

Abstract
Alveologenesis, the final step of lung development, is characterized by the formation of millions of alveolar septa that constitute the vast gas-exchange surface area. The genetic network driving alveologenesis is poorly understood compared with earlier steps in lung development. FGF signaling through receptors Fgfr3 and Fgfr4 is crucial for alveologenesis, but the mechanisms through which they mediate this process remain unclear. Here we show that in Fgfr3;Fgfr4 (Fgfr3;4) global mutant mice, alveolar simplification is first observed at the onset of alveologenesis at postnatal day 3. This is preceded by disorganization of elastin, indicating defects in the extracellular matrix (ECM). Although Fgfr3 and Fgfr4 are expressed in the mesenchyme and epithelium, inactivation in the mesenchyme, but not the epithelium, recapitulated the defects. Expression analysis of components of the elastogenesis machinery revealed that Mfap5 (also known as Magp2), which encodes an elastin-microfibril bridging factor, is upregulated in Fgfr3;4 mutants. Mfap5 mutation in the Fgfr3;4 mutant background partially attenuated the alveologenesis defects. These data demonstrate that, during normal lung maturation, FGF signaling restricts expression of the elastogenic machinery in the lung mesenchyme to control orderly formation of the elastin ECM, thereby driving alveolar septa formation to increase the gas-exchange surface.
KEY WORDS: FGF signaling, Lung development, Alveologenesis, Elastin extracellular matrix, Mouse
Highlighted Article: Fgfr3 and Fgfr4 are required in mouse postnatal lung development, where they regulate elastin fiber organization during alveologenesis via mesenchymal expression of the elastin cross-linking factor MFAP5.
INTRODUCTION
Bronchopulmonary dysplasia (BPD) is a pulmonary disease that affects ∼12,000 premature births per year in the USA. In BPD infants, truncation of gestation disrupts late lung development, of which alveologenesis is the final step. Alveologenesis is one of the least understood aspects of lung development. Gaining knowledge of the basic developmental mechanisms that drive normal alveologenesis is the foundation for understanding BPD pathogenesis.
In mice, alveologenesis initiates after birth. At birth, the alveolar region in the periphery of the lung is composed of millions of saccules, also called primary septa (Morrisey and Hogan, 2010). Although these saccules can support gas exchange during the first few days of life, they only account for ∼5% of the alveolar surface area in an adult mouse (Mund et al., 2008). The drastic expansion of surface area occurs through alveologenesis, which initiates at about postnatal day (P) 3 and continues until ∼P36 (Mund et al., 2008). During this period, new septal ridges (also called secondary septa) arise from the walls of the saccule to subdivide the alveolar lumen (Branchfield et al., 2016). Repeated division and subsequent growth of the alveoli generates 95% of the final gas-exchange surface area in the adult lung (Mund et al., 2008). Hence, disruption of alveologenesis would severely compromise respiration for life.
Septal ridges are intricate structures with multiple cell types that interact intimately among themselves as well as with the extracellular matrix (ECM). In the epithelium, alveolar type I and type II cells line the lumen and serve as an interface between the air and the elaborate underlying capillary network. In the mesenchyme, a key cell type is the myofibroblasts, which have been implicated as a driver for septal ridge formation (Boström et al., 1996; Lindahl et al., 1997). They are also the primary producers of elastin, a major component of the alveolar ECM that is essential for the proper development of the alveoli (Wendel et al., 2000).
Formation of the elastin ECM is an elaborate process with multiple steps (Cirulis et al., 2008; Wagenseil and Mecham, 2007; Yeo et al., 2011). It initiates with aggregation of the elastin monomer tropoelastin, facilitated by lysyl oxidase (LOX) enzymes (Cirulis et al., 2008; Wagenseil and Mecham, 2007). These aggregates are then transported extracellularly to be deposited on a pre-existing microfibril network composed primarily of fibrillins (FBNs). This is facilitated by fibulins (FBLNs) and microfibril-associated glycoproteins (MAGPs). These elastin aggregates are then further crosslinked by LOX to form elastin fibers. Loss of key elastin ECM assembly genes, including Loxl1, Fbln5 and Fbn1, results in postnatal alveolar simplification (Charbonneau et al., 2010; Liu et al., 2004; Neptune et al., 2003; Yanagisawa et al., 2002, 2009).
Signaling pathways have also been implicated in alveologenesis. For example, fibroblast growth factor (FGF) receptor 3 (Fgfr3) and Fgfr4 are expressed throughout the epithelium and mesenchyme of the developing mammalian lung, with their expression peaking during saccular and alveolar stages (Boucherat et al., 2007; Mariani et al., 2002; Weinstein et al., 1998). Recent studies have also implicated the FGF pathway in BPD. BPD biopsy samples show reduced FGF10 expression (Benjamin et al., 2007). A polymorphism in FGFR4 is significantly associated with this disorder in humans (Benjamin et al., 2007; Rezvani et al., 2013). Exposure of newborn mice to hyperoxia, an established BPD model, led to decreased expression of Fgfr3 and Fgfr4 (Park et al., 2007). These data together indicate that disruption of FGF signaling may contribute to BPD.
While much is known about the mechanism of FGF function during prenatal lung branching morphogenesis, relatively little is known about its role in postnatal alveologenesis (Abler et al., 2009; Bellusci et al., 1997; Perl et al., 2003; Srisuma et al., 2010; Volckaert and De Langhe, 2014; Weinstein et al., 1998; White et al., 2006). Loss of either Fgfr3 or Fgfr4 individually does not disrupt lung development, whereas the combined global loss of both genes disrupts secondary septa formation starting after birth (Srisuma et al., 2010; Weinstein et al., 1998). In the Fgfr3;Fgfr4 double mutant (hereafter Fgfr3;4), there is an increase in the elastin expression level as well as staining in the alveolar region starting at P8 (Srisuma et al., 2010; Weinstein et al., 1998). Further studies using cultured primary cells from Fgfr3;4 mutant lungs led to the conclusion that increased production of IGF1 from the epithelium is responsible for the increase in elastin expression. However, the mechanism of FGFR3 and FGFR4 action in elastogenesis has not been tested in vivo.
In this study, we dissected the primary role of Fgfr3 and Fgfr4 in vivo by narrowing down the earliest time point of histological defects to P3. We found that while the elastin level has not yet increased, there is a clear disorganization of the elastin pattern prior to histological defects. Unexpectedly, it is the mesenchyme-specific inactivation, but not the epithelial inactivation, of Fgfr3 and Fgfr4 that recapitulates the defects. We uncovered in vivo genetic evidence to support that MFAP5 (also known as MAGP2), a factor that deposits elastin on the microfibrils, is regulated by FGF signaling, and in turn serves as a likely mediator of FGFR3 and FGFR4 control of alveologenesis.
RESULTS
Fgfr3;4 mutant lungs exhibit simplification at the onset of alveologenesis
In previous studies of Fgfr3;4 mutant mouse lungs (Srisuma et al., 2010; Weinstein et al., 1998), the earliest reported phenotype was at P8, exhibiting clear alveolar simplification. To address the primary mechanism of Fgfr3 and Fgfr4 in alveologenesis, we traced the simplification phenotype to its earliest origin. Quantification by mean linear intercept (MLI) analysis indicated that at P8 and P3, the mutant lungs had a 60% and 20% increase in relative MLI, respectively (P=0.002 and P=0.037, respectively; n=4 for each stage; Fig. 1A-D,G). However, at P2, the MLI of Fgfr3;4 mutant lungs was not statistically different from that of the controls (P=0.60, n=3; Fig. 1E-G).
Fig. 1.
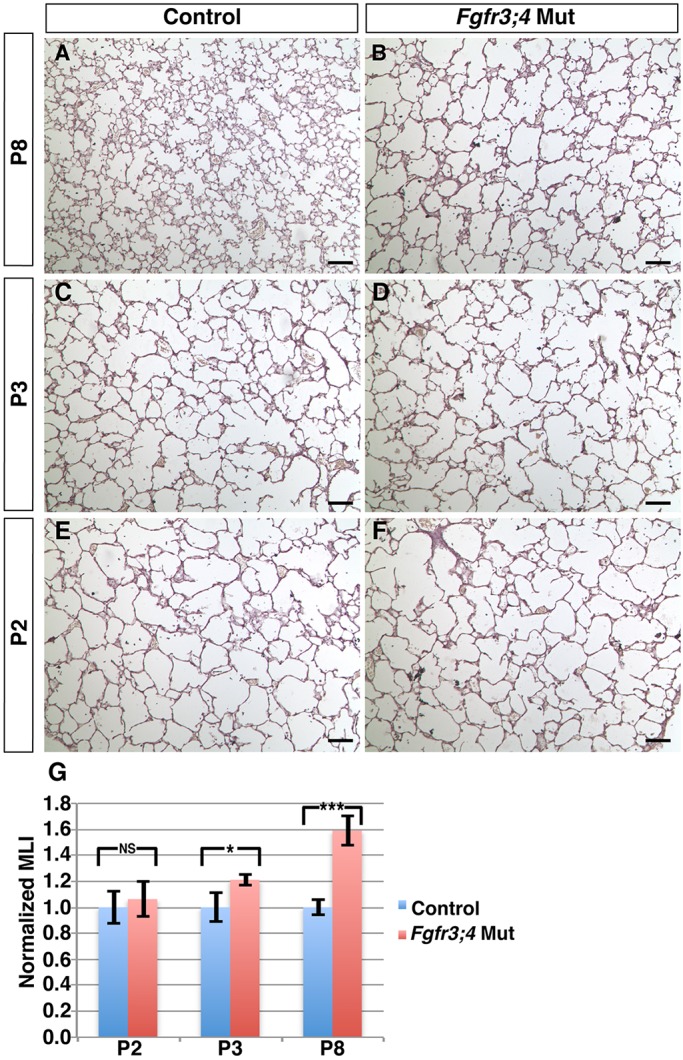
Fgfr3;4 mutant lungs develop simplified alveolar structure during early postnatal lung development. (A-F) Representative H&E-stained sections from the alveolar regions of control (A,C,E) and mutant (B,D,F) mouse lungs at P2, P3 and P8. (G) Quantification of alveolar simplification by MLI. Fgfr3;4 mutant lungs showed a statistically significant increase in relative MLI compared with control at both P8 (***P=0.002, n=4) and P3 (*P=0.037, n=4), but not at P2 (P=0.60, n=3). Mean±s.e.m. Scale bars: 100 μm.
It is known that loss of Fgfr3 leads to reduced skeleton, raising the possibility that the lung defect might be secondary to reduced chest cavity size (Colvin et al., 1996; Deng et al., 1996; Srisuma et al., 2010). However, we found that alveolar simplification at P3 is prior to any change in overall body size/weight, suggesting that the onset of alveolar simplification is not secondary to a decrease in the size of the mutant mice (Fig. S1A). Together, these data demonstrate that Fgfr3 and Fgfr4 function is required for postnatal lung development starting at the onset of alveologenesis.
Fgfr3;4 mutant lungs show increased intensity of transgelin staining in myofibroblasts
To determine the cellular mechanism underlying alveolar simplification starting at P3, we searched for changes prior to this stage. Previously, it was shown that there is a decrease in cell proliferation in Fgfr3;4 mutant lungs at P8 (Srisuma et al., 2010). However, no change in cell proliferation was observed at either P2 or P3 by phosphorylated histone H3 (pHis) antibody staining (Fig. S2A-E). Furthermore, there is no change in cell death rate based on cleaved caspase 3 staining (Fig. S2F-I). These results suggest that the initial alveolar simplification in the Fgfr3;4 mutant lung is not due to a disruption in cell proliferation or cell death.
We next addressed whether any alveolar cell types are affected in the mutant. Neither of the epithelial cell types, alveolar type 1 and type 2 cells (AEC1 and AEC2), appeared altered at P2 or P8 (Fig. S3A-D). Similarly, alveolar endothelial cells remained proportional to other cell types at P2 and P8 (Fig. S3E-H).
Two major cell types in the mesenchyme are myofibroblasts and lipofibroblasts. The myofibroblast lineage is a crucial driver of alveologenesis, as the absence of myofibroblasts leads to severe alveolar simplification in the Pdgfa mutant (Boström et al., 1996; Lindahl et al., 1997). Lipofibroblasts have been viewed as the alternative lineage to myofibroblasts (Branchfield et al., 2016; McGowan and McCoy, 2014). Pdgfra+ cells are proposed to give rise to both lineages (Ntokou et al., 2015; Rehan and Torday, 2003). Quantification of Pdgfra+ cell number by mating a PdgfraH2bGFP/+ nuclear reporter allele into the mutant or control background indicated that there is no difference in the total number of labeled cells in the mutant lungs compared with control at P3 (Fig. S4A-C) (Hamilton et al., 2003). Staining of the lipofibroblast lineage using anti-ADRP (PLIN2) antibody showed a decrease at P3 (Fig. S4D-F). Immunostaining revealed that there is an increase in the opposing myofibroblast cell marker transgelin (TAGLN; also known as SM22α) at both P3 and P8 (Fig. 2A-D) (Srisuma et al., 2010). However, TAGLN staining in the mutant was the same as in the control at P2, suggesting that the increase in TAGLN staining arises at the same time as alveolar simplification (at P3), but does not precede it (Fig. 2C-F), and therefore is unlikely to be the primary cause of alveolar simplification. Western blotting showed that not only at P2, but also at P3, the TAGLN protein level is unaltered (Fig. 2G,H). This suggests that FGFR3 and FGFR4 regulate TAGLN organization, rather than expression level. Finally, as assessed by anti-IsoB4 antibody staining, there is no difference in the number of macrophages in the mutant lung compared with the control at P3 (Fig. S4G-I).
Fig. 2.
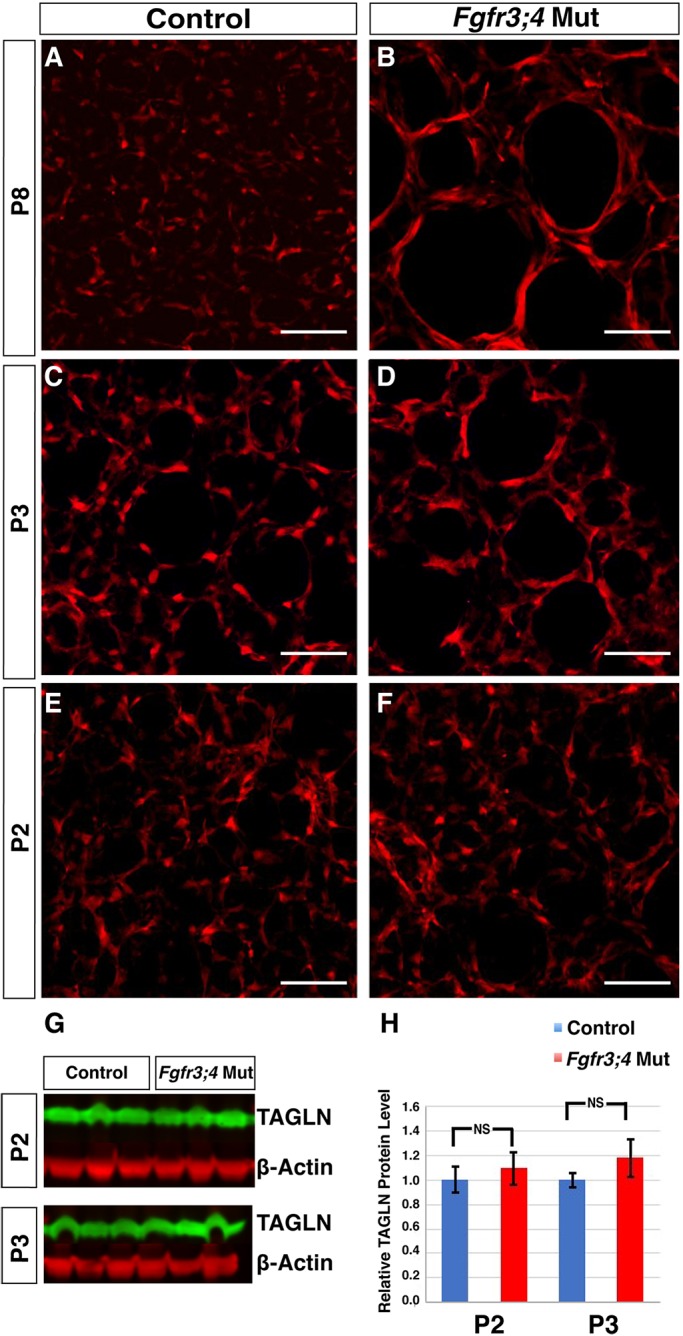
Myofibroblasts show stretched morphology in the Fgfr3;4 mutant lung. (A-F) Reconstructed 50 μm confocal z-stacks of immunofluorescent staining for TAGLN (red) in the alveolar region of control and mutant lungs at P8, P3 and P2. At P8 and P3, Fgfr3;4 mutant lungs displayed increased TAGLN staining, and a stretched myofibroblast morphology (A-D). However, at P2, the staining in the mutant is indistinguishable from that of control lungs (E,F). Scale bars: 100 μm. (G,H) Western blot analysis indicates that the TAGLN (green) level is unaltered in the mutant compared with control at both P2 (P=0.38, n=3) and P3 (P=0.17, n=3). β-actin (red) was used as a control. Mean±s.e.m.
Fgfr3;4 mutant lungs show ECM disorganization prior to structural simplification
To search for the primary cause of alveolar simplification, we assayed ECM organization. Strong evidence indicates that precise organization of the alveolar ECM, including elastin, is essential for alveolar development (Neptune et al., 2003; Wendel et al., 2000; Yanagisawa et al., 2009; Yeo et al., 2011). Previous data show that the elastin staining intensity in two-dimensional (2D) sections is increased in Fgfr3;4 mutant lungs starting at P8 and there is an increase in its transcript level after P12 (Srisuma et al., 2010; Weinstein et al., 1998). To more precisely assess elastin organization, we used confocal microscopy to examine the three-dimensional (3D) architecture (Branchfield et al., 2016). In control lungs at P8, z-stack images showed that elastin fibers are preferentially localized to the alveolar entrance rings that rim the top of the alveolar walls. However, in Fgfr3;4 mutant lungs, there were numerous fibers running through other regions of the alveolar wall (Fig. 3A,B). Elastin disorganization was also detected at P3, and more importantly at P2, prior to alveolar simplification (Fig. 3C-F). Interestingly, although ectopic elastin fibers were observed in the Fgfr3;4 mutant lungs, there was no difference in the protein level of the elastin monomer, tropoelastin, in the mutant lungs at P2 (Fig. 3G). These data together suggest that the primary role of Fgfr3 and Fgfr4 is to control elastin fiber organization immediately preceding the onset of alveologenesis.
Fig. 3.
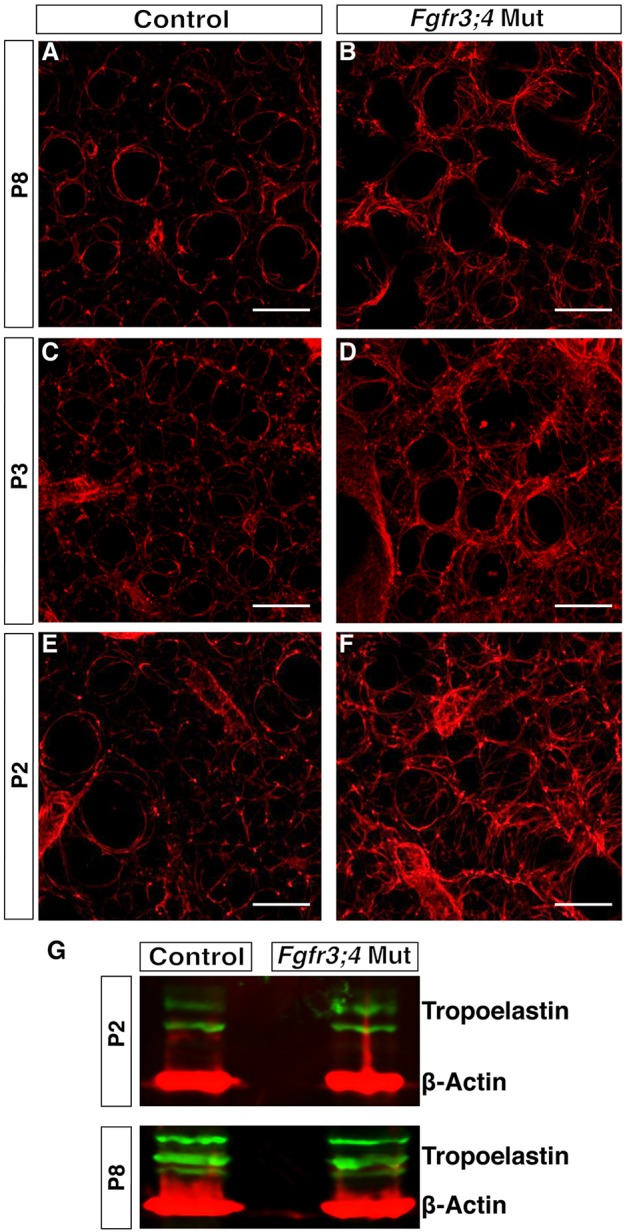
The elastin ECM is disorganized in the Fgfr3;4 mutant lung. (A-F) Reconstructed 50 μm z-stacks of immunofluorescent staining for elastin (red) in the alveolar region of control and mutant lungs at P8, P3 and P2. At all three stages, elastin staining in the Fgfr3;4 mutant lung is disorganized compared with the control. Scale bars: 100 μm. (G) Western blot analysis indicates that the monomer tropoelastin (green) level is slightly increased at P8, but normal at P2, in the mutant compared with control. β-actin (red) was used as control.
Fgfr3 and Fgfr4 expression is required in the lung mesenchyme for alveologenesis
To determine the role of Fgfr3 and Fgfr4 in the regulation of elastin organization, we addressed the tissue-specific requirement for these genes. FGFR3 and FGFR4 proteins have been detected in all cells in the lung parenchyma (Boucherat et al., 2007; Mariani et al., 2002; Weinstein et al., 1998). We confirmed this at P2, when anti-FGFR3 antibody staining showed that FGFR3 is widely expressed in both the epithelium and mesenchyme in the alveolar region (Fig. S5A-C). Prior data from in vitro analysis suggest that Fgfr3 and Fgfr4 function in the epithelium to control the signal for elastin expression (Srisuma et al., 2010). To test this in vivo, and address the lineage-specific requirement, we performed conditional inactivation in each of the cell layers. To conditionally inactivate Fgfr3 and Fgfr4 in the epithelium, we used epithelial-specific Nkx2-1-cre instead of the commonly used Shhcre because the latter is closely linked to Fgfr3 on the same chromosome (Harris et al., 2006; Xu et al., 2008). For inactivation in the mesenchyme, we used Dermo1cre (Dermo1 is also known as Twist2) for its early and widespread activity in mesenchymal cells (Yu et al., 2003). We combined each of these cre lines with a conditional Fgfr3fl allele (Su et al., 2010) and the global Fgfr4 knockout allele, generating Nkx2-1-cre;Fgfr3fl/−;Fgfr4−/− (hereafter Nkx2-1cre;Fgfr3;4) or Dermo1cre;Fgfr3fl/−;Fgfr4−/− (hereafter Dermo1cre;Fgfr3;4) mutant mice.
We found that Nkx2-1cre;Fgfr3;4 lungs were histologically indistinguishable from controls at both P8 and P28 (Fig. 4A-C, Fig. S5D-E). Furthermore, there was no change in either TAGLN or elastin staining at P8 (Fig. 5A,B,E,F). To ensure that the absence of a lung phenotype in the Nkx2-1cre;Fgfr3;4 conditional mutant is not caused by ineffective recombination, we first tested the activity of Nkx2-1-cre by breeding it to a RosaEYFP reporter allele and found that it is active throughout the alveolar epithelium (Fig. S5F-H). qRT-PCR analysis of Fgfr3 using whole lungs confirmed a clear transcript reduction as compared with the heterozygous control, albeit with substantial transcripts remaining, which is likely to be due to unperturbed Fgfr3 expression in the mesenchyme (Fig. 4D). Together, these data indicate that Fgfr3 and Fgfr4 are not required in the epithelium for alveolar development.
Fig. 4.

Fgfr3 and Fgfr4 activity is required in the mesenchyme but not the epithelium for alveologenesis. (A,B,E,F) Representative H&E-stained sections from the alveolar regions of P8 Cre control (A,E) and Nkx2-1cre;Fgfr3;4 (B) or Dermo1cre;Fgfr3;4 (F) conditional mutant lungs. Nkx2-1cre;Fgfr3;4 conditional lungs displayed normal alveoli (A,B), whereas Dermo1cre;Fgfr3;4 conditional lungs displayed simplified alveoli. Scale bars: 100 μm. (C,G) MLI analysis indicated no change in the Nkx2-1cre;Fgfr3;4 mutant compared with control (C; P=0.95, n=3) and a statistically significant increase in Dermo1cre;Fgfr3;4 conditional lungs compared with control (G; **P=0.001, n=4). (D,H) qRT-PCR analysis indicated a statistically significant decrease of Fgfr3 expression in conditional mutant lungs compared with control (D, *P=0.02, n=3 for Nkx2-1cre;Fgfr3;4; H, **P=0.002, n=3 for Dermo1cre;Fgfr3;4). Mean±s.e.m.
Fig. 5.
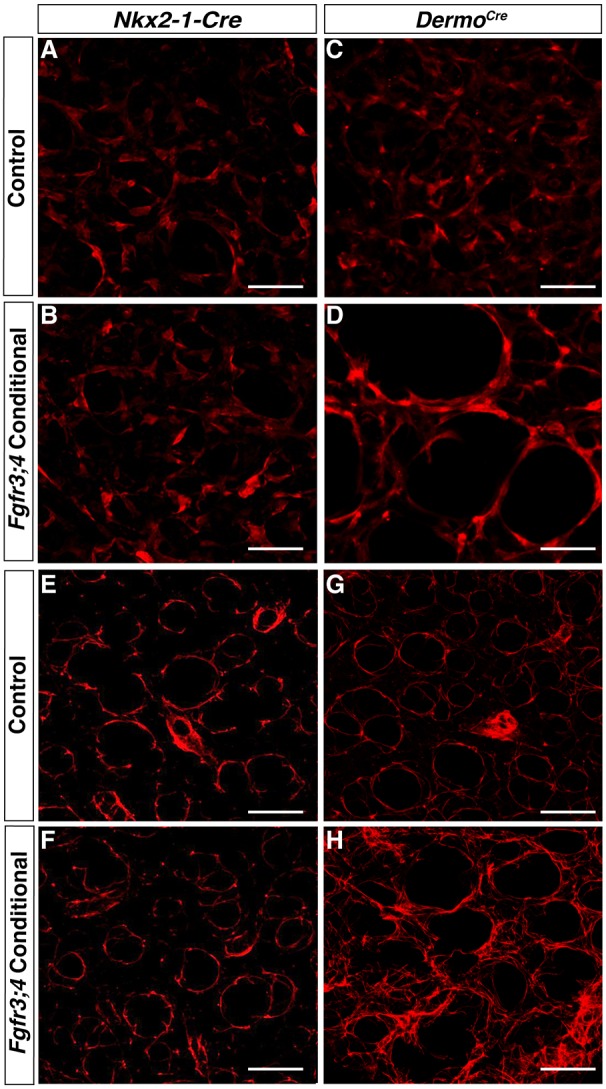
Loss of Fgfr3 and Fgfr4 activity in the mesenchyme leads to stretched myofibroblasts and disorganized elastin ECM. (A-D) Reconstructed 50 μm z-stacks of immunofluorescent staining for TAGLN (red) in the alveolar region of P8 Cre control (A,C) and Nkx2-1cre;Fgfr3;4 (B) or Dermo1cre;Fgfr3;4 (D) conditional mutant lungs. TAGLN-positive myofibroblasts were unaltered in the Nkx2-1cre;Fgfr3;4 conditional lung compared with control (A,B), but were enlarged in the Dermo1cre;Fgfr3;4 conditional lung compared with control (C,D). The latter phenotype is reminiscent of that seen in the Fgfr3;4 global mutant. (E-H) Reconstructed 50 μm z-stacks of immunofluorescent staining for elastin in the alveolar region of P8 Cre control (E,G) and Nkx2-1cre;Fgfr3;4 (F) or Dermo1cre;Fgfr3;4 (H) conditional mutant lungs. In the Nkx2-1cre;Fgfr3;4 conditional lung, the elastin fibers are primarily found around the alveolar entrance ring, similar to the control (E,F). In the Dermo1cre;Fgfr3;4 conditional lung, elastin is disorganized with multiple fibers running through the alveolar walls (G,H). This phenotype is reminiscent of that seen in the Fgfr3;4 global mutant. Scale bars: 100 μm.
By contrast, Dermo1cre;Fgfr3;4 conditional lungs exhibited simplified alveoli at P8 (Fig. 4E,F). Quantification showed an ∼50% increase in MLI at P8 (P=0.001, n=4; Fig. 4G), similar to the MLI increase in global Fgfr3;4 mutants at the same stage (Fig. 1G). In addition, there is disorganized TAGLN and elastin staining in Dermo1cre;Fgfr3;4 conditional lungs at P8, similar to that in global Fgfr3;4 mutant lungs (Fig. 5C,D,G,H). qRT-PCR analysis of Fgfr3 using whole lungs of mutant versus control confirmed a similar degree of transcript reduction in Dermo1cre;Fgfr3;4 as in the Nkx2-1cre;Fgfr3;4 mutant (Fig. 4H).
As Dermo1cre is also active in the skeleton to recombine Fgfr3fl, resulting in reduced body size (Wang et al., 2001; Yu et al., 2003), we wanted to ensure that the alveolar simplification observed in the Dermo1cre;Fgfr3;4 mutants is due to a direct requirement for these genes in the lung mesenchyme. We used the Tbx4-rtTA;tetO-cre combination to specifically drive gene deletion in the lung mesenchyme, and not in other tissues (Zhang et al., 2013). We found that in Tbx4-rtTA;tetO-cre;Fgfr3fl/fl;Fgfr4−/− (hereafter Tbx4rtTA;cre;Fgfr3;4) mutant lungs, prenatal induction of Cre activity led to a 40% increase in MLI compared with controls at P8 (P=0.0001, n=4; Fig. S6A-C). qRT-PCR analysis for Fgfr3 using whole lungs of mutants versus controls confirmed a similar degree of transcript reduction in Tbx4rtTA;cre;Fgfr3;4 as in the Nkx2-1cre;Fgfr3;4 and Dermo1cre;Fgfr3;4 mutants (Fig. S6D). Together, these data provide in vivo evidence that Fgfr3 and Fgfr4 are required in the mesenchyme, but not in the epithelium, for alveologenesis, and that this is likely to be through regulating elastin organization.
Elastin fiber formation in the Fgfr3;4 mutant lung
To determine the mechanism underlying the elastin disorganization phenotype, we used qRT-PCR to examine the expression of key components of the elastogenesis machinery at P2, prior to alveolar simplification (Shifren and Mecham, 2006; Srisuma et al., 2010; Vrhovski and Weiss, 1998; Yeo et al., 2011). We found three genes with altered transcript levels in the Fgfr3;4 mutant lung as compared with the control, and all were increased: insulin-like growth factor 1 (Igf1), fibrillin 2 (Fbn2) and microfibrillar-associated protein 5 (Mfap5) (Fig. 6A).
Fig. 6.

Fgfr3;4 mutant lungs display an increase in Mfap5, Igf1 and Fbn2 expression. (A) qRT-PCR of genes involved in elastogenesis. The expression of Mfap5, Igf1 and Fbn2 was increased in the Fgfr3;4 mutant lung at P2 (*P<0.05, n=3 each). Mean±s.e.m. (B-D) Representative immunofluorescent staining for MFAP5 (green), the myofibroblast marker αSMA (red) and nuclei (DAPI, blue) at P7. Scale bars: 100 μm. (E) Western analysis indicates that MFAP5 (green) is also increased at the protein level in Fgfr3;4 mutants as compared with the control at both P2 and P8. β-actin (red) was used as a loading control.
A previous study of the Fgfr3;4 mutant showed an increase in Igf1 at P8, and that recombinant IGF1 drives upregulation of tropoelastin expression in cultured cells, supporting the possibility that this might in turn lead to the alveolar simplification observed in the Fgfr3;4 mutant (Srisuma et al., 2010). To determine whether the increase in Igf1 expression contributes to alveolar simplification in the Fgfr3;4 mutant lung in vivo, we inactivated Igf1 in either the epithelium or the mesenchyme in the Fgfr3;4 mutant background. This was achieved by combining an Igf1 floxed allele with Nkx2-1-cre or Dermo1cre in the Fgfr3;4 null mutant background, generating Nkx2-1-cre;Fgfr3−/−;Fgfr4−/−;Igf1fl/+ or Dermo1cre;Fgfr3−/−;Fgfr4−/−;Igf1fl/+ mutants. We retained one copy of the Igf1 allele as homozygous Igf1 mutants exhibit an alveolar defect on their own (Liu et al., 1993; Powell-Braxton et al., 1993). We found that with either Cre driver, alveolar simplification of the Fgfr3;4 mutant lung was not attenuated (Fig. S7A-H). There is also no attenuation of the elastin disorganization phenotype. Together, these results suggest that the increase in Igf1 is not a significant in vivo contributor to the alveolar simplification phenotype in the Fgfr3;4 mutant. To ensure that the absence of a lung phenotype in the Nkx2-1-cre;Fgfr3−/−;Fgfr4−/−;Igf1fl/+ and Dermo1cre;Fgfr3−/−;Fgfr4−/−;Igf1fl/+ mutants is not caused by ineffective inactivation of Igf1, we used qRT-PCR to quantify the Igf1 expression level. The results showed that recombining one allele of Igf1 with Nkx2-1-cre or Dermo1cre resulted in a 36% or 42% reduction, respectively, of Igf1 transcripts compared with controls, suggesting robust recombination (Fig. S7I).
Fbn2 encodes proteins that form the microfibril network, on which the elastin ECM is deposited (Yeo et al., 2011). Previous work has demonstrated that mutations in fibrillin genes cause alveolar simplification in mice and Marfan syndrome in humans (Canadas et al., 2010; Carta et al., 2006; Neptune et al., 2003). To examine whether there is a change in microfibril patterning that could underlie alveolar simplification in Fgfr3;4 mutant lung, we performed immunohistochemical staining for both FBN1 and FBN2 in P2 lungs (Charbonneau et al., 2003; Reinhardt et al., 1996). We found no appreciable difference in the organization of FBN1 or FBN2 in the mutant lung, in contrast to the elastin disorganization that is observed at this stage (Fig. S8). The normal pattern of the microfibril network suggests that the change in Fbn2 expression is unlikely to be a major cause of the alveolar simplification phenotype.
MFAP5 has been shown to control the deposition of elastin onto the microfibril network (Lemaire et al., 2007). In the normal lung, MFAP5 is expressed in the mesenchyme and partially colocalizes with the alveolar myofibroblast marker alpha smooth muscle actin (αSMA) (Fig. 6B-D). In addition to an increase in Mfap5 transcript level in Fgfr3;4 mutant lungs, there was also an increase in the MFAP5 protein level at both P2 and P8 (Fig. 6E). It has been shown that overexpression of Mfap5 in cultured cells leads to disorganized elastin fiber formation (Lemaire et al., 2007), raising the possibility that the increase in Mfap5 expression might cause a similar elastin phenotype in vivo, which in turn leads to alveolar simplification in the Fgfr3;4 mutant lung. To test this possibility, we introduced a Mfap5 knockout allele into the Fgfr3;4 mutant background, generating Mfap5−/−;Fgfr3−/−;Fgfr4−/− (hereafter Mfap5;Fgfr3;4) triple-mutant mice. It has been shown that Mfap5−/− by itself does not lead to an alveolar phenotype (Combs et al., 2013). We found that the alveolar simplification of the Fgfr3;4 mutant lung was attenuated in the Mfap5;Fgfr3;4 lung at P8, reversing the MLI increase from 77% in Fgfr3;4 to 42% in Mfap5;Fgfr3;4 as compared with a normal control (Fig. 7A-D). The disorganized elastin phenotype was also attenuated in Mfap5;Fgfr3;4 as compared with Fgfr3;4 mutant lungs at P8 as well as at P2 (Fig. 7E-J). Together, these in vivo results indicate that, although not the sole factor, the increase in Mfap5 is a significant contributor to alveolar simplification in the Fgfr3;4 mutant.
Fig. 7.

Introduction of an Mfap5 knockout allele into the Fgfr3;4 mutant background attenuates alveolar simplification. (A-C) Representative H&E-stained sections from the alveolar regions of P8 Fgfr3;4 mutant and Mfap5;Fgfr3;4 mutant lungs. (D) MLI analysis indicates a statistically significant decrease in Mfap5;Fgfr3;4 lungs compared with Fgfr3;4 mutant lungs (control versus Mfap5+/−;Fgfr3;4, ***P=0.0001; control versus Mfap5−/−;Fgfr3;4, **P=0.006; Mfap5+/−;Fgfr3;4 versus Mfap5−/−;Fgfr3;4, **P=0.002; n=4 each group). (E-J) Representative immunofluorescently stained section for elastin (red) in the alveolar region of P8 and P2 normal control, Fgfr3;4 mutant and Mfap5;Fgfr3;4 lungs. There is rescue to longer and more organized elastin fibers in Mfap5;Fgfr3;4 compared with Fgfr3;4 mutant lungs. Scale bars: 100 μm.
DISCUSSION
In this study, we investigated the in vivo mechanism of Fgfr3 and Fgfr4 function in postnatal lung development. We found that the alveolar simplification phenotype in Fgfr3;4 mutants is first detected three days after birth, and that this phenotype is preceded by disorganization of the alveolar elastin ECM. These defects were recapitulated by inactivating Fgfr3 and Fgfr4 in the mesenchyme, but not the epithelium. Excess MFAP5, an elastin cross-linking factor, is in part responsible for these defects, as reduction of MFAP5 level in the Fgfr3;4 mutant background not only attenuated the elastin disorganization phenotype, but also partially reversed the alveolar simplification defect. These in vivo findings demonstrate that FGFR3 and FGFR4 function in mesenchymal cells to control postnatal alveolar elastin organization, thereby driving the orderly process of alveologenesis.
An array of mutants in FGF ligands or receptors exhibit phenotypes at multiple stages of lung development, suggestive of diverse roles of this signaling pathway (Abler et al., 2009; Al Alam et al., 2015; Chao et al., 2017; Hokuto et al., 2003; Usui et al., 2004; Yi et al., 2009; Yin et al., 2011; Yu et al., 2010). The majority of these mutants affect prenatal developmental steps. In comparison, the Fgfr3;4 mutants are unique in that the lung phenotypes manifest only after birth in the early postnatal period, reflecting requirements for these receptors in the last step of lung development (Fig. 1). Based on ligand-receptor binding specificity and their pattern of expression, FGF18 has been proposed to be the primary ligand for FGFR3 and FGFR4. However, Fgf18 null mutants die at birth with reduced alveolar space and thickened mesenchyme, precluding study at the postnatal stages when the Fgfr3;4 mutant phenotypes are seen (Usui et al., 2004). This lethality at birth might relate to the possibility that FGF18 may, in addition, bind FGFR2. Furthermore, it remains possible that other FGF ligands also act through FGFR3 and FGFR4 to control alveologenesis.
Our lineage-specific inactivation of Fgfr3 and Fgfr4 demonstrates that they are required in the mesenchyme, but not the epithelium, for alveoli formation. This is consistent with their likely role in elastogenesis, as most of the factors that drive elastin formation are produced by fibroblasts (Boström et al., 1996; Boucherat et al., 2007; Lindahl et al., 1997). Interestingly, it has been reported that the epithelial activity of FGFR3 is essential for tumorigenesis in the adult (Yin et al., 2013). Therefore, although Fgfr3 and Fgfr4 activity in the epithelium is not required for early postnatal lung development, it is linked to the pathogenesis of adult lung diseases.
Our findings suggest that the primary role of Fgfr3 and Fgfr4 is to control the orderly formation of elastin fibers. We propose that, in a normal lung, FGFR3 and FGFR4 restrict the expression of MFAP5, among other elastogenesis factors. Analysis of the Mfap5 regulatory region identified multiple FGF-regulated SP1 and SP8 transcription factor binding sites within 20 kb upstream of the transcription start site, suggesting possible mode of regulation. Once transcribed and translated, MFAP5 is then deposited in orderly spaced positions along the microfibril, acting as distributed ‘seeds' to direct elastin polymerization onto the microfibril (Fig. 8A). Once seeded, elastin fibers grow to underlie the septal ridges, and likely stabilize these new gas-exchange surfaces. In the Fgfr3;4 mutant, MFAP5 expression is no longer restricted. The increased MFAP5 coats the microfibrils more densely, allowing initiation of elastin polymerization at many more sites, resulting in shorter and disorganized fibers that run through the walls of the saccule (Fig. 8B). This ‘mat' of elastin network might offer all-directional recoil. This change in mechanical properties would interfere with the ability of contractile myofibroblasts to form new septal ridges, resulting in simplified alveoli. It is also possible that a change in elastin would alter lung compliance, which would in turn increase alveolar size when inflated. We speculate that a similar mechanism could be at play in BPD, where a decrease in FGF signaling may lead to disorganization of elastin, thereby disrupting the normal process of alveologenesis.
Fig. 8.
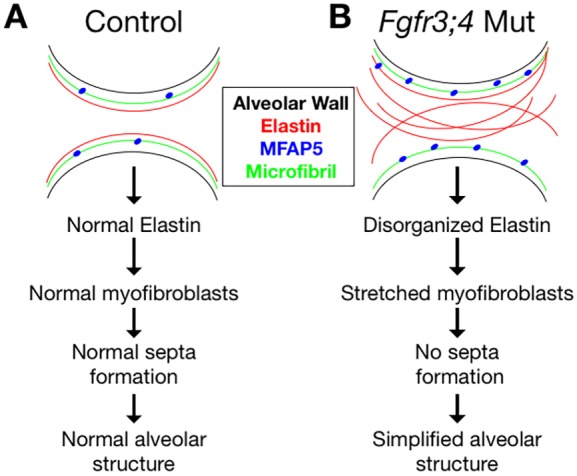
Model of the role of FGFR3 and FGFR4 in alveologenesis. (A) In the control lung, MFAP5 molecules (blue dots) are regularly spaced and act as seeds to facilitate crosslinking of elastin molecules onto the microfibrils in an orderly manner, forming long elastin fibers that line the alveolar entrance ring. This deposition of elastin supports normal morphology and function of myofibroblasts, which drives septa formation, leading to expansion of the gas-exchange surface area. (B) In the Fgfr3;4 mutant lung, the increase in MFAP5 leads to an increase in crosslinking sites of elastin onto the microfibrils, leading to more and shorter elastin fibers running through the walls of the primary alveoli. The disorganized elastin network leads to stretched myofibroblasts and disrupts septa formation, resulting in alveolar simplification.
MATERIALS AND METHODS
Mice
Mice were housed and all experimental procedures were performed in an American Association for Accreditation of Laboratory Animal Care-accredited laboratory animal facility at the University of Wisconsin. This study was approved by the University of Wisconsin and UCSD Animal Care and Use Committee and conformed to the Guide for the Care and Use of Laboratory Animals. Pups were dissected postnatally, with the date of birth being marked as postnatal day (P) 0. Mutant alleles were described previously: Fgfr3− (Deng et al., 1996), Fgfr3fl (Su et al., 2010), Fgfr4− (Weinstein et al., 1998), PdgfraGFP (Hamilton et al., 2003), Mfap5 (Magp2)− (Combs et al., 2013), Dermo1cre (Šošić et al., 2003) and Nkx2-1-cre (Xu et al., 2008); primers used for genotyping can be found in the original publications describing these lines. Control mice were Fgfr3+/−;Fgfr4−/− for the global knockouts and cre;Fgfr3fl/+;Fgfr4−/− (Cre control) for the conditional knockouts.
Histology
For histological analysis, lungs were inflation fixed with 4% paraformaldehyde (PFA), equilibrated into 30% sucrose, embedded in OCT (Sakura, 4583) and sectioned at 5 μm. Sections were stained using a standard Hematoxylin and Eosin (H&E) protocol. Quantification of lung simplification was performed using the mean linear intercept (MLI) method, as previously described (Chen et al., 2012). Three evenly spaced sections from at least three lungs were used to calculate the MLI for mutant and control samples at every stage. Statistical analysis was carried out using Student's t-test. All experiments have been repeated using multiple litters.
Immunofluorescent staining
For immunofluorescent staining, lungs were inflation fixed overnight in 4% PFA. The lungs were cut into ∼75 μm thick sections using a vibratome and stained using a standard immunofluorescence staining protocol (Yi et al., 2009). From each vibratome section, a 50 μm z-stack was taken using a Zeiss 510 confocal laser scanning microscope and then processed into a 2D image using ImageJ. The primary antibodies used were: SFTPC (Seven Hills, WRAB-76994; 1:200), TAGLN (Millipore, MABT167; 1:200), elastin (gift from Dr Robert Mecham, Washington University; 1:400), FBN1 (gift from Dr Lynn Sakai, Oregon Health and Science University; 1:200), FBN2 (gift from Dr Lynn Sakai, 1:200), ADRP (Abcam, ab52356; 1:200), IsoB4 (Sigma, L2895; 1:200), MFAP5 (LifeSpan BioSciences, LS-C423907; 1:200) and FGFR3 (gift from Dr David Ornitz, Washington University; 1:200).
Quantitative RT-PCR
Total RNA was extracted from tissues using Trizol (Invitrogen, 15596-018) and an RNeasy Micro RNA extraction kit (Qiagen). RNA was reverse-transcribed using the Superscript III first-strand synthesis system (Invitrogen). Quantitative PCR was performed using SYBR Green (Applied Biosystems). Three technical and three biological replicates were performed for each gene, and statistical analysis of the results was carried out using Student's t-test. Primers used for qPCR are listed in Table S1.
Western blot analysis
Total protein was collected from at least three control and three mutant mice. Protein samples were homogenized on a Qiagen TissueLyser II and were run on an 8-15% SDS-PAGE gel and transferred to PVDF membrane. The PVDF membrane was blocked in 5% dried milk powder in TBST (with 0.1% Tween 20) for at least 1 h, incubated in the primary antibody overnight, and then incubated with the secondary antibody for 1 h. Quantification was carried out using the Image Studio Lite software system (LI-COR Biosciences). Primary antibodies were MFAP5 (gift from Dr Robert Mecham; 1:1000), tropoelastin (Abcam, ab21600; 1:500) and β-actin (Abcam, ab8226; 1:4000). Protein levels were normalized to β-actin. Three biological replicates were performed for each antibody.
Supplementary Material
Acknowledgements
We thank Dr Lynn Sakai for the generous gift of anti-fibrillin 1 and anti-fibrillin 2 antibodies; Dr Chuxia Deng and Dr David Ornitz for sharing mouse lines; Dr Sebastian Preissl of the UCSD Epigenomic Center for advice on transcription factor binding site analysis; and members of the X.S. laboratory for constructive discussions and reading of the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Investigation: R.L., J.C.H.; Resources: L.C., R.P.M.; Data curation: R.L., J.C.H., X.S.; Writing - original draft: R.L., J.C.H.; Writing - review & editing: R.L., J.C.H., L.C., R.P.M., X.S.; Supervision: X.S.; Funding acquisition: X.S.
Funding
This work was supported by an American Heart Association predoctoral fellowship (11PRE5540006) and a National Institutes of Health predoctoral training grant to the University of Wisconsin Genetics Program (T32 GM007133) to J.C.H.; National Heart, Lung, and Blood Institute grants (RO1 HL113870, RO1 HL097134, RO1 HL122406) and a Wisconsin Partnership Program Grant (2897) from the School of Medicine and Public Health, University of Wisconsin-Madison to X.S.; and National Institutes of Health grants (HL53325 and HL105314) to R.P.M. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.149443.supplemental
References
- Abler L. L., Mansour S. L. and Sun X. (2009). Conditional gene inactivation reveals roles for Fgf10 and Fgfr2 in establishing a normal pattern of epithelial branching in the mouse lung. Dev. Dyn. 238, 1999-2013. 10.1002/dvdy.22032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Alam D., El Agha E., Sakurai R., Kheirollahi V., Moiseenko A., Danopoulos S., Shrestha A., Schmoldt C., Quantius J., Herold S. et al. (2015). Evidence for the involvement of fibroblast growth factor 10 in lipofibroblast formation during embryonic lung development. Development 142, 4139-4150. 10.1242/dev.109173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellusci S., Grindley J., Emoto H., Itoh N. and Hogan B. L. (1997). Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development 124, 4867-4878. [DOI] [PubMed] [Google Scholar]
- Benjamin J. T., Smith R. J., Halloran B. A., Day T. J., Kelly D. R. and Prince L. S. (2007). FGF-10 is decreased in bronchopulmonary dysplasia and suppressed by Toll-like receptor activation. Am. J. Physiol. Lung Cell. Mol. Physiol. 292, L550-L558. 10.1152/ajplung.00329.2006 [DOI] [PubMed] [Google Scholar]
- Boström H., Willetts K., Pekny M., Levéen P., Lindahl P., Hedstrand H., Pekna M., Hellström M., Gebre-Medhin S., Schalling M. et al. (1996). PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 85, 863-873. 10.1016/S0092-8674(00)81270-2 [DOI] [PubMed] [Google Scholar]
- Boucherat O., Franco-Montoya M.-L., Thibault C., Incitti R., Chailley-Heu B., Delacourt C. and Bourbon J. R. (2007). Gene expression profiling in lung fibroblasts reveals new players in alveolarization. Physiol. Genomics 32, 128-141. 10.1152/physiolgenomics.00108.2007 [DOI] [PubMed] [Google Scholar]
- Branchfield K., Li R., Lungova V., Verheyden J. M., McCulley D. and Sun X. (2016). A three-dimensional study of alveologenesis in mouse lung. Dev. Biol. 409, 429-441. 10.1016/j.ydbio.2015.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canadas V., Vilacosta I., Bruna I. and Fuster V. (2010). Marfan syndrome. Part 1: pathophysiology and diagnosis. Nat. Rev. Cardiol. 7, 256-265. 10.1038/nrcardio.2010.30 [DOI] [PubMed] [Google Scholar]
- Carta L., Pereira L., Arteaga-Solis E., Lee-Arteaga S. Y., Lenart B., Starcher B., Merkel C. A., Sukoyan M., Kerkis A., Hazeki N. et al. (2006). Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J. Biol. Chem. 281, 8016-8023. 10.1074/jbc.M511599200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao C.-M., Yahya F., Moiseenko A., Tiozzo C., Shrestha A., Ahmadvand N., El Agha E., Quantius J., Dilai S., Kheirollahi V. et al. (2017). Fgf10 deficiency is causative for lethality in a mouse model of bronchopulmonary dysplasia. J. Pathol. 241, 91-103. 10.1002/path.4834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbonneau N. L., Dzamba B. J., Ono R. N., Keene D. R., Corson G. M., Reinhardt D. P. and Sakai L. Y. (2003). Fibrillins can co-assemble in fibrils, but fibrillin fibril composition displays cell-specific differences. J. Biol. Chem. 278, 2740-2749. 10.1074/jbc.M209201200 [DOI] [PubMed] [Google Scholar]
- Charbonneau N. L., Carlson E. J., Tufa S., Sengle G., Manalo E. C., Carlberg V. M., Ramirez F., Keene D. R. and Sakai L. Y. (2010). In vivo studies of mutant fibrillin-1 microfibrils. J. Biol. Chem. 285, 24943-24955. 10.1074/jbc.M110.130021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Acciani T., Le Cras T., Lutzko C. and Perl A.-K. T. (2012). Dynamic regulation of platelet-derived growth factor receptor alpha expression in alveolar fibroblasts during realveolarization. Am. J. Respir. Cell Mol. Biol. 47, 517-527. 10.1165/rcmb.2012-0030OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulis J. T., Bellingham C. M., Davis E. C., Hubmacher D., Reinhardt D. P., Mecham R. P. and Keeley F. W. (2008). Fibrillins, fibulins, and matrix-associated glycoprotein modulate the kinetics and morphology of in vitro self-assembly of a recombinant elastin-like polypeptide. Biochemistry 47, 12601-12613. 10.1021/bi8005384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin J. S., Bohne B. A., Harding G. W., McEwen D. G. and Ornitz D. M. (1996). Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat. Genet. 12, 390-397. 10.1038/ng0496-390 [DOI] [PubMed] [Google Scholar]
- Combs M. D., Knutsen R. H., Broekelmann T. J., Toennies H. M., Brett T. J., Miller C. A., Kober D. L., Craft C. S., Atkinson J. J., Shipley J. M. et al. (2013). Microfibril-associated glycoprotein 2 (MAGP2) loss of function has pleiotropic effects in vivo. J. Biol. Chem. 288, 28869-28880. 10.1074/jbc.M113.497727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C., Wynshaw-Boris A., Zhou F., Kuo A. and Leder P. (1996). Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell 84, 911-921. 10.1016/S0092-8674(00)81069-7 [DOI] [PubMed] [Google Scholar]
- Hamilton T. G., Klinghoffer R. A., Corrin P. D. and Soriano P. (2003). Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Mol. Cell. Biol. 23, 4013-4025. 10.1128/MCB.23.11.4013-4025.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris K. S., Zhang Z., McManus M. T., Harfe B. D. and Sun X. (2006). Dicer function is essential for lung epithelium morphogenesis. Proc. Natl. Acad. Sci. USA 103, 2208-2213. 10.1073/pnas.0510839103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokuto I., Perl A.-K. T. and Whitsett J. A. (2003). Prenatal, but not postnatal, inhibition of fibroblast growth factor receptor signaling causes emphysema. J. Biol. Chem. 278, 415-421. 10.1074/jbc.M208328200 [DOI] [PubMed] [Google Scholar]
- Lemaire R., Bayle J., Mecham R. P. and Lafyatis R. (2007). Microfibril-associated MAGP-2 stimulates elastic fiber assembly. J. Biol. Chem. 282, 800-808. 10.1074/jbc.M609692200 [DOI] [PubMed] [Google Scholar]
- Lindahl P., Karlsson L., Hellstrom M., Gebre-Medhin S., Willetts K., Heath J. K. and Betsholtz C. (1997). Alveogenesis failure in PDGF-A-deficient mice is coupled to lack of distal spreading of alveolar smooth muscle cell progenitors during lung development. Development 124, 3943-3953. [DOI] [PubMed] [Google Scholar]
- Liu J. P., Baker J., Perkins A. S., Robertson E. J. and Efstratiadis A. (1993). Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 75, 59-72. 10.1016/S0092-8674(05)80084-4 [DOI] [PubMed] [Google Scholar]
- Liu X., Zhao Y., Gao J., Pawlyk B., Starcher B., Spencer J. A., Yanagisawa H., Zuo J. and Li T. (2004). Elastic fiber homeostasis requires lysyl oxidase-like 1 protein. Nat. Genet. 36, 178-182. 10.1038/ng1297 [DOI] [PubMed] [Google Scholar]
- Mariani T. J., Reed J. J. and Shapiro S. D. (2002). Expression profiling of the developing mouse lung: insights into the establishment of the extracellular matrix. Am. J. Respir. Cell Mol. Biol. 26, 541-548. 10.1165/ajrcmb.26.5.2001-00080c [DOI] [PubMed] [Google Scholar]
- McGowan S. E. and McCoy D. M. (2014). Regulation of fibroblast lipid storage and myofibroblast phenotypes during alveolar septation in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 307, L618-L631. 10.1152/ajplung.00144.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrisey E. E. and Hogan B. L. M. (2010). Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev. Cell 18, 8-23. 10.1016/j.devcel.2009.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mund S. I., Stampanoni M. and Schittny J. C. (2008). Developmental alveolarization of the mouse lung. Dev. Dyn. 237, 2108-2116. 10.1002/dvdy.21633 [DOI] [PubMed] [Google Scholar]
- Neptune E. R., Frischmeyer P. A., Arking D. E., Myers L., Bunton T. E., Gayraud B., Ramirez F., Sakai L. Y. and Dietz H. C. (2003). Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat. Genet. 33, 407-411. 10.1038/ng1116 [DOI] [PubMed] [Google Scholar]
- Ntokou A., Klein F., Dontireddy D., Becker S., Bellusci S., Richardson W. D., Szibor M., Braun T., Morty R. E., Seeger W. et al. (2015). Characterization of the platelet-derived growth factor receptor-alpha-positive cell lineage during murine late lung development. Am. J. Physiol. Lung Cell. Mol. Physiol. 309, L942-L958. 10.1152/ajplung.00272.2014 [DOI] [PubMed] [Google Scholar]
- Park M. S., Rieger-Fackeldey E., Schanbacher B. L., Cook A. C., Bauer J. A., Rogers L. K., Hansen T. N., Welty S. E. and Smith C. V. (2007). Altered expressions of fibroblast growth factor receptors and alveolarization in neonatal mice exposed to 85% oxygen. Pediatr. Res. 62, 652-657. 10.1203/PDR.0b013e318159af61 [DOI] [PubMed] [Google Scholar]
- Perl A.-K. T., Hokuto I., Impagnatiello M.-A., Christofori G. and Whitsett J. A. (2003). Temporal effects of Sprouty on lung morphogenesis. Dev. Biol. 258, 154-168. 10.1016/S0012-1606(03)00106-4 [DOI] [PubMed] [Google Scholar]
- Powell-Braxton L., Hollingshead P., Warburton C., Dowd M., Pitts-Meek S., Dalton D., Gillett N. and Stewart T. A. (1993). IGF-I is required for normal embryonic growth in mice. Genes Dev. 7, 2609-2617. 10.1101/gad.7.12b.2609 [DOI] [PubMed] [Google Scholar]
- Rehan V. K. and Torday J. S. (2003). Hyperoxia augments pulmonary lipofibroblast-to-myofibroblast transdifferentiation. Cell Biochem. Biophys. 38, 239-250. 10.1385/CBB:38:3:239 [DOI] [PubMed] [Google Scholar]
- Reinhardt D. P., Keene D. R., Corson G. M., Pöschl E., Bächinger H. P., Gambee J. E. and Sakai L. Y. (1996). Fibrillin-1: organization in microfibrils and structural properties. J. Mol. Biol. 258, 104-116. 10.1006/jmbi.1996.0237 [DOI] [PubMed] [Google Scholar]
- Rezvani M., Wilde J., Vitt P., Mailaparambil B., Grychtol R., Krueger M. and Heinzmann A. (2013). Association of a FGFR-4 gene polymorphism with bronchopulmonary dysplasia and neonatal respiratory distress. Dis. Markers 35, 633-640. 10.1155/2013/932356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifren A. and Mecham R. P. (2006). The stumbling block in lung repair of emphysema: elastic fiber assembly. Proc. Am. Thorac. Soc. 3, 428-433. 10.1513/pats.200601-009AW [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šošić D., Richardson J. A., Yu K., Ornitz D. M. and Olson E. N. (2003). Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell 112, 169-180. 10.1016/S0092-8674(03)00002-3 [DOI] [PubMed] [Google Scholar]
- Srisuma S., Bhattacharya S., Simon D. M., Solleti S. K., Tyagi S., Starcher B. and Mariani T. J. (2010). Fibroblast growth factor receptors control epithelial-mesenchymal interactions necessary for alveolar elastogenesis. Am. J. Respir. Crit. Care Med. 181, 838-850. 10.1164/rccm.200904-0544OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su N., Xu X., Li C., He Q., Zhao L., Li C., Chen S., Luo F., Yi L., Du X. et al. (2010). Generation of Fgfr3 conditional knockout mice. Int. J. Biol. Sci. 6, 327-332. 10.7150/ijbs.6.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui H., Shibayama M., Ohbayashi N., Konishi M., Takada S. and Itoh N. (2004). Fgf18 is required for embryonic lung alveolar development. Biochem. Biophys. Res. Commun. 322, 887-892. 10.1016/j.bbrc.2004.07.198 [DOI] [PubMed] [Google Scholar]
- Volckaert T. and De Langhe S. (2014). Lung epithelial stem cells and their niches: Fgf10 takes center stage. Fibrogen. Tissue Repair 7, 8 10.1186/1755-1536-7-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrhovski B. and Weiss A. S. (1998). Biochemistry of tropoelastin. Eur. J. Biochem. 258, 1-18. 10.1046/j.1432-1327.1998.2580001.x [DOI] [PubMed] [Google Scholar]
- Wagenseil J. E. and Mecham R. P. (2007). New insights into elastic fiber assembly. Birth Defects Res. C Embryo Today 81, 229-240. 10.1002/bdrc.20111 [DOI] [PubMed] [Google Scholar]
- Wang Q., Green R. P., Zhao G. and Ornitz D. M. (2001). Differential regulation of endochondral bone growth and joint development by FGFR1 and FGFR3 tyrosine kinase domains. Development 128, 3867-3876. [DOI] [PubMed] [Google Scholar]
- Weinstein M., Xu X., Ohyama K. and Deng C. X. (1998). FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development 125, 3615-3623. [DOI] [PubMed] [Google Scholar]
- Wendel D. P., Taylor D. G., Albertine K. H., Keating M. T. and Li D. Y. (2000). Impaired distal airway development in mice lacking elastin. Am. J. Respir. Cell Mol. Biol. 23, 320-326. 10.1165/ajrcmb.23.3.3906 [DOI] [PubMed] [Google Scholar]
- White A. C., Xu J., Yin Y., Smith C., Schmid G. and Ornitz D. M. (2006). FGF9 and SHH signaling coordinate lung growth and development through regulation of distinct mesenchymal domains. Development 133, 1507-1517. 10.1242/dev.02313 [DOI] [PubMed] [Google Scholar]
- Xu Q., Tam M. and Anderson S. A. (2008). Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. J. Comp. Neurol. 506, 16-29. 10.1002/cne.21529 [DOI] [PubMed] [Google Scholar]
- Yanagisawa H., Davis E. C., Starcher B. C., Ouchi T., Yanagisawa M., Richardson J. A. and Olson E. N. (2002). Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature 415, 168-171. 10.1038/415168a [DOI] [PubMed] [Google Scholar]
- Yanagisawa H., Schluterman M. K. and Brekken R. A. (2009). Fibulin-5, an integrin-binding matricellular protein: its function in development and disease. J. Cell Commun. Signal. 3, 337-347. 10.1007/s12079-009-0065-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G. C., Keeley F. W. and Weiss A. S. (2011). Coacervation of tropoelastin. Adv. Colloid Interface Sci. 167, 94-103. 10.1016/j.cis.2010.10.003 [DOI] [PubMed] [Google Scholar]
- Yi L., Domyan E. T., Lewandoski M. and Sun X. (2009). Fibroblast growth factor 9 signaling inhibits airway smooth muscle differentiation in mouse lung. Dev. Dyn. 238, 123-137. 10.1002/dvdy.21831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y., Wang F. and Ornitz D. M. (2011). Mesothelial- and epithelial-derived FGF9 have distinct functions in the regulation of lung development. Development 138, 3169-3177. 10.1242/dev.065110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y., Betsuyaku T., Garbow J. R., Miao J., Govindan R. and Ornitz D. M. (2013). Rapid induction of lung adenocarcinoma by fibroblast growth factor 9 signaling through FGF receptor 3. Cancer Res. 73, 5730-5741. 10.1158/0008-5472.CAN-13-0495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K., Xu J., Liu Z., Sosic D., Shao J., Olson E. N., Towler D. A. and Ornitz D. M. (2003). Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130, 3063-3074. 10.1242/dev.00491 [DOI] [PubMed] [Google Scholar]
- Yu S., Poe B., Schwarz M., Elliot S. A., Albertine K. H., Fenton S., Garg V. and Moon A. M. (2010). Fetal and postnatal lung defects reveal a novel and required role for Fgf8 in lung development. Dev. Biol. 347, 92-108. 10.1016/j.ydbio.2010.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Menke D. B., Jiang M., Chen H., Warburton D., Turcatel G., Lu C.-H., Xu W., Luo Y. and Shi W. (2013). Spatial-temporal targeting of lung-specific mesenchyme by a Tbx4 enhancer. BMC Biol. 11, 111 10.1186/1741-7007-11-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.