our ICG-001 signature. Correspondingly, we found that ICG-001 completely suppressed the ability of the UM cells to migrate in vitro. The exact role of these GAGE proteins in UM cell invasion and migration remains to be fully elucidated.
Histologic presence of looping vasculogenic mimicry patterns is observed frequently in UM.59 PKC-driven PI3K/Akt pathway and the Wnt signaling pathway recently have been shown to be important players in vasculogenic mimicry in cancer.60–63 Previously, one UM clinical study reported increased presence of the Wnt ligand and the Wnt signaling molecules in primary UM.64 Our results indicated that ICG-001 exerts inhibitory effects on the Wnt pathway. Strikingly, ICG-001 also was a potent suppressor of many epithelial-related genes, such as CDH1, CITED1, EMP1, EMP3, SDCBP, and SPARC (Fig. 6), which have been shown by Harbour et al.19 to be upregulated in Class 2 UMs.
The Wnt signaling pathway also has been implicated in regulation of melanocyte differentiation.65 In particular, this is thought to be achieved in conjunction with p300.26,66 A recent study found that ICG-001 can induce differentiation and pigmentation of cutaneous melanoma cells.36 Another report in colon cancer has shown that c-Myc can function together with p300/β-catenin to drive a differentiation program in ICG-001-treated cells.26,27,66 In our current study, we did not observe a melanocytic differentiation program in ICG-001-treated UM cells, as there was no increase in DCT or TYR mRNA expression. Although we observed an increase in the mRNA levels of cMyc upon ICG-001 treatment, further gene set enrichment analysis suggested that most of the cMyc targets genes were not induced. Strikingly, ICG-001-induced transcriptional changes were similar to those observed upon inhibition of the EP300 (p300) via either siRNA or the small molecule inhibitor C646. Our failure to observe an induction of a differentiation program in ICG-001-treated UM cells may be due to suppression of p300 signaling by ICG-001.
ICG-001 treatment suppressed multiple kinase signaling pathways, including the MAPK/ERK/MEK and the mTOR signaling cascades. Still, their role in the ICG-001-induced cytotoxicity in UM must be clarified further. ICG-001 treatment resulted in inhibition of tumor growth and improved overall survival in a murine xenograft UM model. The xenograft UM model we used is one of the most ubiquitous preclinical models that allows for easy, fast, and accurate measurement of the tumor burden. It incorporates the interaction of the transplanted melanoma cells with the host blood and lymphatic vessels and the study of the drug response in vivo in a rapid and efficient manner. Unfortunately, this particular model rarely develops systemic metastasis and, thus, survival is determined not by metastatic burden but by local tumor growth surpassing the threshold for animal discomfort. The strong antitumor effect of ICG-001 in this murine UM model indicates promising preclinical efficacy that can be translated in the clinic. The formulation of ICG-001 that we used is not adequately soluble in water and, therefore, it was administered intratumorally. A water-soluble version of ICG-001 (PRI-724) that can be administered systemically to patients currently is in clinical trials for solid and hematopoietic malignancies (NCT01606579; NCT01764477).
In summary, we provided, to our knowledge, the first series of evidence that ICG-001 has potent anticancer activity against UM cells in vitro and in vivo. Collectively, our results suggested that ICG-001 is a promising therapeutic agent against UM through inhibition of the cell cycle and several signaling pathways. These data strongly support the rationale for using ICG-001 or its derivatives in future clinical trials in patients with UM.
Supplementary Material
Acknowledgments
The authors thank Martine J. Jager (Leiden University Medical Center, Leiden, Netherlands), Bruce R. Ksander (Schepens Eye Research Institute, Boston, USA), and Demetrios Vavvas (Massachusetts Eye and Ear Infirmary and Schepens Eye Research Institute, Harvard Medical School, Boston, MA, USA) for generously providing UM cell lines and Asha Gowda and David Medina for technical support.
Supported by Merit Review Award 1I01BX001372 (V.P.) from the United States Department of Veterans Affairs, National Institutes of Health (NIH, Bethesda, MD, USA; 5T32CA174647-03 [S.K.]), and Cancer Prevention Research Institute of Texas (CPRIT) Grant RP120348 to the Proteomics and Metabolomics Core Facility at BCM. The authors alone are responsible for the content and writing of this paper.
Disclosure: S. Kaochar, None; J. Dong, None; M. Torres, None; K. Rajapakshe, None; F. Nikolos, None; C.M. Davis, None; E.A. Ehli, None; C. Coarfa, None; N. Mitsiades, None; V. Poulaki. None
References
- 1. Bedikian AY. . Metastatic uveal melanoma therapy: current options. Int Ophthalmol Clin. 2006; 46: 151– 166. [DOI] [PubMed] [Google Scholar]
- 2. Collaborative Ocular Melanoma Study Group The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma: V. Twelve-year mortality rates and prognostic factors: COMS report No. 28. Arch Ophthalmol. 2006; 124: 1684– 1693. [DOI] [PubMed] [Google Scholar]
- 3. Marshall E, Romaniuk C, Ghaneh P,et al. . MRI in the detection of hepatic metastases from high-risk uveal melanoma: a prospective study in 188 patients. Br J Ophthalmol. 2013; 97: 159– 163. [DOI] [PubMed] [Google Scholar]
- 4. Spagnolo F, Caltabiano G, Queirolo P. . Uveal melanoma. Cancer Treat Rev. 2012; 38: 549– 553. [DOI] [PubMed] [Google Scholar]
- 5. Kujala E, Makitie T, Kivela T. . Very long-term prognosis of patients with malignant uveal melanoma. Invest Ophthalmol Vis Sci. 2003; 44: 4651– 4659. [DOI] [PubMed] [Google Scholar]
- 6. Collaborative Ocular Melanoma Study Group. . Assessment of metastatic disease status at death in 435 patients with large choroidal melanoma in the Collaborative Ocular Melanoma Study (COMS): COMS report no. 15. Arch Ophthalmol. 2001; 119: 670– 676. [DOI] [PubMed] [Google Scholar]
- 7. Augsburger JJ, Correa ZM, Shaikh AH. . Effectiveness of treatments for metastatic uveal melanoma. Am J Ophthalmol. 2009; 148: 119– 127. [DOI] [PubMed] [Google Scholar]
- 8. Buder K, Gesierich A, Gelbrich G, Goebeler M. . Systemic treatment of metastatic uveal melanoma: review of literature and future perspectives. Cancer Med. 2013; 2: 674– 686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Onken MD, Worley LA, Long MD,et al. . Oncogenic mutations in GNAQ occur early in uveal melanoma. Invest Ophthalmol Vis Sci. 2008; 49: 5230– 5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Van Raamsdonk CD, Bezrookove V, Green G,et al. . Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009; 457: 599– 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Becher R, Korn WM, Prescher G. . Use of fluorescence in situ hybridization and comparative genomic hybridization in the cytogenetic analysis of testicular germ cell tumors and uveal melanomas. Cancer Genet Cytogenet. 1997; 93: 22– 28. [DOI] [PubMed] [Google Scholar]
- 12. Sisley K, Parsons MA, Garnham J,et al. . Association of specific chromosome alterations with tumour phenotype in posterior uveal melanoma. Br J Cancer. 2000; 82: 330– 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parrella P, Sidransky D, Merbs SL. . Allelotype of posterior uveal melanoma: implications for a bifurcated tumor progression pathway. Cancer Res. 1999; 59: 3032– 3037. [PubMed] [Google Scholar]
- 14. Griffin CA, Long PP, Schachat AP. . Trisomy 6p in an ocular melanoma. Cancer Genet Cytogenet. 1988; 32: 129– 132. [DOI] [PubMed] [Google Scholar]
- 15. Kilic E, van Gils W, Lodder E,et al. . Clinical and cytogenetic analyses in uveal melanoma. Invest Ophthalmol Vis Sci. 2006; 47: 3703– 3707. [DOI] [PubMed] [Google Scholar]
- 16. Coupland SE, Lake SL, Zeschnigk M, Damato BE. . Molecular pathology of uveal melanoma. Eye (Lond). 2013; 27: 230– 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Onken MD, Worley LA, Ehlers JP, Harbour JW. . Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004; 64: 7205– 7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tschentscher F, Husing J, Holter T,et al. . Tumor classification based on gene expression profiling shows that uveal melanomas with and without monosomy 3 represent two distinct entities. Cancer Res. 2003; 63: 2578– 2584. [PubMed] [Google Scholar]
- 19. Onken MD, Ehlers JP, Worley LA, Makita J, Yokota Y, Harbour JW. . Functional gene expression analysis uncovers phenotypic switch in aggressive uveal melanomas. Cancer Res. 2006; 66: 4602– 4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Field MG, Decatur CL, Kurtenbach S,et al. . PRAME as an independent biomarker for metastasis in uveal melanoma. Clin Cancer Res. 2016; 22: 1234– 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gezgin G, Luk SJ, Cao J,et al. . PRAME as a potential target for immunotherapy in metastatic uveal melanoma. JAMA Ophthalmol. 2017; 135: 541– 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carvajal RD, Schwartz GK, Tezel T, Marr B, Francis JH, Nathan PD. . Metastatic disease from uveal melanoma: treatment options and future prospects. Br J Ophthalmol. 2017; 101: 38– 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Emami KH, Nguyen C, Ma H,et al. . A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc Natl Acad Sci U S A. 2004; 101: 12682– 12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takebe N, Miele L, Harris PJ,et al. . Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015; 12: 445– 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arensman MD, Telesca D, Lay AR,et al. . The CREB-binding protein inhibitor ICG-001 suppresses pancreatic cancer growth. Mol Cancer Ther. 2014; 13: 2303– 2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma H, Nguyen C, Lee KS, Kahn M. . Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene. 2005; 24: 3619– 3631. [DOI] [PubMed] [Google Scholar]
- 27. Eguchi M, Nguyen C, Lee SC, Kahn M. . ICG-001, a novel small molecule regulator of TCF/beta-catenin transcription. Med Chem. 2005; 1: 467– 472. [DOI] [PubMed] [Google Scholar]
- 28. De Waard-Siebinga I, Blom DJ, Griffioen M,et al. . Establishment and characterization of an uveal-melanoma cell line. Int J Cancer. 1995; 62: 155– 161. [DOI] [PubMed] [Google Scholar]
- 29. Jager MJ, Magner JA, Ksander BR, Dubovy SR. . Uveal melanoma cell lines: where do they come from? (An American Ophthalmological Society Thesis). Trans Am Ophthalmol Soc. 2016; 114: T5. [PMC free article] [PubMed] [Google Scholar]
- 30. Blom DJ, Schurmans LR, De Waard-Siebinga I, De Wolff-Rouendaal D, Keunen JE, Jager MJ. . HLA expression in a primary uveal melanoma, its cell line, and four of its metastases. Br J Ophthalmol. 1997; 81: 989– 993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ksander BR, Rubsamen PE, Olsen KR, Cousins SW, Streilein JW. . Studies of tumor-infiltrating lymphocytes from a human choroidal melanoma. Invest Ophthalmol Vis Sci. 1991; 32: 3198– 3208. [PubMed] [Google Scholar]
- 32. Chen PW, Murray TG, Uno T, Salgaller ML, Reddy R, Ksander BR. . Expression of MAGE genes in ocular melanoma during progression from primary to metastatic disease. Clin Exp Metastasis. 1997; 15: 509– 518. [DOI] [PubMed] [Google Scholar]
- 33. Mitsiades N, Chew SA, He B,et al. . Genotype-dependent sensitivity of uveal melanoma cell lines to inhibition of B-Raf, MEK, and Akt kinases: rationale for personalized therapy. Invest Ophthalmol Vis Sci. 2011; 52: 7248– 7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Griewank KG, Yu X, Khalili J,et al. . Genetic and molecular characterization of uveal melanoma cell lines. Pigment Cell Melanoma Res. 2012; 25: 182– 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grigson ER, Ozerova M, Pisklakova A, Liu H, Sullivan DM, Nefedova Y. . Canonical Wnt pathway inhibitor ICG–001 induces cytotoxicity of multiple myeloma cells in Wnt-independent manner. PLoS One. 2015; 10: e0117693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim KI, Jeong DS, Jung EC, Lee JH, Kim CD, Yoon TJ. . Wnt/beta-catenin signaling inhibitor ICG-001 enhances pigmentation of cultured melanoma cells. J Dermatol Sci. 2016; 84: 160– 168. [DOI] [PubMed] [Google Scholar]
- 37. Miyabayashi T, Teo JL, Yamamoto M, McMillan M, Nguyen C, Kahn M. . Wnt/beta-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc Natl Acad Sci U S A. 2007; 104: 5668– 5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Delmore JE, Issa GC, Lemieux ME,et al. . BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011; 146: 904– 917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Feng Q, Zhang Z, Shea MJ,et al. . An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014; 24: 809– 819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Evans PM, Chen X, Zhang W, Liu C. . KLF4 interacts with beta-catenin/TCF4 and blocks p300/CBP recruitment by beta-catenin. Mol Cell Biol. 2010; 30: 372– 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Helmke BM, Polychronidis M, Benner A, Thome M, Arribas J, Deichmann M. . Melanoma metastasis is associated with enhanced expression of the syntenin gene. Oncol Rep. 2004; 12: 221– 228. [PubMed] [Google Scholar]
- 42. Boukerche H, Su ZZ, Emdad L, Sarkar D, Fisher PB. . mda-9/Syntenin regulates the metastatic phenotype in human melanoma cells by activating nuclear factor-kappaB. Cancer Res. 2007; 67: 1812– 1822. [DOI] [PubMed] [Google Scholar]
- 43. Boukerche H, Su ZZ, Prevot C, Sarkar D, Fisher PB. . mda-9/Syntenin promotes metastasis in human melanoma cells by activating c-Src. Proc Natl Acad Sci U S A. 2008; 105: 15914– 15919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gangemi R, Mirisola V, Barisione G,et al. . Mda-9/syntenin is expressed in uveal melanoma and correlates with metastatic progression. PLoS One. 2012; 7: e29989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kadkol SS, Lin AY, Barak V,et al. . Osteopontin expression and serum levels in metastatic uveal melanoma: a pilot study. Invest Ophthalmol Vis Sci. 2006; 47: 802– 806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Alonso SR, Tracey L, Ortiz P,et al. . A high-throughput study in melanoma identifies epithelial-mesenchymal transition as a major determinant of metastasis. Cancer Res. 2007; 67: 3450– 3460. [DOI] [PubMed] [Google Scholar]
- 47. Winnepenninckx V, Lazar V, Michiels S,et al. . Gene expression profiling of primary cutaneous melanoma and clinical outcome. J Natl Cancer Inst. 2006; 98: 472– 482. [DOI] [PubMed] [Google Scholar]
- 48. Komatsubara KM, Manson DK, Carvajal RD. . Selumetinib for the treatment of metastatic uveal melanoma: past and future perspectives. Future Oncol. 2016; 12: 1331– 1344. [DOI] [PubMed] [Google Scholar]
- 49. Carvajal RD, Schwartz GK, Mann H, Smith I, Nathan PD. . Study design and rationale for a randomised, placebo-controlled, double-blind study to assess the efficacy of selumetinib (AZD6244; ARRY-142886) in combination with dacarbazine in patients with metastatic uveal melanoma (SUMIT). BMC Cancer. 2015; 15: 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carvajal RD, Sosman JA, Quevedo JF,et al. . Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA. 2014; 311: 2397– 2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Luke JJ, Callahan MK, Postow MA,et al. . Clinical activity of ipilimumab for metastatic uveal melanoma: a retrospective review of the Dana-Farber Cancer Institute, Massachusetts General Hospital, Memorial Sloan-Kettering Cancer Center, and University Hospital of Lausanne experience. Cancer. 2013; 119: 3687– 3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Buzzacco DM, Abdel-Rahman MH, Park S, Davidorf F, Olencki T, Cebulla CM. . Long-term survivors with metastatic uveal melanoma. Open Ophthalmol J. 2012; 6: 49– 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Topcu-Yilmaz P, Kiratli H, Saglam A, Soylemezoglu F, Hascelik G. . Correlation of clinicopathological parameters with HGF, c-Met, EGFR, and IGF-1R expression in uveal melanoma. Melanoma Res. 2010; 20: 126– 132. [DOI] [PubMed] [Google Scholar]
- 54. All-Ericsson C, Girnita L, Seregard S, Bartolazzi A, Jager MJ, Larsson O. . Insulin-like growth factor-1 receptor in uveal melanoma: a predictor for metastatic disease and a potential therapeutic target. Invest Ophthalmol Vis Sci. 2002; 43: 1– 8. [PubMed] [Google Scholar]
- 55. Economou MA, Andersson S, Vasilcanu D,et al. . Oral picropodophyllin (PPP) is well tolerated in vivo and inhibits IGF-1R expression and growth of uveal melanoma. Invest Ophthalmol Vis Sci. 2008; 49: 2337– 2342. [DOI] [PubMed] [Google Scholar]
- 56. Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. . Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev. 2002; 188: 22– 32. [DOI] [PubMed] [Google Scholar]
- 57. Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. . Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005; 5: 615– 625. [DOI] [PubMed] [Google Scholar]
- 58. Caballero OL, Cohen T, Gurung S,et al. . Effects of CT-Xp gene knock down in melanoma cell lines. Oncotarget. 2013; 4: 531– 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Folberg R, Pe'er J, Gruman LM,et al. . The morphologic characteristics of tumor blood vessels as a marker of tumor progression in primary human uveal melanoma: a matched case-control study. Hum Pathol. 1992; 23: 1298– 1305. [DOI] [PubMed] [Google Scholar]
- 60. McDonald SL, Silver A. . The opposing roles of Wnt-5a in cancer. Br J Cancer. 2009; 101: 209– 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Qi H, Sun B, Zhao X,et al. . Wnt5a promotes vasculogenic mimicry and epithelial-mesenchymal transition via protein kinase Calpha in epithelial ovarian cancer. Oncol Rep. 2014; 32: 771– 779. [DOI] [PubMed] [Google Scholar]
- 62. Qi L, Song W, Liu Z, Zhao X, Cao W, Sun B. . Wnt3a promotes the vasculogenic mimicry formation of colon cancer via Wnt/beta-catenin signaling. Int J Mol Sci. 2015; 16: 18564– 18579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yao L, Sun B, Zhao X,et al. . Overexpression of Wnt5a promotes angiogenesis in NSCLC. Biomed Res Int. 2014; 2014: 832562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zuidervaart W, Pavey S, van Nieuwpoort FA,et al. . Expression of Wnt5a and its downstream effector beta-catenin in uveal melanoma. Melanoma Res. 2007; 17: 380– 386. [DOI] [PubMed] [Google Scholar]
- 65. Bellei B, Pitisci A, Catricala C, Larue L, Picardo M. . Wnt/beta-catenin signaling is stimulated by alpha-melanocyte-stimulating hormone in melanoma and melanocyte cells: implication in cell differentiation. Pigment Cell Melanoma Res. 2011; 24: 309– 325. [DOI] [PubMed] [Google Scholar]
- 66. Rieger ME, Zhou B, Solomon N,et al. . p300/beta-Catenin interactions regulate adult progenitor cell differentiation downstream of WNT5a/protein kinase C (PKC). J Biol Chem. 2016; 291: 6569– 6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.