

Abstract
In this article, the first-principles prediction of enthalpies of formation is demonstrated for 669 polycyclic aromatic hydrocarbon (PAH) compounds and a number of related functionalized molecules. It is shown that by extrapolating density functional theory calculations to a large basis set limit and then applying a group based correction scheme that good results may be obtained. Specifically, a mean unsigned deviation and root mean squared deviation from the experimental enthalpies of formation data of 5.0 and 6.4 kJ/mol, respectively, are obtained using this scheme. This computational scheme is economical to compute and straightforward to apply, while yielding results of reasonable reliability. The results are also compared for a smaller set of molecules to the predictions given by the G3B3 and G3MP2B3 variants of the Gaussian-3 model chemistry with a mean unsigned deviation and root mean squared deviation from the experimental enthalpies of formation of 4.5 and 4.8 kJ/mol, respectively.
Graphical abstract
INTRODUCTION
Background
Polycyclic aromatic hydrocarbons (PAHs) are a class of substances that have been associated with a number of acute and chronic health effects. Additionally, PAHs are suspected carcinogens, mutagens, and teratogens. PAHs occur in the environment due to a number of natural sources including forest fires, volcanoes, and as a component of crude oil. However, their primary source is overwhelmingly anthropogenic. PAHs are created through a number of processes including incomplete combustion of fossil fuels, high-temperature cooking of foods, and burning of municipal refuse and in petroleum based products such as coal tar and asphalt. PAHs in the atmosphere frequently occur as particles attached to dust or as components of soot (soots generally contain a mixture of PAHs). Due to their low solubility in water, PAHs tend to accumulate in soils and on lake and river beds, rather than as a significant water contaminant. Unfortunately, this type of accumulation makes PAH pollutants rather long-lived and difficult to remove from the environment. A number of aquatic species are known to concentrate PAHs in their tissues.
Due to the environmental and health risks associated with PAHs, and because the largest source of PAH molecules is combustion from human activities, it is important to develop a thorough understanding of their chemistry. Thermodynamics quantities are important for understanding the formation and growth of PAHs and are of particular value in modeling studies. These large molecules are important precursors for the formation of soot particles during the combustion of hydrocarbons fuels under fuel-rich conditions. Many detailed chemical kinetic models have been developed to describe the growth of PAHs to predict their concentrations and to aid in predicting sooting behavior. The relative stabilities of these molecules and their derivatives influence the rate of formation and consumption of PAHs, and ultimately affect the rate of soot formation. Many of the important reactions in the formation of initial PAH species, and the subsequent growth of larger PAHs, involve chemically activated steps, such as additions/eliminations, isomerizations (e.g., H atom migrations), cyclizations (ring growth), and β bond scissions (bond breaking reactions in radicals) that are often strongly temperature and pressure dependent. Among the various thermodynamic quantities that may be computed, the enthalpy of formation is of particular importance in characterizing their thermal stability and is thus the subject of the present study.
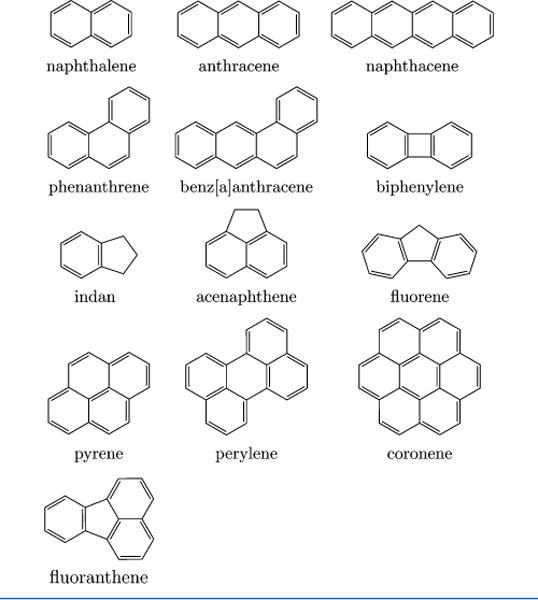
PAHs are characterized by possessing more than two fused aromatic benzene rings and by having no non-hydrogen substituents.1 Several examples of small PAHs are given in Table 1 showing aromatic and other rings fused in different manners. This figure gives a hint at the diversity of possible PAH structures.
Table 1.
Structures of Representative Polycyclic Aromatic Hydrocarbon Compounds (PAHs)
|
The purpose of the present article is to survey the current state of thermochemical data on PAHs from experiment, group additivity, and computational chemistry, and to provide a comprehensive and consistent set of predicted values for the heat of formation of PAHs. Prediction of PAH values will rely on computational chemistry methods with reasonable cost and on group additivity correction schemes to account for systematic errors in those methods.
The remainder of this article is organized as follows. A survey of relevant literature is given in the remainder of this section. The details of the ab initio calculations performed for this work and the procedures and equations necessary for producing enthalpies of formation are presented in the following section. The results of these calculations are compared to experimental data, and to computational and empirical estimates, in the next section. Finally, the value of the present approach in light of previous work is discussed.
Survey of Related Prior Work
Although there has been much work over many years investigating the stability of PAHs using computational methods (as well as experimental measurements), many of these studies have been limited in scope to a few species, have employed quantum chemical methods at lower levels of theory, have not performed a full review of experimental and computational values in the literature, and/or have not made adequate comparisons between the computed and experimental values. The current work significantly expands on the set of PAHs previously studied computationally and uses a range of quantum chemical methods to provide a measure of uncertainties in the computational methods and to facilitate better comparison with experimental data. The thermochemical data (enthalpies of formation) for PAHs are also compiled and briefly evaluated. (For a more detailed evaluation on a more limited set of PAHs, the reader is referred to a recent work by Chickos and coworkers.2) In the present work, quantum chemical calculations are used as a screening tool to identify species where there are significant differences between experimentally derived enthalpies of formation and the corresponding computed values. In these cases, a further examination of both the experimental data and computational values (and methods) is warranted.
A general overview of related prior work is now provided. There are many excellent references (cited below) that provide a better overview of many important background subjects than this article can realistically achieve, on the topics of aromaticity, thermochemical data, group additivity, quantum chemical methods, and detailed chemical kinetic models. These areas will be briefly surveyed before reiterating the objectives of the present work and fully discussing the present methodology and results.
PAH Structures
Polycyclic aromatic hydrocarbons (PAHs) are defined as having more than two fused benzene rings and no non-hydrogen substitutions. However, on a practical level PAHs are formed and grow via many reactions involving smaller PAHs that have unsaturated substituents.3–8 The inclusion of hydrocarbon-substituted PAHs in this study is in recognition of this consideration.
There are several types of fusion that occur in PAHs, and we now briefly discuss these and the related common terminology.1 Several examples of different types of PAH structures are provided in Table 1, showing different types of ring fusion. PAHs that have adjacent rings with two adjacent atoms in common are termed “ortho-fused”, whereas those that have rings with two adjacent atoms in common with two or more rings are termed “ortho- and peri-fused”.
The simplest example of an ortho-fused compound is naphthalene, resulting from the fusion of two benzene rings at two common atoms (fused on one face; see Table 1). Naphthalene has eight peripheral aromatic carbon sites (CbH) terminated by hydrogen atoms, and two fused aromatic carbon sites (Cf), each having two rings in common. The simplest example of a compound with ortho- and peri-fused carbon atoms is pyrene, resulting from the ortho- and peri-fusion of four benzene rings where two or more adjacent bonds are involved (fused on two faces). Pyrene has ten peripheral (CbH) sites, four fused (Cf) sites, and two interior ortho- and peri-fused (Cp) sites.
There are different types of PAH structures (see Table 1 for a number of examples). Polyacenes such as anthracene and naphthacene have ortho-fused benzene rings in a linear arrangement where the next benzene ring is fused on the opposite or “b” face from the previous ring. For example, anthracene could be named as benzo[b]naphthalene. Poly-aphenes are ortho-fused PAHs, such as phenanthrene, and have the next benzene ring fused on an adjacent face that is at 120° (the “a” face) from the previous ring. For example, phenanthrene could be named as benzo[a]naphthalene. As discussed above, ortho- and peri-fused PAHs occur when the aromatic rings are fused on two or more faces; pyrene, perylene, and coronene are representative structures.
PAHs can also involve fused structures where the fused ring involves rings other than six-membered benzene rings. Indan (Table 1), for example, can alternately be named benzocyclopentane or cyclopentabenzene, indicative of a cyclopentene ring fused on a single face (ortho-fused) to a benzene ring. These are sometimes termed as cycloalkane or cycloalkene rings that are fused by being attached or joined to the aromatic rings. Such rings can also be ortho- and peri-fused, as in acenaphthene, where a five-membered ring is created by a –CH2CH2– bridge between the 1 and 8 positions on naphthalene. Compounds with this type of bridge fusion are termed ace-ylenes. Larger representative examples of multifused PAHs with cycloalkane or cycloalkene fused rings are biphenylene, fluorene, and fluoranthene (Table 1), which might be systematically named as benzocyclobuttabenzene, benzocyclopentabenzene, and benzocyclopenta[de]-naphthalene.
As there are a large number of PAH structures that have been optimized in this study, a Web site9 has been created to disseminate this information. The Web site contains data such as names, molecular formulas, 2-D structures, and 3-D structures (that may be viewed on the Web site as well as downloaded as Cartesian coordinates).
Aromaticity
The stability and resonance stabilization energies of polycyclic aromatic hydrocarbons (PAHs) have been the subject of considerable theoretical interest for many years. The earliest work using molecular orbital calculations dates over half a century to work in the 1950s to 1960s by Pariser and Parr,10 by Pople,11 and by Dewar and co-workers.12 These early calculations implemented and further developed the concepts of electron delocalization energies and molecular orbital theory that were formulated by Hückel13 in the 1930s that are, in turn, built on the foundational concepts of chemical valence (Frankland, 1852),14 of structural connectivity (Kekulé, 1858),15 and of aromaticity (Kekulé, 1865).16
There are a number of excellent recent review articles and books on aromaticity, electron delocalization, and resonance stabilization energies, including those by Schleyer,17 Poater et al.,18 Curaski,19 Merino et al.,20 and Aihara.21 The reviews by Schleyer and co-workers22,23 and by Matta and Hernandez-Trujillo24 are very informative and helpful. In a recent work, Cappel et al.25 studied conjugative and hyperconjungative stabilization effects in various conjugated species.
A significant number of current methodologies and discussions in the area of aromaticity derive from work published in 1967 by Polansky and Derflinger,26 who pioneered useful characterizations for quantifying aromaticity in molecules. For data derived from experiment, much of the quantitative knowledge regarding the stability of PAHs is influenced in a number of ways by the work of Kistiakowsky and co-workers in the 1930s who examined the enthalpies of hydrogenation of a variety of unsaturated compounds.27–29 Turner and co-workers30–32 in an extensive series about three decades ago measured the enthalpies of hydrogenation for different classes of compounds. These systematic studies have been continued in more recent times through the extensive work of Roth and co-workers33,34 and by Rogers and coworkers.35–38 A summary of enthalpies of hydrogenation is given by Jensen.39
Sources of Thermochemical Data
A variety of sources of thermochemical data were used in tabulating the experimental enthalpies of formation of the PAHs and reference hydrocarbons studied in this work. These sources included: Pedley et al.,40 Cox and Pilcher,41 the NIST Chemistry Webbook,42 the reports of the IUPAC Commission on Chemical Kinetics,43,44 and the thermochemical tables produced by Gurvich et al.,45 by the JANAF Working Group,46 by the Thermodynamics Research Center,47 and by Burcat.48,49
Additionally, Slayden and Liebman50 have reviewed thermochemical data for PAH species. Thermochemical functions for cyclic hydrocarbons and cyclopentadiene derivatives have been provided by Dorofeeva et al.51,52 and Karni et al.,53 respectively. Of extreme value is the extensive compilation of enthalpies of hydrogenation for unsaturated molecules including PAHs that is given in the widely cited work by Roth et al.33
Chickos et al. have provided an extensive compilation of experimental enthalpies of vaporization and sublimation54,55 for a wide range of compounds and have developed group additivity based methods for estimating enthalpies of vaporization and sublimation.56,57 Sabbah et al.58 have reviewed experimental data and methods for determining enthalpies of vaporization and sublimation. A text book review of enthalpies of vaporization has been provided by Majer and Svoboda.59
The most reliable and self-consistent set of thermochemical data for species relevant to combustion employs the Active Thermochemical Tables (ATcT) approach pioneered about 10 years ago by Ruscic and co-workers.43 The ATcT approach simultaneously considers all species in a thermochemical network through a set of enthalpies of reactions for all reactions involving the species, providing a self-consistent set of constraints that can be minimized. The concept of free energy minimization in chemical systems was first discussed in depth 90 years ago in the seminal work by Lewis and Randall.60 This work was revised 50 years ago by Pitzer and Brewer.61 Computer optimization of thermochemical data was first extensively used in the 1970s and 1980s by Pedley and coworkers in the CATCH tables62,63 at the National Bureau of Standards in producing the NBS Tables,64–68 and by the CODATA Thermodynamics Task Group.69 Optimization of thermochemical free energy relationships has also been used to produce evaluated thermochemistry in biochemical reaction systems.70–72 Although this thermodynamic network methodology has not been applied yet to large molecules such as PAHs, the data produced in this effort are important in determining enthalpies of formation of smaller fundamental hydrocarbon reference species, which are needed for benchmarking ab initio calculations on PAHs, where empirical corrections are needed to generate accurate values.
Empirical Approaches and Quantum Chemistry
Group additivity is an empirical method for predicting thermochemical properties of compounds as sums of the properties of their component parts that has been very successful in predicting data for aliphatic hydrocarbons and other species. This method was initially developed and utilized by Benson and co-workers.73–77
Stein et al. used group additivity techniques to predict thermochemical properties of PAHs.78,79 Alberty and Reif80 estimated the enthalpies of formation (and other standard thermodynamic quantities) of a number of PAH molecules using Benson group additivity73 with various group values.75,78,79 Several articles expanded on this method to correct for deficiencies in the model. Armitage and Bird81 and Moiseeva and co-workers gave updated values for a few critical groups, leading to small improvements in accuracy for PAHs82 and for compounds with five-membered rings.83 Herndon et al.84 compared the performance of group additivity with molecular mechanics and semiempirical quantum chemistry methods on a set of 11 polycyclic benzenoid aromatic hydrocarbons to experimental values, finding that the results were generally in good agreement. Armitage and Bird81 extended the use of group additivity to predict the stability of very large PAH species such as fullerenes (cage-like fused rings). Benzenoid molecules, including radicals of these species, were studied by Wang and Frenklach using the semiempirical AM1 Hamiltonian.85 Heats of formation and heats of sublimation were estimated using a quantitative structure–property relationship (QSPR) model that was derived from a 3-dimensional quantitative structure–activity relationship (QSAR) known as comparative molecular field analysis (CoMFA) by Welsh et al.86 Their results were in very good agreement with experimental results for a small set molecules on which the model was tested. Welsh et al. also used group additivity to estimate enthalpies of sublimation of PAHs.
Yu et al.87 developed a method for estimating thermochemical properties, including heats of formation, of PAHs based on density functional theory (DFT) calculations. Their model, called bond-centered group additivity (BCGA) used 20 structural parameters and yielded average errors (note that this is not the uncertainty) of less than 3 kcal/mol (≈12.5 kJ/mol) over a set of 107 molecules.
Other methods have also been used to examine PAH properties and reaction pathways. Using molecular mechanics methods, Allinger et al.88 studied the stability of five-membered rings, which are important precursors and intermediates in PAH formation.
Both ab initio and DFT methods have been used to calculate physical and chemical properties for PAHs. Given its low computational cost and ability to replicate geometric parameters and vibrational frequencies with reasonable accuracy, B3LYP is widely employed for computing molecular structures, vibrational frequencies, and enthalpies of formation in large molecules with many heavy atoms (non-hydrogen) such as PAHs. To produce more accurate results, the selected method generally requires a large basis set and a good treatment of electron correlation. The B3LYP method includes LYP correlation, giving it an advantage over approaches such as Hartree–Fock, which are uncorrelated methods. Composite methods such as the complete basis set (CBS) method and the Gaussian-n model chemistries use a balanced set of calculations to converge the one-particle (basis set) and n-particle (correlation) expansions, producing high-quality results at an additional computational expense.
Several studies have used advanced group additivity approaches in conjunction with quantum chemical methods, in studying PAHs. Sivaramakrishnan et al.89 used the hybrid DFT method B3LYP90,91 to calculate enthalpies of formation of a significant number of PAHs and substituted-aromatic species and developed a detailed group additivity method for reproducing enthalpies of formation to within about 8 kJ/mol. This work built on earlier B3LYP and the group additivity formalism by Yu et al.87 mentioned above. These studies extended the development of group additivity values for large unsaturated hydrocarbons by Sumathi and Green92 using G293 and CBS-Q94 methods. Sabbe et al.95 used the CBS-QB3 method96 in conjunction with group additivity values applied to a large set of aliphatic hydrocarbons, including cyclic species, and a number of substituted-benzene derivatives. Their group additivity scheme was able to reproduce the enthalpies of formation for the substituted aromatics reportedly within 0.7 kJ/mol. These deviations are comparable to the uncertainties of the experimental values that are on the order of 2 kJ/mol. Herndon100 and co-workers have extensively used group additivity approaches in conjunction with quantum chemical methods. They used the semiempirical AM1 method,97 the DFT method B3LYP,90,91 and the ab initio MP2 theory98 to determine enthalpies of formation for many common PAHs.84,99,100 Marsh and Wornat101 used the semiempirical AM1 method to compute thermochemical functions for a number of PAHs (indene, fluoranthene, pyrene, coronone, fluorene) and their cyclopenta-fused derivatives. Pope and Howard102 employed group additivity in conjunction with the molecular mechanics method MM3103 and the semiempirical MOPAC methods MNDO,104 AM1,97 and PM3105 to study the stability of common PAHs, as well as the fullerenes.
Kassaee et al. used B3LYP to compute thermochemical functions for substituted benzene106 and naphthalenes;107 comparable systematic studies of PAHs have been completed by Wiberg108 and Pogodin and Agranat.109,110 Van Speybroeck et al.111 computed bond dissociation energies (BDE) for aromatic species including PAHs. Papas et al.112 studied the radicals of linear PAHs (naphthalene through pentacene) and found computed electron affinities to be in good agreement with experimental values. Reaction pathways and important intermediates in the pyrolysis of cyclopentadiene were studied by Wang et al. using various DFT methods.113 In a more recent work, Hemelsoet et al.114 have used B3P86 and other DFT methods to predict C–H and C–C bond dissociation energies in PAHs. They also employed the G3(MP2)-RAD method115 to computed BDEs for the smaller (benzene through anthracene) molecules.
In an extensive series of studies, Schulman and co-workers used ab initio (HF, MP2) and density functional methods (B3LYP) to compute enthalpies of formation for PAH species116,117 including benz[e]pyrene, coronene, benz[ghi]-perylene (HF),118 pyracyclene and biphenylene (MP2),119 coronene and benz[ghi]perylene (MP2, B3LYP),120 and helicenes and phenacenes (B3LYP).121
Work by Li et al.122 using the G2 method found a correlation between computed and experimental values for enthalpies of formation and the number of double bonds in the molecules. Notario et al. employed the G2 and G2(MP2) methods93 for predicting enthalpies of formation of hydrocarbons including aromatic species123 and in the case of linear polyacenes.124 Cheung et al.125 used the G2 and CBS-Q methods to predict the relative stabilities of the various isomers of benzene. A recent systematic study using high level ab initio composite model chemistries is found in the work of Bond,126 where enthalpies of formation for hydrocarbons, substituted-hydrocarbons, and derivatives, including for a number of PAH species, were computed using a number of different methods (variations of G2, G3, and CBS-Q).
The G3B3 and G3MP2B3 methods have been used with success by Burcat48 and Janoschek and Rossi127 to generate data for species of interest to combustion. Rogers and McLafferty have conducted a series of studies using the G3MP2 method128 to explore the stability of a number of substituted benzenes,129 strained conjugated molecules,130 and triquinacene.131 Melius and co-workers developed and utilized the BAC (bond additivity correction) method132,133 with ab initio calculations to predict enthalpies of formation of important hydrocarbon species, and have recently extended this method to utilize G3B3 energies.134 Fishtik et al.135 employed the G3 method127 and found it to be accurate, not requiring any empirical corrections for the molecules studied. A database of thermochemical properties for PAHs has been created by Blanquart and Pitsch, consisting of 46 molecules with sizes ranging from benzene to coronene (C24H12).136. The values were derived from G3MP2B3 calculations and employed corrections for hindered rotors as well as group based corrections. Their group-corrected G3MP2B3 showed a deviation from experiment of 2.3 kJ/mol for 8 PAH molecules based on 10 groups (the uncorrected deviation was 22.1 kJ/mol).
Rayne and Forest have reported a set of G4(MP2) calculations on PAHs and substituted PAHs.137 The present results are compared to these to the greatest extent possible. Dorofeeva has questioned the methodology used in this work because the computed values are systematically different than other published values.138 In a recent study, Zauer139 computed enthalpies of formation for 139 PAH compounds using the MINDO, MNDO, AM1, and PM3 semiempirical quantum chemistry methods. The study showed deviations from experiment of at least 13 kJ/mol, and employed a correction scheme using a linear expression in the enthalpy of formation.
Review of Literature
An evaluation of the experimental thermochemical data found in the literature was performed for the PAH species and reference molecules, drawing upon the body of work cited above. The present work significantly expands on the set of PAHs and level of theory employed (in most cases) in earlier works (cited above) on PAHs using other quantum chemical methods and group additivity approaches.
A critical evaluation of experimental data for 63 PAH molecules covering enthalpies of combustion, enthalpies of formation in the condensed state, enthalpies of sublimation, enthalpies of vaporization, and enthalpies of fusion has been carried out by Roux et al.2 Importantly, this work provides gas-phase enthalpies of formation for PAHs at 298.15 K. The values in this reference may be considered to be the best currently available, and each of the molecules included in that study has been computed in the present study, with the recommendation of Roux et al.2 being used to evaluate the quality of the results of the present study. Roux et al.2 have provided critically evaluated thermochemical data for PAHs. In particular, species where substantial disagreement exists between experimentally determined and theoretically calculated enthalpies of formation were identified. In this paper, the possibility that either the experimental value or the computed value must be in error is discussed. It is noted that it is important to consider the thermochemical quantities, both experimental and calculated, for each species relative to data for similar molecules with more well-established values; for example, considering the enthalpies of formation of methylbenzene (toluene) and the methylnaph-thalenes relative to that for benzene and naphthalene, and considering the enthalpies of hydrogenation of ethenylbenzene (styrene) and ethynylbenzene (phenylacetylene) relative to that for propene and propyne.
When gas-phase enthalpies of formation were unavailable, but condensed-phase enthalpies of formation and vaporization or sublimation enthalpies could be found, gas-phase values were computed from these data, as indicated in the tables below. In a few cases, estimated enthalpies of vaporization were employed, derived from the empirical relationship , where nC is the number of carbon atoms in the molecule. This relationship was determined by fitting experimental enthalpies of vaporization for a representative series of about 20 C6–C13 aromatic species. An uncertainty (2σ) of 3 kJ/mol was estimated from the fit.
CALCULATIONS
Selection of Molecules
A set of 983 molecules was created for the present work. This set included 660 PAH compounds (including benzene) taken from NIST Special Publication 922: Polycyclic Aromatic Hydrocarbon Structure Index (SP922).140 Although the focus of NIST SP922 is a model for computing retention indices for gas and liquid chromatography, it serves as a very nice source of PAH molecules for the present work. An additional 5 PAH parent compounds that did not appear in SP922 were added (the others were already included) as well as 4 other PAHs. The compound 5-methylchrysene was added from the 15 + 1 EU PAH list (Commission Regulation (EC) No 1881/2006), and 11 fluorinated PAHs from a commercial catalog were included.141,a The list also contains 98 substituted benzenes, 36 benzene radicals (including substituted benzenes), 14 PAH radicals, and 56 substituted PAHs (including radical substituted). Finally, the list contains 109 aliphatic hydrocarbons used to enhance the fitting of the group correction (described below), including n-alkanes, branched alkanes, alkenes, and alkynes.
Density Functional Theory and ab Initio Calculations
Calculations on the full set of 983 molecules were carried out at the B3LYP/cc-pVDZ level of theory. Each molecular structure was fully optimized at this level of theory and calculations of the vibrational frequencies were used to ensure that each structure was a minimum-energy structure on the potential energy surface and to provide (within the ideal gas, rigid rotor, harmonic oscillator approximations) the values of the zero-point energy, enthalpy increment, and enthalpy for each molecule. Additional energy calculations were carried out at the B3LYP/cc-pVTZ//cc-pVDZ level of theory for use in an energy extrapolation scheme described below. Finally, energy calculations at the B3LYP/cc-pV6Z//cc-pVDZ level of theory were carried out for a set of 16 PAH compounds for calibrating the extrapolation scheme (described below). The choice of computational method (B3LYP) was motivated by the reliability of this functional and its common use by many practitioners, as well as its reasonable computational expense, particularly when applied to larger molecules. All B3LYP calculations were carried out using the GAMESS computational chemistry package.142,143
It is noted that the largest PAH considered in this study has a molecular formula of C38H22, and that it is desirable that the extrapolation model accommodate even larger molecules without a prohibitive computational requirement. These considerations have informed the choice of method and basis set described above.
Ab initio calculations using the G3//B3LYP (hereafter G3B3) and G3(MP2)//B3LYP (hereafter G3MP2B3)144 variants of the Gaussian-3145 model chemistry were carried out on a subset of the compounds described above. Carrying out G3B3 or G3MP2B3 calculations on the full set of 983 molecules was not practical due to limitations in computational time and scratch storage space, making such calculations difficult or impossible on the larger molecules in the set. The largest molecule computed with the G3MP2B3 methods was coronene (C24H12). All G3B3 and G3MP2B3 calculations were carried out using the Gaussian 09 computational chemistry package.146 The G3B3 and G3MP2B3 quantum chemical methods147 were used to compute molecular structures, vibrational frequencies, and molecular energies. (Note that optimized geometries and vibrational frequencies are computed at the B3LYP/6-31g(d) level of theory in the G3B3 and G3MP2B3 methods.) These composite methods are model chemistries that compute total molecular energies from the sums and differences of a set of ab initio calculations using different levels of electron correlation and different basis sets. The G3B3 and G3MP2B3 methods include empirical corrections to the computed ab initio total energies of about 3.4 and 4.2 kJ/mol per electron in each valence bond, respectively. The “B3” in the methods denotes that the hybrid density functional theory (DFT) method B3LYP using 6-31G(d) basis sets to compute molecular geometries and frequencies rather than employing HF and MP2 optimizations as done in the standard G3 and G3MP2 methods; the B3LYP geometries have been shown to be generally more reliable (especially in the case of spin-contaminated radicals) and correlate well with higher level QCISD or CCSD optimizations.147
The G3MP2B3 method was used to compute enthalpies of formation for about 120 PAHs and other substituted aromatics, and the more computationally expensive G3B3 method was also applied to about 40–50 of the smaller molecules. The newer G4 method was not used as the present authors already had an extensive computational database of G3 based calculations. Later in this paper, a comparison is made to results from G3, G4, and other methods by other workers. In short, the different model chemistry methods, after applying (different) systematic corrections, produce the same enthalpies of formation for the PAHs (and aliphatic hydrocarbons) within several kJ/mol and are not statistically significant. The computed enthalpies of formation are compared to experimental values for about 80 molecules. Enthalpies of formation were also computed using the G3MP2B3 and G3B3 model chemistries for about 60 aliphatic hydrocarbons, including saturated and unsaturated species, both acyclic and cyclic, for use as reference values. It was found that the average deviation between experimental enthalpies of formation and computed values was about 3–6 kJ/mol for the G3B3 and 4–8 kJ/mol for the G3MP2B3 method depending on the test set (class of molecules).
In this work, enthalpies of formation for the molecules were computed using atomization energies taken from the CODATA recommendations148 as presented in Table 2. Note that the more recent values due to Ruscic and co-workers149 have a significantly lower uncertainty. These values are 217.9979 ± 0.0001 kJ/mol for H, 716.880 ± 0.054 kJ/mol for C, and 79.393 ± 0.053 kJ/mol for F. The older CODATA values were used in this publication for a variety of reasons, but the use of the more recent values is recommended for future work. Zero point vibrational energies (ZPEs) were computed using scaled B3LYP/6-31G(d) (scaling factor = 0.96)150 and B3LYP/cc-pVDZ (scaling factor = 0.97)151 frequencies. Thermal corrections to the enthalpy were computed using harmonic oscillator partition functions and treating torsional modes as rigid rotors (not as hindered rotors).
Table 2.
Values of the Atomic Enthalpy of Formation (ΔfH°(298 K), kJ/mol) and Atomic Enthalpy Increment (Hinc = H°(298K) − H°(0K), kJ/mol) Taken from the CODATA Recommendation148,171,a
| H | C | F | |
|---|---|---|---|
| ΔfH° | 217.998(±0.006) | 716.68(±0.45) | 79.38(±0.30) |
| Hinc = H°(298K) − H°(0K) | 6.197(±0.001) | 6.536(±0.001) | 6.518(±0.001) |
Uncertainties are given in parentheses. These values were used to compute the enthalpies of formation as described in the text.
Chemical Group Based Corrections
The concept of using group based values to compute thermodynamic quantities goes back to the pioneering work of Benson and Buss.73 They showed that various thermodynamic quantities could be computed with good accuracy by considering a molecule as a collection of atoms, chemical bonds, or chemical groups. Briefly, the idea of group additivity is to identify chemical groups within a molecule and sum their corresponding contributions to the property of interest. For example, normal alkanes CH3–(CH2)n–CH3 can be considered to be composed of two types of groups: methyl groups –CH3 and methylene groups –CH2–. The enthalpy of formation of these molecules, or other physical or chemical properties, could be estimated using group additivity as the sum of values for each of the groups. A slightly more complicated molecule, 1-pentene (CH3CH2CH2CH=CH2), can be considered to be composed of five groups: two sp2-hybridized double-bonded groups –CH= and =CH2 and three sp3-hybridized single-bonded groups –CH3, –CH2–, and –CH2—C=. The latter group is a modified –CH2– group because it is adjacent to an unsaturated site (Cd). In group additivity, there are other types of modified groups that are typically employed such as a correction for alkyl groups on the same side of a double bond, e.g., (Z)-2-butene; corrections for ortho-, meta-, and para-substitution; corrections for steric interactions in branched molecules (repulsion between gauche alkyl groups); and corrections for ring strain (e.g., cyclobutane and cyclopentane have ring strain corrections of 110 and 26 kJ/mol, respectively). This approach has been used by many authors for a variety of purposes. Naturally, some authors have used a group additivity approach as the basis for the correction of values to minimize the error versus experimental data. An early example of this approach has been given by Wiberg for various hydrocarbon compounds.152 Another example is seen in a paper by Wang and Frenklach85 in which they used group based methods to correct a series of AM1 calculations of the enthalpies of formation for substituted benzenes and benzene radicals.
The present article uses group based empirical corrections, and the particular scheme used is now described for the B3LYP set of calculations. The carbon atoms in each molecular structure were classified into one of 14 groups on the basis of their chemical environment. The full list of groups and their descriptions are given in Table 3. These groups are illustrated in Figure 1. The identification of groups within a molecule was aided by the chemical informatics algorithms implemented in the OpenBabel package,153,154 which provides a number of useful algorithms including those to detect chemical bonds and bond orders, perceive ring structures, and determine aromaticity. Optimization of the group error correction values was performed using a linear least-squares approach. It was found that subtracting 6 from the number of benzylic carbon atoms (CbH) reduced the mean unsigned error by about 2 kJ/mol, and this modification was retained in the final algorithm. The full expression for the group based error correction (εcorr) for the B3LYP set of calculations is
| (1) |
where nx represents the number of groups of type x present in the molecule and f(x) represents the group correction value for group x. Optimized group correction values are presented in Table 3.
Table 3.
Base Group Names, Optimized Values (kJ/mol) of Parameters, and Descriptions Used in the Group based Error Correction Scheme Used in the B3LYP based Scheme
| base group | optimized value | description |
|---|---|---|
| CH3 | −1.0506 | terminal methyl group (primary), –CH3 |
| CH2 | 5.5183 | sp3 methylene group (secondary), –CH2– |
| CH2d | 6.3631 | sp3 methylene group adjacent to sp2 group, –CH2–C= |
| CCH | 20.1479 | sp3 methylidyne group (tertiary), –CH< |
| CCC | 36.2218 | methanetetrayl group (quaternary), >C< |
| CdH2 | −1.0962 | terminal methylene group, =CH2 |
| CdH | 3.2000 | sp2 alkene group bonded to a single sp3 group, –CH= |
| CdC | 12.1747 | sp2 isoalkene group bonded to two sp3 groups, >C= |
| CtH | 0.0574 | terminal triple-bonded terminal carbon, ≡CH |
| Ct | 1.5573 | triple-bonded carbon, ≡C– |
| CbH | 3.2202 | aromatic carbon terminated by hydrogen |
| Cb | 9.6256 | aromatic carbon terminated by carbon |
| Cf | 7.9813 | fused aromatic carbon connected to one Cf or Cp group |
| Cp | 9.8124 | pericondensed aromatic carbon (interior) connected to two Cf or Cp groups |
Figure 1.

Atom-centered groups needed for describing PAH molecules included in this special publication. 1-Methylpyrene and 2-methyl-1-vinyl-1,2,3,4-tetrahydronaphthalene are depicted with group identifiers indicated.
The correction term εcorr is added to the calculated enthalpy of formation to obtain the corrected enthalpy of formation
| (2) |
Extrapolation of the Energy
One of the primary sources of error when enthalpies of formation are calculated comes from the molecular energy due to the use of a small basis set. Indeed, initial predictions of enthalpies of formation based solely on the results at the B3LYP/cc-pVDZ level of theory showed poor agreement with the available experimental data, as seen in Table 4. Calculations with sufficiently large basis sets are too computationally resource intensive to be practical. To overcome these limitations, schemes whereby the energy computed using two or more smaller basis sets is extrapolated to the result of a larger basis set calculations were examined. A number of such schemes exist such as those due to Feller,155 Halkier et al.,156 and Truhlar.157 Among these, the method due to Truhlar was found to be the most appropriate for the present work.
Table 4.
Statistical Descriptors of the Errors (kJ/mol) in the Uncorrected (uncorr) and Corrected (corr) Enthalpies of Formation at 298 K Derived from the B3LYP Resultsa
| cc-pVDZ
|
cc-pVTZ
|
extrapolated
|
||||
|---|---|---|---|---|---|---|
| uncorr | corr | uncorr | corr | uncorr | corr | |
| MSD | 138.2 | 4.8 | 34.0 | 0.2 | 16.1 | −0.7 |
| MUD | 138.2 | 14.5 | 36.0 | 3.7 | 23.4 | 4.8 |
| δmin | 25.2 | 0.3 | 0.7 | 0.0 | 0.0 | 0.0 |
| δmax | 338.8 | 71.7 | 152.2 | 15.6 | 123.2 | 17.7 |
| RMSD | 151.4 | 19.0 | 47.2 | 5.1 | 32.3 | 6.1 |
Statistics include the mean unsigned deviation (MUD), the mean signed deviation (MSD), the minimum (δmin) and maximum (δmax) absolute deviations, and the root mean square deviation (RMSD). Results are presented for the cc-pVDZ and cc-pVTZ basis sets, and for the large basis set extrapolated from them.
The basis set extrapolation method of Truhlar157 builds on an observation made by Halkier et al.156 that the optimal extrapolation coefficient be obtained by minimization of the mean unsigned error versus the best estimate of the basis set limit, rather than fitting calculations made with three or more basis sets. Truhlar’s method used the root-mean-square deviation (RMSD), as is also used in the present work. The basis of the method is to write the total energy as the sum of a Hartree–Fock (i.e., uncorrelated) energy and a correlation energy,
| (3) |
and assume that each of the contributions to the total energy reaches its basis set limiting value via a power law functional dependence
| (4) |
where λ represents either the Hartree–Fock or the correlation energy in the previous equation and X is an integer related to the basis set. The exponent α may be selected in such a way as to produce an optimal fit to the data. When the cc-pVDZ (X = 2) and cc-pVTZ (X = 3) basis sets are used for extrapolation, the formula for the extrapolated energy may be written157
| (5) |
where the exponents α and β are optimized separately. Truhlar found that extrapolating results based on the cc-pVDZ and cc-pVTZ basis sets produced a lower RMSD from the complete basis set limit than the corresponding calculation made with the cc-pV6Z basis set at a significantly reduced computational cost. Truhlar states that the motivation for the scheme was economical, which aligns well with the needs of the present work.
It has been noted156 that including extrapolated results computed with the cc-pVDZ basis set (as opposed to only using larger basis sets from the same family) increases the mean error. The paper by Truhlar deliberately used the cc-pVDZ and cc-pVTZ basis sets, obtaining good results but did not address whether this scheme used with the cc-pVTZ and cc-pVQZ basis sets (for example) would produce markedly better results. Due to the number of C atoms in many of the molecules considered in this work, the scheme using the cc-pVDZ and cc-pVTZ basis sets was retained to keep the computational time reasonable. However, this does not preclude the possibility that better results would be obtained if the cc-pVDZ results were omitted and larger basis sets were included.
Thus, the scheme due to Truhlar157 has been implemented in the present work as described in the following. The contributions to the energy from the exchange and correlation parts of the B3LYP functional are separated as follows
| (6) |
where EB3 is the energy from the Becke three-parameter exchange term,90 and ELYP is the energy from the Lee–Yang–Parr correlation term.91 These energetic contributions were used in the following formula for the extrapolation of the B3LYP energy to a large basis set limit (in the present case, the cc-pV6Z basis set)
| (7) |
Fitting was accomplished by subtracting the cc-pV6Z energy values from the cc-pVDZ and cc-pVTZ energy values and minimizing the fitting error using a Levenberg–Marquardt algorithm.158,159 The optimized coefficients (α, β) produced using this scheme are presented in Table 5.
Table 5.
Optimized Values of α and β Used in the Energy Extrapolation, Eq 7, of the B3LYP based Scheme
| α | 3.9807 |
| β | 0.5542 |
Enthalpies of Formation
Computation of enthalpies of formation from ab initio results has been addressed by a number of authors.160,161 The fundamental steps in this computation are given here. The enthalpy of formation for a molecule may be expressed as the difference between a thermodynamic term and the atomization energy
| (8) |
The thermodynamic term (H0) is computed as the sum of the atomic enthalpies of formation, the molecular zero-point energy, and the molecular enthalpy increment, minus the sum of atomic enthalpy increments
| (9) |
Values for the atomic enthalpies of formation, , for atom i at 0 K and enthalpy increment, Hinc, are given in Table 2. The atomization energy of a molecule may be computed by summing the energies of each atom in the molecule and subtracting the molecular energy obtained from the ab initio calculation (Etot)
| (10) |
where i is an index that runs over all N atoms in the molecule, and Ei is the energy of atom i. Values for the atomic energies are given in Table 6. The values of the cc-pV6Z atomic energies were used in the calculations of the enthalpies of formation.
Table 6.
Values of the Atomic Energies (Given in Eh) Computed Using the B3LYP Functional and the Stated Basis Seta
| atom | cc-pVDZ | cc-pVTZ | cc-pV6Z |
|---|---|---|---|
| H | −0.497859 | −0.498765 | −0.499053 |
| C | −37.829103 | −37.835471 | −37.838510 |
| F | −99.691370 | −99.727135 | −99.740409 |
These values were used to compute the enthalpies of formation as described in the text.
Computation of enthalpies of formation in the Gaussian-x model chemistries proceeds along very similar lines. The interested reader is directed to the work of Curtiss et al. for additional details.161
Corrections for Gaussian-3 Values
When the underlying ab initio data are of higher quality, and in particular when the error of the method is more regular, a significantly simpler error correction scheme may be employed. The G3B3 and G3MP2B3 values of the enthalpy of formation reported in this work employ a simpler correction scheme. Thus, the expression for the corrected enthalpy of formation becomes
| (11) |
where λ represents either the G3B3 or the G3MP2B3 enthalpy of formation, but the correction factor is now written as
| (12) |
where nCb is the number of benzene-like (unfused) aromatic carbon atoms, nCf is the number of fused aromatic carbon atoms, nCd is the number of sp2-hybridized carbon atoms, nCt is the number of sp-hybridized carbon atoms, and f (Cb), f (Cf), f (Cd), and f (Ct) are the corresponding correction factors. Optimal values for the correction factors for the G3MP2B3 and G3B3 methods are presented in Table 7.
Table 7.
Values of Correction Factors (kJ/mol) Used To Correct G3MP2B3 Results
| method | Cb | Cf | Cd | Ct |
|---|---|---|---|---|
| G3MP2B3 | 1.25 | 0.93 | 1.00 | 1.20 |
Statistical Descriptors
To facilitate assessment of the results, several statistical measures of data quality (compared to experiment) are used. The deviation δ is defined as the difference between the experimental value and the computed value
| (13) |
The minimum and maximum (absolute) deviations are defined as
| (14) |
| (15) |
The mean signed deviation (MSD) and mean unsigned deviation (MUD) are computed as
| (16) |
| (17) |
It should be noted that, although the MUD is a commonly used metric for comparing calculated results to experimental ones, the MUD is not the same as the uncertainty. The work of Ruscic covers this topic in some detail.162 In particular, the uncertainty in thermodynamic values such as the enthalpies of formation reported here is the 95% confidence interval (u95%), which is twice the standard deviation (|sigma). The MUD is approximately 2.5 times smaller than u95%. Thus, care should be used when the values from this publication are combined with other values with conventional uncertainties. Finally, the root-mean-square deviation (RMSD) is defined
| (18) |
Values of these statistical descriptors are given in Table 4.
RESULTS
Having described the various calculations in the preceding section, the results of these calculations are now considered. Several different comparisons will be made. The first set of comparisons involves intercomparison between the present results and previous results from estimation (e.g., group additivity) and computational techniques. As the amount of experimental data on enthalpies of formation for PAHs is somewhat limited, it is reasonable to examine how the present results fare against other predictions. Also, as the expense of group additivity calculations and some of the quantum chemistry calculations used in the preset work is rather modest, a great deal of thermochemistry of PAHs can be obtained where experiments have not been performed. This comparison will also permit examination of the advantages and shortcomings of the various predictive methods.
The second set of comparisons, and perhaps the most meaningful, involves comparison to experimental data. Though agreement with experimental data may be regarded at the “gold standard” by which the present results should be judged, the reality is a bit more complicated. It will be seen that some of the experimental data are likely to be incorrect for some reason. In this way, the computational predictions for the enthalpies of formation serve as a screening tool by which some erroneous values may be identified. Nevertheless, the agreement between the present results and the available experimental data will firmly establish the reliability of the present results, implying a similar performance for the predicted results.
Comparison to Predicted Values
In the Introduction, a number of previous studies of enthalpy of formation in PAHs were referenced. These studies used a group additivity based method,79–84,86 semiempirical methods,85,139 and quantum chemistry.87,136,137 Summary statistics for the results from these calculations compared to the present extrapolated and corrected B3LYP results are given in Table 8, and the individual values are given in Tables 9, 10, and 11. It may be noted that the present results are in generally good agreement with root-mean-square deviations (RMSD) less than about 6 kJ/mol for all but two studies. The mean unsigned deviations (MUD) are larger, with only three studies having a MUD less than 10 kJ/mol, and all but two having a MUD less than 21 kJ/mol. Given the diversity of the methods employed, this agreement is very reasonable.
Table 8.
Statistical Measures (kJ/mol) of the Deviations of Various Predictions of the Enthalpy of Formation at 298 K versus the Present Results for the B3LYP based Schemea
| reference | n | MUD | RMSD | citation |
|---|---|---|---|---|
| Stein | 26 | 20.70 | 5.78 | 78, 79 |
| Alberty | 19 | 13.10 | 3.97 | 80 |
| Moiseeva | 24 | 17.36 | 4.51 | 82, 83 |
| Herndon | 105 | 15.03 | 2.14 | 84 |
| Armitage | 25 | 15.16 | 3.84 | 81 |
| Wang | 32 | 5.15 | 1.09 | 85 |
| Welsh | 21 | 27.80 | 11.96 | 86 |
| Yu | 23 | 8.32 | 2.19 | 87 |
| Blanquart | 9 | 8.12 | 3.07 | 136 |
| Rayne | 25 | 15.20 | 3.40 | 137 |
| Zauer | 50 | 33.05 | 9.63 | 139 |
The table gives the number of data, n, used in computing the mean unsigned deviation, MUD, and the root mean square deviation, RMSD.
Table 9.
Comparison of Enthalpies of Formation (kJ/mol) Predicted in Previous Studies to B3LYP Results and to Experiment Where Available (Further Discussion in Text)
| molecule | CAS registry no. | Alberty80 | Blanquart136 | Welsh86 | Rayne137 | present | experiment |
|---|---|---|---|---|---|---|---|
| 1-methylnaphthalene | 90-12-0 | 103.8 | 113.5 | 116.9(±2.7) | |||
| 1H-benz[e]indene | 232-54-2 | 209.8 | 224.7 | ||||
| 1H-benz[f]indene | 268-40-6 | 209.6 | 220.4 | ||||
| 1H-cyclopent[b]anthracene | 259-06-3 | 282.0 | 300.5 | ||||
| 1H-cyclopenta[l]phenanthrene | 235-92-7 | 261.3 | 290.1 | ||||
| 1H-phenalene | 203-80-5 | 192.2 | 205.5 | ||||
| 1,2-dihydrobenz[j]aceanthrylene | 479-23-2 | 260.0 | 289.4 | ||||
| 1,4-diethenylbenzene | 935-14-8 | 546.9 | 552.6 | ||||
| 2,3-benzofluorene | 243-17-4 | 230.0 | 244.8 | ||||
| 2-methylnaphthalene | 91-57-6 | 102.9 | 108.7 | 116.1(±2.6) | |||
| 3,4-dihydrocyclopenta[cd]pyrene | 25732-74-5 | 213.3 | 343.4 | ||||
| 17H-cyclopenta[a]phenanthrene | 219-08-9 | 261.5 | 287.5 | ||||
| aceanthrylene | 202-03-9 | 316.3 | 339.8 | ||||
| acenaphthene | 83-32-9 | 154.0 | 143.3 | 150.7 | 156.8(±3.1) | ||
| acenaphthylene | 208-96-8 | 244.2 | 176.9 | 245.9 | 259.8 | 263.2(±3.7) | |
| anthracene | 120-12-7 | 218.3 | 230.1 | 226.4 | 210.8 | 222.6 | 230.9(±3.7) |
| benz[a]anthracene | 56-55-3 | 276.9 | 254.7 | 277.1 | 290.3(±6) | ||
| benzene | 71-43-2 | 82.8 | 83.0 | 75.2 | 83.2(±0.3) | ||
| benzo[a]fluorene | 238-84-6 | 258.5 | 228.3 | 246.5 | |||
| benzo[a]naphthacene | 226-88-0 | 344.7 | 362.9 | ||||
| benzo[a]pyrene | 50-32-8 | 314.9 | 296.0 | ||||
| benzo[b]chrysene | 214-17-5 | 335.5 | 347.0 | ||||
| benzo[b]fluoranthene | 205-99-2 | 305.8 | 332.2 | ||||
| benzo[b]triphenylene | 215-58-7 | 326.3 | 348.0 | ||||
| benzo[c]chrysene | 194-69-4 | 339.0 | 362.2 | ||||
| benzo[c]phenanthrene | 195-19-7 | 280.5 | 292.4 | 295.3 | |||
| benzo[e]pyrene | 192-97-2 | 335.3 | 289.9 | ||||
| benzo[ghi] fluoranthene | 203-12-3 | 335.5 | 364.8 | ||||
| biphenyl | 92-52-4 | 165.7 | 174.2 | 180.3(±3.3) | |||
| biphenylene | 259-79-0 | 191.9 | 403.1 | 410.9 | 417.2(±1.9) | ||
| chrysene | 218-01-9 | 267.7 | 259.9 | 275.6 | 271.1 | 268.5(±2.8) | |
| coronene | 191-07-1 | 292.4 | 352.8 | 296.7 | 300.9(±9.9) | ||
| dibenz[a,c]anthracene | 215-58-7 | 357.2 | 348.0 | ||||
| dibenz[a,h]anthracene | 53-70-3 | 335.5 | 335.0 | ||||
| dibenz[a,j]anthracene | 224-41-9 | 335.5 | 336.3 | ||||
| dibenzo[b,g]phenanthrene | 195-06-2 | 348.3 | 371.5 | ||||
| fluoranthene | 206-44-0 | 284.7 | 262.6 | 277.9 | 282.4(±2.8) | ||
| fluorene | 86-73-7 | 167.0 | 172.2 | 179.6 | 179.4(±3) | ||
| naphthacene | 92-24-0 | 286.1 | 297.0 | 310.5 | 340.7(±3.9) | ||
| naphthalene | 91-20-3 | 150.6 | 148.8 | 158.4 | 137.1 | 141.0 | 150.6(±1.6) |
| pentacene | 135-48-8 | 353.9 | 401.3 | ||||
| pentaphene | 222-93-5 | 344.7 | 349.3 | ||||
| perylene | 198-55-0 | 306.1 | 330.0 | 319.2 | 317.4(±3.5) | ||
| phenanthrene | 85-01-8 | 209.1 | 201.8 | 207.0 | 187.8 | 202.7 | 201.4(±3.5) |
| picene | 213-46-7 | 326.3 | 336.9 | ||||
| pyrene | 129-00-0 | 226.1 | 225.8 | 203.4 | 221.3 | 225.5(±4.3) | |
| triphenylene | 217-59-4 | 258.5 | 276.1 | 275.1 | 270.1(±3.1) |
Table 10.
Comparison of Enthalpies of Formation (kJ/mol) Predicted in Previous Studies to B3LYP Results and to Experiment Where Available (Further Discussion in Text)
| molecule | CAS registry no. | Stein78,79 | Moiseeva82,83 | Armitage81 | Yu87 | present | experiment |
|---|---|---|---|---|---|---|---|
| acenaphthalene | 208-96-8 | 254.8 | 242.7 | 259.8 | |||
| acenaphthylene | 208-96-8 | 218.4 | 259.8 | 263.2(±3.7) | |||
| anthanthrene | 191-26-4 | 310.5 | 335.1 | 310.5 | 341.0 | 323.0 | |
| anthracene | 120-12-7 | 218.4 | 231.0 | 218.4 | 230.5 | 222.6 | 230.9(±3.7) |
| benz[a]aceanthrylene | 203-33-8 | 356.5 | 352.3 | 364.4 | 363.5 | ||
| benz[a]acephenanthrylene | 192-28-9 | 344.8 | 381.6 | 386.6 | 394.0 | ||
| benz[a]anthracene | 56-55-3 | 277.0 | 288.3 | 277.0 | 275.7 | 277.1 | 290.3(±6) |
| benzene | 71-43-2 | 82.8 | 69.5 | 82.8 | 82.4 | 75.2 | 83.2(±0.3) |
| benzo[a]coronene | 190-70-5 | 329.7 | 372.0 | 374.9 | 371.8 | ||
| benzo[a]pyrene | 50-32-8 | 289.1 | 296.0 | ||||
| benzo[c]chrysene | 194-69-4 | 331.0 | 344.8 | 362.2 | |||
| benzo[c]phenanthrene | 195-19-7 | 273.6 | 276.6 | 272.4 | 279.7 | 295.3 | |
| benzo[e]pyrene | 192-97-2 | 280.3 | 293.7 | 279.9 | 291.6 | 289.9 | |
| benzo[ghi] fluoranthene | 203-12-3 | 356.9 | 360.2 | 365.7 | 364.8 | ||
| benzo[k] fluoranthene | 207-08-9 | 254.4 | 369.9 | 356.9 | 339.0 | ||
| chrysene | 218-01-9 | 267.8 | 262.3 | 267.8 | 266.9 | 271.1 | 268.5(±2.8) |
| corannulene | 5821-51-2 | 428.4 | 459.4 | 498.1 | |||
| coronene | 191-07-1 | 322.6 | 295.8 | 322.6 | 318.0 | 296.7 | 300.9(±9.9) |
| cyclopenta[cd]perylene | 189-01-5 | 420.1 | 389.1 | 423.4 | 429.5 | ||
| dibenz[a,c]anthracene | 215-58-7 | 326.4 | 348.0 | ||||
| dibenz[a,j]anthracene | 224-41-9 | 335.6 | 336.3 | ||||
| dibenzo[a,h]pyrene | 189-64-0 | 348.1 | 375.6 | ||||
| dibenzo[a,i]pyrene | 189-55-9 | 348.1 | 366.0 | ||||
| dibenzo[a,1]pyrene | 191-30-0 | 356.5 | 393.3 | ||||
| fluoranthene | 206-44-0 | 289.1 | 289.1 | 270.3 | 277.9 | 282.4(±2.8) | |
| indeno[5,6,7,1-pqra]perylene | 96915-18-3 | 388.3 | 410.5 | 425.9 | 419.8 | ||
| naphthacene | 92-24-0 | 311.7 | 286.2 | 314.6 | 310.5 | 340.7(±3.9) | |
| naphthalene | 91-20-3 | 150.6 | 150.2 | 150.6 | 151.5 | 141.0 | 150.6(±1.6) |
| ovalene | 190-26-1 | 414.6 | 376.7 | 414.6 | 418.4 | 404.5 | |
| pentacene | 135-48-8 | 354.0 | 401.3 | ||||
| perylene | 198-55-0 | 280.3 | 293.7 | 279.9 | 306.7 | 319.2 | 317.4(±3.5) |
| phenanthrene | 85-01-8 | 211.3 | 207.5 | 209.2 | 200.8 | 202.7 | 201.4(±3.5) |
| pyrene | 129-00-0 | 231.0 | 259.8 | 230.5 | 241.0 | 221.3 | 225.5(±4.3) |
| triphenylene | 217-59-4 | 258.6 | 241.4 | 258.6 | 273.2 | 275.1 | 270.1(±3.1) |
Table 11.
Comparison of Enthalpies of Formation (kJ/mol) Predicted in Previous Studies to B3LYP Results and to Experiment Where Available (Further Discussion in Text)
| molecule | CAS registry no. | Zauer139 | Wang3 | Herndon99 | present | experiment | |
|---|---|---|---|---|---|---|---|
| 1-ethyl-2-methylbenzene | 611-14-3 | 1.3 | 0.3 | 1.2(±1.2) | |||
| 1-ethyl-3-methylbenzene | 620-14-4 | −2.9 | −1.6 | −1.9(±1.2) | |||
| 1-ethyl-4-methylbenzene | 622-96-8 | −2.9 | −5.8 | −3.3(±1.4) | |||
| 1-methylnaphthalene | 90-12-0 | 120.5 | 113.5 | 116.9(±2.7) | |||
| 1H-benz[e]indene | 232-54-2 | 371.25 | 224.7 | ||||
| 1H-phenalene | 203-80-5 | 179.1 | 205.5 | ||||
| 1,2-dimethylbenzene | 95-47-6 | 20.1 | 15.0 | 19(±1.1) | |||
| 1,2,3-trimethylbenzene | 526-73-8 | −7.5 | −6.9 | −9.6(±1.3) | |||
| 1,2,4-trimethylbenzene | 95-63-6 | −13 | −15.5 | −13.9(±1.1) | |||
| 1,3-dimethylbenzene | 108-38-3 | 17.6 | 13.3 | 17.2(±0.8) | |||
| 1,3,5-trimethylbenzene | 108-67-8 | −15.5 | −17.5 | −15.9(±1.3) | |||
| 1,4-dimethylbenzene | 106-42-3 | 17.6 | 13.6 | 17.9(±1.0) | |||
| 1,4-diphenylbenzene | 92-94-4 | 258.3 | 272.4 | 284.4(±3.8) | |||
| 1,8-dimethylnaphthalene | 569-41-5 | 111.3 | 114.0 | 108.8(±3.0) | |||
| 2-methylnaphthalene | 91-57-6 | 116.3 | 108.7 | 116.1(±2.6) | |||
| [6]helicene | 187-83-7 | 693.4 | 471.7 | ||||
| acenaphthene | 83-32-9 | 162.5 | 150.7 | 156.8(±3.1) | |||
| acenaphthylene | 208-96-8 | 286.2 | 259.8 | 259.8 | 263.2(±3.7) | ||
| anthanthrene | 191-26-4 | 354.4 | 323.0 | ||||
| anthra[1,2-a]anthracene | 195-00-6 | 451.24 | 448.2 | ||||
| anthra[2,1,9-qra]naphthacene | 189-52-6 | 484.67 | 458.8 | ||||
| anthracene | 120-12-7 | 221.1 | 231.4 | 229.7 | 222.6 | 230.9(±3.7) | |
| azulene | 275-51-4 | 299.6 | 271.2 | ||||
| benz[a]anthracene | 56-55-3 | 277.1 | 282.8 | 285.98 | 277.1 | 290.3(±6.0) | |
| benz[mno]indeno[1,7,6,5-cdef]chrysene | 96915-21-8 | 453.8 | 443.9 | ||||
| benz[mno]indeno[5,6,7,1-defg]chrysene | 96915-20-7 | 506.7 | 444.6 | ||||
| benzene | 71-43-2 | 83.7 | 79.04 | 75.2 | 83.2(±0.3) | ||
| benzo[a]naphth[2,1-j]anthracene | 58029-41-7 | 429.19 | 428.8 | ||||
| benzo[a]naphthacene | 226-88-0 | 362.0 | 373.59 | 362.9 | |||
| benzo[a]naphtho[2,1,8-hij]naphthacene | 190-05-6 | 457.48 | 431.1 | ||||
| benzo[a]pentacene | 239-98-5 | 467.69 | 451.2 | ||||
| benzo[a]pentaphene | 7689-57-8 | 419.99 | 408.2 | ||||
| benzo[a]perylene | 191-85-5 | 433.09 | 428.0 | ||||
| benzo[a]picene | 58029-45-1 | 424.68 | 428.5 | ||||
| benzo[a]pyrene | 50-32-8 | 310.9 | 296.0 | ||||
| benzo[b]chrysene | 214-17-5 | 348.1 | 356.1 | 347.0 | |||
| benzo[b]fluoranthene | 205-99-2 | 386.2 | 332.2 | ||||
| benzo[b]naphthacene | 135-48-8 | 402.8 | 401.3 | ||||
| benzo[b]perylene | 197-70-6 | 394.47 | 384.3 | ||||
| benzo[b]picene | 217-42-5 | 421.04 | 413.9 | ||||
| benzo[b]triphenylene | 215-58-7 | 358.65 | 348.0 | ||||
| benzo[c]chrysene | 194-69-4 | 353.7 | 358.11 | 362.2 | |||
| benzo[c]naphtho[2,1-p ]chrysene | 27798-46-5 | 586.8 | 559.1 | ||||
| benzo[c]pentaphene | 222-54-8 | 419.99 | 407.7 | ||||
| benzo[c]phenanthrene | 195-19-7 | 326.2 | 295.0 | 292.29 | 295.3 | ||
| benzo[c]picene | 217-37-8 | 409.53 | 403.8 | ||||
| benzo[def]chrysene | 50-32-8 | 318.32 | 296.0 | ||||
| benzo[e]pyrene | 192-97-2 | 298.1 | 282.4 | 306.06 | 289.9 | ||
| benzo[f]picene | 58029-47-3 | 426.56 | 434.3 | ||||
| benzo[ghi]cyclopenta[cd]perylene | 190-88-5 | 474.4 | 417.2 | ||||
| benzo[ghi]perylene | 191-24-2 | 289.5 | 326.1 | 301.2 | |||
| benzo[g]chrysene | 196-78-1 | 354.8 | 362.17 | 368.8 | |||
| benzo[h]pentaphene | 214-91-5 | 429.74 | 419.0 | ||||
| benzo[j]fluoranthene | 205-82-3 | 389.7 | 352.0 | ||||
| benzo[k]fluoranthene | 207-08-9 | 376.6 | 339.0 | ||||
| benzo[mno]naphtho[1,2-c]chrysene | 120835-49-6 | 466.26 | 453.0 | ||||
| benzo[pqr]naphtho[8,1,2-bcd]perylene | 190-71-6 | 419.9 | 382.8 | ||||
| benzo[pqr]picene | 189-96-8 | 380.87 | 359.4 | ||||
| benzo[rsf]pentaphene | 189-55-9 | 389.74 | 366.0 | ||||
| benzo[s]picene | 31540-94-0 | 442.21 | 474.5 | ||||
| benzo[vwx]hexaphene | 2828-72-0 | 470.37 | 443.8 | ||||
| biphenyl | 92-52-4 | 165.1 | 179.9 | 174.2 | 180.3(±3.3) | ||
| biphenylene | 259-79-0 | 430.9 | 410.9 | 417.2(±1.9) | |||
| cholanthrene | 479-23-2 | 286.8 | 289.4 | ||||
| chrysene | 218-01-9 | 269.7 | 274.1 | 275.73 | 271.1 | 268.5(±2.8) | |
| coronene | 191-07-1 | 414.4 | 286.6 | 336.48 | 296.7 | 300.9(±9.9) | |
| dibenz[a,j]anthracene | 224-41-9 | 344.89 | 336.3 | ||||
| dibenzo[a,c]naphthacene | 216-00-2 | 444.05 | 431.7 | ||||
| dibenzo[a,f]perylene | 191-29-7 | 547.81 | 555.4 | ||||
| dibenzo[a,j]naphthacene | 227-04-3 | 427.81 | 415.1 | ||||
| dibenzo[a,j]perylene | 191-87-7 | 539.53 | 553.0 | ||||
| dibenzo[a,l]naphthacene | 226-86-8 | 427.81 | 416.2 | ||||
| dibenzo[a,l]pentacene | 227-09-8 | 525.4 | 503.1 | ||||
| dibenzo[a,n]perylene | 191-81-1 | 495.64 | 489.9 | ||||
| dibenzo[a,o]perylene | 190-36-3 | 526.85 | 532.9 | ||||
| dibenzo[a,pqr]picene | 120835-40-7 | 463.8 | 452.2 | ||||
| dibenzo[a,rst]pentaphene | 120835-51-0 | 469.28 | 455.0 | ||||
| dibenzo[b,ghi]perylene | 5869-30-7 | 396.22 | 373.1 | ||||
| dibenzo[b,g]chrysene | 53156-67-5 | 442.79 | 446.0 | ||||
| dibenzo[b,g]phenanthrene | 195-06-2 | 365.6 | 371.5 | ||||
| dibenzo[b,k]chrysene | 217-54-9 | 436.14 | 425.4 | ||||
| dibenzo[b,l]chrysene | 58029-38-2 | 436.14 | 438.0 | ||||
| dibenzo[b,pqr]perylene | 190-95-4 | 393.38 | 373.7 | ||||
| dibenzo[b,p]chrysene | 58029-42-8 | 430.9 | 436.18 | 441.0 | |||
| dibenzo[c,g]chrysene | 53156-66-4 | 429.03 | 455.0 | ||||
| dibenzo[c,g]phenanthrene | 188-52-3 | 360.58 | 385.5 | ||||
| dibenzo[c,l]chrysene | 42850-69-1 | 439.82 | 461.7 | ||||
| dibenzo[c,mno]chrysene | 196-28-1 | 395.97 | 383.9 | ||||
| dibenzo[c,pqr]picene | 120835-44-1 | 448.65 | 427.6 | ||||
| dibenzo[c,p]chrysene | 196-52-1 | 442.21 | 464.3 | ||||
| dibenzo[def,mno]chrysene | 191-26-4 | 361.04 | 323.0 | ||||
| dibenzo[def,p]chrysene | 191-30-0 | 396.06 | 393.3 | ||||
| dibenzo[de,ij]pentaphene | 120835-46-3 | 509.19 | 487.8 | ||||
| dibenzo[de,kl]pentaphene | 83786-06-5 | 574.97 | 536.1 | ||||
| dibenzo[de,mn]naphthacene | 214-63-1 | 446.64 | 421.4 | ||||
| dibenzo[de,qr]naphthacene | 193-09-9 | 377.77 | 362.6 | ||||
| dibenzo[de,qr]pentacene | 120835-53-2 | 509.19 | 483.4 | ||||
| dibenzo[de,st]pentacene | 14147-38-7 | 471.08 | 458.5 | ||||
| dibenzo[de,wv]pentaphene | 120835-48-5 | 462.71 | 462.3 | ||||
| dibenzo[fg,ij]pentaphene | 197-69-3 | 457.02 | 449.8 | ||||
| dibenzo[fg,op]naphthacene | 192-51-8 | 371. | 363.0 | ||||
| dibenzo[fg,qr]pentacene | 197-74-0 | 457.02 | 448.2 | ||||
| dibenzo[g,p]chrysene | 191-68-4 | 439.36 | 479.7 | ||||
| dibenzo[h,rst]pentaphene | 192-47-2 | 447.1 | 435.0 | ||||
| dibenzo[pq,wv]pentaphene | 137593-97-6 | 592.54 | 569.1 | ||||
| ethylbenzene | 100-41-4 | 30.1 | 24.8 | 29.8(±0.8) | |||
| ethynylbenzene | 536-74-3 | 308.8 | 314.1 | 306.6(±1.7) | |||
| fluoranthene | 206-44-0 | 312.4 | 277.9 | 282.4(±2.8) | |||
| fluorene | 86-73-7 | 189.8 | 179.6 | 179.4(±3.0) | |||
| heptacene | 258-38-8 | 691.5 | 586.9 | ||||
| hexacene | 258-31-1 | 492.6 | 517.52 | 493.7 | |||
| hexaphene | 222-78-6 | 448.94 | 432.6 | ||||
| indene | 95-13-6 | 158.4 | 156.4 | ||||
| indeno[5,6,7,1-pqra]perylene | 96915-18-3 | 497.3 | 419.8 | ||||
| naphthacene | 92-24-0 | 308.8 | 320.91 | 310.5 | 340.7(±3.9) | ||
| naphthalene | 91-20-3 | 139.5 | 149.8 | 146.77 | 141.0 | 150.6(±1.6) | |
| naphtho[1,2,3,4-ghi]perylene | 190-84-1 | 404.09 | 382.0 | ||||
| naphtho[1,2,3,4-rst]pentaphene | 191-20-8 | 487.31 | 505.1 | ||||
| naphtho[1,2-a]naphthacene | 58029-39-3 | 460.16 | 456.9 | ||||
| naphtho[1,2-b]chrysene | 220-77-9 | 414.05 | 404.3 | ||||
| naphtho[1,2-b]triphenylene | 215-26-9 | 419.24 | 405.8 | ||||
| naphtho[1,2-c]chrysene | 58029-46-2 | 424.68 | 429.7 | ||||
| naphtho[1,2-g]chrysene | 191-67-3 | 434.76 | 460.3 | ||||
| naphtho[2,1,8-def]picene | 120835-39-4 | 452.33 | 450.9 | ||||
| naphtho[2,1,8-fgh]pentaphene | 19301-88-3 | 448.65 | 435.6 | ||||
| naphtho[2,1-a]naphthacene | 220-82-6 | 445.01 | 434.0 | ||||
| naphtho[2,1-b]chrysene | 58029-43-9 | 414.05 | 404.9 | ||||
| naphtho[2,1-b]perylene | 120835-43-0 | 477.39 | 474.0 | ||||
| naphtho[2,1-c]chrysene | 58029-44-0 | 427.14 | 450.9 | ||||
| naphtho[2,3-c]chrysene | 58029-37-1 | 436.14 | 437.1 | ||||
| naphtho[2,3-g]chrysene | 196-64-5 | 436.18 | 441.3 | ||||
| ovalene | 190-26-1 | 759.1 | 404.5 | ||||
| pentacene | 135-48-8 | 417.4 | 401.3 | ||||
| pentaphene | 222-93-5 | 350.5 | 359.99 | 349.3 | |||
| perylene | 198-55-0 | 317.5 | 304.6 | 331.92 | 319.2 | 317.4(±3.5) | |
| phenanthrene | 85-01-8 | 201.1 | 207.5 | 209.37 | 202.7 | 201.4(±3.5) | |
| phenanthro[1,10,9,8-opqra]perylene | 190-39-6 | 507.6 | 467.0 | ||||
| phenanthro[1,2,3,4-def]chrysene | 137570-58-2 | 461.87 | 458.5 | ||||
| phenanthro[3,4-c]chrysene | 31124-69-3 | 501.75 | 537.6 | ||||
| phenanthro[4,3-a]anthracene | 58029-40-6 | 438.61 | 461.2 | ||||
| phenyl | 2396-01-2 | 328.9 | 326.6 | 337.3(±0.6) | |||
| picene | 213-46-7 | 341.6 | 342.96 | 336.9 | |||
| pyranthrene | 191-13-9 | 495.3 | 458.1 | ||||
| pyrene | 129-00-0 | 237.2 | 225.5 | 242.17 | 221.3 | 225.5(±4.3) | |
| styrene | 100-42-5 | 148.1 | 142.1 | 146.9(±1.0) | |||
| toluene | 108-88-3 | 50.6 | 44.1 | 50.1(±1.1) | |||
| tribenzo[a,hi,mn]naphthacene | 54961-30-7 | 438.73 | 421.2 | ||||
| tribenzo[b,defp]chrysene | 66032-75-9 | 472.16 | 470.6 | ||||
| tribenzo[c,g,mno]chrysene | 108650-10-8 | 459.45 | 477.2 | ||||
| triphenylene | 217-59-4 | 267.2 | 270.3 | 286.06 | 275.1 | 270.1(±3.1) |
Table S1 compares the enthalpies of formation computed using the G3MP2B3 and G3B3 methods for 51 aromatic compounds. It may be observed that the G3MP2B3 values are consistently lower than the G3B3 values and can be adjusted to agree with the G3B3 values within about 0.6 kJ/mol (1 standard deviation) by applying corrections of 1.35 kJ/mol per CbH site, 0.85 kJ/mol per other aromatic carbon sites, 1.24 kJ/mol per Cd (double-bonded carbon), and 0.87 per Ct (triple-bonded carbon).
Table 12 compares the enthalpies of formation computed using the G3MP2B3 and G3B3 methods for 80 unsaturated aliphatic compounds. It was observed that the G3MP2B3 values for this set of molecules could be adjusted to agree with the G3B3 values within about 0.3 kJ/mol (1 standard deviation) by applying corrections of 1.06 kJ/mol per Cd (double-bonded carbon), and 1.09 kJ/mol per Ct (triple-bonded carbon). We note that for the allenes (e.g., propadiene, 1,2-butadiene) we computed the correction per double bond (not per atom). Thus, for example, the same correction is applied to 1,3-butadiene and 1,2-butadiene.
Table 12.
Values of the Enthalpy of Formation (kJ/mol) Computed Using the G3MP2B3 and G3B3 Model Chemistries for Non-PAH Molecules
|
|
|||||
|---|---|---|---|---|---|
| name | formula | CAS registry no. | G3MP2B3 | G3B3 | |
| Alk-1-enes | |||||
| ethene | C2H4 | 74-85-1 | 49.4 | 51.5 | |
| propene | C3H6 | 115-07-1 | 17.8 | 20.0 | |
| but-1-ene | C4H8 | 106-98-9 | −1.8 | 0.5 | |
| pent-1-ene | C5H10 | 109-67-1 | −23.3 | −20.9 | |
| hex-1-ene | C6H12 | 592-41-6 | −44.7 | −42.1 | |
| Branched Alk-1-enes | |||||
| 3-methylbut-1-ene | C5H10 | 563-45-1 | −30.3 | −28.1 | |
| 3-methylpent-1-ene | C6H12 | 760-20-3 | −52.4 | −50.0 | |
| 3-methylhex-1-ene | C7H14 | 3404-61-3 | −73.7 | −71.2 | |
| 4-methylpent-1-ene | C6H12 | 691-37-2 | −50.8 | −48.1 | |
| 3-ethylpent-1-ene | C7H14 | 4038-04-4 | −74.0 | −71.6 | |
| 3,3-dimethylpent-1-ene | C7H14 | 3404-73-7 | −82.4 | −79.8 | |
| Alk-2-enes | |||||
| (E)-but-2-ene | C4H8 | 624-64-6 | −12.2 | −10.3 | |
| (E)-pent-2-ene | C5H10 | 646-04-8 | −32.2 | −30.3 | |
| (E)-hex-2-ene | C6H12 | 4050-45-7 | −53.9 | −51.8 | |
| (E)-hex-3-ene | C6H12 | 13269-52-8 | −52.3 | −50.3 | |
| (E)-4-methylpent-2-ene | C6H12 | 674-76-0 | −61.0 | −59.2 | |
| (Z)-but-2-ene | C4H8 | 590-18-1 | −7.3 | −5.0 | |
| (Z)-pent-2-ene | C5H10 | 627-20-3 | −26.9 | −24.5 | |
| (Z)-hex-2-ene | C6H12 | 7688-21-3 | −48.8 | −46.2 | |
| (Z)-hex-3-ene | C6H12 | 7642-09-3 | −46.6 | −44.2 | |
| Branched Alk-2-enes | |||||
| (Z)-4-methylpent-2-ene | C6H12 | 691-38-3 | −55.8 | −53.5 | |
| Isoalkenes | |||||
| 2-methylprop-1-ene | C4H8 | 115-11-7 | −18.1 | −16.1 | |
| 2-methylbut-1-ene | C5H10 | 563-46-2 | −36.0 | −33.9 | |
| 2-methylpent-1-ene | C6H12 | 763-29-1 | −57.6 | −55.4 | |
| 2-ethylbut-1-ene | C6H12 | 760-21-4 | −54.2 | −52.1 | |
| Isoalk-2-enes | |||||
| 2-methylbut-2-ene | C5H10 | 513-35-9 | −41.7 | −39.7 | |
| 2-methylpent-2-ene | C6H12 | 625-27-4 | −61.9 | −59.9 | |
| (Z)-3-methylpent-2-ene | C6H12 | 922-62-3 | −60.3 | −58.3 | |
| (E)-3-methylpent-2-ene | C6H12 | 616-12-6 | −60.0 | −58.0 | |
| Alk-1-ynes | |||||
| ethyne | C2H2 | 74-86-2 | 225.0 | 227.4 | |
| propyne | C3H4 | 74-99-7 | 182.4 | 184.5 | |
| but-1-yne | C4H6 | 107-00-6 | 163.8 | 165.9 | |
| pent-1-yne | C5H8 | 627-19-0 | 142.1 | 144.4 | |
| Alk-2-ynes | |||||
| but-2-yne | C4H6 | 503-17-3 | 145.3 | 147.0 | |
| pent-2-yne | C5H8 | 627-21-4 | 126.2 | 128.1 | |
| isohexyne | C6H10 | 7154-75-8 | 113.9 | 116.4 | |
| Branched Alk-1-ynes | |||||
| isopentyne | C5H8 | 598-23-2 | 136.6 | 138.8 | |
| tert-hexyne | C6H10 | 917-92-0 | 103.5 | 105.5 | |
| Unconjugated Alkadienes | |||||
| penta-1,4-diene | C5H8 | 591-93-5 | 100.5 | 105.5 | |
| hexa-1,5-diene | C6H10 | 592-42-7 | 78.8 | 84.1 | |
| 1,3 Conjugated Alkadienes | |||||
| (E)-buta-1,3-diene | C4H6 | 106-99-0 | 105.9 | 110.9 | |
| (E)-penta-1,3-diene | C5H8 | 2004-70-8 | 73.9 | 78.6 | |
| (Z)-buta-1,3-diene | C4H6 | 106-99-0 | 118.3 | 123.5 | |
| (Z)-penta-1,3-diene | C5H8 | 1574-41-0 | 91.4 | 96.6 | |
| Branched 1,3 Conjugated Alkadienes | |||||
| 2-methylbuta-1,3-diene | C5H8 | 78-79-5 | 83.2 | 87.9 | |
| 2,3-dimethylbuta-1,3-diene | C6H10 | 513-81-5 | 40.4 | 45.5 | |
| 2,4 Conjugated Alkadienes | |||||
| (E,Z)-hexa-2,4-diene | C6H10 | 5194-50-3 | 47.5 | 52.2 | |
| Conjugated Alkenynes | |||||
| butenyne | C4H4 | 689-97-4 | 283.4 | 288.0 | |
| (E)-pent-3-en-1-yne | C5H6 | 2004-69-5 | 248.1 | 252.8 | |
| (Z)-pent-3-en-1-yne | C5H6 | 1574-40-9 | 249.8 | 254.0 | |
| pent-1-en-3-yne | C5H6 | 646-05-9 | 242.9 | 247.1 | |
| 3-methylbut-1-en-3-yne | C5H6 | 248.5 | 252.7 | ||
| Unconjugated Alkenynes | |||||
| pent-1-en-4-yne | C5H6 | 871-28-3 | 270.2 | 275.3 | |
| Unconjugated Alkadiynes | |||||
| penta-1,4-diyne | C5H4 | 24442-69-1 | 447.0 | 452.1 | |
| hexa-1,5-diyne | C6H6 | 628-16-0 | 412.3 | 417.5 | |
| Conjugated Alkadiynes | |||||
| butadiyne | C4H2 | 460-12-8 | 454.5 | 458.4 | |
| penta-1,3-diyne | C5H4 | 1033-27-7 | 408.4 | 411.9 | |
| Alkatrienes | |||||
| (E)-hexa-1,3,5-triene | C6H8 | 821-07-8 | 158.9 | 166.6 | |
| (Z)-hexa-1,3,5-triene | C6H8 | 2612-46-6 | 165.1 | 173.0 | |
| Diallenes | |||||
| propadiene | C3H4 | 463-49-0 | 183.8 | 188.1 | |
| buta-1,2-diene | C4H6 | 590-19-2 | 157.3 | 161.3 | |
| penta-1,2-diene | C5H8 | 591-95-7 | 135.5 | 139.8 | |
| penta-2,3-diene | C5H8 | 591-96-8 | 130.3 | 134.0 | |
| hexa-2,3-diene | C6H10 | 592-49-4 | 109.4 | 113.2 | |
| 3-methylpenta-1,2-diene | C6H10 | 7417-48-3 | 105.2 | 108.9 | |
| 4-methylpenta-1,2-diene | C6H10 | 13643-05-5 | 107.0 | 111.3 | |
| Cum-Allenes | |||||
| penta-1,2,3-triene | C5H6 | 62018-46-6 | 282.7 | 288.4 | |
| penta-1,2,4-triene | C5H6 | 10563-01-6 | 246.0 | 252.7 | |
| butatriene | C4H4 | 2873-50-9 | 313.9 | 319.9 | |
| (E)-hexa-2,3,4-triene | C6H8 | 59660-65-0 | 252.6 | 258.0 | |
| (E)-hexa-1,2,4,5-tetraene | C6H6 | 29776-96-3 | 387.9 | 396.3 | |
| pentatetraene | C5H4 | 21986-03-8 | 437.4 | 444.6 | |
| hexapentaene | C6H4 | 13703-38-3 | 560.0 | 568.4 | |
| Allenynes | |||||
| penta-1,2-dien-4-yne | C5H4 | 33555-85-0 | 427.1 | 433.5 | |
| hexa-1,2-dien-4-yne | C6H6 | 34783-10-3 | 384.3 | 390.3 | |
| hexa-1,2-dien-5-yne | C6H6 | 33142-15-3 | 413.5 | 420.6 | |
| hexa-1,2,3-trien-5-yne | C6H4 | 895126-88-2 | 551.8 | 559.8 | |
| Alkendiynes | |||||
| (E)-hex-3-en-1,5-diyne | C6H4 | 16668-68-1 | 516.3 | 523.2 | |
| (Z)-hex-3-en-1,5-diyne | C6H4 | 16668-67-0 | 517.1 | 524.3 | |
| Alkatriynes | |||||
| hexatriyne | C6H2 | 3161-99-7 | 679.0 | 684.3 | |
The G3MP2B3 method is a significantly less expensive calculation than the G3B3 method. It uses a single MP2 calculation to approximate the composite total energies in the G3B3 method that are determined from a set of MP2, MP4, and QCISD(T) energies using different basis sets. The good agreement here (after systematic corrections) suggests that not only does the G3MP2B3 method perform adequately well relative to the G3B3 method but also both methods can likely produce accurate values because little difference is observed (after correction) between two different methods—one approximate and the other more exact. Thus, in the following tables, we present only (corrected) enthalpies of formation from the G3MP2B3 method, because our analysis here shows that it is unnecessary to use the much more computationally expensive G3B3 method.
Comparison to Experimental Data
B3LYP
Experimental data are available for 49 PAHs and substituted PAHs, and for 52 benzene and substituted benzene compounds. Experimental data for an additional 81 alkanes, alkenes, and alkynes were used to make the fitting procedure more reliable and to assess the quality of the computational scheme. In total, up to 171 data were used for fitting and evaluation purposes. These values may be found in Table 13. This table also serves as a summary of the available experimental and review data available for the compounds used in the present study, and thus in some cases more than one value is given for a compound.
Table 13.
Experimental and Review Values of Enthalpy of Formation (Including Uncertainties in Parentheses Where Available) at 298 Ka
| molecule | formula | CAS registry no. |
|
method | reference | |
|---|---|---|---|---|---|---|
| methane | C1H4 | 74-82-8 | −74.5(±0.6) | review | 48 | |
| −74.520(±0.054) | network | 172 | ||||
| ethane | C2H6 | 74-84-0 | −84.4(±0.4) | review | 173 | |
| −83.8(±0.2) | review | 48 | ||||
| −83.91(±0.14) | network | 172 | ||||
| propane | C3H8 | 74-98-6 | −104.7(±0.6) | review | 48 | |
| butane | C4H10 | 106-97-8 | −125.9(±0.4) | review | 48 | |
| pentane | C5H12 | 109-66-0 | −146.8(±0.6) | calorim | 174 | |
| hexane | C6H14 | 110-54-3 | −167.2(±0.8) | calorim | 175 | |
| heptane | C7H16 | 142-82-5 | −187.8(±0.8) | calorim | 175 | |
| isobutane | C4H10 | 75-28-5 | −134.4(±0.4) | review | 48 | |
| isopentane | C5H12 | 78-78-4 | −153.7(±0.6) | calorim | 174 | |
| 2-methylpentane | C6H14 | 107-83-5 | −174.3(±1) | calorim | 175 | |
| 2-methylhexane | C7H16 | 591-76-4 | −195(±1.3) | calorim | 175 | |
| 3-methylpentane | C6H14 | 96-14-0 | −171.6(±1) | calorim | 175 | |
| 2,4-dimethylpentane | C7H16 | 108-08-7 | −202.1(±1) | calorim | 175 | |
| 3,3-dimethylpentane | C7H16 | 562-49-2 | −201.5 | calorim | 175 | |
| neopentane | C5H12 | 463-82-1 | −167.9(±0.6) | calorim | 174 | |
| 2,2-dimethylbutane | C6H14 | 75-83-2 | −185.6(±1) | calorim | 175 | |
| 2,2-dimethylpentane | C7H16 | 590-35-2 | −206.2(±1.3) | calorim | 175 | |
| ethene | C2H4 | 74-85-1 | 52.6(±0.2) | review | 48 | |
| 52.45(±0.13) | network | 172 | ||||
| propene | C3H6 | 115-07-1 | 20.2(±0.4) | review | 48 | |
| but-1-ene | C4H8 | 106-98-9 | 0(±0.5) | review | 48 | |
| −0.6(±0.8) | calorim | 176 | ||||
| pent-1-ene | C5H10 | 109-67-1 | −21.3 | heat hydrog | 177 | |
| −17.1(±0.4) | equil | 178 | ||||
| hex-1-ene | C6H12 | 592-41-6 | −42.1(±1.2) | heat hydrog | 177 | |
| −42.1 | heat hydrog | 179 | ||||
| −41.5(±1.2) | heat hydrog | 35 | ||||
| hept-1-ene | C7H14 | 592-76-7 | −62.3 | calorim | 181 | |
| 3-methylbut-1-ene | C5H10 | 563-45-1 | −27.7(±1.2) | calorim | 180 | |
| 3-methylpent-1-ene | C6H12 | 760-20-3 | −47(±1.1) | heat hydrog | 182 | |
| 3-ethylpent-1-ene | C7H14 | 4038-04-4 | −69.5(±2) | heat hydrog | 183 | |
| 4-methylpent-1-ene | C6H12 | 691-37-2 | −49.4(±0.7) | calorim | 184 | |
| 3-methylhex-1-ene | C7H14 | 3404-61-3 | −68.2(±1.5) | heat hydrog | 183 | |
| 2-methylhex-2-ene | C7H14 | 2738-19-4 | −87.8(±1.4) | review | 185 | |
| 3,3-dimethylbut-1-ene | C6H12 | 558-37-2 | −59.7(±2) | heat hydrog | 182 | |
| 3,3-dimethylpent-1-ene | C7H14 | 3404-73-7 | −78.5(±1.7) | heat hydrog | 183 | |
| 2,4-dimethylhex-2-ene | C8H16 | 14255-2-3−3 | −104.9(±2.1) | review | 185 | |
| (E)-but-2-ene | C4H8 | 624-64-6 | −11.2(±0.5) | review | 48 | |
| −10.8(±1) | calorim | 176 | ||||
| (Z)-but-2-ene | C4H8 | 590-18-1 | −7.3(±0.5) | review | 48 | |
| −7.7(±1.3) | calorim | 176 | ||||
| (E)-pent-2-ene | C5H10 | 646-04-8 | −33.1(±1.3) | heat hydrog | 186 | |
| (Z)-pent-2-ene | C5H10 | 627-20-3 | −28(±0.8) | heat hydrog | 186 | |
| (E)-4-methyl-pent-2-ene | C6H12 | 674-76-0 | −60.1(±1.5) | heat hydrog | 182 | |
| (Z)-4-methyl-pent-2-ene | C6H12 | 691-38-3 | −57.9(±1.4) | heat hydrog | 182 | |
| (E)-hex-2-ene | C6H12 | 4050-45-7 | −51.7(±2) | equil | 187 | |
| (Z)-hex-2-ene | C6H12 | 7688-21-3 | −47.9(±2) | equil | 187 | |
| (E)-hex-3-ene | C6H12 | 13269-52-8 | −49.3(±1.1) | equil | 187 | |
| (Z)-hex-3-ene | C6H12 | 7642-09-3 | −46.9(±2) | equil | 187 | |
| 2-methylprop-1-ene | C4H8 | 115-11-7 | −17.5(±0.5) | review | 48 | |
| −16.9(±0.9) | review | 185 | ||||
| 2-methylbut-2-ene | C5H10 | 513-35-9 | −41.5(±0.88) | equil | 188 | |
| −41.8(±1.1) | review | 185 | ||||
| 2-methylbut-1-ene | C5H10 | 563-46-2 | −35.1(±0.8) | equil | 188 | |
| −35.3(±1) | review | 185 | ||||
| 2-ethylbut-1-ene | C6H12 | 760-21-4 | −56.1(±0.9) | calorim | 188 | |
| 2-methylpent-1-ene | C6H12 | 763-29-1 | −58(±1.1) | heat hydrog | 182 | |
| −59.4(±1.3) | review | 185 | ||||
| (E)-3-methyl-pent-2-ene | C6H12 | 616-12-6 | −63.5(±0.9) | calorim | 188 | |
| 2-methylpent-2-ene | C6H12 | 625-27-4 | −62.7(±1.2) | heat hydrog | 182 | |
| −63.2(±1.5) | review | 185 | ||||
| (Z)-3-methylpent-2-ene | C6H12 | 922-62-3 | −61.9(±0.9) | calorim | 188 | |
| 2,3-dimethylbut-1-ene | C6H12 | 563-78-0 | −62.6(±1.3) | review | 185 | |
| 2,3-dimethyl-ent-1-ene | C7H14 | 3404-72-6 | −90.2(±1.4) | review | 185 | |
| ethyne | C2H2 | 74-86-2 | 226.7(±0.8) | calorim | 189 | |
| 228.27(±0.13) | network | 172 | ||||
| propyne | C3H4 | 74-99-7 | 185.4(±0.9) | calorim | 189 | |
| but-1-yne | C4H6 | 107-00-6 | 166.1(±2.1) | calorim | 189 | |
| 165.2(±0.9) | calorim | 176 | ||||
| pent-1-yne | C5H8 | 627-19-0 | 144.3(±2.1) | calorim | 189 | |
| hex-1-yne | C6H10 | 693-02-7 | 122.3(±1.2) | heat hydrog | 190 | |
| hept-1-yne | C7H12 | 628-71-7 | 103.8(±2.6) | heat hydrog | 190 | |
| isopentyne | C5H8 | 598-23-2 | 136.4(±2.1) | calorim | 189 | |
| tert-hexyne | C6H10 | 917-92-0 | 106.1 | calorim | 191 | |
| but-2-yne | C4H6 | 503-17-3 | 148(±1.5) | calorim | 189 | |
| 145.1(±1) | calorim | 176 | ||||
| pent-2-yne | C5H8 | 627-21-4 | 128.9(±2.1) | calorim | 189 | |
| hex-2-yne | C6H10 | 764-35-2 | 107.7(±2.4) | heat hydrog | 190 | |
| hept-2-yne | C7H12 | 1119-65-9 | 84.8(±2.2) | heat hydrog | 190 | |
| hept-3-yne | C7H12 | 2586-89-2 | 82.8(±2.4) | heat hydrog | 190 | |
| penta-1,4-diene | C5H8 | 591-93-5 | 106.3(±1.3) | calorim | 192 | |
| hexa-1,5-diene | C6H10 | 592-42-7 | 85(±2) | heat hydrog | 193 | |
| (E)-buta-1,3-diene | C4H6 | 106-99-0 | 108.8(±0.8) | calorim | 176 | |
| (E)-penta-1,3-diene | C5H8 | 2004-70-8 | 75.77(±0.7) | calorim | 192 | |
| (Z)-penta-1,3-diene | C5H8 | 1574-41-0 | 82.72(±0.9) | calorim | 192 | |
| (E)-hexa-1,3-diene | C6H10 | 20237-34-7 | 54.(±2) | heat hydrog | 193 | |
| (Z)-hexa-1,3-diene | C6H10 | 14596-92-0 | 59(±2) | heat hydrog | 193 | |
| (Z)-hexa-1,4-diene | C6H10 | 7318-67-4 | 77(±2) | heat hydrog | 193 | |
| 2-methylbuta-1,3-diene | C5H8 | 78-79-5 | 75.7(±1) | calorim | 192 | |
| 2,3-dimethylbuta-1,3-diene | C6H10 | 513-81-5 | 56.4(±1.2) | heat hydrog | 177 | |
| 2-ethylbuta-1,3-diene | C6H10 | 3404-63-5 | 63.6 | heat hydrog | 194 | |
| (E,E)-hexa-2,4-diene | C6H10 | 5194-51-4 | 44(±2) | heat hydrog | 193 | |
| (E,Z)-hexa-2,4-diene | C6H10 | 5194-50-3 | 48(±2) | heat hydrog | 193 | |
| (E)-hexa-1,3,5-triene | C6H8 | 821-07-8 | 168(±3) | heat hydrog | 193 | |
| (Z)-hexa-1,3,5-triene | C6H8 | 2612-46-6 | 172(±3) | heat hydrog | 193 | |
| 1-buten-3-yne | C4H4 | 689-97-4 | 295(±3) | heat hydrog | 194 | |
| 2-methylbut-1-en-3-yne | C5H6 | 78-80-8 | 259(±1.3) | calorim | 195 | |
| (E)-pent-3-en-1-yne | C5H6 | 2004-69-5 | 259(±3) | heat hydrog | 194 | |
| (Z)-pent-3-en-1-yne | C5H6 | 1574-40-9 | 258.(±3) | heat hydrog | 194 | |
| (E)-hex-3-en-1,5-diyne | C6H4 | 16668-68-1 | 538(±3) | heat hydrog | 194 | |
| (Z)-hex-3-en-1,5-diyne | C6H4 | 16668-67-0 | 541.8(±3) | heat hydrog | 194 | |
| ethenylbenzene | C8H8 | 100-42-5 | 146.9(±1) | calorim | 196 | |
| (E)-propen-1-ylbenzene | C9H10 | 873-66-5 | 117.2 | calorim | 197 | |
| (Z)-propen-1-ylbenzene | C9H10 | 766-90-5 | 121.4 | calorim | 197 | |
| propen-2-ylbenzene | C9H10 | 98-83-9 | 118.3(±1.4) | equil | 198 | |
| propen-3-ylbenzene | C9H10 | 300-57-2 | 133.8(±1.1) | heat hydr | 199 | |
| isopropenylbenzene | C9H10 | 98-83-9 | 113.8(±2.1) | review | 185 | |
| 2-methylpropen-1-ylbenzene | C10H12 | 768-49-0 | 86.1(±2.1) | review | 185 | |
| benzene | C6H6 | 71-43-2 | 82.9(±0.9) | review | 2 | |
| 82.9(±0.5) | calorim | 196 | ||||
| 82.9(±0.9) | calorim | 200 | ||||
| 82.8(±0.9) | calorim | 201 | ||||
| 83.2(±0.3) | network | 202 | ||||
| toluene | C7H8 | 108-88-3 | 50.1(±1.1) | review | 2 | |
| 50(±0.6) | calorim | 200 | ||||
| 49.9(±1.1) | calorim | 201 | ||||
| ethylbenzene | C8H10 | 100-41-4 | 29.8(±0.8) | calorim | 200 | |
| prop-1-ylbenzene | C9H12 | 103-65-1 | 7.8(±0.8) | calorim | 200 | |
| prop-2-ylbenzene | C9H12 | 98-82-8 | 3.9(±1.1) | calorim | 200 | |
| but-1-ylbenzene | C10H14 | 104-51-8 | −13.8(±1.3) | calorim | 200 | |
| but-2-ylbenzene | C10H14 | 135-98-8 | −17.4(±1.4) | calorim | 200 | |
| isobutylbenzene | C10H14 | 538-93-2 | −21.5(±1.4) | calorim | 200 | |
| tert-butylbenzene | C10H14 | 98-06-6 | −22.7(±1.4) | calorim | 200 | |
| 1,2-dimethylbenzene | C8H10 | 95-47-6 | 19.(±1.1) | calorim | 200 | |
| 17.7 | calorim | 203 | ||||
| 1,3-dimethylbenzene | C8H10 | 108-38-3 | 17.2(±0.8) | calorim | 200 | |
| 14.9 | calorim | 203 | ||||
| 1,4-dimethylbenzene | C8H10 | 106-42-3 | 17.9(±1) | calorim | 200 | |
| 16.5 | calorim | 203 | ||||
| 1-ethyl-2-methylbenzene | C9H12 | 611-14-3 | 1.2(±1.2) | calorim | 196 | |
| 1-ethyl-3-methylbenzene | C9H12 | 620-14-4 | −1.9(±1.2) | calorim | 204 | |
| 1-ethyl-4-methylbenzene | C9H12 | 622-96-8 | −3.3(±1.5) | calorim | 204 | |
| 1,2-diethylbenzene | C10H14 | 135-01-3 | −19.5(±2.2) | calorim | 200 | |
| 1,3-diethylbenzene | C10H14 | 141-93-5 | −21.6(±2.2) | calorim | 200 | |
| 1,4-diethylbenzene | C10H14 | 105-05-5 | −22.1(±2.2) | calorim | 200 | |
| 1,2,3-trimethylbenzene | C9H12 | 526-73-8 | −9.6(±1.3) | calorim | 200 | |
| 1,2,4-trimethylbenzene | C9H12 | 95-63-6 | −13.9(±1.1) | calorim | 200 | |
| 1,3,5-trimethylbenzene | C9H12 | 108-67-8 | −15.9(±1.3) | calorim | 204 | |
| ethynylbenzene | C8H6 | 536-74-3 | 306.6(±1.7) | heat hydrog | 169 | |
| propyn-1-ylbenzene | C9H8 | 673-32-5 | 268.2(±2.2) | heat hydrog | 169 | |
| butyn-1-ylbenzene | C10H10 | 622-76-4 | 248.6(±1) | heat hydrog | 169 | |
| benzyne | C6H4 | 462-80-6 | 446(±13) | ion | 205 | |
| m-benzyne | C6H4 | 1828-89-3 | 490(±10) | ion | [206 | |
| 511(±13) | ion | [205 | ||||
| p-benzyne | C6H4 | 3355-34-8 | 540(±10) | ion | 206 | |
| 575(±14) | ion | 205 | ||||
| cyclopropylbenzene | C9H10 | 873-49-4 | 150.7(±1) | calorim | 207 | |
| 1-cyclopropyl-2-methylbenzene | C10H12 | 27546-46-9 | 125.5(±2.2) | calorim | 208 | |
| cyclohexylbenzene | C12H16 | 827-52-1 | −16.7(±1.5) | calorim | 209 | |
| phenylbenzene | C12H10 | 92-52-4 | 180.3(±3.3) | review | 2 | |
| 182(±0.7) | calorim | 210 | ||||
| benzylbenzene | C13H12 | 101-81-5 | 162.3(±2.3) | review | 2 | |
| 164.8(±1.6) | calorim | 211 | ||||
| 2-phenyltoluene | C13H12 | 643-58-3 | 152.8(±1.5) | calorim | 211 | |
| 3-phenyltoluene | C13H12 | 643-93-6 | 152.5(±8) | calorim | 212 | |
| 4-phenyltoluene | C13H12 | 644-08-6 | 138.2(±2.9) | calorim | 213 | |
| phenylethylbenzene | C14H14 | 103-29-7 | 135.6(±1.3) | review | 2 | |
| (E)-1,2-diphenylethene | C14H12 | 103-30-0 | 233.7(±2) | calorim | 214 | |
| (Z)-1,2-diphenylethene | C14H12 | 645-49-8 | 245.9(±1.3) | calorim | 215 | |
| diphenylethyne | C14H10 | 501-65-5 | 385(±2.7) | heat hydrog | 169 | |
| 407.5(±1.6) | calorim | 216 | ||||
| 1,2-diphenylbenzene | C18H14 | 84-15-1 | 282.8(±3.2) | review | 2 | |
| 1,3-diphenylbenzene | C18H14 | 92-06-8 | 280(±3.9) | review | 2 | |
| 1,4-diphenylbenzene | C18H14 | 92-94-4 | 284.4(±3.8) | review | 2 | |
| 279(±5) | calorim | 217 | ||||
| triphenylmethane | C19H16 | 519-73-3 | 276.1(±4.1) | review | 2 | |
| 268(±4) | calorim | 218 | ||||
| phenyl | C6H5 | 2396-01-2 | 330.1(±3.3) | review | 219 | |
| 337(±2.5) | equil | 220 | ||||
| 338(±3) | review | 221 | ||||
| 339(±8) | review | 222 | ||||
| 339.7(±2.5) | ion | 223 | ||||
| 337.3(±0.6) | network | 202 | ||||
| 1,2-dihydronaphthalene | C10H10 | 447-53-0 | 124.8(±3.3) | heat hydrog | 224 | |
| 1,4-dihydronaphthalene | C10H10 | 612-17-9 | 137.5(±3.2) | heat hydrog | 224 | |
| 1,2,3,4-tetrahydronaphthalene | C10H10 | 119-64-2 | 24(±3.2) | calorim | 225 | |
| trans-decalin | C10H18 | 493-02-7 | −182.2(±2.3) | calorim | 226 | |
| cis-decalin | C10H18 | 493-01-6 | −169.2(±2.3) | calorim | 226 | |
| benzocyclobutene | C8H6 | 4026-23-7 | 406.(±17) | ion | 227 | |
| benzocyclobutane | C8H8 | 694-87-1 | 199.4(±0.9) | calorim | 228 | |
| naphthalene | C10H8 | 91-20-3 | 150.6(±1.6) | review | 2 | |
| 150.6(±1.1) | calorim | 229 | ||||
| 1-methylnaphthalene | C11H10 | 90-12-0 | 116.9(±2.7) | calorim | 226 | |
| 2-methylnaphthalene | C11H10 | 91-57-6 | 116.1(±2.6) | calorim | 226 | |
| 1,8-dimethylnaphthalene | C12H12 | 569-41-5 | 108.8(±3) | calorim | 230 | |
| 2,3-dimethylnaphthalene | C12H12 | 581-40-8 | 79.9(±2) | calorim | 231 | |
| 76.1(±2) | calorim | 232 | ||||
| 2,6-dimethylnaphthalene | C12H12 | 581-42-0 | 78.7(±2.5) | calorim | 231 | |
| 2,7-dimethylnaphthalene | C12H12 | 582-16-1 | 79.5(±0.6) | calorim | 233 | |
| 1-ethyl-8-methylnaphthalene | C13H14 | 61886-71-3 | 98.1(±1.5) | calorim | 234 | |
| 1,4,5,8-tetramethylnaphthalene | C14H16 | 2717-39-7 | 81.6(±3.6) | calorim | 230 | |
| indan | C9H10 | 496-11-7 | 60.7(±1.5) | calorim | 235 | |
| 60.9(±2.1) | review | 2 | ||||
| 1,1-dimethyl-2,3-dihydro-1H-indene | C11H14 | 4912-92-9 | −1.6(±2) | calorim | 235 | |
| 4,6-dimethylindan | C11H14 | 1685-82-1 | −5.8(±1.7) | calorim | 235 | |
| 4,7-dimethylindan | C11H14 | 6682-71-9 | −7.4(±1.7) | calorim | 235 | |
| indene | C9H8 | 95-13-6 | 161.2(±2.3) | review | 2 | |
| 163.3(±1.6) | calorim | 236 | ||||
| 156.7 | equil | 237 | ||||
| anthracene | C14H10 | 120-12-7 | 230.9(±3.7) | review | 2 | |
| 230.8(±4.6) | calorim | 229 | ||||
| phenanthrene | C14H10 | 85-01-8 | 201.4(±3.5) | review | 2 | |
| 201.7(±2.9) | calorim | 238 | ||||
| 201.2(±4.7) | calorim | 239 | ||||
| 206.9(±4.6) | calorim | 229 | ||||
| biphenylene | C12H8 | 259-79-0 | 417.2(±1.9) | review | 2 | |
| 420.4(±1.9) | calorim | 240 | ||||
| acenaphthylene | C12H8 | 208-96-8 | 263.2(±3.7) | review | 2 | |
| 263.8(±3.4) | heat hydrog | 241 | ||||
| 258.2(±5.9) | calorim | 242 | ||||
| acenaphthene | C12H10 | 83-32-9 | 156.8(±3.1) | review | 2 | |
| 156.5(±3.8) | calorim | 242 | ||||
| 155.9(±2.5) | calorim | 241 | ||||
| fluorene | C13H10 | 86-73-7 | 176.7(±3.1) | review | 2 | |
| 175(±1.5) | calorim | 243 | ||||
| 179.4(±3) | calorim | 244 | ||||
| 9-methylfluorene | C14H12 | 2523-37-7 | 148(±1.1) | calorim | 243 | |
| cyclopropa[b]naphthalene | C11H8 | 286-85-1 | 435(±5) | calorim | 245 | |
| naphthacene | C18H12 | 92-24-0 | 340.7(±3.9) | review | 2 | |
| 331.6(±4.4) | calorim | 238 | ||||
| 238.1 | calorim | 166 | ||||
| benz[a]anthracene | C18H12 | 56-55-3 | 290.3(±6) | review | 2 | |
| 294(±5) | calorim | 246 | ||||
| chrysene | C18H12 | 218-01-9 | 268.5(±2.8) | review | 2 | |
| 263(±5) | calorim | 246 | ||||
| benzo[c]phenanthrene | C18H12 | 195-19-7 | 295.3(±9.1) | review | 2 | |
| 291(±5) | calorim | 246 | ||||
| pyracyclene | C14H8 | 187-78-0 | 408.6(±5) | review | 2 | |
| 411.5(±6.2) | calorim | 241 | ||||
| 419.2(±6.2) | heat hydrog | 241 | ||||
| 409.3(±6.2) | calorim | 247 | ||||
| pyracene | C14H12 | 567-79-3 | 174.1(±5.1) | review | 2 | |
| 174.3(±5.3) | calorim | 241 | ||||
| pyrene | C16H10 | 129-00-0 | 225.5(±4.3) | review | 2 | |
| 225.7(±1.3) | calorim | 248 | ||||
| fluoranthene | C16H10 | 206-44-0 | 291.4(±4) | review | 2 | |
| 292(±2.2) | calorim | 242 | ||||
| 282.4(±2.8) | calorim | 168 | ||||
| triphenylene | C18H12 | 217-59-4 | 270.1(±3.1) | review | 2 | |
| 272(±4) | calorim | 246 | ||||
| 5,12-dihydrotetracene | C18H14 | 959-02-4 | 224.9 | review | 2 | |
| 227(±4) | calorim | 246 | ||||
| perylene | C20H12 | 198-55-0 | 317.4(±3.5) | review | 2 | |
| 309(±5) | calorim | 249 | ||||
| 319.4(±2.2) | calorim | 250 | ||||
| benzo[a]pyrene | C20H12 | 50-32-8 | 296.9(±5.5) | review | 2 | |
| benzo[e]pyrene | C20H12 | 192-97-2 | 330.7(±9.2) | review | 2 | |
| benzo[k]fluoranthene | C20H12 | 207-08-9 | 306.2(±6.2) | review | 2 | |
| benzo[b]triphenylene | C22H14 | 215-58-7 | 331(±11) | review | 2 | |
| dibenz[a,h]anthracene | C22H14 | 53-70-3 | 328(±11) | review | 2 | |
| corannulene | C20H10 | 5821-51-2 | 458.7(±9.1) | review | 2 | |
| 460.6(±6.5) | calorim | 250 | ||||
| coronene | C24H12 | 191-07-1 | 300.9(±9.9) | review | 2 | |
| 307.5(±9.8) | calorim | 250 | ||||
| 285.5(±7.3) | calorim | 251 | ||||
| 294.9(±11.1) | calorim | 251 | ||||
| 286.1(±11.4) | calorim | 252 | ||||
| 279.4(±5.3) | calorim | 253 | ||||
| 296.2(±8.8) | review | present work | ||||
| triquinacene | C10H10 | 6053-74-3 | 224(±4.2) | heat hydrog | 254 | |
| azulene | C10H8 | 275-51-4 | 308 | heat hydrog | 255 | |
| 280 | calorim | 256 | ||||
| 6,6-diphenylfulvene | C18H14 | 2175-90-8 | 402(±15) | calorim | 257 |
Units are kJ/mol. Experimental types are denoted “review” (consensus value), “calorim” (calorimetry), “heat hydrog” (heat of hydrogenation), “ion” (ion cycles), “equil” (equilibrium), and “network” (thermochemical network).
The previous studies mentioned above,79–87,136,137,139 are compared to the experimental values individually in Tables 9, 10, and 11, and in summary form in Table 14. One can see slightly better agreement with experiment that with the present extrapolated corrected B3LYP results for most of the studies, though the results of Welsh et al.86 show larger deviations, and the results of Rayne and Forest137 and Zauer139 still show larger deviations as above.
Table 14.
Statistical Measures (kJ/mol) of the Accuracy of Various Predictions of the Enthalpy of Formation at 298 K versus Experimenta
| reference | n | MUD | RMSD | citation |
|---|---|---|---|---|
| Stein | 11 | 14.28 | 6.06 | 78, 79 |
| Alberty | 8 | 12.60 | 7.41 | 80 |
| Moiseeva | 12 | 12.61 | 5.06 | 82, 83 |
| Herndon | 11 | 11.90 | 4.61 | 84 |
| Armitage | 12 | 13.91 | 6.13 | 81 |
| Wang | 29 | 2.71 | 0.86 | 85 |
| Welsh | 14 | 33.68 | 17.97 | 86 |
| Yu | 12 | 9.35 | 3.73 | 87 |
| Blanquart | 9 | 5.67 | 2.81 | 136 |
| Rayne | 13 | 17.32 | 5.25 | 137 |
| Zauer | 17 | 17.73 | 7.50 | 139 |
| B3LYP | 21 | 5.1 | 6.7 |
The table gives the number of data, n, used in computing the mean unsigned deviation, MUD, and the root mean square deviation, RMSD.
In the case of the extrapolated and corrected B3LYP-derived enthalpies of formation, a set of 171 data were used to adjust the parameters of the group additivity based error correction scheme. The optimized group parameters are given in Table 3. A histogram plot of the deviations in the corrected B3LYP enthalpy of formation values with respect to the experimental data used in the fitting process is shown in Figure 2. From this figure, the reliability of the B3LYP scheme is clearly seen, with most errors less than 5 kJ/mol and all errors less than 20 kJ/mol. The corrections to the enthalpies of formation for the extrapolated B3LYP calculations were examined in more detail by the class of compound.
Figure 2.

Histogram plot of deviations of corrected enthalpies of formation derived from B3LYP calculations versus experiment.
Predictions of enthalpies of formation are made for 810 compounds using the extrapolated and corrected B3LYP scheme. The presentation of the results is split into four tables. In Table 15, the B3LYP results for PAHs are compared to experimental values, and in Table 16 the same results for non-PAH molecules are presented. In these tables, the uncorrected and corrected values of the enthalpy of formation are given so that the magnitude of the correction is evident, and the error in the corrected value with respect to the experimental value is given. As a measure of the overall quality of the B3LYP scheme, a mean unsigned deviation of 5.1 kJ/mol and a root-mean-square deviation of 6.7 kJ/mol may be seen for the PAH compounds presented in Table 15. In Table S2, predictions (i.e., no experimental data are available) of the enthalpy are made for PAH compounds, and in Table S3 the same results are presented for non-PAH molecules.
Table 15.
Predicted (B3LYP, Uncorrected, and Corrected) and Experimental Enthalpies of Formation (kJ/mol) for PAH Molecules and Error
| molecule | formula | CAS registry no. |
|
|
|
error | |||
|---|---|---|---|---|---|---|---|---|---|
| acenaphthene | C12H10 | 83-32-9 | 196.5 | 150.7 | 156.8(±3.1) | −6.1 | |||
| acenaphthylene | C12H8 | 208-96-8 | 300.8 | 259.8 | 263.2(±3.7) | −3.2 | |||
| anthracene | C14H10 | 120-12-7 | 265.1 | 222.6 | 230.9(±3.7) | −8.3 | |||
| benz[a]anthracene | C18H12 | 56-55-3 | 340.8 | 277.1 | 290.3(±6.0) | −13.2 | |||
| benzene | C6H6 | 71-43-2 | 75.2 | 75.2 | 83.2(±0.3) | −8.0 | |||
| benzo[a]pyrene | C20H12 | 50-32-8 | 379.0 | 296.0 | 296.9(±5.5) | −0.9 | |||
| benzo[b]triphenylene | C22H14 | 215-58-7 | 432.9 | 348.0 | 331.0(±11) | 17.0 | |||
| benzo[c]phenanthrene | C18H12 | 195-19-7 | 359.1 | 295.3 | 295.3(±9.1) | 0.0 | |||
| benzocyclobutene | C8H6 | 4026-23-7 | 427.9 | 409.8 | 406.0(±17) | 3.8 | |||
| biphenylene | C12H8 | 259-79-0 | 447.3 | 410.9 | 417.2(±1.9) | −6.3 | |||
| chrysene | C18H12 | 218−01−9 | 334.9 | 271.1 | 268.5(±2.8) | 2.6 | |||
| coronene | C24H12 | 191−07−1 | 418.1 | 296.7 | 300.9(±9.9) | 3.3 | |||
| corannulene | C20H10 | 5821−51−2 | 594.2 | 498.5 | 458.7 | 39.8 | |||
| dibenz[a,h]anthracene | C22H14 | 53−70−3 | 420.0 | 335.0 | 328.0 | 7.0 | |||
| fluoranthene | C16H10 | 206−44−0 | 343.8 | 277.9 | 282.4 | −4.5 | |||
| fluorene | C13H10 | 86-73-7 | 227.8 | 179.6 | 179.4 | 0.2 | |||
| indene | C9H8 | 95-13-6 | 180.6 | 156.4 | 161.2 | −4.8 | |||
| naphthacene | C18H12 | 92-24-0 | 374.0 | 310.5 | 340.7 | −30.2 | |||
| naphthalene | C10H8 | 91-20-3 | 162.3 | 141.0 | 150.6 | −9.6 | |||
| perylene | C20H12 | 198-55-0 | 402.2 | 319.2 | 317.4 | 1.8 | |||
| phenanthrene | C14H10 | 85-01-8 | 245.2 | 202.7 | 201.4 | 1.3 | |||
| pyracene | C14H12 | 567-79-3 | 261.6 | 191.4 | 174.1 | 17.3 | |||
| pyracyclene | C14H8 | 187-78-0 | 498.9 | 438.2 | 408.6 | 29.6 | |||
| pyrene | C16H10 | 129-00-0 | 283.0 | 221.3 | 225.5 | −4.2 | |||
| triphenylene | C18H12 | 217-59-4 | 338.8 | 275.1 | 270.1 | 5.0 |
Table 16.
Predicted (B3LYP, Uncorrected and Corrected) and Experimental Enthalpies of Formation (kJ/mol) for Non-PAH Molecules and Error
| molecule | formula | CAS registry no. |
|
|
|
error | |||
|---|---|---|---|---|---|---|---|---|---|
| 1-buten-3-yne | C4H4 | 689-97-4 | 274.2 | 290.6 | 295.0(±3) | −4.4 | |||
| 1-cyclopropyl-2-methylbenzene | C10H12 | 27546-46-9 | 152.3 | 111.1 | 125.5(±2.2) | −14.4 | |||
| 1-ethyl-2-methylbenzene | C9H12 | 611-14-3 | 14.9 | 0.3 | 1.2(±1.2) | −0.9 | |||
| 1-ethyl-3-methylbenzene | C9H12 | 620-14-4 | 13.0 | −1.6 | −1.9(±1.2) | 0.3 | |||
| 1-ethyl-4-methylbenzene | C9H12 | 622-96-8 | 8.8 | −5.8 | −3.3(±1.5) | −2.5 | |||
| 1-ethyl-8-methylnaphthalene | C13H14 | 61886-71-3 | 141.2 | 105.4 | 98.1(±1.5) | 7.3 | |||
| 1-methylnaphthalene | C11H10 | 90-12-0 | 139.4 | 113.5 | 116.9(±2.7) | −3.4 | |||
| 1,1-dimethyl-2,3-dihydro-1H-indene | C11H14 | 4912-92-9 | 48.8 | −7.2 | −1.6(±2) | −5.6 | |||
| 1,2-diethylbenzene | C10H14 | 135-01-3 | 3.7 | −16.1 | −19.5(±2.2) | 3.4 | |||
| 1,2-dihydronaphthalene | C10H10 | 447-53-0 | 154.0 | 124.7 | 124.8(±3.3) | −0.1 | |||
| 1,2-dimethylbenzene | C8H10 | 95-47-6 | 24.4 | 15.0 | 19.0(±1.1) | −4.0 | |||
| 1,2-diphenylbenzene | C18H14 | 84-15-1 | 345.0 | 290.5 | 282.8(±3.2) | 7.7 | |||
| (E)-1,2-diphenylethene | C14H12 | 103-30-0 | 264.7 | 228.6 | 233.7(±2) | −5.1 | |||
| (Z)-1,2-diphenylethene | C14H12 | 645-49-8 | 285.8 | 249.7 | 245.9(±1.3) | 3.8 | |||
| 1,2,3-trimethylbenzene | C9H12 | 526-73-8 | 7.2 | −6.9 | −9.6(±1.3) | 2.7 | |||
| 1,2,3,4-tetrahydronaphthalene | C10H12 | 119-64-2 | 59.0 | 25.8 | 24.0(±3.2) | 1.8 | |||
| 1,2,4-trimethylbenzene | C9H12 | 95-63-6 | −1.3 | −15.5 | −13.9(±1.1) | −1.6 | |||
| 1,3-diethylbenzene | C10H14 | 141-93-5 | −5.4 | −25.2 | −21.6(±2.2) | −3.6 | |||
| 1,3-dimethylbenzene | C8H10 | 108-38-3 | 22.8 | 13.3 | 17.2(±0.8) | −3.9 | |||
| 1,3-diphenylbenzene | C18H14 | 92-06-8 | 328.1 | 273.6 | 280.0(±3.9) | −6.4 | |||
| 1,3,5-trimethylbenzene | C9H12 | 108-67-8 | −3.4 | −17.5 | −15.9(±1.3) | −1.6 | |||
| 1,4-diethylbenzene | C10H14 | 105-05-5 | −5.4 | −25.2 | −22.1(±2.2) | −3.1 | |||
| 1,4-dihydronaphthalene | C10H10 | 612-17-9 | 166.1 | 136.0 | 137.5(±3.2) | −1.5 | |||
| 1,4-dimethylbenzene | C8H10 | 106-42-3 | 23.0 | 13.6 | 17.9(±1) | −4.3 | |||
| 1,4-diphenylbenzene | C18H14 | 92-94-4 | 326.9 | 272.4 | 284.4(±3.8) | −12.0 | |||
| 1,4,5,8-tetramethylnaphthalene | C14H16 | 2717-39-7 | 139.4 | 99.3 | 81.6(±3.6) | 17.7 | |||
| 1,8-dimethylnaphthalene | C12H12 | 569-41-5 | 144.7 | 114.0 | 108.8(±3) | 5.2 | |||
| 2-ethylbut-1-ene | C6H12 | 760-21-4 | −52.1 | −53.2 | −56.1(±0.9) | 2.9 | |||
| 2-ethylbuta-1,3-diene | C6H10 | 3404-63-5 | 56.1 | 58.2 | 63.6 | −5.4 | |||
| 2-methylbut-1-en-3-yne | C5H6 | 78-80-8 | 244.7 | 253.5 | 259.0(±1.3) | −5.5 | |||
| 2-methylbut-1-ene | C5H10 | 563-46-2 | −41.7 | −36.9 | −35.3(±0.8) | −1.6 | |||
| 2-methylbut-2-ene | C5H10 | 513-35-9 | −51.3 | −43.5 | −41.5(±0.88) | −2.0 | |||
| 2-methylbuta-1,3-diene | C5H8 | 78-79-5 | 77.6 | 85.7 | 75.7(±1) | 10.0 | |||
| 2-methylhexane | C7H16 | 591-76-4 | −186.6 | −199.4 | −195.0(±1.3) | −4.4 | |||
| 2-methylnaphthalene | C11H10 | 91-57-6 | 134.7 | 108.7 | 116.1(±2.6) | −7.4 | |||
| 2-methylpent-1-ene | C6H12 | 763-29-1 | −45.7 | −46.0 | −58.0(±1.1) | 12.0 | |||
| 2-methylpent-2-ene | C6H12 | 625-27-4 | −53.8 | −51.9 | −63.2(±1.2) | 11.3 | |||
| 2-methylpentane | C6H14 | 107-83-5 | −170.3 | −177.8 | −174.3(±1) | −3.5 | |||
| 2-methylprop-1-ene | C4H8 | 115-11-7 | −31.0 | −20.2 | −16.9(±0.5) | −3.3 | |||
| 2-phenyltoluene | C13H12 | 643-58-3 | 186.3 | 154.3 | 152.8(±1.5) | 1.5 | |||
| 2,2-dimethylbutane | C6H14 | 75-83-2 | −170.4 | −187.6 | −185.6(±1) | −2.0 | |||
| 2,2-dimethylpentane | C7H16 | 590-35-2 | −184.7 | −207.1 | −206.2(±1.3) | −0.9 | |||
| 2,3-dimethylbuta-1,3-diene | C6H10 | 513-81-5 | 44.3 | 44.8 | 56.4(±1.2) | −11.6 | |||
| 2,3-dimethylnaphthalene | C12H12 | 581-40-8 | 110.1 | 79.5 | 79.9(±2) | −0.4 | |||
| 2,4-dimethylpentane | C7H16 | 108-08-7 | −185.3 | −206.0 | −202.1(±1) | −3.9 | |||
| 2,6-dimethylnaphthalene | C12H12 | 581-42-0 | 106.6 | 76.0 | 78.7(±2.5) | −2.7 | |||
| 2,7-dimethylnaphthalene | C12H12 | 582-16-1 | 106.7 | 76.0 | 79.5(±0.6) | −3.5 | |||
| 3-ethylpent-1-ene | C7H14 | 4038-04-4 | −46.9 | −57.1 | −69.5(±2) | 12.4 | |||
| 3-methylbut-1-ene | C5H10 | 563-45-1 | −22.9 | −22.8 | −27.7(±1.2) | 4.9 | |||
| 3-methylhex-1-ene | C7H14 | 3404-61-3 | −48.9 | −59.2 | −68.2(±1.5) | 9.0 | |||
| 3-methylpent-1-ene | C6H12 | 760-20-3 | −35.0 | −40.0 | −47.0(±1.1) | 7.0 | |||
| (E)-3-methylpent-2-ene | C6H12 | 616-12-6 | −72.7 | −70.8 | −63.5(±0.9) | −7.3 | |||
| (Z)-3-methylpent-2-ene | C6H12 | 922-62-3 | −61.5 | −59.6 | −61.9(±0.9) | 2.3 | |||
| 3-methylpentane | C6H14 | 96-14-0 | −166.4 | −173.9 | −171.6(±1) | −2.3 | |||
| 3-phenyltoluene | C13H12 | 643-93-6 | 175.3 | 143.3 | 152.5(±8) | −9.2 | |||
| 3,3-dimethylbut-1-ene | C6H12 | 558-37-2 | −36.1 | −50.7 | −59.7(±2) | 9.0 | |||
| 3,3-dimethylpent-1-ene | C7H14 | 3404-73-7 | −43.6 | −63.5 | −78.5(±1.7) | 15.0 | |||
| 3,3-dimethylpentane | C7H16 | 562-49-2 | −184.3 | −206.6 | −201.5 | −5.1 | |||
| 4-methylpent-1-ene | C6H12 | 691-37-2 | −46.8 | −52.7 | −49.4(±0.7) | −3.3 | |||
| (E)-4-methylpent-2-ene | C6H12 | 674-76-0 | −50.0 | −52.8 | −60.1(±1.5) | 7.3 | |||
| (Z)-4-methylpent-2-ene | C6H12 | 691-38-3 | −42.9 | −45.7 | −57.9(±1.4) | 12.2 | |||
| 4-phenyltoluene | C13H12 | 644-08-6 | 174.8 | 142.9 | 138.2(±2.9) | 4.7 | |||
| 4,6-dimethylindan | C11H14 | 1685-82-1 | 33.3 | −4.2 | −5.8(±1.7) | 1.6 | |||
| 4,7-dimethylindan | C11H14 | 6682-71-9 | 32.4 | −5.1 | −7.4(±1.7) | 2.3 | |||
| 5,12-dihydrotetracene | C18H14 | 959-02-4 | 302.0 | 227.4 | 224.9 | 2.5 | |||
| 6,6-diphenylfulvene | C18H14 | 2175-90-8 | 474.9 | 409.2 | 402.0(±15) | 7.2 | |||
| 9-methylfluorene | C14H12 | 2523-37-7 | 212.3 | 150.9 | 148.0(±1.1) | 2.9 | |||
| benzocyclobutane | C8H8 | 694-87-1 | 212.9 | 190.0 | 199.4(±0.9) | −9.4 | |||
| benzylbenzene | C13H12 | 101-81-5 | 195.7 | 160.0 | 162.3(±2.3) | −2.3 | |||
| but-1-ene | C4H8 | 106-98-9 | −14.7 | −2.2 | 0.0(±0.5) | −2.2 | |||
| but-1-ylbenzene | C10H14 | 104-51-8 | 2.6 | −17.7 | −13.8(±1.3) | −3.9 | |||
| but-1-yne | C4H6 | 107-00-6 | 156.2 | 170.1 | 166.1(±2.1) | 4.0 | |||
| (E)-but-2-ene | C4H8 | 624-64-6 | −20.3 | −4.8 | −11.2(±0.5) | 6.4 | |||
| (Z)-but-2-ene | C4H8 | 590-18-1 | −22.8 | −7.3 | −7.3(±0.5) | −0.0 | |||
| but-2-ylbenzene | C10H14 | 135-98-8 | 8.6 | −19.6 | −17.4(±1.4) | −2.2 | |||
| but-2-yne | C4H6 | 503-17-3 | 129.1 | 147.9 | 148.0(±1.5) | −0.1 | |||
| (E)-buta-1,3-diene | C4H6 | 106-99-0 | 91.8 | 107.5 | 108.8(±0.8) | −1.3 | |||
| buta-1,3-diyne | C4H2 | 460-12-8 | 450.4 | 467.5 | 464.0(±5) | 3.5 | |||
| butane | C4H10 | 106-97-8 | −140.1 | −129.4 | −125.9(±0.4) | −3.5 | |||
| butyn-1-ylbenzene | C10H10 | 622-76-4 | 261.8 | 249.6 | 248.6(±1) | 1.0 | |||
| cyclohexylbenzene | C12H16 | 827-52-1 | 36.8 | −15.2 | −16.7(±1.5) | 1.5 | |||
| cyclopropa[b]naphthalene | C11H8 | 286-85-1 | 459.9 | 421.0 | 435.0(±5) | −14.0 | |||
| diphenylethyne | C14H10 | 501-65-5 | 421.8 | 389.0 | 385.0(±2.7) | 4.0 | |||
| ethane | C2H6 | 74-84-0 | −107.0 | −85.9 | −83.91(±0.14) | −1.99 | |||
| ethene | C2H4 | 74-85-1 | 31.9 | 53.3 | 52.45(±0.13) | 0.85 | |||
| ethenylbenzene | C8H8 | 100-42-5 | 149.5 | 142.1 | 146.9(±1) | −4.8 | |||
| ethylbenzene | C8H10 | 100-41-4 | 34.7 | 24.8 | 29.8(±0.8) | −5.0 | |||
| ethyne | C2H2 | 74-86-2 | 215.9 | 235.2 | 226.7(±0.8) | 8.5 | |||
| ethynylbenzene | C8H6 | 536-74-3 | 320.8 | 314.1 | 306.6(±1.7) | 7.5 | |||
| hept-1-ene | C7H14 | 592-76-7 | −62.6 | −65.8 | −62.3(±1.5) | −3.5 | |||
| hept-1-yne | C7H12 | 628-71-7 | 107.3 | 105.6 | 103.8(±2.6) | 1.8 | |||
| hept-2-yne | C7H12 | 1119-65-9 | 82.3 | 85.5 | 84.8(±2.2) | 0.7 | |||
| hept-3-yne | C7H12 | 2586-89-2 | 82.8 | 86.0 | 82.8(±2.4) | 3.2 | |||
| heptane | C7H16 | 142-82-5 | −187.8 | −192.6 | −187.8(±0.8) | −4.8 | |||
| hex-1-ene | C6H12 | 592-41-6 | −46.8 | −44.8 | −42.1(±1.2) | −2.7 | |||
| hex-1-yne | C6H10 | 693-02-7 | 123.4 | 127.0 | 122.3(±1.2) | 4.7 | |||
| (E)-hex-2-ene | C6H12 | 4050-45-7 | −60.6 | −56.3 | −51.7(±2.0) | −4.6 | |||
| (Z)-hex-2-ene | C6H12 | 7688-21-3 | −53.3 | −49.0 | −47.9(±2.0) | −1.1 | |||
| hex-2-yne | C6H10 | 764-35-2 | 98.6 | 107.0 | 107.7(±2.4) | −0.7 | |||
| (E)-hex-3-en-1,5-diyne | C6H4 | 16668-68-1 | 524.1 | 535.5 | 538.0(±3.0) | −2.5 | |||
| (Z)-hex-3-en-1,5-diyne | C6H4 | 16668-67-0 | 516.1 | 527.5 | 541.8(±3.0) | −14.3 | |||
| (E)-hex-3-ene | C6H12 | 13269-52-8 | −46.2 | −42.7 | −49.3(±1.1) | 6.6 | |||
| (Z)-hex-3-ene | C6H12 | 7642-09-3 | −51.3 | −47.9 | −46.9(±2.4) | −1.0 | |||
| (E)-hexa-1,3-diene | C6H10 | 20237-34-7 | 60.9 | 67.7 | 54.0(±2.0) | 13.7 | |||
| (Z)-hexa-1,3-diene | C6H10 | 14596-92-0 | 53.0 | 59.8 | 59.0(±2.0) | 0.8 | |||
| (E)-hexa-1,3,5-triene | C6H8 | 821-07-8 | 146.9 | 157.0 | 168.0(±3.0) | −11.0 | |||
| (Z)-hexa-1,3,5-triene | C6H8 | 2612-46-6 | 156.2 | 166.3 | 172.0(±3.0) | −5.7 | |||
| (Z)-hexa-1,4-diene | C6H10 | 7318-67-4 | 73.8 | 80.6 | 77.0(±2.0) | 3.6 | |||
| hexa-1,5-diene | C6H10 | 592-42-7 | 78.3 | 82.0 | 85.0(±2.0) | −3.0 | |||
| (E,E)-hexa-2,4-diene | C6H10 | 5194-51-4 | 29.6 | 39.5 | 44.0(±2.0) | −4.5 | |||
| (E,Z)-hexa-2,4-diene | C6H10 | 5194-50-3 | 36.9 | 46.7 | 48.0(±2.0) | −1.3 | |||
| hexane | C6H14 | 110-54-3 | −171.7 | −171.3 | −167.2(±0.8) | −4.1 | |||
| indan | C9H10 | 496-11-7 | 84.3 | 56.3 | 60.9(±2.1) | −4.6 | |||
| isobutane | C4H10 | 75-28-5 | −143.7 | −140.9 | −134.4(±0.4) | −6.5 | |||
| isobutylbenzene | C10H14 | 538-93-2 | 1.0 | −27.2 | −21.5(±1.4) | −5.7 | |||
| isopentane | C5H12 | 78-78-4 | −155.4 | −157.7 | −153.7(±0.6) | −4.0 | |||
| isopentyne | C5H8 | 598-23-2 | 138.0 | 138.8 | 136.4(±2.1) | 2.4 | |||
| m-benzyne | C6H4 | 1828-89-3 | 521.3 | 482.8 | 490.0(±10) | −7.2 | |||
| neopentane | C5H12 | 463-82-1 | −163.6 | −175.6 | −167.9(±0.6) | −7.7 | |||
| pent-1-ene | C5H10 | 109-67-1 | −30.9 | −23.7 | −21.3(±2.7) | −2.4 | |||
| pent-1-yne | C5H8 | 627-19-0 | 139.5 | 148.2 | 144.3(±2.1) | 3.9 | |||
| (E)-pent-2-ene | C5H10 | 646-04-8 | −44.1 | −34.6 | −33.1(±1.3) | −1.5 | |||
| (Z)-pent-2-ene | C5H10 | 627-20-3 | −37.3 | −27.8 | −28.0(±0.8) | 0.2 | |||
| pent-2-yne | C5H8 | 627-21-4 | 114.4 | 128.0 | 128.9(±2.1) | −0.9 | |||
| (E)-pent-3-en-1-yne | C5H6 | 2004-69-5 | 240.8 | 254.2 | 259.0(±3.0) | −4.8 | |||
| (Z)-pent-3-en-1-yne | C5H6 | 1574-40-9 | 240.6 | 254.0 | 258.0(±3.0) | −4.0 | |||
| (E)-penta-1,3-diene | C5H8 | 2004-70-8 | 60.3 | 73.1 | 75.8(±0.7) | −2.7 | |||
| (Z)-penta-1,3-diene | C5H8 | 1574-41-0 | 82.9 | 95.7 | 82.7(±0.9) | 13.0 | |||
| penta-1,4-diene | C5H8 | 591-93-5 | 95.0 | 104.7 | 106.3(±1.3) | −1.6 | |||
| pentane | C5H12 | 109-66-0 | −156.0 | −150.4 | −146.8(±0.6) | −3.6 | |||
| phenyl | C6H5 | 2396-01-2 | 326.6 | 326.6 | 337.0(±2.5) | −10.4 | |||
| phenylbenzene | C12H10 | 92-52-4 | 201.5 | 174.2 | 180.3(±3.3) | −6.1 | |||
| phenylethylbenzene | C14H14 | 103-29-7 | 176.0 | 135.1 | 135.6(±1.3) | −0.5 | |||
| prop-1-ylbenzene | C9H12 | 103-65-1 | 20.7 | 5.6 | 7.8(±0.8) | −2.2 | |||
| prop-2-ylbenzene | C9H12 | 98-82-8 | 20.6 | −2.4 | 3.9(±1.1) | −6.3 | |||
| propane | C3H8 | 74-98-6 | −124.0 | −108.0 | −104.7(±0.6) | −3.3 | |||
| (E)-propen-1-ylbenzene | C9H10 | 873-66-5 | 130.7 | 120.4 | 117.2(±10) | 3.2 | |||
| (Z)-propen-1-ylbenzene | C9H10 | 766-90-5 | 119.0 | 108.7 | 121.4(±10) | −12.7 | |||
| propen-2-ylbenzene | C9H10 | 98-83-9 | 128.7 | 113.7 | 118.3(±1.4) | −4.6 | |||
| propen-3-ylbenzene | C9H10 | 300-57-2 | 144.4 | 131.0 | 133.8(±1.1) | −2.8 | |||
| propene | C3H6 | 115-07-1 | 0.2 | 18.6 | 20.2(±0.4) | −1.6 | |||
| propyn-1-ylbenzene | C9H8 | 673-32-5 | 276.4 | 269.4 | 268.2(±2.2) | 1.2 | |||
| propyne | C3H4 | 74-99-7 | 170.6 | 189.7 | 185.4(±0.9) | 4.3 | |||
| tert-butylbenzene | C10H14 | 98-06-6 | 12.7 | −25.0 | −22.7(±1.4) | −2.3 | |||
| tert-hexyne | C6H10 | 917-92-0 | 119.7 | 105.7 | 106.1(±1.5) | −0.4 | |||
| toluene | C7H8 | 108-88-3 | 48.9 | 44.1 | 50.1(±1.1) | −6.0 | |||
| trans-decalin | C10H18 | 493-02-7 | −112.6 | −175.7 | −182.2(±2.3) | 6.5 | |||
| triphenylmethane | C19H16 | 519-73-3 | 353.0 | 278.4 | 276.1(±4.1) | 2.3 |
G3MP2B3
In Figure 3, the corrections for the aliphatic species are examined. For each molecule, a value of 7.35 kJ/mol per methylene group (–CH2–) was subtracted from the difference between for the calculated extrapolated G3MP2B3 value and the literature experimental value. The residuals are plotted in Figure 3 as a function of the number of –CH2– groups in the molecule and correspond to a sum of the corrections for the remaining groups in the molecule. For example, the residuals for the n-alkanes (bottom curve at about −19 kJ/mol) correspond to the sum of the corrections for two CH3– groups, the residuals for the 1-alkenes (second to bottom curve at about −14 kJ/mol) correspond to the sum of the corrections for a CH3–, a ≡CH, and a =CH2 group, and the residuals for the tert-alkanes (top curve at about +22 kJ/mol) correspond to the sum of the corrections for four terminal CH3– groups plus one tertiary group –C(C)(C)–. The standard deviation for all of these groups (difference between the individual points and the corresponding line) was about 0.8 kJ/mol, which is comparable to the average uncertainty (about 1.1 kJ/mol) in the experimental values for this group of molecules.
Figure 3.
Plot of group corrections by chemical class for aliphatic compounds for G3MP2B3 results.
In Figure 4, corrections for the PAH species for each type of aromatic carbon are studied. We found a correction of −1.28 kJ/mol for each CbH group (benzene has six). Using the ortho-fused PAHs (e.g., naphthalene, anthracene), the residuals show that each Cf ortho-fused group (naphthalene has two) has a correction of about 16.4 kJ/mol. Using the ortho- and peri-fused PAHs (e.g., pyrene, coronene), the residuals show a correction of about 13.2 kJ/mol for each Cp group (pyrene has two). Using PAHs where one aromatic ring is “joined” or “linked” to another (e.g., biphenylene, fluoranthene in Table 1), we find a correction of about 10.8 kJ/mol for each “Cj” group (biphenylene has four). The resultant standard deviation for all these classes of PAHs is about 3 kJ/mol, which is comparable to the average uncertainties in the experimental values.
Figure 4.
Plot of group corrections by chemical class for PAHs for G3MP2B3 results.
Alkyl-substituted benzene compounds (e.g., 1,3-dimethyl-benzene, prop-1-ylbenzene) and benzocycloalkanes (e.g., indan, 1,4-dihydronaphthalene) were considered and group values (shown in Figure 4) of about 14.9 kJ/mol per CbC group (aromatic group terminated by carbon atom) and 4.1 kJ/mol per “–CH2(Cb)–” group (a “–CH2–” group connected to an aromatic carbon) were found. The resultant standard deviation is about 2 kJ/mol, which is comparable to the average uncertainties in the experimental values.
In short, our analysis shows that the group additivity approach for correcting the extrapolated B3LYP computed enthalpies of formation works very well for the aliphatic compounds, the PAHs, and the substituted PAHs. The standard deviation between our corrected G3MP2B3 results and the experimental results for our limited training set focusing on simple classes was on the order of 3 kJ/mol, whereas for all of the molecules, many with complicated functionalities, the standard deviation was on the order of 6 kJ/mol.
It was found that the G3B3 method accurately computes energies with little systematic differences for molecules with well-established heats of formation (that is, deviations between calculations and experiment were within experimental uncertainties). This was found to hold for a range of hydrocarbons including acyclic aliphatic (alkanes, alkenes, alkynes, etc.) and cyclic aliphatic (cycloalkanes, cycloalkenes, etc.) hydrocarbons. Although the data were fewer for substituted aromatic hydrocarbons and PAH species, small differences were found between enthalpies of formation calculated using the G3B3 method and experimentally derived values. Use of the G3MP2B3 method for unsaturated aliphatic hydrocarbons and aromatics, however, produced computed enthalpies of formation that were consistently lower than those derived from experimental measurements (i.e., from enthalpies of combustion, heats of sublimation, etc.). The differences were found to be small but systematic and on the order of (1.0 to 1.5) kJ/mol per carbon atom.
The G3B3 method is a composite ab initio model chemistry method applicable to a wide range of molecules with reported average errors163,164 of about 3–6 kJ/mol depending on the test set of molecules. The G3B3 method, however, is practically limited (at present) to PAH molecules with 12–18 carbon atoms (depending upon whether symmetry can be imposed) due to computer memory, scratch disk space limitations, and computational time. As a result, it is necessary to compute energies for larger molecules using a less computationally expensive method. The G3MP2B3 method uses a single MP2 calculation to approximate the composite total energies in the G3B3 method that are determined from a set of MP2, MP4, and QCISD(T) energies using different basis sets. The G3MP2B3 method itself is practically limited (at present) to PAH species with up to about 16–24 carbon atoms, depending on symmetry.
Data from G3MP2B3 calculations are presented in Table 17 (benzenoid and PAH compounds) and Table 18 (other compounds). Additional data from G3MP2B3 calculations for which no experimental data are available are given in Table S4, where some comparison is made to nonexperimental literature values.
Table 17.
Corrected Values of the Enthalpy of Formation (kJ/mol) Computed Using the G3MP2B3 model chemistry
| name | formula | CAS registry no. |
|
G3MP2B3 | residual | |
|---|---|---|---|---|---|---|
| Benzene/Alkyls | ||||||
| benzene | C6H6 | 71-43-2 | 82.9 ± 0.9 | 84.5 | 1.6 | |
| toluene | C7H8 | 108-88-3 | 50.1 ± 1.1 | 52.1 | 2.0 | |
| ethylbenzene | C8H10 | 100-41-4 | 29.8 ± 0.8 | 30.3 | 0.5 | |
| prop-1-ylbenzene | C9H12 | 103-65-1 | 7.8 ± 0.8 | 7.7 | −0.1 | |
| prop-2-ylbenzene | C9H12 | 98-82-8 | 3.9 | 3.1 | −0.8 | |
| but-1-ylbenzene | C10H14 | 104-51-8 | −13.8 ± 1.3 | −13.7 | 0.1 | |
| but-2-ylbenzene | C10H14 | 135-98-8 | −17.4 ± 1.4 | −16.9 | 0.5 | |
| isobutylbenzene | C10H14 | 538-93-2 | −21.5 ± 1.4 | −23.2 | −1.7 | |
| tert-butylbenzene | C10H14 | 98-06-6 | −22.7 ± 1.4 | −25.1 | −2.4 | |
| Benzene/Alkyls-Multi | ||||||
| 1,2-dimethylbenzene | C8H10 | 95-47-6 | 19.0 ± 1.1 | 18.9 | −0.1 | |
| 1,3-dimethylbenzene | C8H10 | 108-38-3 | 17.2 ± 0.8 | 17.5 | 0.3 | |
| 1,4-dimethylbenzene | C8H10 | 106-42-3 | 17.9 ± 1.0 | 17.9 | 0.0 | |
| 1-ethyl-2-methylbenzene | C9H12 | 611-14-3 | 1.2 ± 1.2 | −0.7 | −1.9 | |
| 1-ethyl-3-methylbenzene | C9H12 | 620-14-4 | −1.9 ± 1.2 | −2.9 | −1.0 | |
| 1-ethyl-4-methylbenzene | C9H12 | 622-96-8 | −3.3 ± 1.5 | −1.5 | 1.9 | |
| 1,2-diethylbenzene | C10H14 | 135-01-3 | −19.5 ± 2.2 | −21.7 | −2.2 | |
| 1,3-diethylbenzene | C10H14 | 141-93-5 | −21.6 ± 2.2 | −24.1 | −2.5 | |
| 1,4-diethylbenzene | C10H14 | 105-05-5 | −22.1 ± 2.2 | −23.2 | −1.1 | |
| 1,2,3-trimethylbenzene | C9H12 | 526-73-8 | −9.6 ± 1.3 | −8.8 | 0.8 | |
| 1,2,4-trimethylbenzene | C9H12 | 95-63-6 | −13.9 ± 1.1 | −15.3 | −1.4 | |
| 1,3,5-trimethylbenzene | C9H12 | 108-67-8 | −15.9 ± 1.3 | −14.9 | 1.0 | |
| Benzene/Cycloalkyls | ||||||
| 1-cyclopropyl-2-methylbenzene | C10H12 | 27546-46-9 | 125.5 ± 2.2 | 126.8 | 1.3 | |
| cyclohexylbenzene | C12H16 | 827-52-1 | −16.7 ± 1.5 | −19.8 | −3.1 | |
| Naphthalene/Alkyls | ||||||
| naphthalene | C10H8 | 91-20-3 | 150.6 ± 1.6 | 149.4 | −1.2 | |
| 1-methylnaphthalene | C11H10 | 90-12-0 | 116.9 ± 2.7 | 116.7 | −0.2 | |
| 2-methylnaphthalene | C11H10 | 91-57-6 | 116.1 ± 2.6 | 115.6 | −0.5 | |
| 1-ethylnaphthalene | C12H12 | 1127-76-0 | 98.0 ± 5.0 | 97.1 | −0.9 | |
| 1,8-dimethylnaphthalene | C12H12 | 569-41-5 | 108.8 ± 3.0 | 109.5 | 0.7 | |
| 2,3-dimethylnaphthalene | C12H12 | 581-40-8 | 79.9 ± 2.0 | 82.3 | 2.4 | |
| 2,6-dimethylnaphthalne | C12H12 | 581-42-0 | 78.7 ± 2.5 | 82.0 | 3.3 | |
| 2,7-dimethylnaphthalene | C12H12 | 582-16-1 | 79.5 ± 0.6 | 82.0 | 2.5 | |
| 1-ethyl-8-methylnaphthalene | C13H14 | 61886-71-3 | 98.1 ± 1.5 | 96.5 | −1.6 | |
| 1,4,5,8-tetramethylnaphthalene | C14H16 | 2717-39-7 | 81.6 ± 3.6 | 80.7 | −0.9 | |
| Naphthalene/Hydro | ||||||
| 1,2-dihydronaphthalene | C10H10 | 447-53-0 | 124.8 ± 3.3 | 124.4 | −0.4 | |
| 1,4-dihydronaphthalene | C10H10 | 612-17-9 | 137.5 ± 3.2 | 136.8 | −0.7 | |
| 1,2,3,4-tetrahydronaphthalene | C10H12 | 119-64-2 | 24.0 ± 3.2 | 22.9 | −1.1 | |
| trans-decalin | C10H18 | 493-02-7 | −182.2 ± 2.3 | −186.2 | −4.0 | |
| Benzene/Benzocyclo | ||||||
| benzocyclobutane | C8H8 | 694-87-1 | 199.4 ± 0.9 | 199.8 | 0.4 | |
| indan | C9H10 | 496-11-7 | 60.9 ± 2.1 | 59.5 | −1.5 | |
| 4,6-dimethylindan | C11H14 | 1685-82-1 | −5.8 ± 1.7 | −6.4 | −0.6 | |
| benzocyclobutene | C8H6 | 4026-23-7 | 406.0 ± 17.0 | 408.3 | 2.3 | |
| indene | C9H8 | 95-13-6 | 161.2 ± 2.3 | 159.8 | −1.4 | |
| Naphthalene/Naphthacyclo | ||||||
| cyclopropa[b]naphthalene | C11H8 | 286-85-1 | 435.0 ± 5.0 | 440.4 | 5.4 | |
| acenaphthylene | C12H8 | 208-96-8 | 263.2 ± 3.7 | 260.9 | −2.3 | |
| acenaphthene | C12H10 | 83-32-9 | 156.8 ± 3.1 | 155.7 | −1.1 | |
| Benzene/Alkenyls | ||||||
| ethenylbenzene | C8H8 | 100-42-5 | 146.9 ± 1.0 | 148.4 | 1.5 | |
| (E)-propen-1-ylbenzene | C9H10 | 873-66-5 | 117.2 | 117.0 | −0.2 | |
| (Z)-propen-1-ylbenzene | C9H10 | 766-90-5 | 121.4 | 124.2 | 2.8 | |
| propen-2-ylbenzene | C9H10 | 98-83-9 | 118.3 ± 1.4 | 117.0 | −1.3 | |
| propen-3-ylbenzene | C9H10 | 300-57-2 | 133.8 ± 1.1 | 134.3 | 0.5 | |
| Benzene/Phenyl | ||||||
| phenylbenzene | C12H10 | 92-52-4 | 180.3 ± 3.3 | 178.1 | −2.2 | |
| benzylbenzene | C13H12 | 101-81-5 | 162.3 ± 2.3 | 163.6 | 1.3 | |
| 2-phenyltoluene | C13H12 | 643-58-3 | 152.8 ± 1.5 | 149.7 | −3.1 | |
| phenylethylbenzene | C14H14 | 103-29-7 | 135.6 ± 1.3 | 139.9 | 4.3 | |
| PAH/Misc | ||||||
| biphenylene | C12H8 | 259-79-0 | 417.2 ± 1.9 | 415.8 | −1.4 | |
| anthracene | C14H10 | 120-12-7 | 230.9 ± 3.7 | 229.7 | −1.2 | |
| phenanthrene | C14H10 | 85-01-8 | 201.4 ± 3.5 | 206.6 | 5.2 | |
| pyrene | C16H10 | 129-00-0 | 225.5 ± 4.3 | 226.5 | 1.0 | |
| chrysene | C18H12 | 218-01-9 | 268.5 ± 2.8 | 269.2 | 0.7 | |
| benzo[c]phenanthrene | C18H12 | 195-19-7 | 295.3 ± 9.1 | 291.1 | −4.2 | |
| perylene | C20H12 | 198-55-0 | 317.4 ± 3.5 | 315.6 | −1.8 | |
| 6,6-diphenylfulvene | C18H14 | 2175-90-8 | 402.0 ± 15.0 | 396.0 | −6.0 | |
| fluoranthene | C16H10 | 206-44-0 | 291.4 ± 4.0 | 282.4 | −9.0 | |
| coronene | C24H12 | 191-07-1 | 300.9 ± 9.9 | 294.9 | −6.0 | |
| fluorene | C13H10 | 86-73-7 | 176.7 ± 3.1 | 179.4 | 2.4 | |
| Deviations (>3σ Low) | ||||||
| 3-phenyltoluene | C13H12 | 643-93-6 | 152.5 ± 8.0 | 145.3 | −7.2 | |
| benz[a]anthracene | C18H12 | 56-55-3 | 290.3 ± 6.0 | 279.8 | −10.5 | |
| 1,2-diphenylbenzene | C18H14 | 84-15-1 | 282.8 ± 3.2 | 273.2 | −9.6 | |
| azulene | C10H8 | 275-51-4 | 308.0 | 292.1 | −15.9 | |
| Deviations (>3σ High) | ||||||
| 9-methylfluorene | C14H12 | 2523-37-7 | 148.0 ± 1.1 | 155.0 | 7.0 | |
| 4-phenyltoluene | C13H12 | 644-08-6 | 138.2 ± 2.9 | 145.9 | 7.7 | |
| (E)-1,2-diphenylethene | C14H12 | 103-30-0 | 233.7 ± 2.0 | 239.4 | 5.7 | |
| (Z)-1,2-diphenylethene | C14H12 | 645-49-8 | 245.9 ± 1.3 | 252.9 | 7.0 | |
| ethynylbenzene | C8H6 | 536-74-3 | 306.6 ± 1.7 | 318.2 | 11.6 | |
| propyn-1-ylbenzene | C9H8 | 673-32-5 | 268.2 ± 2.2 | 277.2 | 9.0 | |
| Deviations (Large Low) | ||||||
| naphthacene | C18H12 | 92-24-0 | 340.7 ± 3.9 | 316.2 | −24.5 | |
| corannulene | C20H10 | 5821-51-2 | 458.7 ± 9.1 | 427.0 | −31.7 | |
| triphenylmethane | C19H16 | 519-73-3 | 276.1 ± 4.1 | 212.9 | −55.1 | |
| Deviations (Large High) | ||||||
| pyracyclene | C14H8 | 187-78-0 | 408.6 ± 5.0 | 426.7 | 18.1 | |
| pyracene | C14H12 | 567-79-3 | 174.1 ± 5.1 | 190.2 | 16.1 | |
| diphenylethyne | C14H10 | 501-65-5 | 385.0 ± 2.7 | 405.1 | 20.1 | |
| triquinacene | C10H10 | 6053-74-3 | 224.0 ± 4.2 | 239.3 | 15.3 | |
| benzyne | C6H4 | 462-80-6 | 446.0 ± 13.0 | 456.2 | 10.2 | |
Table 18.
Corrected Values of the Enthalpy of Formation (kJ/mol) Computed Using the G3MP2B3 Model Chemistry
| name | formula | CAS registry no. |
|
G3MP2B3 | residual | |
|---|---|---|---|---|---|---|
| Alkanes, Normal | ||||||
| ethane | C2H6 | 74-84-0 | −84.4 ± 0.4 | −84.3 | 0.1 | |
| propane | C3H8 | 74-98-6 | −104.7 ± 0.6 | −105.0 | −0.3 | |
| butane | C4H10 | 106-97-8 | −125.9 ± 0.4 | −126.1 | −0.2 | |
| pentane | C5H12 | 109-66-0 | −146.8 ± 0.6 | −147.1 | −0.3 | |
| hexane | C6H14 | 110-54-3 | −167.2 ± 0.8 | −168.1 | −0.9 | |
| heptane | C7H16 | 142-82-5 | −187.8 ± 0.8 | −189.2 | −1.4 | |
| Alkanes, Branched | ||||||
| isobutane | C4H10 | 75-28-5 | −134.4 ± 0.4 | −134.4 | 0.0 | |
| isopentane | C5H12 | 78-78-4 | −153.7 ± 0.6 | −153.3 | 0.4 | |
| 2-methylpentane | C6H14 | 107-83-5 | −174.3 ± 1.0 | −174.3 | 0.0 | |
| 2-methylhexane | C7H16 | 591-76-4 | −195.0 ± 1.3 | −195.7 | −0.7 | |
| neopentane | C5H12 | 463-82-1 | −167.9 ± 0.6 | −169.0 | −1.1 | |
| 2,2-dimethylbutane | C6H14 | 75-83-2 | −185.6 ± 1.0 | −185.2 | 0.4 | |
| 2,2-dimethylpentane | C7H16 | 590-35-2 | −206.2 ± 1.3 | −206.4 | −0.2 | |
| Alk-1-enes | ||||||
| ethene | C2H4 | 74-85-1 | 52.6 ± 0.2 | 50.1 | −2.5 | |
| propene | C3H6 | 115-07-1 | 20.2 ± 0.4 | 18.6 | −1.6 | |
| but-1-ene | C4H8 | 106-98-9 | 0.0 ± 0.5 | −0.9 | −0.9 | |
| pent-1-ene | C5H10 | 109-67-1 | −21.3 | −22.4 | −1.1 | |
| hex-1-ene | C6H12 | 592-41-6 | −42.1 ± 1.2 | −43.7 | −1.6 | |
| Alk-1-enes, Branched | ||||||
| 3-methylbut-1-ene | C5H10 | 563-45-1 | −27.7 ± 1.2 | −29.4 | −1.7 | |
| 3-methylpent-1-ene | C6H12 | 760-20-3 | −47.0 ± 1.1 | −51.5 | −4.5 | |
| 3-methylhex-1-ene | C7H14 | 3404-61-3 | −68.2 ± 1.5 | −72.7 | −4.5 | |
| 4-methylpent-1-ene | C6H12 | 691-37-2 | −49.4 ± 0.7 | −49.9 | −0.4 | |
| 3-ethylpent-1-ene | C7H14 | 4038-04-4 | −69.5 ± 2.0 | −73.1 | −3.6 | |
| 3,3-dimethylpent-1-ene | C7H14 | 3404-73-7 | −78.5 ± 1.7 | −81.5 | −3.0 | |
| 3,3-dimethylbut-1-ene | C6H12 | 558-37-2 | −59.7 ± 2.0 | −61.7 | −2.0 | |
| Alk-n-enes, (E) | ||||||
| (E)-but-2-ene | C4H8 | 624-64-6 | −11.2 ± 0.5 | −11.4 | −0.2 | |
| (E)-pent-2-ene | C5H10 | 646-04-8 | −33.1 ± 1.3 | −31.3 | 1.8 | |
| (E)-hex-2-ene | C6H12 | 4050-45-7 | −51.7 ± 2.0 | −53.0 | −1.3 | |
| (E)-hex-3-ene | C6H12 | 13269-52-8 | −49.3 ± 1.1 | −51.4 | −2.1 | |
| (E)-4-methylpent-2-ene | C6H12 | 674-76-0 | −60.1 ± 1.5 | −60.1 | 0.0 | |
| Alk-n-enes, (Z) | ||||||
| (Z)-but-2-ene | C4H8 | 590-18-1 | −7.3 ± 0.5 | −6.5 | 0.8 | |
| (Z)-pent-2-ene | C5H10 | 627-20-3 | −28.0 ± 0.8 | −26.0 | 2.0 | |
| (Z)-hex-2-ene | C6H12 | 7688-21-3 | −47.9 ± 2.0 | −47.8 | 0.1 | |
| (Z)-hex-3-ene | C6H12 | 7642-09-3 | −46.9 ± 2.0 | −45.7 | 1.2 | |
| Isoalkenes | ||||||
| 2-methylprop-1-ene | C4H8 | 115-11-7 | −17.5 ± 0.5 | −17.3 | 0.2 | |
| 2-methylbut-1-ene | C5H10 | 563-46-2 | −35.1 ± 0.8 | −35.1 | 0.0 | |
| 2-methylpent-1-ene | C6H12 | 763-29-1 | −58.0 ± 1.1 | −56.7 | 1.3 | |
| 2-methylbut-2-ene | C5H10 | 513-35-9 | −41.5 ± 0.9 | −40.8 | 0.7 | |
| Alk-1-ynes | ||||||
| ethyne | C2H2 | 74-86-2 | 226.7 ± 0.8 | 227.5 | 0.8 | |
| propyne | C3H4 | 74-99-7 | 185.4 ± 0.9 | 184.9 | −0.5 | |
| but-1-yne | C4H6 | 107-00-6 | 166.1 ± 2.1 | 166.4 | 0.3 | |
| pent-1-yne | C5H8 | 627-19-0 | 144.3 ± 2.1 | 144.7 | 0.4 | |
| Alk-1-ynes, Branched | ||||||
| isopentyne | C5H8 | 598-23-2 | 136.4 ± 2.1 | 139.2 | 2.8 | |
| tert-hexyne | C6H10 | 917-92-0 | 106.1 | 106.1 | 0.0 | |
| Alk-2-ynes | ||||||
| but-2-yne | C4H6 | 503-17-3 | 148.0 ± 1.5 | 147.9 | −0.1 | |
| pent-2-yne | C5H8 | 627-21-4 | 128.9 ± 2.1 | 128.9 | 0.0 | |
| Alkadienes | ||||||
| penta-1,4-diene | C5H8 | 591-93-5 | 106.3 ± 1.3 | 102.8 | −3.5 | |
| hexa-1,5-diene | C6H10 | 592-42-7 | 85.0 ± 2.0 | 81.8 | −3.2 | |
| Alkadienes, Conjugated | ||||||
| (E)-buta-1,3-diene | C4H6 | 106-99-0 | 108.8 ± 0.8 | 108.8 | 0.0 | |
| (E)-penta-1,3-diene | C5H8 | 2004-70-8 | 75.8 ± 0.7 | 76.8 | 1.0 | |
| Alkatrienes | ||||||
| (E)-hexa-1,3,5-triene | C6H8 | 821-07-8 | 168.0 ± 3.0 | 163.9 | −4.1 | |
| (Z)-hexa-1,3,5-triene | C6H8 | 2612-46-6 | 172.0 ± 3.0 | 170.1 | −1.9 | |
| Alkenynes | ||||||
| 1-buten-3-yne | C4H4 | 689-97-4 | 295.0 ± 3.0 | 288.0 | −7.0 | |
| (E)-pent-3-en-1-yne | C5H6 | 2004-69-5 | 259.0 ± 3.0 | 252.7 | −6.3 | |
| (Z)-pent-3-en-1-yne | C5H6 | 1574-40-9 | 258.0 ± 3.0 | 254.4 | −3.6 | |
| 2-methylbut-1-en-3-yne | C5H6 | 78-80-8 | 259.0 ± 1.3 | 253.1 | −5.9 | |
| Alkadiynes | ||||||
| butadiyne | C4H2 | 460-12-8 | 464.0 | 454.5 | −9.5 | |
| 1,5-hexadiyne | C6H6 | 628-16-0 | 416.0 | 412.3 | −3.7 | |
| Alkendiynes | ||||||
| (E)-hex-3-en-1,5-diyne | C6H4 | 16668-68-1 | 538.0 ± 3.0 | 524.8 | −13.2 | |
| (Z)-hex-3-en-1,5-diyne | C6H4 | 16668-67-0 | 541.8 ± 3.0 | 525.6 | −16.2 | |
The G3MP2B3 method was found to be nearly as accurate as the G3B3 method for computing energies for saturated hydrocarbons (unsigned differences of less than 0.3 kJ/mol). For unsaturated species, however, a systematic deviation was found that correlated well with the number of unsaturated sites in the molecule. Slight differences in the correlations (close to being statistically insignificant) were observed for different for different types of unsaturated sites (i.e., alkenes, alkynes, aromatics). Enthalpies of formation for unsaturated species computed using the G3MP2B3 method were consistently lower than those computed using the G3B3 method, on the order of 2 kJ/mol per unsaturated bond.
Aliphatic Hydrocarbons – G3MP2B3 (Corrected)
Enthalpies of formation for the aliphatic hydrocarbons (alkanes, alkenes, and alkynes) were computed using both the G3MP2B3 and G3B3 methods and compared to experimental values. These data are presented in Table 18 for about 60 compounds. Only the G3MP2B3 results are presented here because the differences between the G3MP2B3 and G3B3 values (after corrections) were small (less than 1.0 kJ/mol). This systematic study was used to develop corrections for unsaturated bonds in aliphatic hydrocarbons that could be used in making corrections to alkenyl- and alkynyl-substituted aromatic hydrocarbons.
It is observed that the G3MP2B3 enthalpies of formation are consistently lower than the experimental values. Using linear regression, we found best fit values for coefficients (in kJ/mol) to correct the G3MP2B3 values of 0.69nH, −1.43nC, −0.32nCd, and −0.50nCt, where nH, nC, nCd, and nCt are the number of hydrogen atoms, total number of carbon atoms, number of sp2-hybridized carbon atoms (in double bonds), and number of sp-hybridized carbon atoms (in triple bonds), respectively. Equivalently, one could use corrections of +2.02 kJ/mol and +2.38 kJ/mol per double and triple bond, respectively.
It was found that the enthalpies of formation computed using the G3MP2B3 method after applying systematic corrections agreed well with the experimental values. The average uncertainty in the (quoted) experimental enthalpies of formation for this set of molecules is about 1.3 kJ/mol. The standard deviation of the differences between the corrected G3MP2B3 and the experimental values is 2.1 kJ/mol for the entire set and drops to 1.5 kJ/mol if one excludes those molecules with more than one unsaturated bond.
Inspection of the differences by the class of compound, one can see that the residuals for the alkadienes, alkadiynes, and alkenynes are consistently negative. We note that many of the experimental values were determined from heats of hydrogenation in the liquid phase, and to compute a gas-phase enthalpy of formation it was assumed that heats of hydrogenation in the liquid and gas phases were the same (i.e., identical enthalpies of vaporization for the saturated and unsaturated compounds). This could introduce a small systematic uncertainty—likely less than 1.0–1.5 kJ/mol. We note that the computed (corrected) values are lower than the experimental values and little change is observed whether the G3MP2B3 or G3B3 method is used. This suggests that that there may be systematic uncertainties in the heat of hydrogenation measurements (or systematic defects in the G3 methods), because any higher level calculations would likely only lower than computed enthalpy of formation.
Aromatic Hydrocarbons – G3MP2B3 (Corrected)
In Table 17, we present a list of about 150 aromatic hydrocarbon compounds where we have computed enthalpies of formation using the G3MP2B3 method (and after applying systematic corrections). Using linear regression, we found best fit values for coefficients (in kJ/mol) to correct the G3MP2B3 values of 1.25nCbH, 0.93nCf (or nCp, nCg), 1.00nCd, and 1.20nCt. Here, CbH denotes peripheral aromatic carbon atoms (terminated by hydrogen atoms); Cf, Cp, and Cg denote ortho-fused, ortho- and peri-fused, and fused aromatic carbons contained as part of a non-six-membered ring, respectively. Cd and Ct denote aliphatic carbon atoms part of double and triple bonds, respectively.
We compared the corrected G3MP2B3 values with experimental values. The average (reported) uncertainty in the experimental values was about 2.4–2.8 kJ/mol with most of the uncertainties in the range 1–6 kJ/mol. We found that there were about 60 compounds where the corrected G3MP2B3 values were within about 2.0 kJ/mol (standard deviation) of the experimental values and another approximately 10 compounds that were within about 6 kJ/mol (3 standard deviations) of the experimental values. This can be considered excellent agreement. There were about another 10 compounds where there were large differences between the computed and experimental values. In these cases, there maybe an error in the computed values or in the experimental values.
In summary, we find that the G3MP2B3 method (after applying systematic corrections for the hybridization of each carbon site) can be used to produce enthalpies of formation for both aliphatic and aromatic hydrocarbons, including PAHs and substituted-aromatic hydrocarbons.
DISCUSSION
The results presented in the previous section demonstrate that quantum chemistry can produce good data on the enthalpy of formation for PAH molecules, particularly when empirical corrections such as the group based model are used. Given that the reliability of the present methodology has been established, various uses of the data generated by this methodology may be considered. Three such uses are presented below: the value of predictions of enthalpies of formation where none are available, the limitations of group additivity, and use as a screening tool.
Prediction
Predicted values (for which no experimental determinations are known to be available) are given in Tables S2, S3, and S4. These values may be regarded as the best available values until such time as they are supplanted by experimental data, more accurate calculations, or improved models. As the methodology developed in this article is straightforward and economical to apply, this methodology may be used to produce predictions of enthalpies of formation for any number of PAHs with reasonable accuracy. If and when additional experimental data become available, or improvements are made to some portion of the model, the present data set can be updated to yield more accurate predictions. These predicted values may be used for creating thermodynamic tables for PAHs and in modeling studies. Though it is impossible to place uncertainties on the predicted enthalpies of formation derived in this article, the mean unsigned deviation and the root-mean-square deviation may serve as guides in the assessment of the data quality.
It is unsurprising that the G3MP2B3 results are better than the B3LYP results; the G3 model chemistries have been carefully tuned to produce good thermochemical data. However, when the group additivity based correction is applied to the extrapolated B3LYP results, the quality of these results is significantly improved. When the data in Tables 17 and 18 are compared to experimental values, the mean unsigned deviation (MUD) and root-mean-square deviation (RMSD) are 3.8 and 8.0 kJ/mol, respectively. If the molecules with larger errors in Table 17 (that is the last 21 values, or those in the last four sections) are removed, these values decrease to 1.9 and 3.0 kJ/mol, respectively. For the extrapolated and corrected B3LYP results, these values are 6.8 and 18.3 kJ/mol, respectively (4.5 and 6.2 kJ/mol, respectively, when the same data are removed). If nonbenzenoid/non-PAH data are considered (Table 18), values of the MUD and RMSD are 2.2 and 3.7 kJ/mol, respectively, for the G3MP2B3 data versus 4.6 and 6.2 kJ/mol for the B3LYP results. Similarly, for the benzenoid and PAH compounds (Table 17) with the compounds with larger errors removed as above, values of the MUD and RMSD are 1.7 and 2.1 kJ/mol, respectively, for the G3MP2B3 results and 4.4 and 5.7 kJ/mol, respectively, for the B3LYP results. It is seen then that the deviations of the B3LYP set are approximately twice that of the G3MP2B3 set, a very good result considering that the B3LYP results require significantly less computer time and resources.
Group Additivity Based Empirical Corrections
Thermochemical data for a number of PAH species (and other hydrocarbons) have been estimated using group additivity methods (originally developed by Benson75 and Cohen76,77). For PAHs, an additivity approximation for neighboring groups may not be correct, because resonance stabilization energies are longer range, and ring strains cannot be readily predicted. Thus, such estimates may have high uncertainties, coupled with the fact that thermochemical data for reference species used to develop the groups may be uncertain. For many PAHs, there are significant uncertainties, on the order of 5–15) kJ/mol, in the condensed-phase enthalpies of formation and enthalpies of sublimation used to derived gas-phase enthalpies of formation.
The combination of quantum chemistry methods with group additivity corrections is particularly powerful, as seen in the present work. Many group additivity methods lack terms to account for important chemical effects such as the difference in (E) and (Z) isomers, the difference in ortho-, meta-, and para- substitution on benzene rings, and steric effects. These effects (and others) are all included in the quantum chemistry calculation, obviating the need for specific group additivity terms to account for specific chemistries. If the overall error can be ascribed in whole or in part to systematic deviations of specific chemical groups, then the group additivity model is appropriate for correcting the quantum chemistry results. The quality of the underlying quantum chemistry results will ultimately dictate the limits of the correction. In general, calculations made with larger basis sets, and more accurate or inclusive correlation methods will have smaller and more regular deviations from the correct experimental values. Such high-quality quantum chemistry results will typically result in a better fitting of the correction terms and thus in a more accurate set of results. This was seen in the present study wherein the G3MP2B3 results were corrected with a simpler model (fewer terms) and yielded smaller deviations from experiment.
Ultimately, the procedures used in the present work are limited by the uncertainty of the experimental data used to compute the model corrections. This points to the need for improved data for certain key compounds, and to the need for new measurements on PAH compounds.
Empirically-Corrected Quantum Calculations for Screening
Given the reliability that has been established for the present results, the use of these results as a check on the current experimental value is suggested. During the fitting of the correction term, it became apparent that including certain PAH compounds (e.g., azulene, naphthacene, pyracyclene, and triquinacene) in the fits reduced the overall quality of the results. (Note that an enthalpy of formation for naphthacene of 73.9 kcal/mol has recently been given by Karton and Martin using the W1–F12 ab initio computational thermochemistry method.165) This result strongly suggests that some or all of the compounds may have substantial errors beyond their uncertainty limits, and a reevaluation of the reported enthalpies of formation is warranted. Such analysis might also be applied to new predictions based on group additivity or quantum chemistry techniques.
Molecules with Large Deviations
Computed enthalpies of formation for four sets of molecules are shown in Table 19. The first set of molecules have computed enthalpies of formation (after corrections) that differ significantly from the experimental values, but the lower level B3LYP and the higher level G3MP2B3 agree very well. This suggests (but does not prove) that the experimental values may be in error. Of particular concern are naphthacene, pyracene, and pyracyclene, which are highly prototypical molecules. Naphthacene is a simple molecule in the important basic series benzene, naphthalene, anthracene, and naphthacene. Pyracene and pyracyclene are representative of “ace” (ethylene) bridge aromatic hydrocarbons.
Table 19.
Comparison of Enthalpies of Formation at 298 K (kJ/mol) Computed Using the Corrected G3MP2B3 and Corrected B3LYP Methods
| molecule | formula | CAS registry no. | expt | G3MP2B3 (corr) | dev | B3LYP (corr) | dev |
|---|---|---|---|---|---|---|---|
| Set 1 | |||||||
| naphthacene | C18H12 | 92-24-0 | 340.7(±3.9) | 316.2 | −24.5 | 310.5 | −30.2 |
| benz[a]anthracene | C18H12 | 56-55-3 | 290.3(±6.0) | 279.8 | −10.5 | 277.1 | −13.2 |
| benzo[b]triphenylene | C22H14 | 215-58-7 | 331.0(±11) | 348.0 | 17.0 | ||
| pyracene | C14H12 | 567-79-3 | 174.1(±5.1) | 190.2 | 16.1 | 191.4 | 17.3 |
| pyracyclene | C14H8 | 187-78-0 | 408.6(±5.0) | 426.7 | 18.1 | 438.2 | 29.6 |
| Set 2 | |||||||
| 9-methylfluorene | C14H12 | 2523-37-7 | 148.0(±1.1) | 155.0 | 7.0 | 150.9 | 2.9 |
| (Z)-1,2-diphenylethene | C14H12 | 645-49-8 | 245.9(±1.3) | 252.9 | 7.0 | 245.9 | 3.8 |
| 4-phenyltoluene | C13H12 | 644-08-6 | 138.2(±2.9) | 145.9 | 7.7 | 142.9 | 4.7 |
| diphenylethyne | C14H10 | 501-65-5 | 385.0(±2.7) | 405.1 | 20.1 | 389.0 | 4.0 |
| propyn-1-ylbenzene | C9H8 | 673-32-5 | 268.2(±2.2) | 277.2 | 9.0 | 269.4 | 1.2 |
| ethynylbenzene | C8H6 | 536-74-3 | 306.6(±1.7) | 318.2 | 11.6 | 314.1 | 7.5 |
| Set 3 | |||||||
| 3-phenyltoluene | C13H12 | 643-93-6 | 152.5(±8.0) | 145.3 | −7.2 | 143.3 | −9.2 |
| (E)-1,2-diphenylethene | C14H12 | 103-30-0 | 233.7(±2.0) | 239.4 | 5.7 | 233.7 | −5.1 |
| Set 4 | |||||||
| 1,2-diphenylbenzene | C18H14 | 84-15-1 | 282.8(±3.2) | 273.2 | −9.6 | 290.5 | 7.7 |
| corannulene | C20H10 | 5821-51-2 | 458.7(±9.1) | 427.0 | −31.7 | 498.5 | 39.8 |
The second set of molecules have computed enthalpies of formation (after corrections) from the lower level B3LYP method that agree well with the experimental values (deviations on the order of 4 kJ/mol), but the deviations from experiment for the higher level (and corrected) G3MP2B3 method are somewhat larger (on the order of 10 kJ/mol). This good agreement (for the B3LYP) method suggests that the experimental values are good but raises the question why the higher level G3MP2B3 method has higher deviations particular the alkynyl-substituted compounds (ethynyl- and propynylbenzene).
The third set of molecules have computed enthalpies of formation (after corrections) that differ from the experimental values by a relatively modest amount (6–8 kJ/mol) for both computational methods, but the experimental uncertainties and the deviations suggested that experimental (or computational) values with tighter uncertainties would be in order, and worthy of further experimental or computational studies. Of particular concern are fluoranthene and coronene, which are prototypical molecules. Coronene is a good reference for large PAHs and fluoranthene for five-membered ring fused systems.
The fourth set of molecules have substantial deviations between the computational methods and the experimental values, and further investigations are warranted. Of particular interest is corannulene, a highly strained “bowl-like” molecule, where there is a large difference between the computed methods.
In Table 19, there are a number of molecules with significant differences between the computed and experimentally derived enthalpies of formation. We will discuss several of them here. We believe in some cases that the experimental values may be in error whereas in others the computations may be in error.
There is only one reliable measurement for the enthalpy of formation of naphthacene. This value is about 25 kJ/mol higher than the G3MP2B3 computed value. The computed values for the series benzene, naphthalene, anthracene, and naphthacene appear to follow a simple trend, an incremental value of about 77 kJ/mol per each additional benzo ring with the difference in predicted and computed on the order of about 4–6 kJ/mol. The experimental values also follow this trend, except the value for naphthalene is about 25–30 kJ/mol higher. The consistency between the calculated and the “group additivity” incremental approach suggests that the experimental enthalpy of formation for naphthacene may be in error. There is no reason why an incremental approach should not be able to roughly predict the trend in this homologous series.
There is only one measurement for the enthalpy of formation of benz[a]anthracene and it is about 11 kJ/mol higher than the G3MP2B3 value. There are a number of enthalpies of combustion for similar molecules by Magnus and co-workers.166 One of them is a determination for naphthacene that differs from the other measurement by about 50–60 kJ/mol. In addition, they also measured enthalpies of sublimation for a number of PAHs including benzo[a]anthracene, phenanthrene, triphenylene, and naphthacene that are lower than values measured by other groups by about 8–14 kJ/mol. This is on the order of the difference observed here.
There have been several determinations of the enthalpy of formation of pyracene and pyracyclene. These values are consistently lower than the computed G3MP2B3 value by about 15–20 kJ/mol. For these molecules, we believe that the computations may be in error, in part because the computed energies are higher. The structures of these molecules are influenced by the strain due to five-membered rings in the plane that would prefer to have asymmetric (non-D2h symmetry), puckered, or skewed structures. However, loss of planarity and symmetry would destroy the energy gained by aromatic/delocalized electrons. It is possible that the structures we computed with low-level B3LYP/6-31G(d) calculations may not reflect the true structures. Furthermore, these molecules have a significant number of low frequency and anharmonic modes that are likely coupled to other modes in the molecules.167 The difference between computed and experimental enthalpies of formation could simply be that our simple zero point energy (ZPE) correction using computed harmonic frequencies may be incorrect; a 15 kJ/mol difference corresponds to about 1300 cm−1.
The G3MP2B3 and B3LYP computed enthalpies of formation for corannulene differ substantially form the available experimentally derived values (which appear to be very reliable). Given the high strain in this molecule, it is very possible that the computed geometries may not reflect the true structures and vibrational partition functions.
There are several other molecules in Table 19 where there are some differences between the computed and experimental values. One thing to note is that several of them involve aromatic structures modified by sp2 (triple bond) substitution. Given the conjugation of the unsaturation with the aromatic ring, there may be unusual effects that could be reflected in both the computations and the experimental determinations. Interestingly, the B3LYP computed values seem to be more consistent with the experimental values than the higher-level G3MP2B3 calculations.
Without a detailed, systematic study of these molecules to explore the reasons where there are significant differences, we cannot draw any conclusion and simply propose the possible rational as given above.
Uncertainties in G3MP2B3 (Corrected) Enthalpies of Formation
We now provide a short discussion of the overall uncertainties in the G3MP2B3 (empirically corrected) enthalpies of formation of the PAHs, paying attention to several particular prototypical molecules.
The (corrected) enthalpy of formation for fluoranthene of 282–283 kJ/mol agrees (within about 1 kJ/mol) with the recommended value by Monte et al.168 but differs from that of Roux et al.2 by about 9 kJ/mol (see experimental values in Table 13). Given the uncertainty in the experimental values (about 4 kJ/mol) and the uncertainty in the computed value (estimated 6–8 kJ/mol), overall the agreement between experimental and computed values is good.
We computed G3MP2B3 (corrected) enthalpies of formation for ethynylbenzene and propyn-1-ylbenzene of 308–318 and 267–277 kJ/mol depending on the empirical fit parameters employed, respectively. These values agree within about 2–10 kJ/mol of the experimental values of 306.6 ± 1.7 and 268.2 ± 2.3 kJ/mol, respectively. Consequently, we consider that there is fair-to-good agreement between experimental and computed values. We computed a G3MP2B3 (corrected) enthalpy of formation of about 394–405 kJ/mol for diphenylethyne which is about 10–20 kJ/mol higher (depending on the empirical fit parameters employed) than the experimental value of 385.0 ± 2.7 kJ/mol derived from the heat of hydrogenation measurements by Davis et al.169 and about 0–10 kJ/mol lower than the 404 ± 5 kJ/mol derived from the measurements by Flitcroft and Skinner170 (see experimental values in Table 13). The gas-phase enthalpy of formation for this molecule was derived from the heat of hydrogenation of the liquid and solid phases in the Davis et al.169 and Flitcroft and Skinner170 experiments, respectively, and this procedure using the condensed-phase heats of hydrogenation introduces some uncertainties. The substantial difference between these two experimental values suggests that both the experimental and computational values should be reexamined.
We find that the G3MP2B3 (corrected) enthalpy of formation for benzyne is dependent upon how one treats the unsaturation in the molecule (modified aromatic or a cyclic compound with triple and double bonds). We find computed values that differ from the experimental value of 446 ± 13 kJ/mol by about 2–10 kJ/mol. Given the uncertainty in the experimental value, the agreement is good.
Overall, a good measure of the minimum uncertainty in the computed enthalpies of formation for the PAHs can be estimated from the small (three or four fused rings) PAHs anthracene, phenanthrene, chrysene, benzo[c]phenanthrene, and benzo[a]anthracene. The differences between the calculated and experimental values are −0.3 ± 3.7, +6.1 ± 3.5, +2.5 ± 2.8, −2.4 ± 9.1, and −8.8 ± 6.0 kJ/mol, respectively, where the uncertainties (±) given here are the experimental values. (See Tables 13 and 17 for the specific experimental and computed values.) These data suggest an uncertainty (2σ ≈ 95% coverage) in the enthalpies of formation of the small PAHs of about 3–4 kJ/mol, largely dominated by the uncertainties in the experimental values. Our estimate for the uncertainties (2σ ≈ 95% coverage) in the larger PAHs (e.g., coronene) rises to about 6–9 kJ/mol, due to the combined uncertainties in the experimental values and the fitted empirical corrections to the G3MP2B3 values.
CONCLUSION
In this article, an approach for computing enthalpies of formation for PAH and related compounds from relatively inexpensive ab initio calculations and using a chemical group based correction scheme has been presented. The values computed in this manner are compared to the available experimental data, to the results of higher-level G3B3 and G3MP2B3 calculations, and to other empirical models. The application of an energy extrapolation scheme significantly improved the quality of the results, and the application of a group based correction scheme produces results that are in good agreement with the available experimental data. This extrapolation–correction model is then used to predict enthalpies of formation for 810 compounds for which no experimental data are known to be available (or for which the experimental data was deemed unreliable). The G3B3 and G3MP2B3 methods are interesting in their own right, and the present study clearly establishes their reliability for predicting the thermochemistry of PAHs, particularly after a simple correction is applied. This collection of data represent perhaps the best-known values of the enthalpies of formation for PAH compounds (including substituted PAHs). The extrapolation–correction model is generally applicable to other PAHs and substituted PAH molecules and should be valuable for predicting enthalpies of formation for such compounds with reasonable accuracy.
Supplementary Material
Footnotes
Notes
authors declare no competing financial interest.
Certain commercial equipment, instruments, or materials are identified in this paper to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement by the National Institute of Standards and Technology, nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpca.5b07908.
References
- 1.Moss GP. Nomenclature of Fused and Bridged Fused Ring Systems (IUPAC Recommendations 1998) Pure Appl Chem. 1998;70:143–216. [Google Scholar]
- 2.Roux MV, Temprado M, Chickos JS, Nagano Y. Critically Evaluated Thermochemical Properties of Polycyclic Aromatic Hydrocarbons. J Phys Chem Ref Data. 2008;37:1855–1996. [Google Scholar]
- 3.Wang H, Frenklach M. A Detailed Kinetic Modeling Study of Aromatics Formation in Laminar Premixed Acetylene and Ethylene Flames. Combust Flame. 1997;110:173–221. [Google Scholar]
- 4.Richter H, Mazyar OA, Sumathi R, Green WH, Howard JB, Bozzelli J. Detailed Kinetic Study of the Growth of Small Polycyclic Aromatic Hydrocarbons. 1. 1-Naphthyl Plus Ethyne. J Phys Chem A. 2001;105:1561–1573. [Google Scholar]
- 5.McEnally CS, Pfefferle LD, Atakan B, Kohse-Hoeinghaus K. Studies of Aromatic Hydrocarbon Formation Mechanisms in Flames: Progress Towards Closing the Fuel Gap. Prog Energy Combust Sci. 2006;32:247–294. [Google Scholar]
- 6.Slavinskaya NA, Riedel U, Dworkin SB, Thomson M. Detailed Numerical Modeling of PAH Formation and Growth in Non-Premixed Ethylene and Ethane Flames. Combust Flame. 2012;159:979–995. [Google Scholar]
- 7.Wang H. Formation of Nascent Soot and Other Condensed-Phase Materials in Flames. Proc Combust Inst. 2011;33:41–67. [Google Scholar]
- 8.Elvati P, Violi A. Thermodynamics of Poly-Aromatic Hydrocarbon Clustering and the Effects of Substituted Aliphatic Chains. Proc Combust Inst. 2013;34:1837–1843. [Google Scholar]
- 9.NIST Polycyclic Aromatic Hydrocarbon Structure Index. 2013 http://pah.nist.gov.
- 10.Pariser R, Parr RG. A Semi-Empirical Theory of the Electronic Spectra and Electronic Structure of Complex Unsaturated Molecules. J Chem Phys. 1953;21:466–471. [Google Scholar]
- 11.Pople JA. Electron Interaction in Unsaturated Molecules. Trans Faraday Soc. 1953;49:1375–1385. [Google Scholar]
- 12.Chung ALH, Dewar MJS. Ground States of Conjugated Molecules. I. Semi-Empirical SCF MO Treatment and Its Application to Aromatic Hydrocarbons. J Chem Phys. 1965;42:756–766. doi: 10.1063/1.1696003. [DOI] [PubMed] [Google Scholar]
- 13.Hückel E. Quantum-Theoretical Contributions to the Benzene Problem. I. The Electron Configuration of Benzene and Related Compounds. Eur Phys J A. 1931;70:204–286. [Google Scholar]
- 14.Frankland E. On a New Series of Organic Bodies Containing Metals. Philos Trans R Soc London. 1852;142:417–444. [Google Scholar]
- 15.Kekulè A. Über Die Constitution Und Die Metamorphosen Der Chemischen Verbindungen Und über Die Chemische Natur Des Kohlenstoffs. Annal Chem Pharm. 1858;106:129–159. [Google Scholar]
- 16.Kekulè A. Sur La Constitution Des Substances Aromatiques. Bull Soc Chim Fr. 1865;3:98–111. [Google Scholar]
- 17.Schleyer PvR. Introduction: Delocalization – Pi and Sigma. Chem Rev. 2005;105:3433–3435. [Google Scholar]
- 18.Poater J, Duran M, Solà M, Silvi B. Theoretical Evaluation of Electron Delocalization in Aromatic Molecules by Means of Atoms in Molecules (AIM) and Electron Localization Function (ELF) Topological Approaches. Chem Rev. 2005;105:3911–3947. doi: 10.1021/cr030085x. [DOI] [PubMed] [Google Scholar]
- 19.Cyrasnki MK. Energetic Aspects of Cyclic Pi-Electron Delocalization: Evaluation of the Methods of Estimating Aromatic Stabilization Energies. Chem Rev. 2005;105:3773–3811. doi: 10.1021/cr0300845. [DOI] [PubMed] [Google Scholar]
- 20.Merino G, Vela A, Heine T. Description of Electron Delocalization Via the Analysis of Molecular Fields. Chem Rev. 2005;105:3812–3841. doi: 10.1021/cr030086p. [DOI] [PubMed] [Google Scholar]
- 21.Aihara J. Resonance Energies of Benzenoid Hydrocarbons. J Am Chem Soc. 1977;99:2048–2053. [Google Scholar]
- 22.Schleyer PvR. Introduction: Aromaticity. Chem Rev. 2001;101:1115–1118. doi: 10.1021/cr0103221. [DOI] [PubMed] [Google Scholar]
- 23.Schleyer PvR, Manoharan M, Jiao H, Stahl F. The Acenes: Is There a Relationship Between Aromatic Stabilization and Reactivity? Org Lett. 2001;3:3643–3646. doi: 10.1021/ol016553b. [DOI] [PubMed] [Google Scholar]
- 24.Matta CF, Hernandez-Trujillo J. Bonding in Polycyclic Aromatic Hydrocarbons in Terms of the Electron Density and of Electron Delocalization. J Phys Chem A. 2003;107:7496–7504. [Google Scholar]
- 25.Cappel D, Tüllman S, Krapp A, Frenking G. Direct Estimate of the Conjugative and Hyperconjugative Stabilization in Diynes, Dienes, and Related Compounds. Angew Chem, Int Ed. 2005;44:3617–3620. doi: 10.1002/anie.200500452. [DOI] [PubMed] [Google Scholar]
- 26.Polansky O, Derflinger G. Clar Theory of Local Benzene-like Regions in Condensed Aromatic Compounds. Int J Quantum Chem. 1967;1:379–401. [Google Scholar]
- 27.Kistiakowsky GB, Romeyn HJr, Ruhoff JR, Smith HA, Vaughan WE. Heats of Organic Reactions. I. The Apparatus and the Heat of Hydrogenation of Ethylene. J Am Chem Soc. 1935;57:65. [Google Scholar]
- 28.Kistiakowsky GB, Ruhoff JR, Smith HA, Vaughan WE. Heats of Organic Reactions. IV. Hydrogenation of Some Dienes and of Benzene. J Am Chem Soc. 1936;58:146–153. [Google Scholar]
- 29.Conant JB, Kistiakowsky GB. Energy Changes Involved in the Addition Reactions of Unsaturated Hydrocarbons. Chem Rev. 1937;20:181–194. [Google Scholar]
- 30.Turner RB, Nettleton DE, Perelman M. Heats of Hydrogenation. VI. Heats of Hydrogenation of Some Substituted Ethylenes. J Am Chem Soc. 1958;80:1430–1433. [Google Scholar]
- 31.Turner RB, Goebel P, Mallon BJ, Doering WVE, Coburn JF, Pomerant M. Heats of Hydrogenation. 8. Compounds with 3- and 4-Membered Rings. J Am Chem Soc. 1968;90:4315–4322. [Google Scholar]
- 32.Turner RB, Mallon BJ, Tichy M, Doering WVE, Roth WR, Schroder G. Heats of Hydrogenation. 10. Conjugative Interaction in Cyclic Dienes and Trienes. J Am Chem Soc. 1973;95:8605–8610. [Google Scholar]
- 33.Roth WR, Adamczak O, Breuckmann R, Lennartz HW, Boese R. Resonance Energy Calculation - the MM2ERW Force-Field. Chem Ber. 1991;124:2499–2521. [Google Scholar]
- 34.Roth WR, Klarner FG, Lennartz HW. Heats of Hydrogenation 2. Heats of Hydrogenation of Bicyclo[2.1.0]pent-2-Ene, and Antiaromatic System. Chem Ber. 1992;125:217–224. [Google Scholar]
- 35.Rogers DW, Skanupong S. Heats of Hydrogenation of Sixteen Terminal Monoolefins. The Alternating Effect. J Phys Chem. 1974;78:2569–2572. [Google Scholar]
- 36.Rogers DW, Crooks EL. Enthalpies of Hydrogenation of the Isomers of N-Hexene. J Chem Thermodyn. 1983;15:1087–1092. [Google Scholar]
- 37.Fang W, Rogers DW. Enthalpy of Hydrogenation of the Hexadienes and Cis- and Trans-1,3,5-Hexatriene. J Org Chem. 1992;57:2294–2297. [Google Scholar]
- 38.Caldwell RA, Liebman JF, Rogers DW, Unett DJ. Enthalpies of Hydrogenation and of Formation of 1-Phenyl Cycloalkenes. J Mol Struct. 1997;413:575–578. [Google Scholar]
- 39.Jensen JL. Heats of Hydrogenation: A Brief Summary. Prog Phys Org Chem. 1976;12:189–228. [Google Scholar]
- 40.Pedley JB, Naylor RD, Kirby SP. Thermochemical Data of Organic Compounds. 2nd. Chapman & Hall; London: 1986. [Google Scholar]
- 41.Cox JD, Pilcher G. Thermochemistry of Organic and Organometallic Compounds. Academic Press; New York: p. 1970. [Google Scholar]
- 42.Linstrom PJ, Mallard WG. NIST Chemistry WebBook NIST Standard Reference Database Number 69. National Institute of Standards and Technology; Gaithersburg, MD: Jun, 2005. http://webbook.nist.gov. [Google Scholar]
- 43.Ruscic B, Pinzon RE, Morton ML, von Laszevski G, Bittner SJ, Nijsure SG, Amin KA, Minkoff M, Wagner AF. Introduction to Active Thermochemical Tables: Several ’key’ Enthalpies of Formation Revisited. J Phys Chem A. 2004;108:9979–9997. [Google Scholar]
- 44.Baulch DL, Bowman CT, Cox CJ, Cobos RA, Just T, Kerr JA, Pilling MJ, Stocker D, Troe J, Tsang W, et al. Evaluated Kinetic Data for Combustion Modeling: Supplement II. J Phys Chem Ref Data. 2005;34:757–1397. [Google Scholar]
- 45.Gurvich LV, Veyts IV, Alcock CB. Thermodynamic Properties of Individual Substances. 4th. Vol. 2 Hemisphere Publishing; New York; 1991. [Google Scholar]
- 46.Chase MW. J Phys Chem Ref Data, Monograph. Vol. 2. AIP; Melville, NY: 1998. NIST-JANAF Thermochemical Tables (Fourth Edition) p. 9. [Google Scholar]
- 47.Frenkel M, Hong X, editors. RCW TRC Thermodynamic Tables - Hydrocarbons. NIST; Gaithersburg, MD: 2000. (NSRDS-NIST Report 75–120). [Google Scholar]
- 48.Burcat A, Ruscic B. Third Millennium Ideal Gas and Condensed Phase Thermochemical Database for Combustion with Updates from Active Thermochemical Tables. Argonne National Laboratory; Argonne, IL: 2005. [Google Scholar]
- 49.Ruscic B. Unpublished Work Based on Active Thermochemical Tables As Cited in Burcat and Ruscic. 2005 [Google Scholar]
- 50.Slayden SW, Liebman JF. The Energetics of Aromatic Hydrocarbons: An Experimental Thermochemical Perspective. Chem Rev. 2001;101:1541–1566. doi: 10.1021/cr990324+. [DOI] [PubMed] [Google Scholar]
- 51.Dorofeeva OV, Gurvich LV, Jorish LS. Enthalpies of Sublimation of Organic and Organometallic Compounds 1910–2001. J Phys Chem Ref Data. 1986;15:437. [Google Scholar]
- 52.Dorofeeva OV, Moiseeva NF, Yungman VS, Novikov VP. Ideal Gas Thermodynamic Properties of Biphenyl. Thermochim Acta. 2001;374:7–11. [Google Scholar]
- 53.Karni M, Oref I, Burcat A. Ab Initio Calculations and Ideal Gas Thermodynamic Functions of Cyclopentadiene and Cyclopentadiene Derivatives. J Phys Chem Ref Data. 1991;20:665–683. [Google Scholar]
- 54.Chickos JS, Acree WE., Jr Enthalpies of Sublimation of Organic and Organometallic Compounds. 1910–2001. J Phys Chem Ref Data. 2002;31:537–698. [Google Scholar]
- 55.Chickos JS, Acree WE., Jr Enthalpies of Vaporization of Organic and Organometallic Compounds, 1880–2002. J Phys Chem Ref Data. 2003;32:519–878. [Google Scholar]
- 56.Chickos JS, Hesse DG, Liebman JF. A Group Additivity Approach for the Estimation of Heat Capacities of Organic Liquids and Solids at 298 K. Struct Chem. 1993;4:261–269. [Google Scholar]
- 57.Chickos JS, Acree WE, Jr, Liebman JF. Estimating Solid-Liquid Phase Change Enthalpies and Entropies. J Phys Chem Ref Data. 1999;28:1535–1673. [Google Scholar]
- 58.Sabbah R, Xu-Wu A, Chickos JS, Planas-Leitao ML, Roux MV, Torres LA. Reference Materials for Calorimetry and Differential Scanning Calorimetry. Thermochim Acta. 1999;331:93–204. [Google Scholar]
- 59.Majer V, Svoboda V. Enthalpies of Vaporization of Organic Compounds: A Critical Review and Data Compilation. Blackwell Scientific Publications; Oxford, U.K: 1985. [Google Scholar]
- 60.Lewis G, Randall M. Thermodynamics and the Free Energy of Chemical Substances. McGraw-Hill; New York: 1923. [Google Scholar]
- 61.Pitzer K, Brewer L. Thermodynamics. second. McGraw-Hill; New York: 1961. [Google Scholar]
- 62.Pedley JB. Computer Analysis of Thermochemical Data: CATCH Tables. Elsevier; Amsterdam: 1972. [Google Scholar]
- 63.Barnes K, Pedley JB, Woodbridge BA. New Approach to the Computer Processing of Thermochemical Quantities Illustrated by Data on C7 Bicyclic Hydrocarbons from the Previous Paper. J Chem Thermodyn. 1978;10:929–933. [Google Scholar]
- 64.Garvin D, Parker VB, Wagman DD, Evans WH. Proceedgins of the 5th International CODATa Conference. Pergamon Press; Oxford UK: 1976. pp. 567–575. [Google Scholar]
- 65.Garvin D, Parker VB, Wagman DD, Evans WH. NBSIR Report No 76–1147. NBS; Washington, DC: 1977. A Combined Least Sums and Least Squares Approach to the Evaluation of Thermodynamic Data Networks. [Google Scholar]
- 66.Wagman DD, Schumm RH, Parker V. NBSIR-77–1300. NBS; Washington, DC: 1977. A Computer Assisted Evaluation of the Thermochemical Data of the Compounds of Thorium. [Google Scholar]
- 67.Wagman DD, Evans WH, Parker VB, Schumm RH, Halow I, Bailey SM, Churney KL, Nuttall RL. The NBS Tables of Chemical Thermodynamic Properties. Selected Values for Inorganic and C1 and C2 Organic Substances in SI Units. J Phys Chem Ref Data. 1982;11(suppl 2) [Google Scholar]
- 68.Parker VB, Evans WH, Nuttall RL. The Thermochemical Measurements on Rubidium Compounds: A Comparison of Measured Values with Those Predicted by the NBS Tables of Chemical and Thermodynamic Properties. J Phys Chem Ref Data. 1987;16:7–60. [Google Scholar]
- 69.Cox JD, Drowart J, Hepler LG, Medvedev VA, Wagman DD, Evans WH, Gurvich LV, Pedley JB, Sunner S. CODATA Recommended Key Values for Thermodynamics, 1977 Report of the CODATA Task Group on Key Values for Thermodynamics, 1977. J Chem Thermodyn. 1978;10:903–906. [Google Scholar]
- 70.Goldberg RN, Tewari YB. Thermodynamic and Transport Properties of Carbohydrates and Their Monophosphates: The Pentoses and Hexoses. J Phys Chem Ref Data. 1989;18:809–880. [Google Scholar]
- 71.Miller SL, Smith-Magowan D. The Thermodynamics of the Krebs Cycle and Related Compounds. J Phys Chem Ref Data. 1990;19:1049–1073. [Google Scholar]
- 72.Akers DL, Goldberg RN. BioEqCalc: A Package for Performing Equilibrium Calculations on Biochemical Reactions. Mathematica J. 2001;8:86–113. [Google Scholar]
- 73.Benson SW, Buss JH. Additivity Rules for the Estimation of Molecular Properties. Thermodynamic Properties. J Chem Phys. 1958;29:546–572. [Google Scholar]
- 74.Benson SW, Cruickshank FR, Golden DM, Haugen GR, O’Neal HE, Shaw AS, Rodgers R, Walsh R. Additivity Rules for Estimation of Thermochemical Properties. Chem Rev. 1969;69:279–324. [Google Scholar]
- 75.Benson SW. Thermochemical Kinetics. Wiley; New York: 1976. [Google Scholar]
- 76.Cohen N, Benson S. Estimation of Heats of Formation of Organic Compounds by Additivity Methods. Chem Rev. 1993;93:2419–2438. [Google Scholar]
- 77.Cohen N. Revised Group Additivity Values for Enthalpies of Formation (at 298 K) of Carbon-Hydrogen and Carbon-Hydrogen-Oxygen Compounds. J Phys Chem Ref Data. 1996;25:1411–1481. [Google Scholar]
- 78.Stein SE, Golden DM, Benson SW. Predictive Scheme for Thermochemical Properties of Polycyclic Aromatic Hydrocarbons. J Phys Chem. 1977;81:314. [Google Scholar]
- 79.Stein SE, Fahr A. High-Temperature Stabilities of Hydrocarbons. J Phys Chem. 1985;89:3714. [Google Scholar]
- 80.Alberty RA, Reif AK. Standard Chemical Thermodynamic Properties of Polycyclic Aromatic Hydrocarbons and Their Isomer Groups I. Benzene Series. J Phys Chem Ref Data. 1988;17:241–253. [Google Scholar]
- 81.Armitage DA, Bird CW. Enthalpies of Formation of Fullerenes by the Group Additivity Method. Tetrahedron Lett. 1993;34:5811–5812. [Google Scholar]
- 82.Moiseeva NF, Dorofeeva OV, Jorish VS. Development of Benson Group Additivity Method for Estimation of Ideal Gas Thermodynamic Properties of Polycyclic Aromatic Hydrocarbons. Thermochim Acta. 1989;153:77–85. [Google Scholar]
- 83.Moiseeva NF, Dorofeeva OV. Group Additivity Scheme for Calculating the Chemical Thermodynamic Properties of Gaseous Polycyclic Aromatic Hydrocarbons Containing Five-Membered Rings. Thermochim Acta. 1990;168:179–186. [Google Scholar]
- 84.Herndon WC, Nowak PC, Connor DA, Lin P. Empirical Model Calculations for Thermodynamic and Structural Properties of Condensed Polycyclic Aromatic Hydrocarbons. J Am Chem Soc. 1992;114:41–47. [Google Scholar]
- 85.Wang H, Frenklach M. Enthalpies of Formation of Benzenoid Aromatic Molecules and Radicals. J Phys Chem. 1993;97:3867–3874. [Google Scholar]
- 86.Welsh WJ, Tong WD, Collantes ER, Chickos JS, Gagarin SG. Enthalpies of Sublimation and Formation of Polycyclic Aromatic Hydrocarbons (PAHs) Derived from Comparative Molecular Field Analysis (CoMFA): Application of Moment of Inertia for Molecular Alignment. Thermochim Acta. 1997;290:55–64. [Google Scholar]
- 87.Yu J, Sumathi R, Green WH., Jr Accurate and Efficient Method for Predicting Thermochemistry of Polycyclic Aromatic Hydrocarbons. J Am Chem Soc. 2004;126:12685–12700. doi: 10.1021/ja048333+. [DOI] [PubMed] [Google Scholar]
- 88.Allinger NL, Dodziuk H, Rogers DW, Naik SN. Heats of Hydrogenation and Formation of Some 5-Membered Ring Compounds by Molecular Mechanics Calculations and Direct Measurements. Tetrahedron. 1982;38:1593–1597. [Google Scholar]
- 89.Sivaramakrishnan R, Tranter RS, Brezinsky R. Ring Conserved Isodesmic Reactions: A New Method for Estimating the Heats of Formation of Aromatics and PAHs. J Phys Chem A. 2005;109:1621–1628. doi: 10.1021/jp045076m. [DOI] [PubMed] [Google Scholar]
- 90.Becke AD. A New Mixing of Hartree–Fock and Local Density-Functional Theories. J Chem Phys. 1993;98:1372–1377. [Google Scholar]
- 91.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys Rev B: Condens Matter Mater Phys. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 92.Sumathi R, Green WH., Jr Missing Thermochemical Groups for Large Unsaturated Hydrocarbons: Contrasting Predictions of G2 and CBS-Q. J Phys Chem A. 2002;106:11141–11149. [Google Scholar]
- 93.Curtiss LA, Raghavachari K, Trucks GW, Pople JA. Gaussian-2 Theory for Molecular Energies of 1st Row and 2nd Row Compounds. J Chem Phys. 1991;94:7221–7230. [Google Scholar]
- 94.Ochterski JW, Petersson GA, Montgomery JAJr. A Complete Basis Set Model Chemistry. V. Extensions to Six or More Heavy Atoms. J Chem Phys. 1996;104:2598. [Google Scholar]
- 95.Sabbe MK, Sayes M, Reyniers MF, Marin GB, Speybroeck VV, Waroquier M. Group Additive Values for the Gas-Phase Standard Enthalpy of Formation of Hydrocarbons and Hydrocarbon Radicals. J Phys Chem A. 2005;109:7466–7480. doi: 10.1021/jp050484r. [DOI] [PubMed] [Google Scholar]
- 96.Montgomery JA, Jr, Frisch MJ, Ochterski JW, Petersson GA. A Complete Basis Set Model Chemistry. VII. Use of the Minimum Population Localization Method. J Chem Phys. 2000;112:6532–6542. [Google Scholar]
- 97.Dewar MJS, Zoebisch EG, Healy EF, Stewart JJP. M1: A New General Purpose Quantum Mechanical Molecular Model. J Am Chem Soc. 1985;107:3902–3909. [Google Scholar]
- 98.Head-Gordon M, Pople JA, Frisch MJ. MP2 Energy Evaluation by Direct Methods. Chem Phys Lett. 1988;153:503–506. [Google Scholar]
- 99.Herndon WC, Connor DA, Lin PP. Structure Enthalpy Relationships in Polycyclic Cata-Condensed Aromatic Hydrocarbons. Pure Appl Chem. 1990;62:435–444. [Google Scholar]
- 100.Herndon WC, Biedermann PU, Agranat I. Molecular Structure Parameters and Predictions of Enthalpies of Formation for Catacondensed and Pericondensed Polycyclic Aromatic Hydrocarbons. J Org Chem. 1998;63:7445–7448. doi: 10.1021/jo981280s. [DOI] [PubMed] [Google Scholar]
- 101.Marsh ND, Wornat MJ. Polycyclic Aromatic Hydrocarbons with Five-Membered Rings: Distributions Within Isomer Families in Experiments and Computed Equilibria. J Phys Chem A. 2004;108:5399–5407. [Google Scholar]
- 102.Pope CJ, Howard JB. Thermochemical Properties of Curved PAH and Fullerenes. A Group Additivity Method Compared with MM3(92) and MOPAC Predictions. J Phys Chem. 1995;99:4306–4316. [Google Scholar]
- 103.Allinger NL, Yuh YH, Lii J-H. Molecular Mechanics. The MM3 Force Field for Hydrocarbons. 1. J Am Chem Soc. 1989;111:8551–8565. [Google Scholar]
- 104.Dewar MJS, Thiel W. Ground States of Molecules. 38. The MNDO Method. Approximations and Parameters. J Am Chem Soc. 1977;99:4899. [Google Scholar]
- 105.Stewart JJP. Optimization of Parameters for Semi-Empirical Methods I-Method. J Comput Chem. 1989;10:221. [Google Scholar]
- 106.Kassaee MH, Keffer DJ, Steele WV. A Comparison Between Entropies of Aromatic Compounds from Quantum Mechanical Calculations and Experiment. J Mol Struct: THEOCHEM. 2007;802:23–34. [Google Scholar]
- 107.Kassaee MH, Keffer DJ, Steele WV. Theoretical Calculation of Thermodynamic Properties of Naphthalene, Methylnaphthalenes, and Dimethylnaphthalenes. J Chem Eng Data. 2007;52:1843–1850. [Google Scholar]
- 108.Wiberg KB. Properties of Some Condensed Aromatic Systems. J Org Chem. 1997;62:5720–5727. [Google Scholar]
- 109.Pogodin S, Agranat I. Large PAHs Derived from Benzanthrone and Naphthanthrone. A Semiempirical Study. Polycyclic Aromat Compd. 2001;18:247–263. [Google Scholar]
- 110.Pogodin S, Agranat I. Overcrowding Motifs in Large PAHs. An Ab Initio Study. J Org Chem. 2002;67:265–270. doi: 10.1021/jo0107251. [DOI] [PubMed] [Google Scholar]
- 111.van Speybroeck V, Marin GB, Waroquier M. Hydrocarbon Bond Dissociation Enthalpies: From Substituted Aromatics to Polyaromatics. ChemPhysChem. 2006;7:2205–2214. doi: 10.1002/cphc.200600161. [DOI] [PubMed] [Google Scholar]
- 112.Papas BN, Wang SY, DeYonker NJ, Woodcock HL, Schaefer HF. Naphthalenyl, Anthracenyl, Tetracenyl, and Pentacenyl Radicals and Their Anions. J Phys Chem A. 2003;107:6311–6316. [Google Scholar]
- 113.Wang D, Violi A, Kim DH, Mullholland JA. Formation of Naphthalene, Indene, and Benzene from Cyclopentadiene Pyrolysis: A DFT Study. J Phys Chem A. 2006;110:4719–4725. doi: 10.1021/jp053628a. [DOI] [PubMed] [Google Scholar]
- 114.Hemelsoet K, Speybroeck VV, Waroquier M. Bond Dissociation Enthalpies of Large Aromatic Carbon-Centered Radicals. J Phys Chem A. 2008;112:13566–13573. doi: 10.1021/jp801551c. [DOI] [PubMed] [Google Scholar]
- 115.Henry DJ, Sullivan MB, Radom L. G3-RAD and G3X-RAD: Modified Gaussian-3 (G3) and Gaussian-3X (G3X) Procedures for Radical Thermochemistry. J Chem Phys. 2003;118:4849–4860. [Google Scholar]
- 116.Schulman JM, Peck RC, Disch RL. Ab Initio Heats of Formation of Medium-Sized Hydrocarbons. 11. The Benzoid Aromatics. J Am Chem Soc. 1989;111:5675. [Google Scholar]
- 117.Peck RC, Schulman JM, Disch RL. Ab Initio Heats of Formation of Medium-Sized Hydrocarbons. 12. 6-31G(d) Studies of Benzenoid Aromatics. J Phys Chem. 1990;94:6637–6641. [Google Scholar]
- 118.Disch RL, Schulman JM, Peck RC. Ab Initio Heats of Formation of Medium Sized Hydrocarbons. 13. Studies of Benzo[e]-pyrene, Benoz[ghi]perylene, Coronene, and Circumcoronene. J Phys Chem. 1992;96:3998–4002. [Google Scholar]
- 119.Schulman JM, Disch RL. Ab Initio Heats of Formation of Medium Sized Hydrocarbons. 14. Pyracyelene and Biphenylene. J Mol Struct: THEOCHEM. 1992;259:173–179. [Google Scholar]
- 120.Schulman JM, Disch RL. Thermal and Magnetic Properties of Coronene and Related Molecules. J Phys Chem A. 1997;101:9176–9179. [Google Scholar]
- 121.Schulman JM, Disch RL. Aromatic Character of [n]helicenes and [n]phenacene. J Phys Chem A. 1999;103:6669–6672. [Google Scholar]
- 122.Li ZR, Rogers DW, McLaffery FJ, Mandziuk M, Podesenin AV. Ab Initio Calculations of Enthalpies of Hydrogenation, Isomerization, and Formation of Cyclic C6 Hydrocarbons. Benzene Isomers. J Phys Chem A. 1999;103:426–430. [Google Scholar]
- 123.Notario R, Castano O, Abboud JLM, Gomperts R, Frutos LM, Palmeiro R. Organic Thermochemistry at High Ab Initio Levels. 1. A G2(MP2) and G2 Study of Cyclic Saturated and Unsatured Hydrocrbons (including Aromatics) J Org Chem. 1999;64:9011–9014. doi: 10.1021/jo000089r. [DOI] [PubMed] [Google Scholar]
- 124.Notario R, Abboud JLM. Probing the Limits of Resonance Stabilization. The Case of Linear Polyacenes. J Phys Chem A. 1998;102:5290–5297. [Google Scholar]
- 125.Cheung YS, Wong CK, Li WK. Ab Initio Calculations of the Heats of Formation for (CH)6 Isomers. J Mol Struct: THEOCHEM. 1998;454:17–24. [Google Scholar]
- 126.Bond D. Computational Methods in Organic Thermochemistry. 1. Hydrocarbon Enthalpies and Free Energies of Formation. J Org Chem. 2007;72:5555–5566. doi: 10.1021/jo070383k. [DOI] [PubMed] [Google Scholar]
- 127.Janoschek R, Rossi MJ. Thermochemical Properties of Free Radicals from G3MP2B3 Calculations. Int J Chem Kinet. 2002;34:550–560. [Google Scholar]
- 128.Curtiss LA, Raghavachari K, Redfern PC, Rassolov V, Pople JA. Gaussian-3 (G3) Theory for Molecules Containing First and Second-Row Atoms. J Chem Phys. 1998;109:7764–7776. [Google Scholar]
- 129.Rogers DW, McLafferty FJ. The Influence of Substituent Groups on the Resonance Stabilization of Benzene. an Ab Initio Computational Study. J Org Chem. 2001;66:1157–1162. doi: 10.1021/jo001242k. [DOI] [PubMed] [Google Scholar]
- 130.Rogers DW, McLafferty FJ. G3(MP2) Calculations of Enthalpies of Hydrogenation, Isomerization, and Formation of [3]-Radialene and Related Compounds. J Phys Chem A. 2002;106:1054–1059. [Google Scholar]
- 131.Rogers DW, McLafferty FJ. G3(MP2) Calculations of Enthalpies of Hydrogenation, Isomerization, and Formation of Bi- and Tricyclic C8 and C10 Hydrocarbons. The Bicyclo[3.3.0]octenes and Triquinacenes. J Phys Chem A. 2000;104:9356–9361. [Google Scholar]
- 132.Melius CF. In: Chemistry and Physics of Energetic Materials. Bulusu SN, editor. Vol. 309. Kluwer Academic Publishers; Dorderecht, The Netherlands: 1990. p. 21. [Google Scholar]
- 133.Allendorf MD, Medlin M, Besmann TM. Thermodynamics Resource Data Base; Sandia Report SAND 2007-6205. Sandia National Laboratories; Albuquerque, NM: 2007. [Google Scholar]
- 134.Anantharaman B, Melius CF. Bond Additivity Corrections for G3B3 and G3MP2B3 Quantum Chemistry Methods. J Phys Chem A. 2005;109:1734–1747. doi: 10.1021/jp045883l. [DOI] [PubMed] [Google Scholar]
- 135.Fishtik F, Urban D, Wilcox J. The Effect of Stoichiometry on Ab Initio based Thermochemistry Predictions. Chem Phys Lett. 2006;417:185–189. [Google Scholar]
- 136.Blanquart G, Pitsch H. Thermochemical Properties of Polycyclic Aromatic Hydrocarbons (PAH) from G3MP2B3 Calculations. J Phys Chem A. 2007;111:6510–6520. doi: 10.1021/jp068579w. [DOI] [PubMed] [Google Scholar]
- 137.Rayne S, Forest KA. G4MP2 Theoretical Study on the Gas Phase Enthalpies of Formation for Various Polycyclic Aromatic Hydrocarbons (PAHs) and Other C10 Through C20 Unsaturated Hydrocarbons. Nat Proc. 2011 doi: 10.1038/npre.2011.6450.1. [DOI] [Google Scholar]
- 138.Dorofeeva OV. Comments on Estimated Gas Phase Standard Enthalpies of Formation for Organic Compounds Using the Gaussian-4 (G4) and W1BD Theoretical Methods. J Chem Eng Data. 2011;56:682–683. [Google Scholar]
- 139.Zauer EA. Enthalpies of Formation of Polycyclic Aromatic Hydrocarbons. Russ J Gen Chem. 2012;82:1135–1144. [Google Scholar]
- 140.Sander LC, Wise SA. NIST Special Publication 922. NIST; Gaithersburg, MD: 1997. [Google Scholar]
- 141.Chiron AS. 2013 http://chiron.no/
- 142.Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, et al. General Atomic and Molecular Electronic Structure System. J Comput Chem. 1993;14:1347–1363. [Google Scholar]
- 143.Gordon MS, Schmidt MW. In: Theory and Applications of Computational Chemistry: The First Forty Years. Dykstra CE, Frenking G, Kim KS, Scuseria GE, editors. Elsevier; Amsterdam: 2005. pp. 1167–1189. [Google Scholar]
- 144.Baboul AG, Curtiss LA, Redfern PC, Raghavachari K. Gaussian-3 Theory Using Density Functional Geometries and Zero-Point Energies. J Chem Phys. 1999;110:7650–7657. [Google Scholar]
- 145.Curtiss LA, Raghavachari K, Redfern PC, Rassolov V, Pople JA. Gaussian-3 (G3) Theory for Molecules Containing First and Second-Row Atoms. J Chem Phys. 1998;109:7764–7776. [Google Scholar]
- 146.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, et al. Gaussian 09, Revision C.1. Gaussian Inc; Wallingford, CT: 2009. [Google Scholar]
- 147.Baboul AG, Curtiss LA, Redfern PC, Raghavachari K. Gaussian-3 Theory Using Density Functional Geometries and Zero-Point Energies. J Chem Phys. 1999;110:7650–7657. [Google Scholar]
- 148.Cox JD, Wagman DD, Medvedev VA. CODATA Key Values for Thermodynamics. Hemisphere; New York: 1989. [Google Scholar]
- 149.Ruscic B, Feller D, Peterson KA. Active Thermochemical Tables: Dissociation Energies of Several Homonuclear First-Row Diatomics and Related Thermochemical Values. Theor Chem Acc. 2014;133:1415. [Google Scholar]
- 150.Scott AP, Radom L. Harmonic Vibrational Frequencies: An Evaluation of Hartree-Fock, Møller-Plesset, Quadratic Configuration Interaction, Density Functional Theory, and Semiempirical Scale Factors. J Phys Chem. 1996;100:16502–16513. [Google Scholar]
- 151.Irikura KK, Johnson RD, Jr, Kacker RN. Uncertainties in Scaling Factors for Ab Initio Vibrational Frequencies. J Phys Chem A. 2005;109:8430–8433. doi: 10.1021/jp052793n. [DOI] [PubMed] [Google Scholar]
- 152.Wiberg KB. Group Equivalents for Converting Ab Initio Energies to Enthalpies of Formation. J Comput Chem. 1984;5:197–199. [Google Scholar]
- 153.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An Open Chemical Toolbox. J Cheminf. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Open Babel Package, Version 2.3.1. 2013 http://openbabel.org/
- 155.Feller D. Application of Systematic Sequences of Wave Functions to the Water Dimer. J Chem Phys. 1992;96:6104–6114. [Google Scholar]
- 156.Halkier A, Helgaker T, Rgensen PJ, Klopper W, Koch H, Olsen J, Wilson AK. Basis-Set Convergence in Correlated Calculations on Ne, N2, and H2O. Chem Phys Lett. 1998;286:243–252. [Google Scholar]
- 157.Truhlar DG. Basis-Set Extrapolation. Chem Phys Lett. 1998;294:45–48. [Google Scholar]
- 158.Levenberg K. A Method for the Solution of Certain Non-Linear Problems in Least Squares. Q Appl Math. 1944;2:164–168. [Google Scholar]
- 159.Marquardt D. An Algorithm for Least-Squares Estimation of Nonlinear Parameters. J Soc Ind Appl Math. 1963;11:431–441. [Google Scholar]
- 160.Ochterski JW, Petersson GA, Wiberg KB. A Comparison of Model Chemistries. J Am Chem Soc. 1995;117:11299–11308. [Google Scholar]
- 161.Curtiss L, Raghavachari K, Redfern PC, Pople JA. Assessment of Gaussian-2 and Density Functional Theories for the Computation of Enthalpies of Formation. J Chem Phys. 1997;106:1063–1079. [Google Scholar]
- 162.Ruscic B. Uncertainty Quantification in Thermochemistry, Benchmarking Electronic Structure Computations, and Active Thermochemical Tables. Int J Quantum Chem. 2014;114:1097–1101. [Google Scholar]
- 163.Curtiss LA, Raghavachari K, Redfern PC, Pople JA. Assessment of Gaussian-3 and Density Functional Theories for a Larger Experimental Test Set. J Chem Phys. 2000;112:7374–7383. doi: 10.1063/1.2039080. [DOI] [PubMed] [Google Scholar]
- 164.Curtiss LA, Redfern PC, Raghavachari K, Pople JA. Gaussian-3X. G3X Theory: Use of Improved Geometries, Zero-Point Energies, and Hartree-Fock Basis Sets. J Chem Phys. 2001;114:108–117. [Google Scholar]
- 165.Karton A, Martin JML. Explicitly Correlated Wn Theory: W1-F12 and W2-F12. J Chem Phys. 2012;136:124114. doi: 10.1063/1.3697678. [DOI] [PubMed] [Google Scholar]
- 166.Magnus A, Hartmann H, Becker F. Verbrennungswarmen Und Resonanzenergien Von Mehrkernigen Aromatischen Kohlenwas-serstoffen. Z Phys Chem. 1951;197:75–91. [Google Scholar]
- 167.Auerswald J, Engels B, Fischer I, Gerbich T, Herterich J, Krueger A, Lang M, Schmitt HC, Schon C, Walter C. The Electronic Structure of Pyracene: A Spectroscopic and Computational Study. Phys Chem Chem Phys. 2013;15:8151–8161. doi: 10.1039/c3cp44271c. [DOI] [PubMed] [Google Scholar]
- 168.Monte M, Notario R, Pinto S. Thermodynamic Properties of Fluoranthene: An Experimental and Computational Study. J Chem Thermodyn. 2012;49:159–164. [Google Scholar]
- 169.Davis HE, Allinger NL, Rogers DW. Enthalpies of Hydrogenation of Phenylalkynes: Indirect Determination of the Enthalpy of Formation of Diphenylcyclopropenone. J Org Chem. 1985;50:3601–3604. [Google Scholar]
- 170.Flitcroft TL, Skinner HA. Heats of Hydrogenation: Part 2. Acetylene Derivatives. Trans Faraday Soc. 1958;54:47–53. [Google Scholar]
- 171.CODATA Key Values for Thermodynamics. 2013 http://www.codata.org/resources/databases/key1.html.
- 172.Ruscic B. Active Thermochemical Tables: Sequential Bond Dissociation Enthalpies of Methane, Ethane, and Methanol and the Related Thermochemistry. J Phys Chem A. 2015;119:7810–7837. doi: 10.1021/acs.jpca.5b01346. [DOI] [PubMed] [Google Scholar]
- 173.Gurvich LV, Veyts IV, Alcock CB. Thermodynamic Properties of Individual Substances. fourth. Vol. 2 Hemisphere Publishing Corp; New York: 1991. [Google Scholar]
- 174.Good WD. The Enthalpies of Combustion and Formation of the Isomeric Pentanes. J Chem Thermodyn. 1970;2:237–244. [Google Scholar]
- 175.Prosen EJ, Rossini FD. Heats of Combustion and Formation of the Paraffin Hydrocarbons at 25°C. J Res NBS. 1945;34:263–267. [Google Scholar]
- 176.Prosen EJ, Maron FW, Rossini FD. Heats of Combustion, Formation, and Insomerization of Ten C4 Hydrocarbons. J Res NBS. 1951;46:106–112. [Google Scholar]
- 177.Molnar A, Rachford R, Smith GV, Liu R. Heats of Hydrogenation by a Simple and Rapid Flow Calorimetric Method. Appl Catal. 1984;9:219–223. [Google Scholar]
- 178.Wiberg KB, Wasserman DJ, Martin E. Enthalpies of Hydration of Alkenes. 2. The N-Heptenes and N-Pentenes. J Phys Chem. 1984;88:3684–3688. [Google Scholar]
- 179.Bretschneider E, Rogers DW. A New Microcalorimeter: Heats of Hydrogenation of Four Monoolefins. Microchim Acta. 1970;58:482–490. [Google Scholar]
- 180.Rockenfeller JD, Rossini FD. Heats of Combustion, Isomerization, and Formation of Selected C7, C8, and C10 Monoolefin Hydrocarbons. J Phys Chem. 1961;65:267–272. [Google Scholar]
- 181.Good WD, Smith NK. The Enthalpies of Combustion of the Isomeric Pentenes in the Liquid State. A Warning to Combustion Calorimetrists About Sample Drying. J Chem Thermodyn. 1979;11:111–118. [Google Scholar]
- 182.Rogers DW, Crooks E, Dejroongruang K. Enthalpies of Hydrogenation of the Hexenes. J Chem Thermodyn. 1987;19:1209–1215. [Google Scholar]
- 183.Rogers DW, Dejroongruang K. Enthalpies of Hydrogenation of the N-Heptenes and the Methylhexenes. J Chem Thermodyn. 1988;20:675–680. [Google Scholar]
- 184.Steele WV, Chirico RD, Cowell AB, Knipmeyer SE, Nguyen A. Thermodynamic Properties and Ideal-Gas Enthalpies of Formation for 2-Aminoisobutyric Acid (2-Methylalanine), Acetic Acid, (4-Methyl-3-Penten-2-One), 4-Methylpent-1-Ene, 2,2′-Bis-(phenylthio)propane, and Glycidyl Phenyl Ether (1,2-Epoxy-3-Phenoxypropane) J Chem Eng Data. 1997;42:1053–1066. [Google Scholar]
- 185.Abboud J-LM, Jimenez P, Roux MV, Turrion C, Lopez-Mardomingo C, Podosenin A, Rogers DW, Liebman JF. Interrelations of the Energetics of Amides and Alkenes: Enthalpies of Formation of N,N-Dimethyl Derivatives of Pivalamide, 1-Adaman-tylcarboxamide and Benzamide, and of Styrene and Its α-, Trans-β-and β,β-Methylated Derivates. J Phys Org Chem. 1995;8:15–25. [Google Scholar]
- 186.Egger KW, Benson SW. Nitric Oxide and Iodine Catalyzed Isomerization of Olefins. VI. Thermodynamic Data from Equilibrium Studies of the Geometrical and Positional Isomerization of N-Pentenes. J Am Chem Soc. 1966;88:236–240. [Google Scholar]
- 187.Wiberg KB, Wasserman DJ. Enthalpies of Hydration of Alkenes. 1. The N-Hexenes. J Am Chem Soc. 1981;103:6563–6566. [Google Scholar]
- 188.Wiberg KB, Hao S. Enthalpies of Hydration of Alkenes. 4. Formation of Acyclic Tert-Alcohols. J Org Chem. 1991;56:5108–5110. [Google Scholar]
- 189.Wagman DD, Kilpatrick JE, Pitzer KS, Rossini FD. Heats, Equilibrium Constants, and Free Energies of Formation of the Acetylene Hydrocarbons Through the Pentynes, to 1,500 K. J Res NBS. 1945;35:467–496. [Google Scholar]
- 190.Rogers DW, Dagdagan OA, Allinger NL. Heats of Hydrogenation and Formation of Linear Alkynes and a Molecular Mechanics Interpretation. J Am Chem Soc. 1979;101:671–676. [Google Scholar]
- 191.Pimenova SM, Kozina MP, Gal’chenko GL, Siretskaya TV, Takhistov VV, Kuznetsova TS, Proskurnina MV, Kotel’nikova TA. Heats of Combustion and Formation of Some Compounds with Acetylene Bonding. Termodin Org Soedin. 1986:12–16. [Google Scholar]
- 192.Fraser FM, Prosen EJ. Heats of Combustion and Isomerization of Six Pentadienes and Spiropentane. J Res NBS. 1955;54:143–148. [Google Scholar]
- 193.Fang W, Rogers DW. Enthalpy of Hydrogenation of the Hexadienes and Cis- and Trans-1,3,5-Hexatriene. J Org Chem. 1992;57:2294–2297. [Google Scholar]
- 194.Roth WR, Adamczak O, Breuckmann R, Lennartz H-W, Boese R. Die Berechnung Von Resonanzenergien; Das MM2ERW-Kraftfeld. Chem Ber. 1991;124:2499–2521. [Google Scholar]
- 195.Lebedeva ND, Ryadnenko VL, Kiseleva NN, Nazarova LF. Enthalpy of Formation of Isopropenylacetylene and Diisopro-penyldiacetylene. Vses Konf Kalorim Rasshir Tezisy Dokl. (7th) 1977;1:91–95. [Google Scholar]
- 196.Prosen EJ, Gilmont R, Rossini FD. Heats of Combustion of Benzene, Toluene, Ethyl-Benzene, O-Xylene, M-Xylene, P-Xylene, N-Propylbenzene, and Styrene. J Res NBS. 1945;34:65–70. [Google Scholar]
- 197.Krall RE, Roberts JD. Strain Variation in the Unsaturated Cyclobutane Ring. Am Chem Soc Div Petrol Chem. 1958;3:63–68. [Google Scholar]
- 198.Guthrie JP. Equilibrium Constants for a Series of Simple Aldol Condensations, and Linear Free Energy Relations with Other Carbonyl Addition Reactions. Can J Chem. 1978;56:962–973. [Google Scholar]
- 199.Rogers DW, McLafferty FJ. A New Hydrogen Calorimeter. Heats of Hydrogenation of Allyl and Vinyl Unsaturation Adjacent to a Ring. Tetrahedron. 1971;27:3765–3775. [Google Scholar]
- 200.Prosen EJ, Johnson WH, Rossini FD. Heats of Combustion and Formation at 25°C of the Alkylbenzenes Through C10H14, and of the Higher Normal Monoalkylbenzenes. J Res NBS. 1946;36:455–461. [Google Scholar]
- 201.Good WD, Smith NK. Enthalpies of Combustion of Toluene, Benzene, Cyclohexane, Cyclohexene, Methylcyclopentane, 1-Methylcyclopentene, and N-Hexane. J Chem Eng Data. 1969;14:102–106. [Google Scholar]
- 202.Stevens WR, Ruscic B, Baer T. Heats of Formation of C6H5·, C6H+5, and C6H5NO by Threshold Photoelectron Photoion Coincidence and Active Thermochemical Tables Analysis. J Phys Chem A. 2010;114:13134–13145. doi: 10.1021/jp107561s. [DOI] [PubMed] [Google Scholar]
- 203.Coops J, Mulder D, Dienske JW, Smittenberg J. The Heats of Combustion of a Number of Hydrocarbons. Rec Trav Chim Pays/Bas. 1946;65:128. [Google Scholar]
- 204.Johnson WH, Prosen EJ, Rossini FD. Heats of Combustion and Isomerization of the Eight C9H12 Alkylbenzenes. J Res NBS. 1945;35:141–146. [Google Scholar]
- 205.Wenthold PG, Squires RR. Biradical Thermochemistry from Collision-Induced Dissociation Threshold Energy Measurements. Absolute Heats of Formation of Ortho-, Meta-, and Para-Benzyne. J Am Chem Soc. 1994;116:6401–6412. doi: 10.1021/ja011630j. [DOI] [PubMed] [Google Scholar]
- 206.Wenthold PG, Paulino JA, Squires RR. The Absolute Heats of Formation of O-,m-, and P-Benzyne. J Am Chem Soc. 1991;113:7414–7415. [Google Scholar]
- 207.Fuchs R, Hallman JH, Perlman MO. Thermochemistry of Conjugation of Simple Cyclopropane Derivatives. Can J Chem. 1982;60:1832–1835. [Google Scholar]
- 208.Kozina MP, Kropivnitskaya AG, Gembitskii PA, Loim NM. Heats of Combustion of Some Vinyl- and Cyclopropylbenzenes. Moscow Univ Chem Bull (Engl Transl) 1969;24:60–62. [Google Scholar]
- 209.Montgomery RL, Rossini FD, Mansson M. Enthalpies of Combustion, Vaporization, and Formation of Phenylbenzene, Cyclohexylbenzene, and Cyclohexylcyclohexane; Enthalpy of Hydrogenation of Certain Aromatic Systems. J Chem Eng Data. 1978;23:125–129. [Google Scholar]
- 210.Chirico RD, Knipmeyer SE, Nguyen A, Steele WV. The Thermodynamic Properties of Biphenyl. J Chem Thermodyn. 1989;21:1307–1331. [Google Scholar]
- 211.Steele WV, Chirico RD, Smith NK. The Standard Enthalpies of Formation of 2-Methylbiphenyl and Diphenylmethane. J Chem Thermodyn. 1995;27:671–678. [Google Scholar]
- 212.Brull L. Sui Calori Di Combustione Di Alcuni Derivati Del Bifenil. Gazz Chim Ital. 1935;65:19–28. [Google Scholar]
- 213.Ribeiro da Silva MAV, Matos MAR, do Rio CMA, Morais VMF. Thermochemical and Theoretical Studies of 4- Methylbiphenyl, 4.4′-Dimethylbiphenyl, 4.4′-Dimethyl-2.2′-Bipyridine. J Chem Soc Faraday Trans. 1997;93:3061. [Google Scholar]
- 214.Lobanov GA, Karmanova LP. Enthalpy of Formation of Some Organic Substances. Izv Vyssh Uchebdn Zaved. 1971;14:865–867. [Google Scholar]
- 215.Yates K, McDonald RS. A Thermochemical Probe into the Mechanism of Electrophilic Addition to Olefins. J Am Chem Soc. 1971;93:6297–6299. [Google Scholar]
- 216.Tkachenko E, Ekaterina S, Varushchenko R, Druzhinina A. Heat Capacity and Thermodynamic Functions of Diphenylacetylene. J Chem Eng Data. 2011;56:4700–4709. [Google Scholar]
- 217.Balepin AA, Lebedev VP, Miroshnichenko EA, Koldobskii GI, Ostovskii VA, Larionov BP, Gidaspov BV, Lebedev YA. Energy Effects in Polyphenylenes and Phenyltetrazoles. Svoistva Veshchestv Str Mol Svoistva Veshchestv Str Mol. 1977:93–98. [Google Scholar]
- 218.Parks GS, West TJ, Naylor BF, Fujii PS, McClaine LA. Thermal Data on Organic Compounds. XXIII. Modern Combustion Data for Fourteen Hydrocarbons and Five Polyhydroxy Alcohols. J Am Chem Soc. 1946;68:2524–2527. [Google Scholar]
- 219.Berkowitz J, Ellison GB, Gutman D. Three Methods to Measure RH Bond Energies. J Phys Chem. 1994;98:2744–2765. [Google Scholar]
- 220.Alecu IM, Gao YD, Hsieh PC, Sand JP, Ors A, McLeod A, Marshall P. Studies of the Kinetics and Thermochemistry of the Forward and Reverse Reaction Cl+C6H6 = HCl+C6H5. J Phys Chem A. 2007;111:3970–3976. doi: 10.1021/jp067212o. [DOI] [PubMed] [Google Scholar]
- 221.Heckmann E, Hippler H, Troe J. High-Temperature Reactions and Thermodynamic Properties of Phenyl Radicals. Symp Combust [Proc] 1996;26:543–550. [Google Scholar]
- 222.Tsang W. In: Energetics of Organic Free Radicals. Martinho-Simoes JF, editor. Blackie Academic and Professional; London: 1996. pp. 22–58. [Google Scholar]
- 223.Davico GE, Bierbaum VM, DePuy CH, Ellison GB, Squires RR. The C-H Bond Energy of Benzene. J Am Chem Soc. 1995;117:2590–2599. [Google Scholar]
- 224.Williams RB. Heats of Catalytic Hydrogenation in Solution. I. Apparatus, Technique, and the Heats of Hydrogenation of Certain Pairs of Stereoisomers. J Am Chem Soc. 1942;64:1395–1404. [Google Scholar]
- 225.Good WD, Lee SH. The Enthalpies of Formation of Selected Naphthalenes, Diphenylmethanes, and Bicyclic Hydrocarbons. J Chem Thermodyn. 1976;8:643–650. [Google Scholar]
- 226.Speros DM, Rossini FD. Heats of Combustion and Formation of Naphthalene, the Two Methylnaphthalenes, Cis and Trans Decahydronaphthalene and Related Compounds. J Phys Chem. 1960;64:1723–1727. [Google Scholar]
- 227.Broadus KM, Kass SR. Benzocyclobutadienyl Anion: Formation and Energetics of an Antiaromatic Molecule. J Org Chem. 2000;65:6566–6571. doi: 10.1021/jo000709o. [DOI] [PubMed] [Google Scholar]
- 228.Roth WR, Biermann M, Dekker H, Jochems R, Mosselman C, Hermann H. Energy Profile of the O-Quinodi-methane-Benzocyclobutene Equilibrium. Chem Ber. 1978;111:3892–3903. [Google Scholar]
- 229.Coleman DJ, Pilcher G. Heats of Combustion of Biphenyl, Bibenzyl, Naphthalene, Anthracene, and Phenanthrene. Trans Faraday Soc. 1966;62:821–827. [Google Scholar]
- 230.Mansson M. A Calorimetric Study of Peri Strain in 1,8-Dimethylnaphthalene and 1,4,5,8-Tetramethylnaphthalene. Acta Chem Scand. 1974;28:677–680. [Google Scholar]
- 231.Good WD. The Enthalpies of Combustion and Formation of 1,8-Dimethylnaphthalene, 2,3-Dimethylnaphthalene, 2,6-Dimethyl-naphthalene, and 2,7-Dimethylnaphthalene. J Chem Thermodyn. 1973;5:715–720. [Google Scholar]
- 232.Colomina M, Jimenez P, Roux MV, Turrion C. Thermochemical Properties of Naphthalene Derivatives. V. Formation Enthalpies of 2,3-Dimethylnaphthalene and 2,3-Dihydroxynaphtha-lene. An Quim. 1979;75:620–624. [Google Scholar]
- 233.Chirico RD, Knipmeyer SE, Nguyen A, Steele WV. The Thermodynamic Properties to the Temperature 700 K of Naphthalene and of 2,7-Dimethylnaphthalene. J Chem Thermodyn. 1993;25:1461–1494. [Google Scholar]
- 234.Smith NK, Gammon BE, Good WD. Thermodynamics of Organic Compounds. Gov Rep Announce Index (US) Avail NTIS AD-A 110430. 1982;82:2252. [Google Scholar]
- 235.Good WD. The Enthalpies of Combustion and Formation of Indan and Seven Alkylindans. J Chem Thermodyn. 1971;3:711–717. [Google Scholar]
- 236.Stull DR, Sinke GC, McDonald RA, Hatton WE, Hildenbrand DL. Thermodynamic Properties of Indane and Indene. Pure Appl Chem. 1961;2:315–322. [Google Scholar]
- 237.Frye CG, Weitkamp AW. Equilibrium Hydrogenations of Multi-Ring Aromatics. J Chem Eng Data. 1969;14:372–376. [Google Scholar]
- 238.Nagano Y. Standard Enthalpies of Formation of Phenanthrene and Naphthacene. J Chem Thermodyn. 2002;34:377–383. [Google Scholar]
- 239.Steele WV, Chirico RD, Nguyen A, Hossenlopp IA, Smith NK. Determination of Ideal-Gas Enthalpies of Formation for Key Compounds. AIChE Symp Ser. 1990;86:138–154. [Google Scholar]
- 240.Good WD. The Enthalpies of Formation of Some Bridged-Ring Polynuclear Aromatic Hydrocarbons. J Chem Thermodyn. 1978;10:553–558. [Google Scholar]
- 241.Diogo HP, Kiyobayashi T, da Piedade MEM, Burlak N, Rogers DW, McMasters D, Persy G, Wirz J, Liebman JF. The Aromaticity of Pyracylene: An Experimental and Computational Study of the Energetics of the Hydrogenation of Acenaphthylene and Pyracylene. J Am Chem Soc. 2002;124:2065–2072. doi: 10.1021/ja012541+. [DOI] [PubMed] [Google Scholar]
- 242.Boyd RH, Christensen RL, Pua R. The Heats of Combustion of Acenaphthene, Acenaphthylene, and Fluoranthene. Strain and Delocalization in Bridged Naphthalenes. J Am Chem Soc. 1965;87:3554–3559. [Google Scholar]
- 243.Rakus K, Verevkin SP, Schatzer J, Beckhaus HD, Ruchardt C. Thermochemistry and Thermal Decomposition of 9,9′-Bifluorenyl and 9,9′-Dimethyl-9,9′-Bifluorenyl - the Stabilization Energy of 9-Fluorenyl Radicals. Chem Ber. 1994;127:1095–1103. [Google Scholar]
- 244.Monte M, Pinto S, Lobo Ferreira A. I. M. C. Fluorene: An Extended Experimental Thermodynamic Study. J Chem Thermodyn. 2012;45:53–58. [Google Scholar]
- 245.Billups WE, Chow WY, Leavell KH, Lewis ES, Margrave JL, Sass RL, Shieh JJ, Werness PG, Wood JL. Structure and Thermochemistry of Benzocyclopropenes. Bond Fixation and Strain Energy. J Am Chem Soc. 1973;95:7878–7880. [Google Scholar]
- 246.Magnus A, Hartmann H, Becker F. Heats of Combustion and Resonance Energies of Polynuclear Aromatic Hydrocarbons. Z Phys Chem (Leipzig) 1951;197:75. [Google Scholar]
- 247.Diogo HP, Persy G, Minas da Piedade ME, Wirz J. The Enthalpy of Formation of Pyracyclene. J Org Chem. 1996;61:6733–6734. doi: 10.1021/jo952182z. [DOI] [PubMed] [Google Scholar]
- 248.Smith NK, Stewart RCJr, Osborn AG, Scott DW. Pyrene: Vapor Pressure, Enthalpy of Combustion, and Chemical Thermodynamic Properties. J Chem Thermodyn. 1980;12:919–926. [Google Scholar]
- 249.Wong WK, Westrum EF., Jr Thermodynamics of Polynuclear Aromatic Molecules. II. Low Temperature Thermal Properties of Perylene, Coronene, and Naphthacene. Mol Cryst Liq Cryst. 1980;61:207–228. [Google Scholar]
- 250.Chickos JS, Webb P, Nichols G, Cheng TKPC, Scott L. The Enthalpy of Vaporization and Sublimation of Corannulene, Coronene, and Perylene at T = 298.15 K. J Chem Thermodyn. 2002;34:1195–1206. [Google Scholar]
- 251.Kiyobayashi T, Nagano Y, Sakiyama M, Yamamoto K, Cheng P-C, Scott LT. Formation Enthalpy of Corannulene: Microbomb Combustion Calorimetry. J Am Chem Soc. 1995;117:3270–3271. [Google Scholar]
- 252.Torres L, Campos M, Martinez M. The Thermochemistry of Coronene Revisited. J Chem Thermodyn. 2009;41:957–965. [Google Scholar]
- 253.Nagano Y. Micro-Combustion Calorimetry of Coronene. J Chem Thermodyn. 2000;32:973–977. [Google Scholar]
- 254.Liebman JF, Paquette LA, Peterson JR, Rogers DW. Is Triquinacene Homoaromatic? A Thermochemical Answer in the Affirmative. J Am Chem Soc. 1986;108:8267–8268. [Google Scholar]
- 255.Roth WR, Bohm M, Lenhartz H-W, Vogel E. Heats of Hydrogenation. Part 5. Resonance Energy of Bridged [10]annulenes. Angew Chem. 1983;95:1011–1012. [Google Scholar]
- 256.Kovats E. Thermochemical Properties of Azulene. Helv Chim Acta. 1955;38:1912–1919. [Google Scholar]
- 257.Day JH, Oestreich C. Fulvenes: III. Heat of Combustion and Resonance Energy. J Org Chem. 1957;22:214–216. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.