

ABSTRACT
Allorecognition is a key factor in Dictyostelium development and sociality. It is mediated by two polymorphic transmembrane proteins, TgrB1 and TgrC1, which contain extracellular immunoglobulin domains. TgrB1 and TgrC1 are necessary and sufficient for allorecognition, and they carry out separate albeit overlapping functions in development, but their mechanism of action is unknown. Here, we show that TgrB1 acts as a receptor with TgrC1 as its ligand in cooperative aggregation and differentiation. The proteins bind each other in a sequence-specific manner; TgrB1 exhibits a cell-autonomous function and TgrC1 acts non-cell-autonomously. The TgrB1 cytoplasmic tail is essential for its function and it becomes phosphorylated upon association with TgrC1. Dominant mutations in TgrB1 activate the receptor function and confer partial ligand independence. These roles in development and sociality suggest that allorecognition is crucial in the integration of individual cells into a coherent organism.
KEY WORDS: Allorecognition, Cooperative differentiation, Receptor-ligand interaction, Dictyostelium, TgrB1, TgrC1
Highlighted Article: Two polymorphic membrane proteins that mediate allorecognition in Dictyostelium function as a ligand–receptor pair and their binding activates a signaling mechanism that is required for cooperative aggregation and differentiation.
INTRODUCTION
Dictyostelium are soil amoebae that prey on bacteria and propagate to generate large masses of vegetative cells. When food is scarce, the amoebae stop replicating their chromosomes and aggregate into multicellular structures. After 24 h, these aggregates form fruiting bodies containing viable spores and dead stalk cells (Kessin, 2001). The aggregative nature of development and the proximity of genetically distinct clones in nature may lead to social conflicts, which can be limited by allorecognition (Fortunato et al., 2003; Ho et al., 2013; Strassmann et al., 2000).
Different strains of Dictyostelium discoideum distinguish kin from non-kin and cooperate preferentially with related strains (Gruenheit et al., 2017; Ostrowski et al., 2008). Allorecognition is mediated by two membrane proteins, TgrB1 and TgrC1, which are encoded by two adjacent genes on chromosome 3. The genes exhibit similar developmental regulation – the mRNAs are first observed at 4–5 h of development, peak around 9 h and decline thereafter (Benabentos et al., 2009; Rosengarten et al., 2015). The proteins have similar primary structures – long extracellular, glycosylated domains that contain several IPT/TIG motifs, a single transmembrane domain near the C-terminus, and a short cytoplasmic tail. TgrC1 was initially described as the cell–cell adhesion glycoprotein gp150 (Gao et al., 1992; Loomis et al., 1983). The relationship between gp150 and tgrC1, which was previously named lagC, was established several years later (Wang et al., 2000). Identification of TgrB1 (formerly named lagB) as the binding partner of TgrC1 resulted from genetic studies (Benabentos et al., 2009; Hirose et al., 2011) and from biochemistry and cell biology analyses (Chen et al., 2013, 2014). Inactivation of tgrC1 results in perturbed cAMP signaling patterns and cell aggregation into loose aggregates, which is followed by repeated cycles of disaggregation and reaggregation (Dynes et al., 1994; Kibler et al., 2003; Sukumaran et al., 1998). Inactivation of tgrB1 leads to developmental attenuation at the loose aggregate stage, followed by asynchronous progression of 10–20% of the cells into gnarled fruiting bodies. Deletion of both genes results in a phenotype that is nearly identical to that of the tgrB1-null strain (Benabentos et al., 2009). The TgrB1 and TgrC1 proteins are located on the plasma membrane and they bind each other through interactions between specific protein domains in trans, across the gap between adjacent cells (Chen et al., 2013). Binding mediates cell–cell adhesion, but recent studies show that TgrB1 and TgrC1 participate in signaling as well (Hirose et al., 2015; Li et al., 2016). TgrC1 proteins form oligomers in cis, on the surface of one cell. When they bind TgrB1 in trans, on the surface of an adjacent cell, the TgrB1 proteins also cluster (Chen et al., 2014).
The coding regions of tgrB1 and tgrC1 are among the most polymorphic in the D. discoideum genome (Benabentos et al., 2009; Ostrowski et al., 2015). Studies of cooperative aggregation between wild isolates of D. discoideum, collected from locations covering much of the natural range, suggested that genetic distance is directly proportional to the degree of segregation between the strains (Ostrowski et al., 2008). The original study utilized polymorphic microsatellite loci to evaluate genetic distance, but later studies showed that the relationship between kinship and cooperation is fully explained by the degree of sequence dissimilarity between the tgrB1 and tgrC1 alleles of the wild strains. Moreover, these studies showed that deletion of tgrB1 and tgrC1 from the laboratory wild-type strain AX4 (tgrB1–tgrC1–) caused segregation from the wild type (Benabentos et al., 2009). Subsequent studies utilized pairs of tgrB1 and tgrC1 alleles from strains QS4, QS31, QS38 and QS45. These were strains from the original study (Ostrowski et al., 2008) that exhibited the highest degree of sequence difference from the laboratory strain AX4 and from each other (Hirose et al., 2011). In those studies, the resident alleles of tgrB1 and tgrC1 were deleted from AX4 and replaced with matching sets of tgrB1 and tgrC1 from one of the wild isolates (e.g. AX4 tgrB1C1QS4 are AX4 cells in which the resident alleles of tgrB1 and tgrC1 have been replaced with a matching pair of alleles from the wild isolate QS4). The wild-isolate alleles allowed these genetically engineered cells to recognize themselves as self. They also recognized the ‘donor’ wild isolates as self and cooperated with them, and they failed to recognize the AX4 parental strain as self. These studies indicated that the presence of a matching pair of tgrB1 and tgrC1 alleles is necessary and sufficient for cooperative aggregation (Hirose et al., 2011). Measurements of protein–protein interactions between pairs of TgrB1 and TgrC1 from four other wild isolates indicate that segregation is inversely proportional to TgrB1–TgrC1 binding (Gruenheit et al., 2017).
Strain segregation is measured by mixing and starving differentially labeled cells (with RFP or GFP) at known proportions. Initially, all strain combinations co-aggregate in response to the chemoattractant cAMP. Strains that carry compatible TgrB1–TgrC1 pairs remain mixed and produce aggregates and fruiting bodies that contain differentially labeled cells at the initial mixing proportions. The behavior of strains that carry incompatible TgrB1–TgrC1 pairs depends on the mixing ratio (Hirose et al., 2015; Ho and Shaulsky, 2015). If an incompatible strain is present at 5% or more of the population, the cells clump with their kin and segregate from the non-kin cells. At lower proportions, the incompatible cells have a lower probability of finding matching partners. These single incompatible cells fail to establish proper polarization and do not participate in the cooperative circular movement that characterizes post-aggregative cells. They also fail to express cell-type-specific markers (e.g. ecmA-GFP, cotB-GFP), which can be quantified by flow cytometry (Hirose et al., 2015).
The transition from unicellular to multicellular development requires interactions between matching TgrB1 and TgrC1. Cells that carry mismatched tgrB1 and tgrC1 alleles behave like the tgrC1-null strain (Li et al., 2015). Genetic screens for suppressors of the tgrB1-tgrC1 mismatch or tgrC1 deletion suggested that TgrB1 and TgrC1 participate in signal transduction and that allorecognition and development are mediated by partly overlapping but distinct pathways (Li et al., 2015, 2016; Wang and Shaulsky, 2015). Some suppressors found by chemical mutagenesis are dominant alleles of tgrB1 (Li et al., 2016), suggesting that TgrB1 may function as a receptor in the signal transduction pathways. We therefore hypothesize that TgrB1 and TgrC1 function as a receptor–ligand pair.
Here, we provide experimental support for the hypothesis by showing that expression of a compatible allele of tgrB1 but not tgrC1 allows rare, incompatible cells to cooperate with the majority. Expressing a compatible allele of tgrC1 but not tgrB1 in the majority cells facilitates cooperation with rare cells. We also show that binding with compatible TgrC1 in trans induces phosphorylation of the TgrB1 cytoplasmic tail and that dominant tgrB1 alleles behave as if the receptor activity of TgrB1 has been constitutively activated with respect to cooperative development and differentiation.
RESULTS
Binding specificity
Compatible TgrB1 and TgrC1 proteins bind each other specifically in vitro (Gruenheit et al., 2017). These studies were done with bacterially expressed proteins and did not include most of the alleles we have employed in our studies (Benabentos et al., 2009; Hirose et al., 2011), so we tested five pairs of tgrB1 and tgrC1 alleles by purifying the extracellular domains of the proteins from a D. discoideum expression system. We incubated 5 different TgrB1 types with five different TgrC1 types and found binding only in matching pairs of TgrB1 and TgrC1 (Fig. S1). These results support the receptor–ligand hypothesis because they show that the presumed binding is indeed specific. They also validate and extend the previous study (Gruenheit et al., 2017).
Receptor function
To test whether TgrB1 and TgrC1 function as a receptor–ligand pair in allorecognition, we utilized two assays – cooperative aggregation and prespore differentiation. When a few cells are mixed with many incompatible cells, they initially co-aggregate with the majority, but then they stop cooperating and segregate into discrete clumps (Hirose et al., 2015). We reasoned that providing the incompatible minority cells with a compatible allorecognition receptor would allow them to mix evenly with the majority cells (Fig. 1A). In the controls, mixing 90% unlabeled AX4 cells with 5% compatible RFP-labeled AX4 cells and 5% incompatible GFP-labeled tgrB1C1QS31 cells resulted in even distribution of the compatible red cells and clumping (segregation) of the incompatible green cells (Fig. 1B). Expression of the AX4 allele of tgrC1 in the minority cells (tgrB1C1QS31tgrC1AX4) had no discernible effect on segregation (Fig. 1C), but expression of the AX4 allele of tgrB1 in the minority cells (tgrB1C1QS31tgrB1AX4) had a marked effect (Fig. 1D). The green and red cells were evenly mixed, suggesting that TgrB1, but not TgrC1, functions as a receptor in the cooperative aggregation aspect of allorecognition. To quantify segregation, we calculated the variance of the red- and green-labeled cell distributions in each image and the respective statistical differences. The results, shown as a dendrogram below the images, support the conclusions (Fig. 1B–D). We note that the tgrB1C1QS31tgrB1AX4 cells began to segregate from the majority cells upon prolonged incubation (Fig. S2).
Fig. 1.
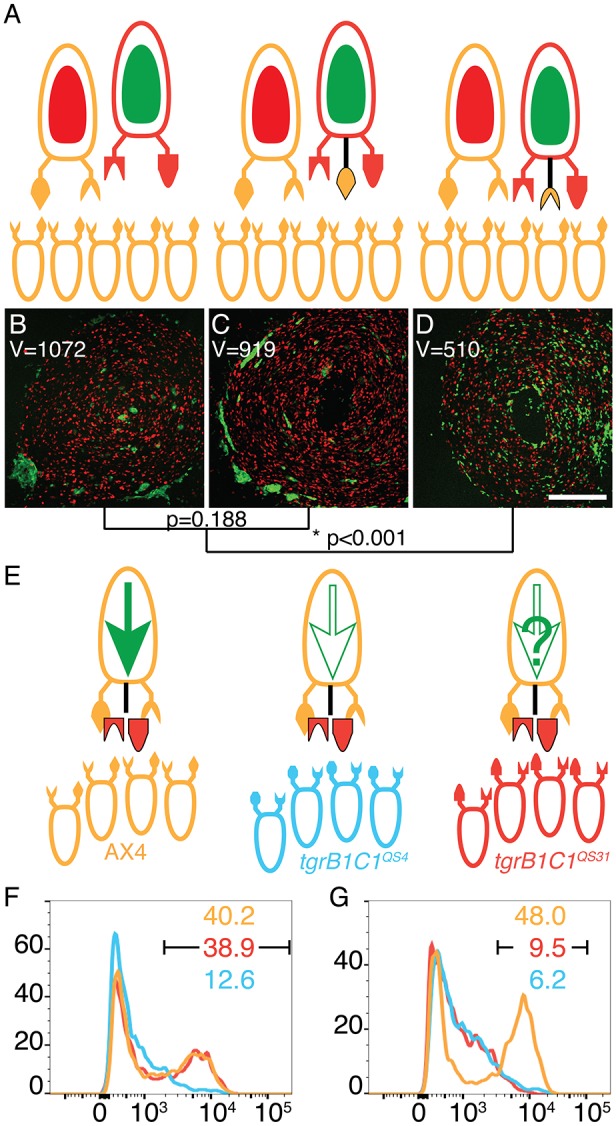
TgrB1 functions as an allotype-specific receptor. (A) Ovoids represent cells, protrusions represent TgrB1 and TgrC1 proteins, and colors represent allotypes: tan, AX4; red, tgrB1C1QS31. Black protrusions represent extra alleles. Internal green and red ovoids represent constitutive GFP and RFP expression, respectively. The larger, single cells on top represent the minority and the smaller cells at the bottom represent the majority strains. (B–D) Cooperative aggregation of mixes of 90% unlabeled AX4 cells with 5% RFP-labeled AX4 (control) and 5% GFP-labeled test cells: tgrB1C1QS31 (B), tgrB1C1QS31tgrC1AX4 (C) and tgrB1C1QS31tgrB1AX4 (D). The spatial distribution variance (V) is shown inside each frame. Comparisons between the variances are shown as a dendrogram below the images with P-values calculated from F-tests; *P<0.001. Scale bar: 0.3 mm. (E) Cooperative differentiation with mixes of 0.2% AX4 cotB-GFP cells expressing an extra allele of either tgrB1QS31 or tgrC1QS31 with 99.8% unlabeled cells of different allotypes: tan, AX4; blue, tgrB1C1QS4; red, tgrB1C1QS31. A full green arrow inside the cell represents cotB-GFP expression; empty arrow indicates no expression. We evaluated GFP fluorescence levels by flow cytometry and plotted the results as histograms for tgrB1QS31 (F) and tgrC1QS31 (G). The x-axes represent fluorescence intensify (arbitrary units) and the y-axes, the number of events. Black bars inside the histograms indicate the GFP-positive populations and the respectively colored numbers indicate the fraction (%) of GFP-positive cells.
To test the cooperative differentiation aspect of allorecognition, we measured expression of the prespore-specific marker cotB-GFP in the minority cells. This assay requires mixing of majority and minority cells at more extreme ratios to ensure that the minority cells will not form small local aggregates in which they would differentiate (Hirose et al., 2015). Because the GFP-positive cells are rare (no more than 0.2% of the population), their presence was quantified by flow cytometry. We mixed 99.8% unlabeled cells with 0.2% cotB-GFP cells that were constructed to express the resident tgrB1 and tgrC1 alleles of AX4 as well as a non-matching test allele from QS31 – either AX4 tgrB1QS31cotB-GFP or AX4 tgrC1QS31cotB-GFP (Hirose et al., 2011). For the majority cells, we used two control strains and one test strain (Fig. 1E). AX4 is a positive control that should support cotB-GFP expression in the minority cells regardless of the additional non-matching test allele, because all the minority cells express tgrB1 and tgrC1 from AX4. The negative control is a strain in which the resident tgrB1 and tgrC1 alleles were replaced with alleles from QS4 (tgrB1C1QS4). This strain should not support cotB-GFP expression in the minority cells, regardless of the non-matching test allele, because its allotype is incompatible with both AX4 and QS31 (Hirose et al., 2011). In the test case, the majority cells are tgrB1C1QS31. We found that when the minority cells carried the tgrB1QS31 allele, they expressed cotB-GFP when mixed with AX4 (positive control, tan), but not with tgrB1C1QS4 (negative control, blue). They expressed cotB-GFP at positive control levels when mixed with tgrB1C1QS31 (Fig. 1F, red). When the minority cells expressed tgrC1QS31, they only expressed cotB-GFP when mixed with AX4 (positive control, tan), but not with either tgrB1C1QS4 or tgrB1C1QS31 (Fig. 1G, blue and red). These results suggest that TgrB1, but not TgrC1, functions as a receptor in the cooperative aggregation and differentiation aspects of allorecognition.
Ligand function
We tested whether TgrC1 functions as a ligand using the same system, but we expressed the test alleles in the majority cells. In the segregation assay, we used both 5% RFP-labeled AX4 and 5% GFP-labeled tgrB1C1QS31 as the minority cells. In the controls, we used 90% unlabeled AX4 or unlabeled tgrB1C1QS31 as the majority (Fig. 2A). In these experiments, we expected the compatible minority cells to be evenly distributed among the unlabeled counterparts and the incompatible minority cells to segregate and become clumped. Indeed, we found that the GFP-labeled tgrB1C1QS31 were clumped when mixed with a majority of unlabeled AX4 cells, whereas the compatible minority RFP-labeled AX4 cells were evenly dispersed (Fig. 2B). The findings were reversed when the majority cells were tgrB1C1QS31 (Fig. 2C). In the test mixes, we used either tgrB1C1QS31tgrB1AX4 or tgrB1C1QS31tgrC1AX4 as the majority (Fig. 2A). We expected the GFP-labeled tgrB1C1QS31 cells to be evenly dispersed in either mix, because they share the majority allotype, and this was indeed the case (Fig. 2D,E). We predicted that the RFP-labeled AX4 cells would be clumped when mixed with a majority of cells carrying the AX4-type receptor, but evenly dispersed among the cells carrying the AX4-type ligand. We found that when the majority cells were tgrB1C1QS31tgrB1AX4, the RFP-labeled AX4 cells were clumped (Fig. 2D), suggesting that TgrB1 does not function as a ligand. When the majority cells were tgrB1C1QS31tgrC1AX4, the RFP-labeled AX4 cells were evenly dispersed (Fig. 2E), suggesting that TgrC1 functions as a ligand. Quantitative analysis of distribution variances shows that the two control experiments (Fig. 2B,C) and the tgrB1C1QS31tgrB1AX4 experiment (Fig. 2D) were similar to one another and the tgrB1C1QS31tgrC1AX4 experiment (Fig. 2E) was significantly different. These results are shown as a dendrogram below the images (Fig. 2B–E).
Fig. 2.

TgrC1 functions as an allotype-specific ligand. (A–E) Cooperative aggregation of mixes of 90% unlabeled cells with 5% RFP-labeled AX4 and 5% GFP-labeled tgrB1C1QS31 cells. The unlabeled majority cells were: AX4 (B), tgrB1C1QS31 (C), tgrB1C1QS31tgrB1AX4 (D) and tgrB1C1QS31tgrC1AX4 (E). The spatial distribution variance (V) is shown inside each frame. Comparisons between the variances are shown as a dendrogram below the images with P-values calculated from F-tests; *P<0.001. Scale bar: 0.2 mm. (F) Cooperative differentiation of mixes of 0.2% AX4 cotB-GFP cells (minority, large cell on top) with 99.8% unlabeled host cells from 4 different strains as illustrated on the right. GFP fluorescence levels were assessed by flow cytometry results plotted as histograms: AX4 (positive control, tan), tgrB1C1QS31 (negative control, red) and tgrB1C1QS31 expressing an extra allele of either tgrB1AX4 (green) or tgrC1AX4 (blue). The x-axis represents fluorescence intensify (arbitrary units) and the y-axis the number of events. The black bar indicates the GFP-positive populations and the respectively colored numbers indicate the fraction (%) of GFP-positive cells.
We also tested whether TgrC1 functions as a ligand in the cooperative differentiation aspect of allorecognition. In this case, the 0.2% minority cotB-GFP-carrying cells were AX4. The majority cells were the same four strains described in Fig. 2A. The flow cytometry data in Fig. 2F show that cotB-GFP was expressed in the positive control mix with AX4 (tan) and was nearly undetectable in the negative control mix with tgrB1C1QS31 (red). Expression was also nearly undetectable in the mix with tgrB1C1QS31tgrB1AX4 (green), but evident in the mix with tgrB1C1QS31tgrC1AX4 (blue, Fig. 2F). These results further support the hypothesis that TgrC1 functions as a ligand and TgrB1 does not.
The role of the TgrB1 cytoplasmic domain
TgrB1 and TgrC1 are single-pass transmembrane glycoproteins whose long, extracellular domains contain IPT/TIG domains. The cytoplasmic domain lengths are 77 amino acids (aa) in TgrB1 and 16 aa in TgrC1 (Benabentos et al., 2009). Since TgrB1 functions as a receptor, its cytoplasmic domain might participate in signal transduction. To test that hypothesis, we tagged the C-terminal domain with an HA-epitope and deleted most of the cytoplasmic domain between amino acids 828 and 902 (Fig. S3A). We generated strains in which the resident tgrB1AX4 allele was replaced with either tgrB1AX4-HA or tgrB1AX4Δ828-902-HA. The developmental morphologies of the parental strain, AX4, (Fig. S3B) and the tgrB1AX4-HA strain (Fig. S3C) were essentially indistinguishable – both formed well-proportioned fruiting bodies at 24 h of development, suggesting that the HA tag did not compromise the protein function. The tgrB1AX4Δ828-902-HA strain did not form fruiting bodies – its development was arrested at the mound stage (Fig. S3D), much like the tgrB1-null strain (Benabentos et al., 2009). The sporulation efficiencies of these strains were consistent with the morphology – the tgrB1AX4-HA strain sporulated with similar efficiency to AX4, whereas the tgrB1AX4Δ828-902-HA strain exhibited reduced sporulation (Fig. S3E), much like the tgrB1-null strain (Benabentos et al., 2009). To test whether the failure of tgrB1AX4Δ828-902-HA to complement the null allele was due to gross protein mislocalization, we fractionated the cells into membranes and soluble fractions (Wang et al., 1999). We found that both TgrB1–HA and TgrB1AX4Δ828-902–HA proteins were associated with the insoluble fraction (Fig. S3F), suggesting that both were membrane localized. We also found that the tgrB1AX4-HA strain expressed cotB-GFP at near-wild-type levels, whereas the tgrB1AX4Δ828-902-HA strain did not express the marker when developed in pure populations (Fig. S3G), further supporting the hypothesis that the cytoplasmic domain is essential for TgrB1 function.
We then tested the activity of the cytoplasmic domain of TgrB1 in cooperative differentiation. We mixed 90% of unlabeled AX4 cells with 5% GFP-labeled AX4 cells (control) and 5% RFP-labeled cells (test) carrying either one of the HA-tagged tgrB1 alleles (Fig. S4A). In both cases, the test cells were evenly mixed with their AX4 counterparts and the respective variances suggest no statistically significant difference (Fig. S4B,C). In the cooperative differentiation assay, the two cotB-GFP-labeled strains behaved differently (Fig. S4D) – cells carrying the intact tgrB1AX4-HA allele expressed cotB-GFP (green) whereas cells carrying the tgrB1AX4Δ828-902-HA deletion allele did not (purple). These results suggest that the cytoplasmic domain of TgrB1 is essential for its function in cooperative differentiation, but may be dispensable for cooperative aggregation. To test the latter possibility more rigorously, we transformed tgrB1AX4-HA and tgrB1AX4Δ828-902-HA into tgrB1C1QS31 and examined cooperative aggregation and differentiation. In the cooperative aggregation assay (Fig. 3A), the RFP-labeled tgrB1C1QS31tgrB1AX4-HA cells (positive control) were evenly dispersed among the majority AX4 cells (Fig. 3B), whereas the RFP-labeled tgrB1C1QS31tgrB1AX4Δ828-902-HA cells segregated from the majority cells (Fig. 3C). The quantitative analysis indicates that the difference was statistically significant. In the cooperative differentiation assay, the tgrB1C1QS31tgrB1AX4-HA cells expressed cotB-GFP as expected (positive control), but the tgrB1C1QS31tgrB1Δ828-902-HA cells did not express the prespore marker (Fig. 3D). These results suggest that the cytoplasmic tail of TgrB1 is required for both activities.
Fig. 3.

The cytoplasmic domain of TgrB1 is required for cooperative aggregation and differentiation. (A–C) Ovoids represent cells, protrusions represent TgrB1 and TgrC1 proteins, and colors represent allotypes: AX4 (tan) and tgrB1C1QS31 (red). Black protrusions represent extra alleles: tgrB1AX4-HA (green) and tgrB1AX4Δ828-902-HA (purple). Cooperative aggregation of mixes of 90% unlabeled AX4 cells with 5% GFP-labeled AX4 and 5% RFP-labeled tgrB1C1QS31tgrB1AX4-HA (B) or tgrB1C1QS31tgrB1AX4Δ828-902-HA (C). The spatial distribution variance (V) is shown inside each frame. A comparison between the variances is shown below the images with a P-value calculated from F-tests; *P<0.000001. Scale bar: 0.3 mm. (D) Cooperative differentiation of mixes of 0.2% cells carrying cotB-GFP with 99.8% unlabeled AX4 cells. Protrusions represent TgrB1 and TgrC1 and the colors represent alleles as above. Full green arrow inside the cells shows cotB-GFP expression; empty arrow indicates no expression. We evaluated GFP fluorescence levels by flow cytometry and plotted the results as histograms. The x-axis represents fluorescence intensify (arbitrary units) and the y-axis the number of events. The black bar indicates the GFP-positive populations and the numbers indicate the respective fractions (%) of GFP-positive cells.
Phosphorylation of TgrB1
The cytoplasmic tails of some receptors become phosphorylated in response to ligand binding, so we tested whether that was also the case for TgrB1. We constructed a TgrB1 expression vector in which the signal peptide was followed by two Myc epitopes (TgrB1myc2AX4), such that the mature protein would carry the epitope tags at the N-terminus (Fig. 4A). Introducing the tagged allele into tgrB1-null cells complemented the developmental defect (data not shown). We developed the cells, collected lysates at different developmental times, and used anti-Myc antibodies conjugated to beads to pull down the tagged TgrB1 protein. Western blot analysis with anti-TgrB1 antibodies revealed that the protein was present between 8 and 20 h of development and analysis with anti-phosphoserine antibodies revealed that TgrB1 was phosphorylated (Fig. 4B). We then analyzed enriched fractions of TgrB1 protein by mass spectrometry and found that serine 845 was phosphorylated (Fig. S5A). We found no evidence for phosphorylation of other amino acids in the cytoplasmic domain. We then mutated serine 845 to alanine and used an allele replacement strategy to test the consequences. We found that phosphorylation of TgrB1S845A was markedly reduced compared with the wild type (Fig. 4C), suggesting that phosphorylation of serine 845 is responsible for the signal we observed. Replacing the wild-type allele with the tgrB1S845A allele had no gross effects on developmental morphology (Fig. S5B–E). We also tested the consequences of replacing wild-type tgrB1 with the tgrB1S845A allele on cooperative aggregation (Fig. S5F). We found that the mutant cells co-aggregated well with a majority of parental AX4 cells (Fig. S5G), segregated well from a majority of incompatible tgrB1C1QS31 cells (Fig. S5H) and behaved distinctively differently compared with minority (5%) tgrB1C1QS31 cells when mixed with the parental AX4 cells (Fig. S5I). Moreover, we tested the effect of the tgrB1S845A allele on differentiation (Fig. S5J) in mixing with compatible AX4 cells and with incompatible tgrB1C1QS31 cells and found that the cotB-GFP reporter was expressed in the former but not in the latter mix (Fig. S5K). These findings suggest that phosphorylation is not essential for TgrB1 function.
Fig. 4.

Phosphorylation of TgrB1 on serine 845. (A) Schematic representation of Myc-tagged TgrB1. Cyan bars represent the signal peptide at the N-terminus and the transmembrane domain near the C-terminus; magenta bars represent the IPT/TIG domains and the yellow bar represents the cytoplasmic domain. Amino acid numbers are shown below the illustration, which is drawn to scale. The triangle near the N-terminus represents an insertion of two Myc epitopes immediately after the signal peptide (not to scale). The black line represents serine 845. (B) Western blot analysis of protein pulled down with anti-Myc antibodies from cells expressing TgrB1myc2AX4 developed for the indicated times (hours). (C) Western blot analysis of cells expressing either TgrB1myc2AX4 (WT) or the mutant TgrB1myc2AX4S845A (S845A) developed for 12 h.
If binding to TgrC1 induces phosphorylation of the matching TgrB1, we should be able to detect phosphorylation only in cells that express matching sets of TgrB1 and TgrC1. To test that hypothesis, we introduced the tgrB1myc2AX4 allele into tgrB1-null cells, where the resulting TgrB1myc2AX4 protein would have a matching TgrC1 protein and would be expected to become phosphorylated (Fig. 5A). We introduced the tgrB1myc2AX4 allele into tgrB1–tgrC1– cells, where the TgrB1 protein does not have any TgrC1 protein to interact with and should therefore not become phosphorylated. We also introduced the tgrB1myc2AX4 allele into the double-gene replacement strains tgrB1C1QS31 and tgrB1C1QS4. In both cases, the cells are expected to develop normally, because they have matching sets of untagged TgrB1 and TgrC1 (Hirose et al., 2015), but the tagged receptor would not have a matching ligand and would therefore remain un-phosphorylated, according to our hypothesis (Fig. 5A). We developed the cells for 12 h, pulled down the tagged TgrB1 protein and analyzed it by western blotting. The anti-TgrB1 antibody revealed TgrB1 in all the samples (Fig. 5B). The anti-phosphoserine antibody revealed TgrB1 phosphorylation in the first case, where a matching TgrC1 was present. When TgrC1 was absent (B1–C1– tgrB1myc2AX4) and when TgrC1 was not matching (B1C1QS31 tgrB1myc2AX4 and B1C1QS4 tgrB1myc2AX4), phosphorylation of the tagged TgrB1 was greatly reduced (Fig. 5B).
Fig. 5.
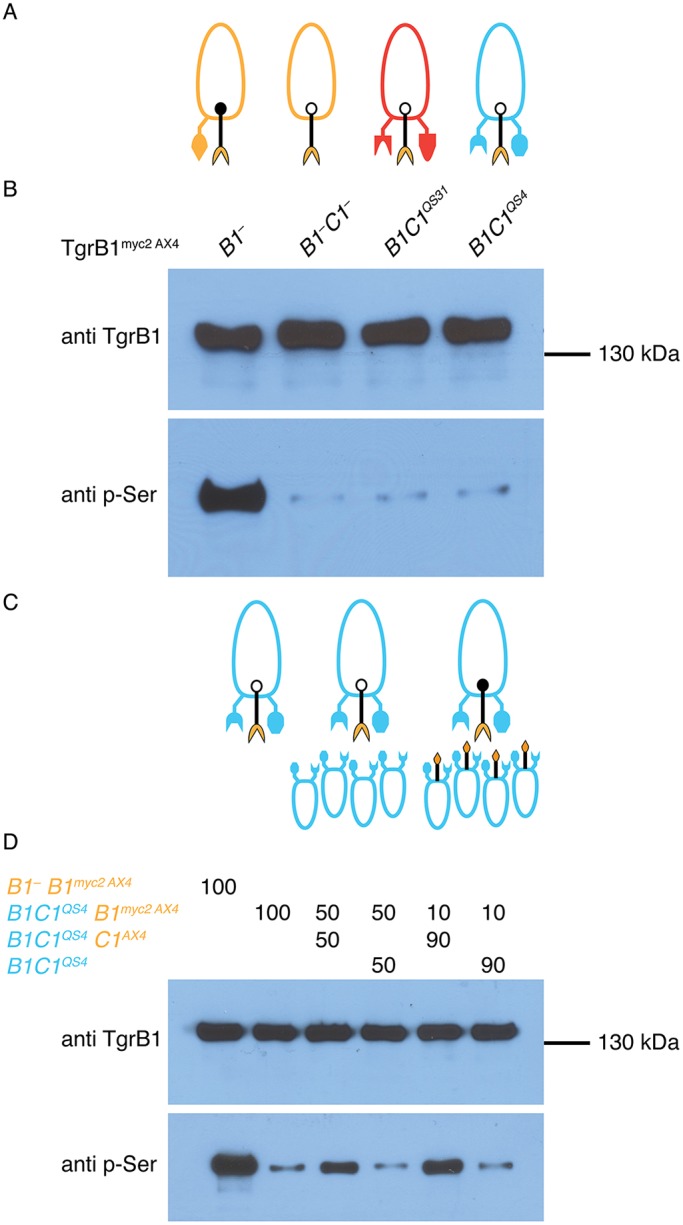
Phosphorylation of TgrB1 depends on association with a matching TgrC1. (A) Ovoids represent cells, protrusions represent TgrB1 and TgrC1 proteins, and colors represent allotypes: AX4 (tan), tgrB1C1QS31 (red), tgrB1C1QS4 (blue). The black protrusions represent the extra TgrB1myc2AX4 protein. A solid dot on TgrB1 represents phosphorylation; open dot indicates no phosphorylation. (B) Western blot analysis of proteins pulled down with anti-Myc antibodies from tgrB1-null (B1–), tgrB1-null, tgrC1-null (B1–C1–), tgrB1C1QS31 (B1C1QS31) and tgrB1C1QS4 (B1C1QS4) cells. (C,D) tgrB1C1QS4tgrB1myc2AX4 cells in a pure population and in mixes with tgrB1C1QS4 or with tgrB1C1QS4tgrC1AX4 (C) were used for western blot analysis (D). The mixing ratios (%) are indicated above the lanes. We included the tgrB1–tgrB1myc2AX4 strain as a positive control (left lane).
To test whether TgrB1myc2AX4 was not phosphorylated in the latter case for reasons other than lack of a matching ligand, we mixed the tgrB1C1QS4tgrB1myc2AX4 cells with tgrB1C1QS4tgrC1AX4 cells at different proportions. In this case, the two cells would develop together because they have matching TgrB1 and TgrC1 proteins (Hirose et al., 2015), and the TgrB1myc2AX4 protein in the test cells would have a matching TgrC1 from the mixing partner. We used mixing with tgrB1C1QS4 and no mixing at all as negative controls (Fig. 5C). Western blot analysis of 12 h cell lysates with anti-TgrB1 antibodies showed that the tagged protein was present in all the samples (Fig. 5D). The anti-phosphoserine antibody showed that TgrB1 was phosphorylated when we mixed the tgrB1C1QS4tgrB1myc2AX4 cells with tgrB1C1QS4tgrC1AX4 cells at either 50:50 or 10:90 ratio (Fig. 5D). When we developed the tgrB1C1QS4tgrB1myc2AX4 cells in a pure population, or mixed with tgrB1C1QS4 cells, phosphorylation of the tagged TgrB1 was greatly reduced. We repeated these experiments with a second set of alleles, using tgrB1C1QS31 instead of tgrB1C1QS4, and found essentially identical results (Fig. S6A,B). We also found that expressing two non-matching copies of TgrC1 in the mixing partner did not induce phosphorylation of TgrB1myc2AX4 in the reporter strain (Fig. S6C,D), suggesting that the effects we observed above were not due to TgrC1 dosage. These results suggest that TgrB1, which is displayed on the membrane of one cell, becomes phosphorylated when it binds a matching TgrC1 protein displayed on the membrane of an adjacent cell.
To test if soluble TgrC1 could induce phosphorylation of TgrB1, we constructed a strain in which tgrB1 and tgrC1 were deleted and tgrB1myc2AX4 was expressed from the tgrB1 promoter (Fig. S6E). These cells expressed the TgrB1 protein during development, but no phosphorylation was observed without additional treatment. When we added soluble recombinant His7–TgrC1 protein, the Myc-tagged TgrB1 protein became phosphorylated. When we also added antibodies against the His tag, we observed increased phosphorylation (Fig. S7). This finding suggested that the antibody, which is divalent, could have induced phosphorylation by clumping the soluble TgrC1 proteins and thus inducing clumping of TgrB1, as previously suggested (Chen et al., 2014). To test that possibility further, we treated the cells with an antibody against the Myc epitope such that the Myc-tagged TgrB1 proteins would be induced to clump without TgrC1 binding. We found that this treatment alone was sufficient to induce TgrB1 phosphorylation (Fig. S6E), suggesting that oligomerization of TgrB1 may be sufficient for activating the receptor function. As controls, we incubated the cells with purified soluble His7–TgrB1 alone or with purified soluble His7–TgrB1 along with the anti His-tag antibodies. In either case, we found no phosphorylation, suggesting that the presence of the antibodies or irrelevant soluble proteins were not the cause of phosphorylation.
Dominant alleles of tgrB1
Previously, we identified mutations in tgrB1 that suppressed the developmental defects caused by an engineered mismatch between tgrB1 and tgrC1 (Li et al., 2016). These mutations are dominant, so we hypothesized that they might have constitutively activated the TgrB1 receptor activity. We chose three alleles to test this hypothesis – two with mutations in the first IPT/TIG domain, G275D and G307D, and one with a mutation in the cytoplasmic domain, L846F (Fig. 6A). We expressed the alleles separately in cells lacking both tgrB1 and tgrC1 (tgrB1–tgrC1–) and tested developmental morphology and sporulation in pure populations.
Fig. 6.
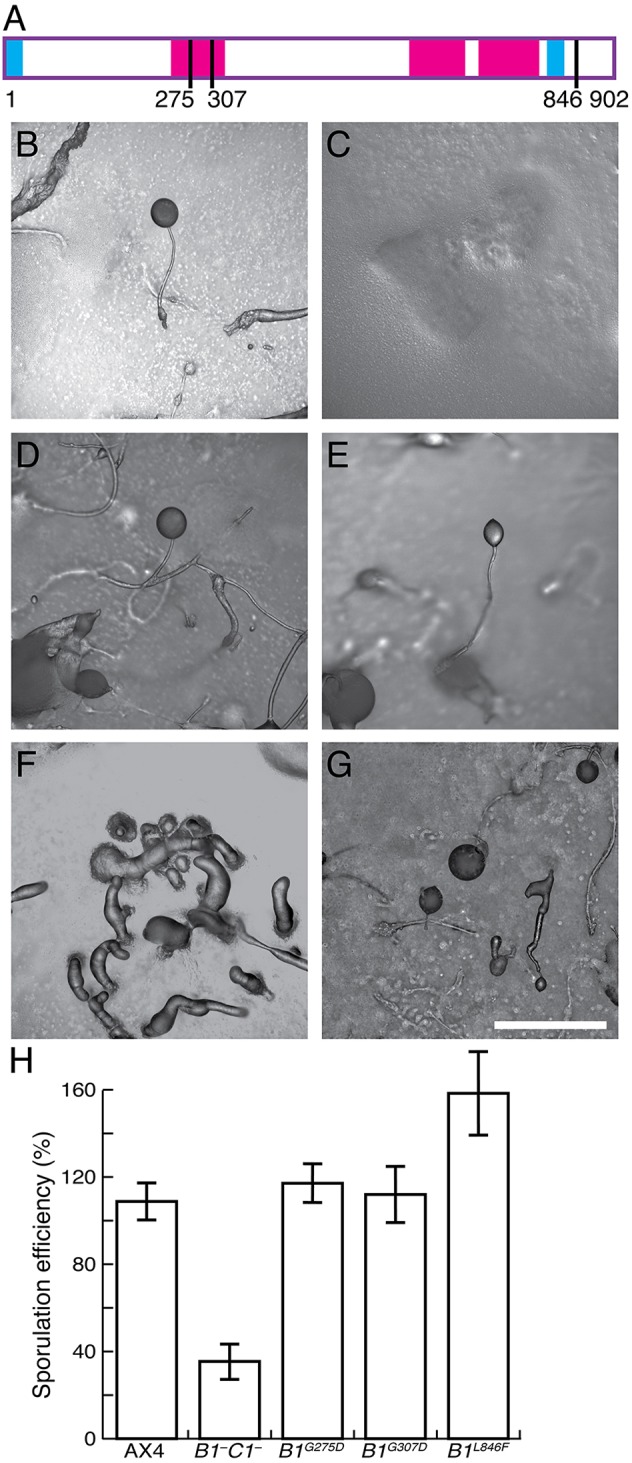
Dominant alleles of tgrB1 bypass the need for tgrC1 in development. (A) Schematic representation of TgrB1. Cyan bars represent the signal peptide at the N-terminus and the transmembrane domain near the C-terminus; magenta bars represent the IPT/TIG domains; black lines represent mutations: G275D, G307D and L846F. Amino acid numbers are shown below the illustration, which is drawn to scale. (B–G) Developmental morphologies of AX4 (B), tgrB1–tgrC1– untransformed (C) or transformed with tgrB1G275D (D), tgrB1G307D (E) and tgrB1L846F (F,G). Scale bar: 0.5 mm. (H) Sporulation efficiencies of the strains as indicated (x-axis) as the percentage of cells that became spores (y-axis); data are means±s.e.m. of three independent replicates.
When Dictyostelium cells grow in association with bacteria on nutrient agar, the amoebae consume the nearby bacteria and form a plaque in the bacterial lawn. As they do so, they starve in the center of the plaque and continue to grow at the edge. Consequently, the amoebae develop in an asynchronous manner, allowing observation of several developmental stages at once. Under these conditions, wild-type AX4 cells exhibited mainly fruiting bodies (Fig. 6B) and the tgrB1–tgrC1– cells exhibited loose aggregates (Fig. 6C), as expected (Hirose et al., 2011). Cells carrying tgrB1G275D (Fig. 6D) or tgrB1G307D (Fig. 6E) developed into fruiting bodies, as seen with AX4. Cells carrying the tgrB1L846F allele developed into clumps of multiple fingers (Fig. 6F), from which small fruiting bodies emerged (Fig. 6G). The sporulation efficiencies of the respective strains were consistent with the morphology (Fig. 6H). AX4 exhibited nearly 100% sporulation efficiency and the tgrB1–tgrC1– cells exhibited much lower sporulation efficiency, as expected (Hirose et al., 2011). The G275D and G307D mutations restored sporulation to wild-type levels, despite the absence of tgrC1 (Li et al., 2016), and the L846F mutation resulted in a somewhat elevated sporulation efficiency (Fig. 6H).
To test the effect of the dominant mutations on cooperative differentiation, we constructed tgrB1–tgrC1– cells that expressed either one of the three dominant tgrB1 alleles and the prespore reporter cotB-GFP. We mixed 0.2% of the cotB-GFP cells with either 99.8% unlabeled AX4, unlabeled tgrB1C1QS31, or unlabeled tgrB1C1QS38. In the control, we used 0.2% AX4 cells carrying the cotB-GFP reporter (Fig. 7A). In the experiment, we used cells carrying one of the mutant tgrB1 alleles and no tgrC1 (Fig. 7B). As expected (Hirose et al., 2015), the controls showed that labeled AX4 cells expressed GFP when mixed with a majority of compatible unlabeled AX4 cells, but not with a majority of incompatible cells (either tgrB1C1QS31 or tgrB1C1QS38) (Fig. 7C). In the experiments (Fig. 7D–F), the minority, activated-tgrB1 cells expressed cotB-GFP at high levels when mixed with AX4 (tgrB1G275D in Fig. 7D, tgrB1G307D in Fig. 7E and tgrB1L846F in Fig. 7F). These cells expressed GFP at intermediate levels upon mixing with a majority of incompatible unlabeled tgrB1C1QS31 or tgrB1C1QS38 cells (Fig. 7D–F). We also confirmed that the parental tgrB1–tgrC1– cotB-GFP cells did not express the prespore reporter when mixed with either one of the three majority strains (Fig. S7). These results suggest that the dominant mutations have partially activated the receptor function of TgrB1 with respect to developmental morphology and cell-type differentiation.
Fig. 7.
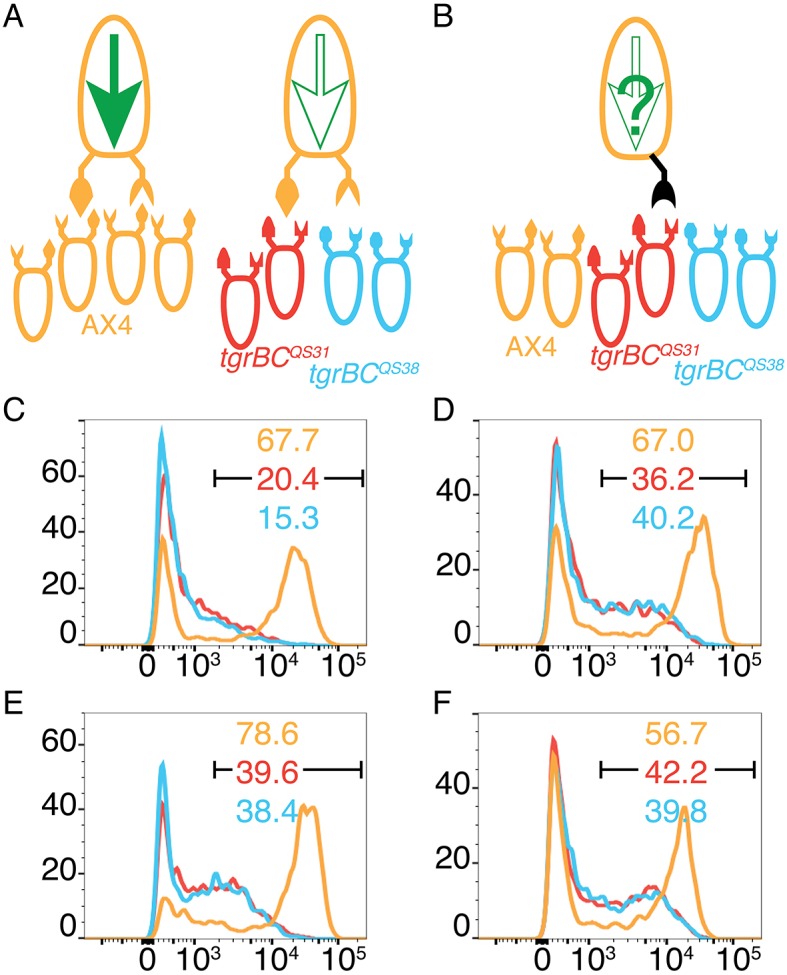
Dominant alleles of tgrB1 activate the receptor function. (A,B) Mixes of 0.2% cells carrying cotB-GFP and 99.8% unlabeled cells of different allotypes: AX4 (tan), tgrB1C1QS31 (red), tgrB1C1QS38 (blue). Ovoids represent cells, protrusions represent TgrB1 and TgrC1 proteins. The larger, single cells on top represent the minority and the smaller, bottom cells represent the majority strains (each tested separately). A full green arrow inside the cell represents cotB-GFP expression; empty arrows indicate no expression. (A) Controls made by mixing with compatible AX4 cells results in cotB-GFP expression and mixing with either one of the incompatible strains results in no expression. (B) The black protrusion represents expression of one of the tgrB1 dominant alleles in the absence of resident tgrB1 or tgrC1. (C–F) GFP fluorescence levels assessed by flow cytometry and plotted as histograms. The x-axes represent fluorescence intensity (arbitrary units) and the y-axes, the number of events. The black bar indicates the GFP-positive populations and the numbers indicate the respective fractions (%) of GFP-positive cells. The majority cells are as indicated by the line colors. The minority cotB-GFP cells carry the dominant tgrB1 alleles: tgrB1G275D (D), tgrB1G307D (E) or tgrB1L846F (F).
DISCUSSION
Allorecognition in D. discoideum is essential for development and differentiation. The expression patterns of tgrB1 and TgrC1, the phenotypes of the null strains and the interactions between incompatible strains suggest that allorecognition is effective during the transition from unicellular to multicellular development (Benabentos et al., 2009; Hirose et al., 2011, 2015; Ho and Shaulsky, 2015). Previous experiments suggested that TgrB1 and TgrC1 are involved in signaling but did not provide mechanistic information. The data shown here suggest that TgrB1 and TgrC1 function as a receptor–ligand pair and that the receptor, TgrB1, mediates signal transduction in both allorecognition and differentiation.
Support for the hypothesis includes biochemical, genetic and cellular evidence. Binding specificity of matching TgrB1 and TgrC1 proteins has been shown with proteins expressed in bacteria (Gruenheit et al., 2017). We tested a different set of alleles and expressed them in D. discoideum to approximate the native protein structure, as these proteins are naturally glycosylated (Gao et al., 1992; Li et al., 2015; Wang et al., 2000). Our finding that matching pairs of TgrB1 and TgrC1 bind in vitro validate the previous study (Gruenheit et al., 2017), which was somewhat inconsistent with another study where extensive refolding of bacterially expressed proteins was required to observe binding (Chen et al., 2013). More importantly, all these studies demonstrate binding specificity, which is essential in receptor–ligand interactions. Our experiments showed no binding between non-matching proteins whereas the bacterially expressed proteins exhibited some cross-binding (Gruenheit et al., 2017). This difference could be the result of the choice of alleles, different experimental conditions or the effect of glycosylation on binding specificity. Although we may have missed weak binding between non-matching proteins, we do not think it has a major function in allorecognition because the respective null strains behave like incompatible strains in chimerae (Benabentos et al., 2009; Hirose et al., 2011; Li et al., 2016).
The most compelling evidence in support of the receptor–ligand hypothesis stems from the merodiploid experiments (most D. discoideum strains are haploid, so transformants that carry two alleles of a given gene are considered merodiploid). Minority incompatible cells fail to cooperate with the majority cells and are unable to differentiate (Hirose et al., 2011, 2015). Expression of the majority-allotype allele of tgrB1 in the minority cells is sufficient to overcome the cooperative aggregation and the differentiation defects of the minority cells. This cell-autonomous property is unique to tgrB1, indicating that TgrB1 is a receptor and TgrC1 is not. The partial segregation of the merodiploid cells observed at late developmental times may be due to cis interactions between TgrB1 proteins, which are induced by trans binding to TgrC1 and lead to protein assembly in membrane patches (Chen et al., 2014). It is possible that the resident TgrB1 and the transgenic TgrB1 compete for cis binding in the merodiploid strains, which might result in compromised protein assembly over time. Alternatively, the merodiploids that carry a matching TgrC1 are recognized as self by the majority cells, but they cannot reciprocate because they lack a matching TgrB1 and are therefore not recognized as self by the neighboring cells. Such lack of reciprocity at the cellular level may lead to segregation over time.
The non-cell-autonomous effect conferred by the tgrC1 merodiploid strain supports the idea that TgrC1 is a ligand. When the majority cells express TgrC1 that is compatible with the minority allotype, the otherwise incompatible minority cells cooperate with the majority and differentiate. This is not the case when the majority cells express the minority-allotype TgrB1. These non-cell-autonomous phenotypes indicate that TgrC1 functions as a ligand and TgrB1 does not.
The TgrB1 cytoplasmic tail (77 aa) is longer than the TgrC1 tail (16 aa), suggesting that it might participate in signal transduction. Indeed, deletion of the TgrB1 cytoplasmic tail largely conferred the same phenotype as deleting the entire coding region even though the modified protein was localized on the plasma membrane. One exception was the finding that the truncated tgrB1 allele mediated even distribution of cells in a mixing experiment. This result is consistent with previous observations that tgrB1-null cells can integrate into multicellular structures better than tgrC1-null cells (Benabentos et al., 2009), so it does not refute the hypothesis that the cytoplasmic tail of TgrB1 is essential for its function. Moreover, the cytoplasmic tail of TgrB1 becomes phosphorylated upon binding to a matching TgrC1 ligand, even though phosphorylation on serine 845 is not essential for the TgrB1 functions we were able to measure. Stimulation of phosphorylation by addition of recombinant soluble TgrC1 and by the addition of antibodies suggests that receptor oligomerization may be sufficient to activate the receptor function of TgrB1. We know neither the role of phosphorylation nor the factors that mediate it, but our observations support the argument that TgrB1 is a receptor.
The TgrB1–TgrC1 system is one of the best-characterized social allorecognition systems (Benabentos et al., 2009; Gruenheit et al., 2017; Hirose et al., 2011, 2015; Ho et al., 2013; Ho and Shaulsky, 2015). The molecular characterization of these proteins as a ligand–receptor pair provides a mechanistic explanation of the system and opens up the field to investigation of the downstream signal transduction pathways. Indeed, the chemical mutagenesis screen for suppressors of the TgrB1–TgrC1 mismatch discovered several putative signal transduction genes, including small GTPases, GTPase activators and guanine-nucleotide exchange factors (Li et al., 2016). Screens that utilized insertional mutagenesis showed that allorecognition and differentiation are mediated by different, albeit somewhat overlapping, signaling pathways (Li et al., 2015; Wang and Shaulsky, 2015). This overlap between development and allorecognition provides an interesting perspective in the context of the evolution of multicellularity.
MATERIALS AND METHODS
Cells and growth conditions
We used Dictyostelium discoideum strain AX4 (Knecht et al., 1986) and its derivatives in all experiments. The strains and their relevant genotypes are listed in Table S1. Unless indicated otherwise, we grew the cells in shaking suspension in HL5 medium at 22°C (Sussman, 1987), we harvested them at the logarithmic growth phase and developed them on filters (Shaulsky and Loomis, 1993) or on agar (Huang et al., 2006) as indicated. We performed strain validation by monitoring developmental phenotypes and red versus green fluorescence when possible, or otherwise by PCR of specific genomic junctions. Sequencing of specific PCR fragments was used when PCR alone was insufficient (e.g. to validate point mutations). Where indicated, we grew the amoebae in association with Klebsiella pneumoniae bacteria (DictyBase strain ID, DBS0305928).
Vectors
Key vectors used in this work are listed in Table S2. To generate the tgrB1 and tgrC1 single-gene merodiploid vectors, we adapted the tgrB1:tgrB1AX4 and tgrC1:tgrC1AX4 vectors from the tgrB1AX4-tgrC1AX4 double-gene merodiploid vector (Hirose et al., 2011) by removing either the tgrC1 or tgrB1 genes, respectively. The tgrB1:tgrB1AX4 vector was generated by cutting the double merodiploid vector with HpaI and NotI restriction endonucleases to remove tgrC1, followed by filling-in with Klenow fragment and self-ligation of the vector. The tgrC1:tgrC1AX4 vector was generated by cutting the double merodiploid vector with AgeI and SpeI restriction endonucleases to remove tgrB1, followed by filling-in with Klenow fragment and self-ligation of the vector. Manipulations of the TgrB1 cytoplasmic tail were done by modifying the tgrB1:tgrB1AX4 vector. To generate the full-length tgrB1-HA allele, we PCR-amplified the 3′ region of tgrB1 with the forward primer, 5′-CGGTGGAGTGGTTACTATCAAT-3′ and the reverse primer which included an HA tag sequence (underlined), 5′-AAAAACTAGTTTAAGCATAATCTGGAACATCATATGGATAATCAGTATGTTCTTTGAAAC-3′. To generate the truncation allele tgrB1Δ828-902-HA, we used the reverse primer: 5′-TTACTAGTTTAAGCATAATCTGGAACATCATATGGATATTTAGCGGCAAATGAAATTAAT-3′. The HA-tagged fragments were placed into tgrB1:tgrB1AX4 between the PstI–SpeI restriction sites. We then cloned the modified alleles into the tgrB1AX4tgrC1AX4 double-gene replacement vector (Hirose et al., 2011) by replacing the respective BglII–SpeI regions.
To express the Myc-tagged TgrB1 protein, we generated a new expression vector, pDMBSr. We removed the Neomycin-resistance cassette and the act15 promoter from pDM304 (Veltman et al., 2009) by digestion with BamHI and SpeI restriction endonucleases and replaced that fragment with a Blasticidin S resistance cassette that we amplified from pLPBLP (Faix et al., 2004) with the primers: plpBSRupBamHI, 5′-AGGTCAGGATCCATTATACGAAGTTATAGATCCTCTAG-3′ and plpBSRdownSalSpe, 5′-GGTCTAACTAGTAGATGAGTCGACAGATCCGAGCTTTCGGGTCAGCTTTATC-3′, after digestion with the same enzymes. We PCR-amplified the upstream region of tgrB1AX4, including the endogenous promoter and the first 22 codons of the ORF with the forward primer B1promoterupSalI, 5′-AGACCTAGTCGACAGTAATATTATTTTCTTTTCCATTTTTATCAATG-3′, and reverse primer B1NterEQKLHindIIIdown, 5′-ACTCAGAAGCTTTTGTTCACTTGATTTAACAAATAAAAATTTACAAAC-3′, digested the PCR product with SalI and HindIII restriction endonucleases and ligated with pDXA–CFP (Knetsch et al., 2002) that was digested with the same enzymes. Then, we digested the plasmid with HindIII and XbaI restriction endonucleases to remove the CFP ORF. We PCR-amplified the rest of the TgrB1 ORF twice with the same reverse primer, SpeIB1stopdown, 5′-CATTCCACTAGTTAATCAGTATGTTCTTTGAAAC-3′ and two different forward primers, B1up+myc1, 5′-AAGTTGATTAGTGAAGAAGATTTAAAATCAAGTTGCTCTTTAAAAGTTGGAAAAATAG-3′, in the first round, and B1up+myc2, 5′-ACTTGCAAGCTTATTAGTGAAGAAGATTTAGAACAAAAGTTGATTAGTGAAGAAGATTTA-3′ in the second round, to add two consecutive Myc epitopes at the N-terminus of the mature protein, after signal-peptide processing. We digested the PCR fragment with HindIII and SpeI restriction endonucleases and ligated with the HindIII- and XbaI-digested plasmid to replace the CFP ORF with the Myc-tagged 3′ end of tgrB1. The resulting predicted amino acid sequence is MKVIYIYLLLLLVCKFLFVKSSEQKLISEEDLEQKLISEEDLKSSCSL where the underlined sequence indicates the tandem Myc epitope and the italicized KSS sequence represent the last 3 amino acids of the TgrB1 signal peptide, which was duplicated to flank the tandem Myc epitope. In the final steps, the Myc-tagged tgrB1 fragment with its endogenous promoter were PCR-amplified with the forward primer B1promoterupSalI and the reverse primer SpeIB1stopdown (see above), digested with SalI and SpeI restriction endonucleases and ligated with pDMBsr that had been digested with the same enzymes to construct pDMBsr tgrB1::ss-myc2TgrB1. We transformed the vector into four strains: tgrB1–, tgrB1–tgrC1–, tgrB1C1QS4 and tgrB1C1QS31.
To express the extracellular domains of TgrC1 for binding experiments in vitro, we used the pDXA-3C vector, where expression is driven by the actin 15 promoter (Knetsch et al., 2002). We PCR-amplified the DNA regions coding for the extracellular domains of five tgrC1 alleles from strains AX4, QS4, QS31, QS37 and QS45 (Benabentos et al., 2009), digested the products with SacI and SpeI and ligated into pDXA-3C between the SacI and XbaI sites. To express the extracellular domains of TgrB1, we modified the expression vector to encode the signal peptide-coding region of tgrC1AX4 (Met1-Pro25) followed by an in-frame His7-tag so the mature protein will carry the His7-tag at the N-terminus after signal-sequence processing. We PCR-amplified the act15::tgrC1AX4 from the abovementioned vector twice with the same forward primer, A15upSalI, 5′-TGAAGCTTGCATGCCTGCAGGTCGACT-3′ and two different reverse primers: C1ss+His1, 5′-ATGGTGATGATGGTGATGATGTGGTGTTGGAGGATTCATTGAATATCCTGA-3′ in the first round, and C1ss+His2KpnI, 5′-GAGCTCGGTACCATGGTGATGATGGTGATGATGTGGTGTTGG-3′ in the second round. We inserted this PCR fragment between the SalI and KpnI restriction sites of pDXA-3C. We then PCR-amplified the extracellular region of tgrB1 without the endogenous signal peptide from strains AX4, QS4, QS31, QS37 and QS45. We double digested the PCR products with SacI (BamHI in the case of AX4) and SpeI and inserted the fragments into the modified pDXA-3C vector between the SacI (BamHI in the case of AX4) and XbaI sites. We transformed the vectors into AX4 cells. In all cases, the tgrB1 and tgrC1 coding regions included only the extracellular domains, without the transmembrane domain or the cytoplasmic tails, so the proteins were secreted into the medium.
Analysis of TgrB1 phosphorylation
We developed cells on non-nutrient agar at a density of 2.5×106 cells/cm2 for 12–14 h. We resuspended them at 2×108 cells/ml in ice-cold cell lysis buffer [40 mM Tris-HCl, pH 7.0, 120 mM NaCl, 0.8% NP-40, 1.0 mM EDTA, 0.1 mM Na3VO4 and 1× protease/phosphatase inhibitor cocktail (Cell Signaling, 5872S)] on a Labquake shaker at 8 rpm for 30 min at 4°C. We precleared the lysates by centrifugation for 15 min at 13,000 g at 4°C and mixed the supernatant with 5 µl anti-Myc affinity gel (Biotool, B23401). We incubated the samples for 45 min at 4°C while rocking as above, then washed the samples three times with ice-cold washing buffer (lysate buffer with 0.2% NP-40 instead of 0.8% NP-40), collected the gel by brief centrifugation, added 100 µl 1× SDS-PAGE loading buffer and boiled for 5 min. We resolved 25 µl of each sample by SDS-PAGE (8% polyacrylamide gel), stained with Coomassie Brilliant Blue and extracted the ∼130 kDa band. Independent samples were analyzed by liquid-chromatography tandem mass spectrometry (LC/MS/MS) at the Taplin Biological Mass Spectrometry Facility at Harvard Medical School and at the Mass Spectrometry Proteomics Core at Baylor College of Medicine.
To test the effects of soluble purified TgrC1 on TgrB1 phosphorylation, we developed tgrB1–tgrC1–tgrB1myc2 cells for 14 h as above. We collected 1.5×108 cells per sample and resuspended the cells in 0.75 ml KK2 buffer containing 5 mM EDTA (KK2-EDTA). We added 20 µg of purified recombinant protein (His7-conjugated TgrB1 or TgrC1) or made no additions, and incubated the cells while rocking at room temperature for 40 min. We then washed the cells twice with 1.2 ml KK2-EDTA and resuspended them again in 0.75 ml KK2-EDTA. We added either mouse-anti-Myc antibodies (Santa Cruz Biotechnology, SC-40, 1:200) to bridge the TgrB1 proteins on the cell membrane or mouse-anti His-tag antibodies (BioLegend Inc., 901502, 1:200) to bridge the soluble recombinant proteins. We incubated the cells for 40 min and washed them as above. Finally, we lysed the cells and pulled down the proteins with Protein A beads or with beads conjugated to anti-Myc antibodies as above.
For western blot analysis, we developed the cells as above for 8–20 h and collected 7.0×107 cells per sample. We lysed the cells and purified myc2TgrB1 on 5 µl anti-Myc affinity gel as above. For testing TgrB1 phosphorylation in chimerae, we mixed the myc2TgrB1 cells at ratios of 1:1 or 1:9 with the indicated strains and then developed them. We collected cells at the indicated times, and treated as above. We resolved 25 µl of each sample by SDS-PAGE (8% polyacrylamide gel) and electro-transferred the proteins onto PVDF membranes. We blocked the membranes with 10% skimmed milk in PBS buffer for analysis with the anti-TgrB1 antibody (1:2500 Chen et al., 2013), or with 3% BSA and 0.1% gelatin in PBS buffer for analysis with rabbit anti-phosphoserine antibody (Abcam, 9332, 1:200). We incubated the membranes for 1 h with the primary antibody, washed three times with PBST, incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, 115-035-146) for 1 h and washed three times with PBST. We visualized the signal with SuperSignal West Pico Chemiluminescent Substrate kit (Thermo Scientific, 34077).
Binding specificity of TgrB1 and TgrC1
We collected cells at the logarithmic growth phase, washed once in PDF buffer (20 mM KCl, 9.2 mM K2HPO4, 15 mM KH2PO4, 1 mM CaCl2, 2.5 mM MgSO4, pH 6.4), resuspended the cells at 2.5×108/ml in PDF buffer and incubated them in shaking suspension at 220 rpm at 22°C for 5 h. We removed the cells by centrifugation at 2755 g for 8 min, collected the supernatant and passed it through a 0.2 µm filter. To pull down the His7–TgrB1 protein, we added 150 µl Ni-NTA-resin (Thermo Scientific) into 10 ml His7–TgrB1-containing sample. We incubated the sample on a rocker platform for 45 min at room temperature and washed three times with PDF buffer. We then added 30 µl of each of the five His7-TgrB1-bound resins to 1.2 ml of each of the five TgrC1-containing samples and incubated on a rocker platform for 45 min at room temperature. We washed the samples three times with PDF buffer, removed the supernatant, added 150 µl 1× SDS-PAGE loading buffer and boiled the samples for 5 min. We used 40 µl of each sample for SDS-PAGE analysis followed by Coomassie Brilliant Blue staining or western blot analysis as above.
Development under agar (quasi two-dimensions)
We grew the cells in shaking suspension in HL-5 with the appropriate antibiotics and harvested them at the logarithmic growth phase. We washed the cells twice with PDF buffer and spread them at a density of 1.2×105 cells/cm2 on 1.5% Noble agar (DIFCO, 214230) plates made in 10 mM potassium phosphate buffer pH 6.5. We cut the agar into 2 cm×2 cm portions and inverted them onto glass-bottom plates (MatTek), so the cells were sandwiched between the glass and the agar. We incubated the plates in a humid chamber at 22°C for 10–12 h. We captured images with a Leica confocal microscope system (TCS-SP5) and with a Nikon inverted microscope system (Eclipse Ti).
Sporulation efficiency
We developed 2.5×107 cells on nitrocellulose filters for 48 h in a humid chamber. We collected the contents of the entire filter in a 50 ml conical test tube and washed once with PDF. To lyse non-spore cells, we incubated the cells in 5 ml of 10 mM EDTA, 0.1% NP40 at 42°C for 45 min. We washed the spores once and resuspended in PDF. We counted the number of spores using a hemocytometer and divided it by 2.5×107 to calculate the sporulation efficiency.
Prespore differentiation and flow cytometry
The relevant strains express RFP constitutively under the actin 15 promoter and sfGFP (Pédelacq et al., 2006) under the prespore-specific cotB promoter, which is a marker for prespore differentiation (Fosnaugh and Loomis, 1993). We mixed a small ratio of the cotB-GFP cells (0.2%) with 99.8% of AX4 or tgrB1QS31tgrC1QS31 cells, plated a total of 2.5×107 cells on a 5 cm nitrocellulose filter and incubated in a humid chamber for 12–14 h. We collected the cells, dissociated them by repeated pipetting and fixed them with 2% paraformaldehyde for 5 min at room temperature. We washed the cells twice with 10 mM potassium phosphate buffer pH 6.5 and analyzed by flow cytometry (LSRFortessa, BD Biosciences) where the test cells were first separated from the unlabeled majority by positive red fluorescence. The gated RFP-positive cells were analyzed for GFP fluorescence intensity.
Sub-cellular fractionation
We developed 1.0×108 cells for 9 h on nitrocellulose filters. We collected the cells, resuspended them in 5 ml of lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM PSMF) and lysed them by three cycles of freezing at −80°C and then thawing. We removed unbroken cells and large debris by centrifugation at 2100 g for 5 min at 4°C. We then fractionated the supernatant by centrifugation at 90,000 g for 30 min at 4°C. We separated the supernatant (soluble) and pellet (membrane) and quantified the amount of protein using a Bio-Rad Protein Assay Dye. We precipitated the proteins by adding 10% trichloroacetic acid and centrifugation at 2100 g for 10 min at 4°C. The protein precipitate was washed with 70% ethanol twice, resuspended in 1× SDS-PAGE sample buffer and boiled for 10 min. We resolved the proteins by SDS-PAGE (6% polyacrylamide gel), electro-transferred onto PVDF membrane and detected the HA-tagged proteins by western blotting with anti-HA monoclonal antibody (Biolegend, 901502, 1:200) as the primary antibody and peroxidase-conjugated AffiPure Goat Anti-Mouse IgG (Jackson ImmunoResearch, 115-035-146) as the secondary antibody. We detected the signal as above.
Segregation tests – image analysis and statistics
We developed mixed populations of cells with different allotypes using the glass-agar sandwich method and recorded images by fluorescence microscopy. The mixes consisted of a majority (90%) of unlabeled cells and a minority of GFP- and RFP-labeled cells (5% each). One of the labeled minority strains was always compatible with the unlabeled majority, serving as a control that would exhibit even distribution. The other labeled minority strain – the test case – was either compatible or incompatible with the majority. If the test cells were compatible, we would expect them to distribute evenly, like the control cells. If they were incompatible, we would expect them to segregate and form clumps within or at the periphery of the larger aggregate (Hirose et al., 2015). This property of spatial distribution variance is quantifiable, and comparison between the variances of the control and the test case indicate whether the distributions are significantly different.
Prior to image quantification, we converted the 8-bit monochromatic images to binary images in the green and red channels. To quantify cell distribution, we divided each image into a 12×12 grid, where each of the 144 squares is a region of interest (ROI). We counted the number of green and red pixels in each ROI and removed empty ROIs from subsequent analysis. We then calculated the proportion of red pixels out of the total pixels in each ROI [pixelred/(pixelred+pixelgreen)] and computed the variance between these values across the entire image. The statistical significance of the difference between images was calculated by an F-test. The size of the ROIs was determined empirically from the images in Fig. 3B,C, such that it generated the smallest P-value in the F-test, and was used on all the other images.
Supplementary Material
Acknowledgements
The authors wish to acknowledge the stimulating discussions and encouragement provided by William F. Loomis, who passed away on June 30, 2016. We thank Xiaoqun Xu for generating the tgrB1 S845A mutation.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: S.H., G.C., A.K., G.S.; Methodology: S.H., G.C., A.K., G.S.; Formal analysis: S.H., G.C., G.S.; Investigation: S.H., G.C.; Resources: S.H., G.C.; Data curation: S.H., G.C.; Writing - original draft: G.S.; Writing - review & editing: S.H., G.C., A.K., G.S.; Visualization: G.S.; Supervision: A.K., G.S.; Project administration: G.S.; Funding acquisition: A.K., G.S.
Funding
This work was supported by grant R35GM118016 from the National Institutes of Health. This work benefited from support by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (P30 AI036211, P30 CA125123 and S10 RR024574) and the expert assistance of Joel M. Sederstrom. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.208975.supplemental
References
- Benabentos R., Hirose S., Sucgang R., Curk T., Katoh M., Ostrowski E. A., Strassmann J. E., Queller D. C., Zupan B., Shaulsky G. et al. (2009). Polymorphic members of the lag gene family mediate kin discrimination in Dictyostelium. Curr. Biol. 19, 567-572. 10.1016/j.cub.2009.02.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G., Wang J., Xu X., Wu X., Piao R. and Siu C.-H. (2013). TgrC1 mediates cell-cell adhesion by interacting with TgrB1 via mutual IPT/TIG domains during development of Dictyostelium discoideum. Biochem. J. 452, 259-269. 10.1042/BJ20121674 [DOI] [PubMed] [Google Scholar]
- Chen G., Xu X., Wu X., Thomson A. and Siu C.-H. (2014). Assembly of the TgrB1-TgrC1 cell adhesion complex during Dictyostelium discoideum development. Biochem. J. 459, 241-249. 10.1042/BJ20131594 [DOI] [PubMed] [Google Scholar]
- Dynes J. L., Clark A. M., Shaulsky G., Kuspa A., Loomis W. F. and Firtel R. A. (1994). LagC is required for cell-cell interactions that are essential for cell-type differentiation in Dictyostelium. Genes Dev. 8, 948-958. 10.1101/gad.8.8.948 [DOI] [PubMed] [Google Scholar]
- Faix J., Kreppel L., Shaulsky G., Schleicher M. and Kimmel A. R. (2004). A rapid and efficient method to generate multiple gene disruptions in Dictyostelium discoideum using a single selectable marker and the Cre-loxP system. Nucleic Acids Res. 32, e143 10.1093/nar/gnh136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortunato A., Strassmann J. E., Santorelli L. and Queller D. C. (2003). Co-occurrence in nature of different clones of the social amoeba, Dictyostelium discoideum. Mol. Ecol. 12, 1031-1038. 10.1046/j.1365-294X.2003.01792.x [DOI] [PubMed] [Google Scholar]
- Fosnaugh K. L. and Loomis W. F. (1993). Enhancer regions responsible for temporal and cell-type-specific expression of a spore coat gene in Dictyostelium. Dev. Biol. 157, 38-48. 10.1006/dbio.1993.1110 [DOI] [PubMed] [Google Scholar]
- Gao E. N., Shier P. and Siu C. H. (1992). Purification and partial characterization of a cell adhesion molecule (gp150) involved in postaggregation stage cell-cell binding in Dictyostelium discoideum. J. Biol. Chem. 267, 9409-9415. [PubMed] [Google Scholar]
- Gruenheit N., Parkinson K., Stewart B., Howie J. A., Wolf J. B. and Thompson C. R. L. (2017). A polychromatic ‘greenbeard’ locus determines patterns of cooperation in a social amoeba. Nat. Commun. 8, 14171 10.1038/ncomms14171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose S., Benabentos R., Ho H.-I., Kuspa A. and Shaulsky G. (2011). Self-recognition in social amoebae is mediated by allelic pairs of tiger genes. Science 333, 467-470. 10.1126/science.1203903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose S., Santhanam B., Katoh-Kurosawa M., Shaulsky G. and Kuspa A. (2015). Allorecognition, via TgrB1 and TgrC1, mediates the transition from unicellularity to multicellularity in the social amoeba Dictyostelium discoideum. Development 142, 3561-3570. 10.1242/dev.123281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho H.-I. and Shaulsky G. (2015). Temporal regulation of kin recognition maintains recognition-cue diversity and suppresses cheating. Nat. Commun. 6, 7144 10.1038/ncomms8144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho H.-I., Hirose S., Kuspa A. and Shaulsky G. (2013). Kin recognition protects cooperators against cheaters. Curr. Biol. 23, 1590-1595. 10.1016/j.cub.2013.06.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang E., Blagg S. L., Keller T., Katoh M., Shaulsky G. and Thompson C. R. L. (2006). bZIP transcription factor interactions regulate DIF responses in Dictyostelium. Development 133, 449-458. 10.1242/dev.02240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessin R. H. (2001). Dictyostelium - Evolution, Cell Biology, and the Development of Multicellularity. Cambridge, UK: Cambridge Univ. Press. [Google Scholar]
- Kibler K., Svetz J., Nguyen T.-L., Shaw C. and Shaulsky G. (2003). A cell-adhesion pathway regulates intercellular communication during Dictyostelium development. Dev. Biol. 264, 506-521. 10.1016/j.ydbio.2003.08.025 [DOI] [PubMed] [Google Scholar]
- Knecht D. A., Cohen S. M., Loomis W. F. and Lodish H. F. (1986). Developmental regulation of Dictyostelium discoideum actin gene fusions carried on low-copy and high-copy transformation vectors. Mol. Cell. Biol. 6, 3973-3983. 10.1128/MCB.6.11.3973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knetsch M. L. W., Tsiavaliaris G., Zimmermann S., Rühl U. and Manstein D. J. (2002). Expression vectors for studying cytoskeletal proteins in Dictyostelium discoideum. J. Muscle Res. Cell Motil. 23, 605-611. 10.1023/A:1024498805771 [DOI] [PubMed] [Google Scholar]
- Li C.-L. F., Chen G., Webb A. N. and Shaulsky G. (2015). Altered N-glycosylation modulates TgrB1- and TgrC1-mediated development but not allorecognition in Dictyostelium. J. Cell Sci. 128, 3990-3996. 10.1242/jcs.172882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.-L. F., Santhanam B., Webb A. N., Zupan B. and Shaulsky G. (2016). Gene discovery by chemical mutagenesis and whole-genome sequencing in Dictyostelium. Genome Res. 26, 1268-1276. 10.1101/gr.205682.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis W. F., Murray B. A., Yee L. and Jongens T. (1983). Adhesion-blocking antibodies prepared against gp150 react with gp80 of Dictyostelium. Exp. Cell Res. 147, 231-234. 10.1016/0014-4827(83)90290-2 [DOI] [PubMed] [Google Scholar]
- Ostrowski E. A., Katoh M., Shaulsky G., Queller D. C. and Strassmann J. E. (2008). Kin discrimination increases with genetic distance in a social amoeba. PLoS Biol. 6, e287 10.1371/journal.pbio.0060287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski E. A., Shen Y., Tian X., Sucgang R., Jiang H., Qu J., Katoh-Kurasawa M., Brock D. A., Dinh C., Lara-Garduno F. et al. (2015). Genomic signatures of cooperation and conflict in the social amoeba. Curr. Biol. 25, 1661-1665. 10.1016/j.cub.2015.04.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pédelacq J.-D., Cabantous S., Tran T., Terwilliger T. C. and Waldo G. S. (2006). Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 24, 79-88. 10.1038/nbt1172 [DOI] [PubMed] [Google Scholar]
- Rosengarten R. D., Santhanam B., Fuller D., Katoh-Kurasawa M., Loomis W. F., Zupan B. and Shaulsky G. (2015). Leaps and lulls in the developmental transcriptome of Dictyostelium discoideum. BMC Genomics 16, 294 10.1186/s12864-015-1491-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulsky G. and Loomis W. F. (1993). Cell type regulation in response to expression of ricin A in Dictyostelium. Dev. Biol. 160, 85-98. 10.1006/dbio.1993.1288 [DOI] [PubMed] [Google Scholar]
- Strassmann J. E., Zhu Y. and Queller D. C. (2000). Altruism and social cheating in the social amoeba Dictyostelium discoideum. Nature 408, 965-967. 10.1038/35050087 [DOI] [PubMed] [Google Scholar]
- Sukumaran S., Brown J. M., Firtel R. A. and McNally J. G. (1998). lagC-null and gbf-null cells define key steps in the morphogenesis of Dictyostelium mounds. Dev. Biol. 200, 16-26. 10.1006/dbio.1998.8934 [DOI] [PubMed] [Google Scholar]
- Sussman M. (1987). Cultivation and synchronous morphogenesis of Dictyostelium under controlled experimental conditions. Meth. Cell Biol. 28, 9-29. 10.1016/S0091-679X(08)61635-0 [DOI] [PubMed] [Google Scholar]
- Veltman D. M., Akar G., Bosgraaf L. and Van Haastert P. J. M. (2009). A new set of small, extrachromosomal expression vectors for Dictyostelium discoideum. Plasmid 61, 110-118. 10.1016/j.plasmid.2008.11.003 [DOI] [PubMed] [Google Scholar]
- Wang Y. and Shaulsky G. (2015). TgrC1 has distinct functions in dictyostelium development and allorecognition. PLoS ONE 10, e0124270 10.1371/journal.pone.0124270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N., Söderbom F., Anjard C., Shaulsky G. and Loomis W. F. (1999). SDF-2 induction of terminal differentiation in Dictyostelium discoideum is mediated by the membrane-spanning sensor kinase DhkA. Mol. Cell. Biol. 19, 4750-4756. 10.1128/MCB.19.7.4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Hou L., Awrey D., Loomis W. F., Firtel R. A. and Siu C.-H. (2000). The membrane glycoprotein gp150 is encoded by the lagC gene and mediates cell-cell adhesion by heterophilic binding during Dictyostelium development. Dev. Biol. 227, 734-745. 10.1006/dbio.2000.9881 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.