

Abstract
Multicentric Castleman’s disease (MCD) is a rare, widespread lymphoproliferative disorder and a life-threatening disease involving hyperactivity of the immune system, excessive proinflammatory cytokine release, immune cell proliferation, and organ system dysfunction. Interleukin-6 (IL-6) is a cytokine that plays a key role in the pathogenesis of MCD, as it is involved in the synthesis of acute-phase reactants and aids in the induction of B-cell proliferation. Siltuximab is an anti-IL-6 chimeric monoclonal antibody that acts as a novel treatment modality to bind to IL-6 with high affinity, thus neutralizing the cytokine bioactivity and inhibiting B-cell proliferation. Clinical trials with siltuximab have shown early clinical promise for patients with MCD for many years, leading to recent US Food and Drug Administration approval as a novel agent for the treatment of MCD. Here, a systematic review was conducted to include 171 cases of MCD patients treated with siltuximab. While traditional treatment methods were able to achieve a 5-year survival rate of only 55%–77%, results of siltuximab treatment demonstrated 5-year survival rates of nearly 96.4% (only 2 deaths reported out of 55 patients with follow-up data). Ultimately, the results from multiple clinical trials have demonstrated that siltuximab is extremely efficacious in alleviating disease symptoms (fatigue, pain, and lymphadenopathy) while simultaneously achieving disease remission, thus extending progression-free survival for years longer than the average 5-year survival rates for MCD.
Keywords: Castleman’s disease, angiofollicular lymph node hyperplasia, giant lymph node hyperplasia, siltuximab, IL-6 receptor
Introduction
Castleman’s disease (CD), also known as angiofollicular lymph node hyperplasia and giant lymph node hyperplasia, is a rare lymphoproliferative disorder that represents a heterogeneous collection of lymphoproliferative processes that all share common lymph node histological features.1 CD can be localized to a single lymph node (unicentric) or widespread (multicentric), with treatment and outcome dependent on the degree of proliferation and spread.1 Multicentric CD (MCD) is a serious, life-threatening disorder that involves hyperactivity of the immune system, excessive proinflammatory cytokine release, immune cell proliferation, and organ system dysfunction.2,3 MCD is associated with other cellular proliferation disorders such as lymphoma, Kaposi’s sarcoma, and POEMS syndrome, and viruses such as human herpes virus 8 (HHV-8) and human immunodeficiency virus (HIV).3
The exact cause of unicentric CD and HHV-8-negative/idiopathic MCD is unknown, and this rare disorder can affect any patient population.2 Symptoms commonly associated with MCD include fatigue and weakness, fever, night sweats, loss of appetite with unintentional weight loss, enlarged lymph nodes, and hepatosplenomegaly.4 The typical symptoms of MCD parallel the presentation of acute lymphomas; thus, the diagnosis of MCD presents a challenge based on clinical presentation alone.1,2 Histologically, MCD is classified into a plasmacytic form, a hyaline-vascular form, or a mixed variation.5 Additionally, MCD can be classified as HHV-8-negative/idiopathic MCD or iMCD, and because treatment modalities vary in efficacy for these two subsets, it is essential to determine HHV-8 status in patients diagnosed with MCD.6,7
CD was first discovered in the 1950s and has largely remained a mystery to the medical community over the last 60 years. There arê6,500–7,700 new cases of CD reported in the USA every year; despite the considerable interest and research devoted to this rare disorder, there still remains no overall consensus on the most effective treatment regimen for iMCD.1,2 Traditional treatment modalities include splenic removal, chemotherapy, systemic corticosteroids, antiviral management of HIV and HHV-8, and thalidomide, but they have limited success in causing remission and overall patient outcomes.8,9 The 5-year survival rate is only 65% for iMCD with traditional treatment.8,10 Although MCD is a very rare disorder, it has a worse mortality rate than many common soft tissue cancers and lymphomas such as stage II colon cancer, stage III breast cancer, and progressive non-Hodgkin’s lymphoma.
Interleukin-6 (IL-6) is a cytokine that plays a key role in the pathogenesis of MCD, as it is involved in the synthesis of acute-phase reactants and aids in the induction of B-cell proliferation.11 IL-6, also a participant in hepcidin synthesis and as such a significant cause of anemia in chronic disease states, is extremely elevated in CD. The elevation of IL-6 is responsible for the proinflammatory state and typical symptoms seen in MCD.11,12 Monoclonal antibody therapy against IL-6 is an emerging, novel treatment modality that has shown significant promise in initial clinical trials for non-Hodgkin’s lymphoma, multiple myeloma, and CD.12 Two anti-IL-6 therapies have been examined for iMCD treatment. Tocilizumab is a recombinant humanized immunoglobulin G1 (IgG1) monoclonal antibody approved in Japan for the treatment of CD. Siltuximab is an anti-IL-6 chimeric monoclonal antibody that binds to IL-6 with high affinity, thus neutralizing the cytokine bioactivity and inhibiting B-cell proliferation.12–14 Though clinical trials with siltuximab have shown early clinical promise for patients with MCD for many years, this drug has only recently received US Food and Drug Administration approval for patients with MCD who are negative for both HIV and HHV-8.12–14
The goal of this present study was to comprehensively analyze the available literature and to evaluate the impact of siltuximab on patient survival outcomes in MCD. This article represents the largest and most exhaustive review of relevant patient data on MCD treatment with siltuximab and seeks to make recommendations for the utilization of this novel therapy to improve outcomes in the treatment of MCD.
Methods
The National Library of Medicine’s PubMed database was systematically searched by both the research team and the school of medicine health sciences library database up to May 2017. The following search terms were used: “Castleman’s disease”, “Castleman disease”, “angiofollicular lymph node hyperplasia”, “giant lymph node hyperplasia”, in combination with “Siltuximab”, “IL-6 receptor”, and “IL-6 drug”. The full text of potentially relevant articles was retrieved for review following initial title and abstract screening. The articles included in this review were chosen based on the following criteria: the paper must be written or translated in the English language, report only cases using primary human subjects, contain original cases or results of clinical testing, and discuss MCD treated primarily with siltuximab. All other articles that did not adhere to these standards were effectively excluded. The methodology of this review was designed to reduce bias in article selection, appraisal, and data extraction and analysis. However, the study is limited by the consistency of the literature in reporting data, even with thorough extraction of available data.
Results
The initial search of PubMed returned 26 articles. Upon screening the titles and abstracts for relevance, the full text of 14 articles was retrieved for review by the research team. After completion of the full-text review, seven articles were excluded because of failure to meet criteria standards. Ultimately, 7 articles were included in this literature review, consisting of 161 total cases of MCD treated primarily with siltuximab. Figure 1 demonstrates the selection process utilized for this review, and Table 1 illustrates the number of cases present in the contributing articles. All included articles were either clinical trials (N=5) or case reports (N=2). The greatest number of cases was reported in the USA (N=75).
Figure 1.
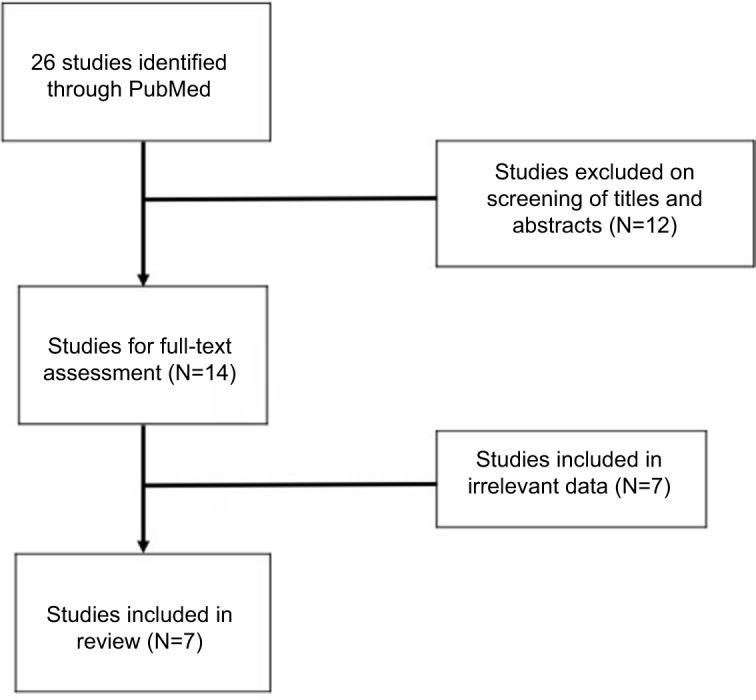
Systematic search of PubMed returned 26 studies. Following review of the titles, abstracts, and full-text, 7 studies were included in the present review.
Table 1.
Number of patients in each contributing article of the review
Clinical presentation
The mean patient age for available patient data was 56.8 years (range 18–78 years) with moderately unequal gender distribution with a total of 89 male patients (58%) and 65 female patients (42%). Patient demographics were reported in most clinical trials and totaled 65 Caucasian patients, 41 Asian patients, and 2 African American patients. The MCD subsets analyzed in the cases included 143 patients with iMCD, 17 patients with unspecified MCD type, and 1 patient with MCD (HHV-8 positive and HIV negative). Summary of the study characteristics and patient demographics is reported in Table 2.
Table 2.
Study characteristics and patient demographics
| Country | Studies (cases) |
| USA | 5 (75) |
| Multinational study | 2 (96) |
| Study design | |
| Clinical trials | 5 (169) |
| Case report | 2 (2) |
| Demographics | N (range) |
| Total patients | 161 |
| Mean age | 56.8 (18–78 years) |
| Ethnicity, (n) | |
| Caucasian | 65 |
| Asian | 41 |
| African American | 2 |
| Gender, (n) | |
| Female | 65 |
| Male | 89 |
Patients’ relevant past medical history included four cases of asthma, two cases of thrombotic thrombocytopenic purpura, two cases of unspecified autoimmune disorder, one case of idiopathic thrombocytopenic purpura, one case of systemic lupus erythematosus, one case of antiphospholipid syndrome, one case of diffuse large B-cell non-Hodgkin’s lymphoma, and one concurrent diagnosis of separate Hodg-kin’s lymphoma and sarcoidosis. In articles that reported symptoms, the most common symptomatic complaints at initial presentation were B-symptoms (N=15), hepatosplenomegaly (N=7), fever (N=5), pleural effusion and/or ascites (N=4), and organ failure (N=2). A majority of patients presented with clear, diffuse lymphadenopathy, and other minor disease symptoms included bone pain (N=1), confusion (N=1), and plasmablastic microlymphoma formation (N=1). Patient characteristics are summarized in Table 3. Patients sought out medical treatment after an average of 4.8 months from symptom start (range 1.3–93.2 months).
Table 3.
Clinical presentation of multicentric Castleman’s disease (N=171)
| N (%) | |
|---|---|
| Symptoms | |
| B-symptoms | 15/25 (60) |
| Hepatosplenomegaly | 7/25 (28) |
| Fever | 5/25 (20) |
| Pleural effusion/ascites | 4/25 (16) |
| Organ failure | 2/25 (8) |
| Bone pain | 1/25 (4) |
| Confusion | 1/25 (4) |
| Plasmablastic microlymphoma | 1/25 (4) |
| Lymph node architecture | |
| Hyaline-vascular variant | 62/152 (40.8) |
| Plasmacytic variant | 54/152 (35.5) |
| Mixed variant | 36/152 (23.7) |
| Past medical history | |
| Asthma | 4/14 (28.6) |
| Thrombotic thrombocytopenic purpura | 2/14 (14.3) |
| Unspecified autoimmune disorder | 2/14 (14.3) |
| Idiopathic thrombocytopenic purpura | 1/14 (7.1) |
| Systemic lupus erythematosus | 1/14 (7.1) |
| Antiphospholipid syndrome | 1/14 (7.1) |
| Diffuse large B-cell lymphoma | 1/14 (7.1) |
| Hodgkin’s lymphoma | 1/14 (7.1) |
| Sarcoidosis | 1/14 (7.1) |
| Mean duration (months) | 4.8 (range 1.3–93.2) |
Workup
Blood chemistries were reported in two of the larger clinical studies, revealing elevated C-reactive protein levels (N=79), symptomatic anemia (N=27), elevated beta-2 microglobulin levels (N=16), elevated serum alkaline phosphatase levels (N=10), elevated erythrocyte sedimentation rate (N=9), hyper IgG levels (N=7), hyper IgA levels (N=6), hypoalbuminemia (N=4), and elevated lactate dehydrogenase levels (N=4). All cases reported elevation of IL-6 grossly greater than normal value ranges, which was consistent with classic iMCD presentation. However, a trend in variation of IL-6 levels was observed in different subtypes of MCD histology, with lower mean IL-6 levels seen in the hyaline-vascular histology subtype compared with the plasmacytic subtype.9 The majority of patients underwent initial lymph node biopsy for diagnostic purposes and for lymph node architecture classification. iMCD can be classified on the basis of lymph node architecture abnormalities as hyaline-vascular, plasmacytic, or mixed type. Histological analysis demonstrated 62 patients with the hyaline-vascular variant, 54 patients with the plasmacytic variant, and 36 patients with the mixed variant architecture.
In the retrospective review of tissue samples by Yu et al,8 bone marrow biopsies were obtained from each patient and analyzed for the first time in the diagnostic workup of this rare disorder. A normal bone marrow biopsy was seen in only 5 of 22 patients’ laboratory reports, with abnormal bone marrow biopsies classified as hypercellular in 9 of 22 cases, hypocellular in 4 cases, and fibrotic in 1 case. Immunostaining data were limited in the literature as was the use of reported diagnostic imaging studies for the evaluation of disease status and progression/remission.
Outcome
The primary goal of the treatment for iMCD is alleviation of disease symptoms in addition to directed therapy aimed at extension of disease-free survival time. All cases reported in this literature review were treated with the novel treatment therapy siltuximab, typically after failing conventional treatment options in previous therapy regimens, with a mean treatment duration of 5.1 years. A total of 27 patients underwent previous treatments that failed to be successful, including rituximab (N=19), systemic corticosteroids (N=16), surgical excision (N=4), thalidomide (N=2), cyclophosphamide (N=1), radiotherapy (N=1), and autologous stem-cell transplantation (N=1). Additional therapeutic measures in concurrence with siltuximab were recorded in a small minority of cases. For example, corticosteroid therapy was given in one case report of a patient with concurrent sarcoidosis, as siltuximab alone was not effective in symptom control.
Of the papers that reported patient follow-up results (N=55), the mean follow-up period was 39 months (range 10.5–64.7 months) with only 2 reported deaths in the reported 55 cases. Overall, the results from siltuximab treatment surpassed any outcomes seen in previous studies of other, traditional treatment modalities. Only two patients were reported to have died during the multiyear-long clinical trial study, which revealed exceptionally better outcomes compared with the average 5-year survival rates of iMCD patients. A total of 47 patients were able to obtain complete remission and/or sustained disease control with siltuximab treatment, while 7 patients achieved partial remission of iMCD. Only five patients failed to attain remission of iMCD but, nonetheless, reported improvement in symptoms and quality of life. One case showed near-complete resolution of lung and mediastinal disease and the patient continued to receive treatment for 3.5 years without serious adverse events. Additionally, the patient regained the 18 kg lost before treatment and was strong enough to engage in 48–64 km of bicycling per week.4
Another important objective that was investigated in the literature was appropriate drug dosing selection for optimal results and minimal adverse side effect profile. In a majority of patient cases (N=139), a dose of 11 mg/kg siltuximab was administered intravenously every 3 weeks. The study by van Rhee et al14 investigated the following varied drug dosing regimens: 3 mg/kg q2w followed by 6 mg/kg dose (N=1), 6 mg/kg q2w followed by 6 mg/kg dose (N=2), 6 mg/kg q1w followed by 6 mg/kg dose (N=3), 9 mg/kg q3w followed by 9 mg/kg dose (N=6), 12 mg/kg q3w followed by 12 mg/kg (N=6), 12 mg/kg q2w followed by 12 mg/kg dose (N=3), and 12 mg/kg weekly followed by 12 mg/kg dose group (N=2). In summary, the study found that administration of 11 mg/kg siltuximab q3w for iMCD patients with elevated baseline, mean C-reactive protein of 17.6 mg/L resulted in rapid C-reactive protein suppression to 1.04 mg/L by cycle 1 day 8 and sustained suppression throughout the treatment period. Ultimately, it was concluded unanimously in the literature that 11–12 mg/kg of siltuximab administered intravenously every 3 weeks is the most effective dose with the least toxic side effect profile for the optimal treatment of iMCD.
Discussion
Though the overall clinical presentation of iMCD is reasonably consistent, this disorder, nonetheless, presents a considerable challenge to the medical community in both timely recognition and treatment efficacy. Although immediate diagnosis is essential to reduce mortality in patients with this disorder, the diagnosis is often delayed because of the rarity of the disease and vague clinical symptoms.2 The typical symptoms of iMCD parallel the presentation of acute lymphomas; thus, the diagnosis of iMCD is unlikely based on clinical presentation alone.1,2 None of the cases reported the use of noninvasive imaging techniques, and at this time the utilization of this technology has no place in initial diagnosis of iMCD.
Lymph node biopsy and blood chemistries, thus, remain the gold standard for diagnosis and should be initiated at first detection of symptoms or lymphadenopathy. In cases that reported blood laboratory reports, a wide variety of blood chemistries were abnormal, including universal elevation of IL-6 seen in all cases. Histologically, iMCD is classified into a plasmacystic form, a hyaline-vascular form, or a mixed variation, and the presence of these unique lymph node architecture types confirmed the diagnosis of iMCD.5
A gold standard treatment regimen does not exist for iMCD, and thus the purpose of this study was to make recommendations for treatment based on the success of siltuximab observed in the current literature. When compared with siltuximab and other novel treatments like tocilizumab, aggressive treatment of iMCD with traditional therapeutic modalities such as splenic removal, corticosteroids, thalidomide, and chemotherapy yields worse survival outcomes. Novel treatment modalities like siltuximab, which are targeted at the elevated IL-6 levels that drive disease pathology, currently offer the only hope for improved treatment and survival outcomes. While traditional treatment methods were able to achieve a 5-year survival rate of only 55%–77%, results of siltuximab treatment demonstrated 5-year survival rates of nearly 96.4% (only 2 deaths reported out of 55 patients). Ultimately, the results from multiple clinical trials have demonstrated that siltuximab, when compared with other treatments, is extremely efficacious in alleviating disease symptoms (fatigue, pain, and lymphadenopathy) while simultaneously achieving disease remission, thus extending progression-free survival for years longer than the average 5-year survival rates for iMCD.
Yu et al described the most recent and comprehensive results from a previous clinical trial of monoclonal anti-IL-6 therapies and analyzed the therapeutic response of 21 patients with iMCD to siltuximab. The results of this large clinical trial concluded that siltuximab had a greater clinically significant proportion of complete remissions for iMCD and longer progression-free survival rates compared with rituximab. The study concluded that siltuximab was associated with significantly better outcomes and that rituximab was no better than traditional chemotherapy or systemic corticosteroid treatment in terms of complete remission results and progression-free survival rates. Siltuximab effectively improved clinical symptoms for iMCD patients while simultaneously controlling disease progression in those patients that failed initial rituximab treatment. Additionally, the analysis of data indicated that mean patient response rate of siltuximab was significantly higher than that of rituximab, corticosteroids, or chemotherapy. The study ultimately found a response rate of 75% for North American patients treated with siltuximab, which was much greater than the 34% response rate that was reported in the first randomized controlled trial.
Patients with iMCD often suffer from anemia of chronic disease caused by the proinflammatory cytokines that drive the symptomatic pathology of this potentially fatal lymphoproliferative syndrome.10 The comorbid anemia state that is frequently present in patients can exacerbate already present disease symptoms.11 Successful treatment of anemia in these patients is only achievable with effective reduction in systemic inflammatory cytokine levels; thus, the mainstay of treatment is aimed at IL-6, C-reactive protein, and erythrocyte sedimentation rate decrease.12,13 In the study analysis by Casper et al,11 siltuximab demonstrated a significant increase in hemoglobin levels (change of >15 g/L) in 61% of patients while simultaneously decreasing hepcidin levels by 47%. Additionally, in the placebo-controlled study of 79 patients with MCD, highly elevated C-reactive protein levels decreased by 92% in only 8 days of siltuximab treatment while remaining high in the placebo group.12 The clinical trial by van Rhee et al13 demonstrates the efficacy of this drug in gaining symptom remission with effective cytokine control and the clinically significant effects obtained through cytokine level depression.
Siltuximab must be continuously taken as a long-term maintenance therapy in order to receive continued clinical remission of iMCD; thus, the safety of such long-term pharmaceutical use must be demonstrated to outweigh any risk of long-term toxicity or adverse drug side effects. The safety profile of siltuximab was investigated by Rhee et al in a Phase II, open-label, multicenter study of the long-term safety of siltuximab.9 The study followed 19 iMCD patients on siltuximab over a 7-year period and reported complete patient survival during this period. Results of this study revealed the following confirmed adverse side effects from medication use: hypertension (N=3), nausea, fatigue, cellulitis (N=2), hypertriglyceridemia (N=8), and hypercholesterolemia (N=19). Other serious side effects that were possibly attributed to siltuximab were leukopenia, lymphopenia, and polycythemia (N=1). The article reported an extensive list of additional adverse effects; however, the study could not confirm the side effect etiology as drug toxicity, disease-associated pathology, or natural aging phenomenon. Therefore, patients who are negative for HIV or HHV-8 and do not already demonstrate some of the major conditions associated with the side effects of this treatment regimen are strongly encouraged to begin siltuximab therapy. This study represents the only clinical trial with concern to siltuximab safety and ultimately concluded that long-term use demonstrated sustained disease-free control without evidence of toxicity, need for discontinuation, and with few serious side effects.
Major limitations of this paper include factors surrounding patient selection bias for the clinical trials. For example, the clinical trials analyzed by this literature review had inclusion criteria that limited the sickest patients from enrolling. Subsequently, patients who were life-threateningly sick or newly diagnosed and relatively asymptomatic were excluded, thus removing groups that did not fall within the middle stage of the disease progression. Additionally, the two deaths described in the study by Yu et al8 did not specify which treatment group these fatalities were ascribed to; thus, there may be no deaths associated with siltuximab treatment. We believe that this delineation fails to be clinically significant because siltuximab treatment has shown to be a statistically significantly more superior treatment than chemotherapy or rituximab in the treatment of iMCD.
Conclusion
iMCD is an uncommon, potentially fatal lymphoproliferative disorder involving hyperactivity of the immune system, excessive proinflammatory cytokine release, immune cell proliferation, and organ system dysfunction. Because IL-6 plays a key role in the pathogenesis of the disease, it is important to research and develop therapeutic agents that target the cytokine. Siltuximab is a novel agent that acts as an anti-IL-6 chimeric monoclonal antibody that binds to IL-6 with high affinity, thus neutralizing the cytokine bioactivity and inhibiting B-cell proliferation. Studies and research trials of the drug show clinical promise, and when compared with traditional therapeutic modalities, the results of siltuximab treatment demonstrated 5-year survival rates of 96.4%. The patient population that may benefit the most from this novel treatment are those who are negative for HIV and HHV-8. This systematic review demonstrates that siltuximab is incredibly efficacious in alleviating the common symptoms of MCD while simultaneously achieving disease remission, thus extending patient survival for years longer than was otherwise achieved for MCD.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Chan K, Lade S, Prince H, Harrison S. Update and new approaches in the treatment of Castleman disease. J Blood Med. 2016;7:145–158. doi: 10.2147/JBM.S60514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garcia A, Cobo M, Peña P. Current diagnosis and treatment of Castle-man’s disease. Rev Clin Esp. 2016;216(3):146–156. doi: 10.1016/j.rce.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 3.Nishimoto N, Kanakura Y, Aozasa K, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106(8):2627–2632. doi: 10.1182/blood-2004-12-4602. [DOI] [PubMed] [Google Scholar]
- 4.Mohammed A, Janku F, Qi M, Kurzrock R. Castleman’s disease and sarcoidosis, a rare association resulting in a “mixed” response: a case report. J Med Case Rep. 2015;9(1):45. doi: 10.1186/s13256-015-0517-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koenig G, Stevens TM, Peker D. Plasmablastic microlymphoma arising in human herpesvirus-8-associated multicentric Castleman disease in a human immunodeficiency virus-seronegative patient with clinical response to anti-interleukin-6 therapy. Histopathology. 2015;67(6):930–932. doi: 10.1111/his.12718. [DOI] [PubMed] [Google Scholar]
- 6.Fajgenbaum, David C, Bagg A, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. 2017;129(12):1646–1657. doi: 10.1182/blood-2016-10-746933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fajgenbaum DC, Liu A, Ruth J, et al. HHV-8-Negative, idiopathic multicentric Castleman disease (iMCD): a description of clinical features and therapeutic options through a systematic literature review. Blood. 2014;124(21):4861. [Google Scholar]
- 8.Yu L, Tu M, Cortes J, et al. Clinical and pathological characteristics of HIV- and HHV-8 negative Castleman disease. Blood Adv. 2017;129(12):1658–1668. doi: 10.1182/blood-2016-11-748855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Rhee F, Casper C, Voorhees P, et al. A phase 2, open-label, multicenter study of the long-term safety of siltuximab (an anti-interleukin-6 monoclonal antibody) in patients with multicentric Castleman disease. Oncotarget. 2015;6(30):30408–3019. doi: 10.18632/oncotarget.4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dispenzieri A, Armitage JO, Loe MJ, et al. The clinical spectrum of Castleman’s disease. Am J Hematol. 2012;87(11):997–1002. doi: 10.1002/ajh.23291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casper C, Chaturvedi S, Munshi N, et al. Analysis of inflammatory and anemia-related biomarkers in a randomized, double-blind, placebo-controlled study of siltuximab (anti-IL6 monoclonal antibody) in patients with multicentric Castleman disease. Clin Cancer Res. 2015;21(19):4294–4304. doi: 10.1158/1078-0432.CCR-15-0134. [DOI] [PubMed] [Google Scholar]
- 12.Mayer CL, Xie L, Bandekar R, et al. Dose selection of siltuximab for multicentric Castleman’s disease. Cancer Chemother Pharmacol. 2015;75(5):1037–1045. doi: 10.1007/s00280-015-2720-0. [DOI] [PubMed] [Google Scholar]
- 13.Van Rhee F, Wong RS, Munshi N, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014;15(9):966–974. doi: 10.1016/S1470-2045(14)70319-5. [DOI] [PubMed] [Google Scholar]
- 14.van Rhee F, Fayad L, Voorhees P, et al. Siltuximab, a novel anti-interleukin-6 monoclonal antibody, for Castleman’s disease. J Clin Oncol. 2010;28(23):3701–3708. doi: 10.1200/JCO.2009.27.2377. [DOI] [PubMed] [Google Scholar]