

Abstract
Expression levels of many microRNAs (miRNAs) change during aging, notably declining globally in a number of organisms and tissues across taxa. However, little is known about the mechanisms or the biological relevance for this change. We investigated the network of genes that controls miRNA transcription and processing during C. elegans aging. We found that miRNA biogenesis genes are highly networked with transcription factors and aging-associated miRNAs. In particular, miR-71, known to influence life span and itself up-regulated during aging, represses alg-1/Argonaute expression post-transcriptionally during aging. Increased ALG-1 abundance in mir-71 loss-of-function mutants led to globally increased miRNA expression. Interestingly, these mutants demonstrated widespread mRNA expression dysregulation and diminished levels of variability both in gene expression and in overall life span. Thus, the progressive molecular decline often thought to be the result of accumulated damage over an organism's life may be partially explained by a miRNA-directed mechanism of age-associated decline.
Keywords: microRNAs, aging, Caenorhabditis elegans, Argonaute, miR-71
INTRODUCTION
Many microRNAs (miRNAs) are dynamically expressed during aging, fulfilling important regulatory roles. A handful of specific miRNAs have been shown to affect normal life span by targeting genetic pathways that are important for normal aging (Boehm and Slack 2005; de Lencastre et al. 2010; Boulias and Horvitz 2012; Lucanic et al. 2013). In particular, miR-71 promotes longevity in the nematode Caenorhabditis elegans by interacting with multiple aging-associated pathways including insulin signaling and germline signaling (de Lencastre et al. 2010; Boulias and Horvitz 2012). However, most miRNAs individually have no apparent effect on normal life span (Boulias and Horvitz 2012).
MiRNAs can have broad, if diffuse, impacts across complex multi-pathway processes: MiRNAs recognize targets through imperfect complementarity allowing individual miRNAs to target multiple mRNAs, and multiple miRNAs to target the same mRNA (Krek et al. 2005; Lewis et al. 2005). Thus, although the most widely studied role of miRNAs is in targeting one or a few specific mRNAs, miRNAs can also modulate and integrate across a large number of biological inputs and outputs. Indeed, miRNAs have been proposed to act as mediators of crosstalk between pathways and as “noise buffers” to canalize specific fates (Hornstein and Shomron 2006; Herranz and Cohen 2010; Inui et al. 2010). It is possible that miRNAs collectively assume roles that impact general gene expression, despite the notable difficulty in assigning clear phenotypes to many individual miRNAs or even miRNA families (Miska et al. 2007; Alvarez-Saavedra and Horvitz 2010).
The regulation of miRNAs during aging is similarly poorly understood. A global decline in miRNA abundance with age has been observed in a number of organisms and tissues (Ibañez-Ventoso et al. 2006; de Lencastre et al. 2010; Noren Hooten et al. 2010; Inukai et al. 2012). This decline was previously linked to age-related decreases in miRNA biogenesis, and could be prevented by life span-extending interventions such as dietary restriction (Mori et al. 2012). Thus, while global changes in miRNA expression are linked to aging, the mechanism of this process remains unclear.
In this study, we investigated the regulatory network surrounding miRNA biogenesis genes, which led us to identify one possible mechanism for the aging-associated decline in miRNA abundance in C. elegans. Our bioinformatically derived network pointed to a central role for alg-1/Argonaute in feedback regulation of global miRNA biogenesis, with many transcription factors and miRNAs putatively targeting this gene. We experimentally demonstrate that miR-71, a particularly highly connected node in the network, regulates alg-1 expression post-transcriptionally through direct binding to the alg-1 3′ untranslated region (UTR). This regulatory relationship affects global miRNA abundance during aging and likely contributes to global alterations in mRNA expression. Further, we find significant changes in gene expression variability and life span variability in the absence of mir-71 gene expression. These results show a previously unexpected complexity to the regulation of miRNA biogenesis, and demonstrate mechanisms by which miRNAs have global impacts on gene expression and phenotypic outcomes during aging.
RESULTS
Building a miRNA-centric gene regulatory network
We attempted to infer the gene regulatory network encompassing aging-associated miRNAs, select transcription factors (TFs), and miRNA biogenesis genes from experimental results and computational predictions: TF-to-gene interactions were retrieved from TF binding data collected during development by the modENCODE project (Gerstein et al. 2010; Zhong et al. 2010; Niu et al. 2011), and miRNA-to-gene interactions were predicted by mirWIP (Hammell et al. 2008). We previously used a similar, though sparser, network to show that miRNAs and TFs regulate each other via direct feedback regulation during aging (Smith-Vikos et al. 2014). We have now expanded this network to investigate the role in aging of miRNA biogenesis genes and their regulators.
The current network comprises 90 TFs for which binding data were available through the modENCODE project, compared with 21 previously (Gerstein et al. 2010; Zhong et al. 2010; Niu et al. 2011); 81 aging-associated miRNAs (the union of those found in Ibañez-Ventoso et al. 2006; de Lencastre et al. 2010; Kato et al. 2011); and eight genes in the canonical C. elegans miRNA biogenesis pathway. The network represents a total of 5780 interactions (5039 TF-to-gene interactions and 741 miRNA-to-gene interactions) (Supplemental Table S1; Supplemental Fig. S1A). Note that miRNA biogenesis genes in this network lack outgoing edges, which would represent the regulatory interaction that they presumably exert on all miRNAs. These edges were omitted because they would otherwise bias the connectivity of the network in a nonproductive manner.
Overall, this network is densely connected and contains many feedback loops (Supplemental Fig. S1B; Supplemental Table S1): Nodes are highly interconnected, with two second-neighbor genes sharing 64.1% of their first-neighbor nodes on average. MiRNA biogenesis genes stand out in this network as some of the most connected genes (despite the omitted outgoing edges) (Supplemental Fig. S1C) and have more unique neighbors than do TFs or miRNAs (69 versus 58 and 58, respectively). Previous studies have reported that non-TF genes are less targeted by miRNAs (Shalgi et al. 2007; Cheng et al. 2011). In our network, miRNA biogenesis genes are targeted by miRNAs to the same extent that TFs are (Wilcoxon rank sum test, P = 0.1513). Conversely, TFs target miRNA biogenesis genes to the same extent that TFs target miRNAs (P = 0.5429. See also Materials and Methods). Additionally, miRNA biogenesis genes are regulated by 1704 interacting miRNA–TF pairs (i.e., TF regulated by a miRNA and vice versa, together forming feedforward loops with the miRNA biogenesis gene), which correspond to over one-third of all TF–miRNA pairs (4789). These findings suggest that miRNA biogenesis genes are key nodes for regulation by both miRNAs and TFs.
The C. elegans homolog of the miRNA-specific Argonaute gene, alg-1, is particularly highly networked: It has the highest number of unique neighbors in the network (72 TFs and 26 miRNAs), the highest number of incoming edges (Fig. 1A; Supplemental Table S2), and the third most densely miRNA-targeted 3′ UTR after daf-16 and daf-2 (Fig. 1B; Supplemental Table S3). The 1550-nt 3′ UTR of alg-1 is considerably longer than that of most other genes in the network, and of C. elegans genes in general, which have an average 3′ UTR length of ∼200 nt (Fig. 1C, alg-1 shown in pink) (Mangone et al. 2010). Since 3′ UTR evolution is driven by avoiding or retaining miRNA regulation (Stark et al. 2005; Cheng et al. 2009), its long 3′ UTR harboring many miRNA target sites suggests that miRNA-mediated regulation of alg-1 is biologically relevant. Additionally, alg-1 has a high betweenness centrality index, reflecting the importance of a node in influencing the interactions of other nodes and modules in the network (Fig. 1D; Supplemental Fig. S1D; Doncheva et al. 2012). While betweenness centrality and the number of neighbors are positively correlated, alg-1 is a clear outlier (Fig. 1D, arrow). Our new finding of the relationship of alg-1 to aging associated miRNAs points to an important role for alg-1 in regulating gene expression during aging and is consistent with the finding that adult-specific knockdown of alg-1 expression affects life span (Kato et al. 2011).
FIGURE 1.

alg-1 is an important node in the regulatory gene network during aging. (A) alg-1 and its first neighbors, ordered by highest connectivity (number of unique edges) from 6 o'clock counterclockwise. alg-1 is shown in the center in red. Seventy-two TFs and 26 miRNAs target alg-1. (Yellow nodes) TFs; (blue nodes) aging-associated miRNAs; (pink edges) miRNA-to-gene regulation; (blue edges) TF-to-gene regulation. (B) alg-1 is targeted by 26 different miRNAs through a total of 69 sites. (Left) Edge width reflects the number of target sites for each miRNA in the alg-1 3′ UTR. Nodes ordered by the number of target sites, as in A. (Right) Numeric representation of graphical data on left. (C) 3′ UTR length versus the number of aging-associated miRNA target sites. MiRNA target site numbers retrieved from mirWIP (Hammell et al. 2008). alg-1 is shown by the pink dot. (Red line) Linear fit. (D) Betweenness centrality distribution of nodes. The table (inset) shows the top 10 genes by betweenness centrality index. (Red line) Power-law fit. See also Supplemental Figure S1 and Supplemental Tables S1–S3.
Our findings of alg-1 are in stark contrast to those for alg-2: No aging-associated miRNAs are predicted to target alg-2 in our network. While alg-2 is also an Argonaute gene implicated in miRNA biogenesis, alg-1 and alg-2 are not completely redundant, preferentially associating with distinct sets of miRNAs during development and demonstrating distinct mutant phenotypes (Bukhari et al. 2012; Vasquez-Rifo et al. 2012). The regulatory relationships shown here may reflect possible molecular functional divergence of these genes.
miR-71 post-transcriptionally regulates alg-1 expression during aging
The long 3′ UTR and the number of miRNAs targeting alg-1 prompted us to investigate miRNA-mediated regulation of alg-1. In particular, we were interested in the potential regulation by miR-71, which has the highest number of predicted target sites for alg-1 (Fig. 1B). Despite shortcomings of miRNA target prediction software, specific miR-71 target sites are consistently found by multiple programs, fall within conserved regions of the nematode genome, and overlap known ALG-1 binding sites in the alg-1 3′ UTR during development (Fig. 2A, arrowheads; Enright et al. 2003; Krek et al. 2005; Lewis et al. 2005; Siepel et al. 2005; Zisoulis et al. 2010). These findings are particularly interesting in light of the known functions of miR-71 during aging. Work in our laboratory and others has shown that miR-71 promotes longevity by mediating signals from several known aging-associated pathways (de Lencastre et al. 2010; Boulias and Horvitz 2012; Lucanic et al. 2013). Within our network, miR-71 is the most connected node with respect to the total number of edges (incoming and outgoing) (Supplemental Table S2), suggesting a key regulatory role for miR-71.
FIGURE 2.

miR-71 directly regulates alg-1 post-transcriptionally. (A) Multiple miRNA target prediction algorithms predict miR-71 target sites in the alg-1 3′ UTR. Black arrowheads indicate miR-71 binding sites that are predicted by >3 algorithms, fall in conserved regions of the nematode genome, and overlap with known ALG-1 binding sites. ALG-1 binding sites (Zisoulis et al. 2010) and PhastCons nematode conservation track (Siepel et al. 2005) retrieved via UCSC Genome Browser (ce6). iCLIP-determined miR-71 binding sites during mid-L4 from Broughton et al. (2016). (B) alg-1 mRNA expression during aging in N2 wild-type background shown as mean ± s.e.m. (n = 2 biological replicates) (normalized to Y45F10D.4 expression). (C–E) alg-1 expression in N2 versus mir-71(n4115) background. (Blue) N2 wild-type or control; (red) mir-71(n4115). (C) alg-1 mRNA expression on Day 3 of adulthood (mean ± s.e.m. [n = 2 biological replicates], normalized to N2 expression levels). (D) Normalized mean GFP reporter expression over whole body at Day 3 of adulthood for control versus mir-71(n4115) background ([*] unpaired t-test, P < 0.01) (mean ± s.d.). (Top) Schematic of GFP reporter construct. miR-71 binding sites indicated by blue boxes. Similarly significantly higher GFP reporter expression was observed in the mir-71(n4115) background on Days 1–9. (E) Protein ALG-1 expression on Days 2 and 5 of adulthood. Protein expression normalized to β-tubulin or actin abundance. (F) (Top) Schematic of 3′ UTR regulation reporter constructs. Control 3′ UTR with wild-type miR-71 binding sites indicated by blue boxes, as in D. Experimental 3′ UTR with mutated miR-71 binding sites indicated by red x. (Bottom) Quantification of reporter expression. Normalized mean GFP signal over whole body at Day 3 of adulthood. Each is average of two integrated strains. GFP reporter expression is significantly higher for mutant 3′ UTR reporter compared with control (unpaired t-test, P < 1 × 10−9). Mean ± s.d. is shown. Similar results observed for Days 1 and 5 of adulthood. See also Supplemental Figure S2.
We next profiled alg-1 expression during aging via qRT-PCR. alg-1 mRNA levels sharply decrease between early- and mid-adulthood (Fig. 2B). This expression profile is consistent with those of published studies (Mori et al. 2012; Walther et al. 2015). Notably, this alg-1 expression profile is opposite to that observed for mir-71 promoter activity: A mir-71-promoter::GFP reporter increases in expression between young to mid-adulthood and declines gradually thereafter (de Lencastre et al. 2010).
We thus investigated whether miR-71 affects alg-1 expression. We found that alg-1 mRNA expression is higher in mir-71(n4115) animals which lack miR-71 expression (Fig. 2C). Similarly, a GFP reporter containing the alg-1 3′ UTR sequence [gfp::alg-1(zaIs5)], demonstrated increased expression throughout aging in mir-71(n4115) compared to a wild-type background (Fig. 2D). ALG-1 protein abundance was also elevated in the mir-71(n4115) strain (Fig. 2E).
To determine if the observed miR-71-mediated regulation was direct, we investigated whether mutating the two putative miR-71 binding sites indicated in Figure 2A would affect expression of the alg-1 GFP reporter construct. The sequences corresponding to these two miRNA seed binding sites were replaced with a sequence determined computationally to not affect binding by other miRNAs. Four independently generated integrated transgenic lines (two mutant, two control) were tested, and the GFP signal was consistently higher in animals carrying reporter constructs with mutated miR-71 binding sites (Fig. 2F). These findings provide evidence that miR-71 regulation of alg-1 is direct and mediated through specific miR-71 binding sites in the alg-1 3′ UTR.
Recently published biochemical data support the direct targeting of alg-1 by miR-71. Pasquinelli and colleagues found ∼5000 reproducible miRNA–target chimeric reads from their iCLIP (individual-nucleotide resolution crosslinking immunoprecipitation) method, corresponding to endogenous miRNA–target interactions (Broughton et al. 2016). Two miR-71 target sites are found in the alg-1 3′ UTR, one overlapping a site we mutated in our reporter construct and the other very near the second site we mutated (Fig. 2A, in red). These findings support our reporter construct results that miR-71 regulates alg-1 through direct interaction with at least one of the two sites we mutated. Additional genome editing in the future would constitute the ultimate confirmation of these interactions.
To directly test for a genetic interaction, we asked whether alg-1 expression affects the short life span of mir-71(n4115) animals (Supplemental Fig. S2A,B). Overexpression of alg-1 via a transgene suppresses the short life span of mir-71(n4115) animals (Supplemental Fig. S2A,C) while not affecting animal development, including brood size (Supplemental Fig. S2D). In addition, alg-1 RNAi, which shortens the life span of wild-type animals (Kato et al. 2011), did not further shorten the life span of mir-71(n4115) animals (Supplemental Fig. S2B). These findings are consistent with alg-1 functioning downstream from mir-71. Taken together with the requirement for alg-1 in miR-71 processing and function (Hammell et al. 2009; Vasquez-Rifo et al. 2012), these findings show that mir-71 and alg-1 form a direct regulatory loop.
miR-71 affects global miRNA abundance during aging
Argonaute proteins have been shown to stabilize mature miRNA expression post-transcriptionally (Diederichs and Haber 2007). We hypothesized that the increased alg-1 expression in mir-71(n4115) mutant animals would result in globally increased miRNA expression. We therefore profiled miRNA expression in wild-type and mir-71(n4115) animals during aging by small RNA-seq (Fig. 3). We chose Day 0 of adulthood as the “young” time point since alg-1 expression drops rapidly soon after (Fig. 2B). Day 5 of adulthood was chosen as the “old” time point based on the mir-71(n4115) animal mean life span which is 8.7 ± 0.1 d at 20°C (de Lencastre et al. 2010); at Day 5, most animals would still be alive even in the short-lived mir-71(n4115) strain. Our laboratory has shown that expression levels of specific genes vary within isogenic populations in correlation with the animals’ eventual life span; for example, animals that express more miR-71 at Day 3 of adulthood eventually live longer lives than do siblings that express less miR-71 (Pincus et al. 2011). By choosing a time point at which most animals are still alive, we avoided the possibility of survivorship bias. In total, 213 miRNAs were detected across twelve samples (miRBase release 20, [Kozomara and Griffiths-Jones 2014]).
FIGURE 3.

mir-71 affects global miRNA abundance. (A) Waterfall plot of miRNA expression fold change during aging in N2 wild-type (blue) versus mir-71(n4115) animals (red). MiRNAs ordered from left to right by magnitude of expression fold change (most positive to most negative) in N2. Corresponding miRNAs are shown in the overlapping position. (B) Histograms of expression fold changes during aging between N2 and mir-71(n4115) animals for microRNAs (top) and mRNAs (bottom). MicroRNA expression shifts globally, with a Spearman's rank correlation coefficient between N2 and mir-71(n4115) of 0.83 (Pearson correlation coefficient = 0.85). mRNA expression does not shift globally but appears to be de-regulated in mir-71(n4115) compared with N2 (Spearman's rank correlation coefficient = 0.55; Pearson correlation coefficient = 0.66). (C) Validation of miRNA expression during aging in N2 versus mir-71(n4115) animals by qRT-PCR. Expression levels were normalized to U18 and respective Day 0/1 time points. (Mean ± s.e.m. n = 2 biological replicates.) Note, y-axis is log scale. See also Supplemental Figure S3 and Supplemental Tables S4–S6.
In agreement with other studies, miRNA abundance in wild-type animals shows a trend of decline in expression with age, with 69% of miRNAs showing a negative expression change (at adjusted P < 0.1, there are 19 up- and 61 down-regulated miRNAs) (Fig. 3A [in blue], 3B top [in blue], and Supplemental Table S4) (Ibañez-Ventoso et al. 2006; de Lencastre et al. 2010; Mori et al. 2012). In aging mir-71(n4115) animals, however, miRNA expression generally increased, with only 37% of miRNAs showing negative expression changes (at adjusted P < 0.1, 25 up- and 18 down-regulated miRNAs) (Fig. 3A [in red], 3B top [in red], and Supplemental Table S5). This is in agreement with the TaqMan qRT-PCR validation of 13 miRNAs (Fig. 3C). The differences in miRNA expression fold change distributions are statistically significantly different (Welch two sample t-test, P = 3.01 × 10−7) (Supplemental Fig. S3A). In contrast, no obvious shift in miRNA abundance was observed in the short-lived daf-16(mu86) animals (Supplemental Fig. S3B), suggesting that increased miRNA abundance is not a general hallmark of a short life span but a direct result of increased alg-1 expression in mir-71(n4115) animals. There are also no other consistently predicted or experimentally detected target sites for miRNAs up-regulated with aging in the alg-1 3′ UTR, suggesting this mechanism to be specific to miR-71.
The widespread shift toward increased miRNA abundance in aging mir-71(n4115) animals is most striking when comparing the significantly differentially expressed miRNAs (adjusted P < 0.1) between control and mutant animals at Day 5. Note that 102 miRNAs are more abundant in the absence of mir-71 expression, whereas the only two miRNAs that were less abundant in the mutant animals were those deleted in mir-71(n4115): miR-71-5p and miR-71-3p (Supplemental Table S6). These findings suggest that miR-71 acts specifically during aging to down-regulate alg-1 expression, which decreases the global output of the miRNA biogenesis pathway.
mRNA expression is de-regulated in the absence of mir-71 expression
MiRNAs are predicted to target a large proportion of the transcriptome (Lewis et al. 2005). Thus, we next examined how the globally shifted miRNA expression affected mRNA expression by conducting RNA-seq on the same samples that were used for small RNA-seq. Unlike the case with miRNAs, there was no global shift in mRNA expression fold change with age between wild-type and mir-71(n4115) animals (Fig. 3B, bottom). Indeed, many of the mRNAs differentially expressed between young and old animals (adjusted P < 0.01; corresponding to ∼10% of detected mRNAs, Supplemental Tables S7, S8) are the same in the mir-71(n4115) and wild-type backgrounds (Fig. 4A). Among the most differentially expressed are collagen-related cuticle genes and major sperm protein genes.
FIGURE 4.

mRNA expression is dysregulated in aging mir-71(n4115) animals. (A) Comparisons of differentially expressed mRNAs during aging between N2 wild-type (blue) and mir-71(n4115) (red) (P < 0.01). (Left) Venn diagram of up-regulated genes during aging. (Right) Venn diagram of down-regulated genes during aging. (B) Enriched GO terms for differentially expressed genes during aging. Overlay of GO terms for genes up-regulated in N2 (dark blue), genes down-regulated in N2 (green), genes up-regulated in mir-71(n4115) (magenta), and genes down-regulated in mir-71(n4115) (cyan). Y-axis is the log of fractional difference between observed numbers of genes vs. expected numbers of genes. Positive values indicate enrichment and negative values indicate depletion. Statistically significant enrichment of GO term in a given experimental condition indicated by asterisk (*) (hypergeometric test, Benjamini–Hochberg adjusted P < 0.001). Image generated in PANTHER (Mi et al. 2013). (C) Adapted waterfall plot of mRNA expression fold change during aging in N2 wild-type (blue) versus mir-71(n4115) (red). Genes ordered from left to right by magnitude of expression fold change (most positive to most negative) in N2. Corresponding genes are shown in the overlapping position. See also Supplemental Tables S7 and S8.
We further investigated what functions these differentially expressed mRNAs represent by testing for Gene Ontology (GO) term enrichment. Most of the enriched GO terms for down-regulated mRNAs overlap between wild-type and mir-71(n4115) animals (Fig. 4B; Supplemental Table S9). These terms included protein and phosphorous metabolic processes and protein/macromolecule modification processes. The GO terms for mRNAs that are up-regulated in old compared to young animals, on the other hand, were distinct between wild-type and mir-71(n4115) animals (Supplemental Table S10). For example, GO terms for mRNAs up-regulated in old mir-71(n4115) animals include terms related to oxidative respiration, which are often enriched in aging studies (McCarroll et al. 2004; Hamilton et al. 2005; Kim 2007). We did not detect enrichment in oxidative respiration in old wild-type animals, however. The genes that correspond to GO terms enriched in mir-71(n4115) versus wild-type are not particularly enriched for miR-71 targets or for targets of down-regulated miRNAs. Thus, the enrichment of these terms may be reflecting, in general, the age-related stress experienced by these short-lived animals rather than specifically from changes in miRNA expression.
While global mRNA expression did not appear at first to be affected by the altered miRNA milieu in mir-71(n4115) animals compared with wild-type animals, we found dramatic differences between the two strains in the specific mRNAs that increase or decrease expression over time (Fig. 4C). Indeed, unlike for miRNAs, there is no discernible correlation between the direction or magnitude of mRNA expression changes through time in wild-type animals versus in mutant animals (Figs. 3B [legend], 4C). These findings are consistent with generally dysregulated mRNA expression in mir-71(n4115) animals, perhaps as a result of globally altered miRNA expression.
mir-71 affects gene expression variability between populations during aging
MiRNAs have been proposed to function as gene expression noise buffers, acting to decrease interindividual variability in gene expression levels (Hornstein and Shomron 2006; Li et al. 2009; Herranz and Cohen 2010; Ebert and Sharp 2012). Thus, we hypothesized that if the miRNA transcriptome were perturbed, as in mir-71(n4115) animals, this may be revealed as alterations in noise buffering. We investigated this by examining gene expression variability between populations in the mRNA-seq data described above. Often, gene expression variability, or “noise,” is measured directly by examining the variance of the distribution of gene expression levels across individual cells/organisms. In this case, however, we had only replicate measures of the mean gene expression level of a population. Fortunately, the variance in the means of replicate samples (i.e., standard error) has a simple relationship to the variance of the underlying sampling distribution. This relation has been used by others to analyze cell-to-cell variation in gene expression with population-level analyses (Anderson et al. 2014). Thus, while our data set represents population averages and contains a small number of replicates (n = 3, each consisting of RNA from approximately 400 individuals), we determined that it is feasible to conduct an exploratory investigation of gene expression variability over aging.
We calculated two different measures of gene expression variability, or “noise,” within our replicate sets (three replicates for each genotype and age), and compared these between N2 wild-type and mir-71(n4115) animals (Fig. 5A; Supplemental Fig. S4A,B) (coefficient of variation [CV] and power law-corrected noise, as in Vallania et al. 2014; see Materials and Methods). Our data suggest that gene expression becomes slightly noisier with increased age, as ∼4% more genes show increases in intersample variability in the Day 5 samples (Fig. 5A, left panel). This small bias is nevertheless statistically significant (binomial test, P = 0.004). Some studies have reported that gene expression becomes more heterogeneous within populations with age (Bahar et al. 2006; Somel et al. 2006), while others have reported results to the contrary (Golden and Melov 2004). Our results suggest while there is a modest increase in gene-expression heterogeneity with age in wild-type animals, it is likely of small magnitude.
FIGURE 5.

Gene expression variability during aging and life span variability is diminished in mir-71(n4115) animals. (A) Histogram of noise difference between Day 5 versus Day 0 (Day 5–Day 0) in N2 (left panel) versus mir-71(n4115) (middle panel) (right panel is composite). Y-axis is shown as density rather than counts. Slightly more genes demonstrate positive gene expression noise difference in N2 (+4%, binomial test P = 0.004). More genes demonstrate negative noise difference in mir-71(n4115) (+109%, binomial test, P < 2.2 × 10−16). (B) Histogram of noise difference between mir-71(n4115) and N2 wild-type [mir-71(n4115) − N2] at Day 0 (left panel) versus Day 5 (middle panel) (right panel is composite). Noise was calculated as (σ2)/(µ1.549275). (C) Life span variability of different strains plotted as CV of life span. Each dot represents a biological replicate experiment. See also Supplemental Figures S4 and S5.
In contrast, mir-71(n4115) animals show a remarkable shift toward decreased gene expression noise with increased age. In this genetic background, more than twice as many genes have decreased intersample variability, in older animals compared to younger, than have increased variability (decreased/increased = 2.09, binomial test, P < 2.2 × 10−16) (Fig. 5A, middle and right panels). This trend toward decreased noise in old mir-71(n4115) animals is consistent across either of the common definitions of gene-expression variability (variance scaled by the mean, or by a power-law fit to the empirical relationship between mean and variance; see Materials and Methods). Furthermore, these trends remain true even when we include additional RNA-seq data sets that were generated independently of the first three biological replicates (Supplemental Fig. S4C,D). This phenomenon of decreased gene expression noise seems to be adulthood-specific, based on our comparison of noise between mir-71(n4115) and wild-type animals at each time point (Fig. 5B).
Further, as determined by fluorescent microscopy, gfp::alg-1 expression variability between individuals declines more steeply with age in mir-71(n4115) animals than in controls (Supplemental Fig. S5C). These findings are consistent with an increased capacity for noise buffering due to increases in global miRNA abundance in aged mir-71(n4115) animals. In contrast, under decreased global miRNA abundance, as has been observed in alg-1 loss-of-function animals (Zisoulis et al. 2010), gene expression noise is greater than in wild-type animals at L4 stage using the same metric (Supplemental Fig. S5D), further supporting the contributions of miRNAs in gene expression variability.
miR-71 is associated with life span variability within populations
Individuals in a population of ostensibly isogenic C. elegans animals grown under identical conditions exhibit a variation of life spans that are correlated with the expression levels of specific miRNA genes like miR-71 (Pincus et al. 2011). Our observations above led us to investigate whether gene expression variability was similarly connected to life span variability. We calculated life span variability as the CV of animal life spans in a population and compared this value between mir-71 mutant and wild-type animals (Fig. 5C; Supplemental Fig. S5A). We consistently observe reduced life span variability in mir-71(n4115) animals compared with wild-type. While we have not explored the gene expression variability of animals that overexpress miR-71, such animals’ life spans are more variable than those of mir-71(n4115) (Supplemental Fig. S5A). Taken together, this evidence suggests a possible link between miR-71 expression variability, gene expression variability, and life span variability.
DISCUSSION
MiRNA-mediated regulation of age-dependent miRNA decline
Many studies in a variety of organisms have reported global miRNA decline during aging (Ibañez-Ventoso et al. 2006; de Lencastre et al. 2010; Noren Hooten et al. 2010; Inukai et al. 2012; Mori et al. 2012). Kahn and colleagues previously showed that expressions of miRNA biogenesis genes also decline with age (Mori et al. 2012), though the specific molecular mechanisms that affect aging-associated decline in miRNA biogenesis genes were not explored. Using a computational approach to inferring gene regulatory networks combined with experimental methods, we found that miRNA biogenesis genes are highly targeted by miRNAs and TFs, many of which have known roles associated with aging. In particular, our findings suggest one mechanism that establishes a central role for miR-71 in down-regulating global miRNA expression during aging, which produces a concomitant increase in mRNA expression variability during aging (Fig. 6). Based on the short life span phenotype of mir-71 loss-of-function animals (de Lencastre et al. 2010), we speculate that failure to decrease miRNA expression during mid-adulthood may be detrimental to normal aging. We propose that one function of miR-71 in promoting normal life span may be to directly repress the expression of alg-1 and thus indirectly decreasing miRNA abundance and/or activity and increasing gene expression noise.
FIGURE 6.
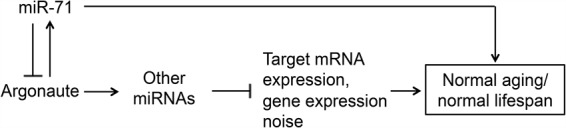
A model for miR-71-mediated regulation of miRNAs. miR-71 is in a composite feedback loop with alg-1. This interaction, by affecting alg-1/Argonaute expression, influences the expression of other miRNAs. Altered miRNA expression seems to in turn affect target mRNA expression and gene expression noise, ultimately affecting longevity.
The life span phenotypes of alg-1 and mir-71 loss-of-function and overexpression mutants cannot be adequately interpreted in the framework of a simple two-factor alg-1–mir-71 feedback system. For example, in a simple two-factor system, alg-1 overexpression would be expected to increase life span; however, this is not what we observe (Supplemental Fig. S2A). It is possible that in the absence of mir-71 expression, the level of alg-1 overexpression is restricted by the concomitantly increased expressions of other miRNAs; this is consistent with our observation of only modest ALG-1 protein expression increase. It is likely that there is a narrow range of optimal alg-1 expression, which is heavily regulated by multiple factors.
miR-71 has been studied extensively in the context of aging. Whereas most miRNAs decline with age, miR-71 is strongly up-regulated during early-to-middle adulthood (Ibañez-Ventoso et al. 2006; de Lencastre et al. 2010; Kato et al. 2011), and it is thought to function specifically in adults on aging-associated pathways to promote longevity (de Lencastre et al. 2010; Pincus et al. 2011; Boulias and Horvitz 2012; Nehammer et al. 2015). miR-71 is expressed throughout development (Kato et al. 2009), and based on its adult-specific activities, it is possible that the signals that regulate it during development and adulthood are distinct. Some aging-associated pathways that lie upstream of miR-71 genetically have been identified and include DNA damage response and calorie restriction; the influences of these upstream regulators are convoluted by the fact that they are often feedback regulated by miR-71 (de Lencastre et al. 2010; Pincus et al. 2011; Smith-Vikos et al. 2014). It is possible that miR-71 functions through different combinations of pathways in different tissues/cell types: Though broadly expressed, miR-71 activity in neurons was shown to be required for germline-mediated longevity (Boulias and Horvitz 2012). It remains to be determined whether the impacts of miR-71–alg-1 interaction are similarly tissue context-dependent. Our network also had many feedback interactions between miR-71 and TFs. Thus, miR-71 regulation appears to involve a complex interplay between multiple inputs and outputs.
Like in C. elegans, the 3′ UTRs of many miRNA-specific Argonaute genes in mammals are long. While miR-71 is not known in mammals, mammalian Argonaute genes also have many predicted miRNA target sites. We thus predict that feedback mechanisms exist in mammals to regulate global miRNA expression via Argonaute, perhaps in the course of aging as in C. elegans, and/or perhaps in novel contexts.
A miRNA-mediated mechanism that affects global gene expression
Our work further suggests that a miRNA-directed mechanism can explain at least some of the aging-associated molecular dysregulation often thought to be the result of accumulated damage over an organism's life. This notion is consistent with that of a recent proteomics study which suggested that observed increases in protein abundance during aging are likely due in part to de-repression of miRNA-mediated regulation (Walther et al. 2015). miR-71 up-regulation in adult life is consistently observed, and while the signals governing miR-71 expression are not known, miR-71 and miRNAs clearly play a role in propelling normal age-associated molecular decline.
miR-71 and gene expression variability
We find evidence of decreased interindividual variability in gene expression in mir-71 mutant populations compared to wild-type. As we have shown that miR-71 acts to decrease miRNA expression later in adulthood, we speculate that the reduced variance among mir-71 mutants is directly due to the global increase in miRNA expression. This is consistent with theoretical and experimental evidence that suggests that higher levels of microRNA targeting can decrease interindividual variability by providing robustness against fluctuations in mRNA levels (Hornstein and Shomron 2006; Osella et al. 2011; Ebert and Sharp 2012; Schmiedel et al. 2015). Essentially, in the presence of miRNA regulation, a higher transcription rate is required to achieve any given steady-state level of a target mRNA than without miRNAs present. With high transcription rates and thus a large transcribed pool, mRNA levels are proportionally less affected by short-timescale fluctuations in transcription due to “bursting” or other stochastic events, or due to fluctuations in intra- or extracellular transactivation signals.
The decrease in variability in life span in mir-71 mutants (and increase in life span variability in mir-71 overexpressers) further suggests that gene-expression variability may have consequences on phenotypic variability. Why might a mechanism such as miR-71, acting to increase interindividual variability, evolve? One possibility is that, as much of the increase in variance occurs in post-reproductive animals, this trait was not under selective pressure. Alternately, interindividual variability may reflect variability in the micro-environments that each individual experiences; with too many miRNAs around (e.g., absent miR-71), individuals may be hampered in their ability to respond to rapidly changing external signals. Finally, phenotypic variability per se may be an adaptive “bet-hedging” strategy, allowing a population of genetically identical individuals to confront an uncertain future by taking on a distribution of phenotypes (Martin 2009).
We take no position on which of these possibilities is most likely. Nevertheless, we have demonstrated a central role for miR-71 in down-regulating global miRNA expression during aging, with a concomitant increase in mRNA expression noise (Fig. 6). We propose that miR-71 promotes normal life span by directly repressing the expression of alg-1 and thus indirectly decreasing miRNA abundance and/or activity, and increasing gene expression noise. Overall, this study demonstrates the far-reaching impacts of miRNAs and miRNA regulation on gene expression during aging.
MATERIALS AND METHODS
Network construction
The network of regulatory factors was constructed as described previously (Smith-Vikos et al. 2014). MiRNA gene coordinates were retrieved from miRBase (release 21) (Kozomara and Griffiths-Jones 2014). A total of 225 TF ChIP-seq data sets, representing 90 independent TFs, were retrieved from the modENCODE project (Gerstein et al. 2010). MiRNA target predictions were retrieved from mirWIP (Hammell et al. 2008). Some TFs were not included in the mirWIP database. Relevant miRNA target sites and TF binding sites were retrieved using a custom Python script. Visualization and quantitative analyses of network topology were performed in Cytoscape (version 3.2.1) (Cline et al. 2007).
To address whether the dense miRNA targeting of miRNA biogenesis genes, matching that of TFs, was an artifact of mirWIP's predictions, we conducted a separate analysis using TargetScan's “most conserved sites” as the list of miRNA target sites (Friedman et al. 2009). We compared miRNA targeting of miRNA biogenesis genes, TFs, and non-TF/non-miRNA biogenesis genes by randomly sampling sets of eight genes (5000 samplings) from TFs and non-TF/non-miRNA biogenesis genes to match the number of miRNA biogenesis genes. Using this approach, miRNA biogenesis genes were significantly more targeted than TFs (P < 2.2−16, binomial test) and non-TF/non-miRNA biogenesis genes (P < 2.2−16, binomial test). A list of C. elegans TFs was retrieved from Reece-Hoyes et al. (2005) (restricting to 583 genes with gene names), and all remaining genes were retrieved from those annotated in WS235 (total 30,379 genes).
Animal strains
All animals were reared at 20°C. The following strains were used in this study: N2 Bristol (wild-type), mir-71(n4115) (MT12993), gfp::alg-1(zaIs5) (CT20), and daf-16(mu86). N2, mir-71(n4115), and daf-16(mu86) strains were provided by the Caenorhabditis Genetics Center; gfp::alg-1(zaIs5) was generated in our laboratory, as described in Chan and Slack (2009). Other strains generated for this study are described below. Hermaphrodites were used in this study.
Animal sample collection
For time course experiments, animals were transferred to culture plates containing 0.1 mg/mL 5-fluorodeoxyuridine shortly after L4/adult molt to prevent eggs from hatching. Young adult animals within 8 h post L4 molt (when most animals do not contain embryos) were collected as Day 0 adults. Animals were randomly assigned to time course sample groups.
Fluorescent microscopy analysis
The gfp::alg-1(CT20) strain was crossed into mir-71(n4115) animals to generate the gfp::alg-1;mir-71 strain. Fluorescent microscopy images were acquired on Days 1, 2, 3, 4, 5, 7, 8, and 9 of adulthood. gfp::alg-1 n = 5, 5, 7, 7, 5, 7, 5, 7, respectively. gfp::alg-1;mir-71 n = 8, 6, 7, 7, 7, 5, 5, 5, respectively.
The mutant and control alg-1 3′ UTR GFP reporter strains were constructed using pBS-F48gfp (Dr. Craig Mello, University of Massachusetts Medical School). This plasmid contains a GFP reporter fused to the 5′ of the alg-1 CDS and is driven by the alg-1 promoter sequence followed by the alg-1 3′ UTR sequence. The alg-1 CDS was omitted from the construct so as to function only as a reporter. To generate mutant alg-1 3′ UTR GFP reporter strains, predicted miR-71 binding site seed sequences (TCTTTC) were changed to CCCCCC via site-directed mutagenesis. The constructs were microinjected into N2 wild-type young adult animals as described in Mello and Fire (1995), integrated, and backcrossed 6× prior to further analysis. Fluorescent microscopy images were acquired on Days 1, 3, and 5 of adulthood. Control 3′ UTR n = 25, 31, 32, respectively. Mutant 3′ UTR n = 30, 44, 45, respectively.
Microscopy images were captured using a Zeiss AxioCam MRm camera on an Axioskop2 Plus microscope using AxioVision software (Release 4.8.2). Acquired GFP images were flat-field- and intensity-corrected (to calibrate between days) using custom software. Mean pixel intensity was taken as the measurement for that animal.
Comparison with iCLIP data
iCLIP findings were reported in ce10/WS220 genome assembly (Broughton et al. 2016). Because previously reported ALG-1 binding sites (Zisoulis et al. 2010) were shown in the ce6/WS190 assembly, we converted the iCLIP miR-71 binding sites to ce6 coordinates using the NCBI Remap tool (https://www.ncbi.nlm.nih.gov/genome/tools/remap).
Western blotting
Extracted protein was probed with anti-ALG-1 (1:5000, Pierce, PA1-031) and anti-β-tubulin (1:5000, Sigma-Aldrich, T4026) primary antibodies followed by goat α-rabbit IgG-HRP (1:5000, Santa Cruz Biotechnology, sc-2004) and goat α-mouse IgG-HRP (1:5000, Santa Cruz Biotechnology, sc-2031) secondary antibodies.
RNA-seq experiments
Young and old (Day 0 and Day 5 of adulthood, respectively) animals were collected in biological triplicate for N2 wild-type and mir-71(n4115) animals. “Day 0” is immediately after L4 molt, and embryos are still largely absent from animals; at Day 5, most embryos have already been laid. Total RNA was extracted from whole animals. For small RNA-seq, RNA libraries were size selected, multiplexed, and run on HiSeq2500 (Illumina) (single-ended, 50-bp reads). For mRNA-seq, RNA was poly(A) selected, multiplexed, and run on HiSeq2500 (single-ended, 75-bp reads) (N = 3); additional, independently generated biological replicate mRNA-seq data sets were run on HiSeq2000 (single-ended, 50-bp reads) (N = 2) (A de Lencastre and FJ Slack, unpubl.).
Sequencing reads were mapped to the C. elegans reference genome (WBcel235 assembly) using STAR (version 2.4.0h) (Dobin et al. 2013). For small RNA-seq, miRNAs were annotated against miRBase release 20 (http://www.mirbase.org/) (Kozomara and Griffiths-Jones 2014) using SeqBuster (Pantano et al. 2010). MiRNAs that were omitted from release 21 were manually removed from further analyses. For mRNA-seq, data processing and analysis was performed primarily using the bcbio-nextgen pipeline (https:// bcbio-nextgen.readthedocs.org/). Differential expression for both small RNA- and mRNA-seq data was tested using DESeq2; P-values were adjusted for multiple testing by Benjamini–Hochberg method (Benjamini and Hochberg 1995; Love et al. 2014). For small RNA-seq, size factor calculation was conducted against the number of sequences excluding those mapping to rRNA and repeat sequences.
GO term enrichment was tested in WebGestalt2 and PANTHER (Duncan et al. 2010; Mi et al. 2013). Enrichment was determined by the hypergeometric test, corrected for multiple testing, with an adjusted P-value threshold of <0.001.
Gene expression variability analysis
The linear regression of variance given expression mean for each gene across biological replicates was used to estimate the exponent value in the power law-like relationship between these values, as in Vallania et al. (2014) (Supplemental Fig. S3B). The average exponent value across the four experimental conditions was used to calculate noise (=σ2/µexponent). For each gene, we then evaluated the changes in noise between old (Day 5) and young (Day 0) animals by subtracting Day 0 noise from Day 5 noise. Similarly, using fold change ratios rather than differences in the noise measures does not change these findings (data not shown). We also examined expression range ([maximum − minimum]/read count) as a proxy for noise; findings were unchanged (data not shown). We also calculated noise including two additional, independently generated, biological replicate RNA-seq data sets (N = 5) (Supplemental Fig. S3C,D).
Life span assays
Animals were reared at 20°C for at least three generations prior to the commencement of egg prep for life span assays. Animals were placed on each plate containing FUdR at Day 0 of adulthood. Experiments were conducted in biological triplicate.
Code availability
Custom Python and R scripts used for data mining and noise analyses are available upon request.
DATA DEPOSITION
Small RNA-seq and mRNA-seq data have been deposited in NCBI's Gene Expression Omnibus (Edgar et al. 2002) and are accessible through GEO Series accession number GSE72234. Microarray data of N2 and alg-1(gk214) animals from Zisoulis et al. (2010) were accessed via GEO (GSE19138).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Rob Mitra (Washington University in St. Louis) for helpful discussions on gene expression variability. We also thank the Bioinformatics Core at Harvard School of Public Health for their help with RNA-seq data processing; computations for this project were run on the Yale Biomedical HPC and Harvard Medical School HPC. We also thank Chanatip Metheetrairut and members of the Slack laboratory for helpful discussions and critical reading of the manuscript. F.J.S. was funded by National Institutes of Health R01 AG033921; Z.P. was funded by National Insitutes of Health R00 AG042487. Some C. elegans strains were provided by the CGC, which is funded by National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440).
Author contributions: S.I. and F.J.S. conceived the project; S.I. performed the investigation; A.D.L. validated the study; S.I. and Z.P. performed the formal analysis; S.I. wrote the original draft; S.I., Z.P., A.D.L., and F.J.S. reviewed and edited the paper; F.J.S. supervised the study and acquired the funding.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.062190.117.
REFERENCES
- Alvarez-Saavedra E, Horvitz HR. 2010. Many families of C. elegans microRNAs are not essential for development or viability. Curr Biol 20: 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MZ, Gerstein AC, Wigen L, Baller JA, Berman J. 2014. Silencing is noisy: population and cell level noise in telomere-adjacent genes is dependent on telomere position and sir2. PLoS Genet 10: e1004436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dollé ME, Calder RB, Chisholm GB, Pollock BH, Klein CA, et al. 2006. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 441: 1011–1014. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B 57: 289–300. [Google Scholar]
- Boehm M, Slack F. 2005. A developmental timing microRNA and its target regulate life span in C. elegans. Science 310: 1954–1957. [DOI] [PubMed] [Google Scholar]
- Boulias K, Horvitz HR. 2012. The C. elegans microRNA mir-71 acts in neurons to promote germline-mediated longevity through regulation of DAF-16/FOXO. Cell Metab 15: 439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton JP, Lovci MT, Huang JL, Yeo GW, Pasquinelli AE. 2016. Pairing beyond the seed supports microRNA targeting specificity. Mol Cell 64: 320–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukhari SI, Vasquez-Rifo A, Gagné D, Paquet ER, Zetka M, Robert C, Masson JY, Simard MJ. 2012. The microRNA pathway controls germ cell proliferation and differentiation in C. elegans. Cell Res 22: 1034–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SP, Slack FJ. 2009. Ribosomal protein RPS-14 modulates let-7 microRNA function in Caenorhabditis elegans. Dev Biol 334: 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Bhardwaj N, Gerstein M. 2009. The relationship between the evolution of microRNA targets and the length of their UTRs. BMC Genomics 10: 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Yan KK, Hwang W, Qian J, Bhardwaj N, Rozowsky J, Lu ZJ, Niu W, Alves P, Kato M, et al. 2011. Construction and analysis of an integrated regulatory network derived from high-throughput sequencing data. PLoS Comput Biol 7: e1002190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, et al. 2007. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2: 2366–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lencastre A, Pincus Z, Zhou K, Kato M, Lee SS, Slack FJ. 2010. MicroRNAs both promote and antagonize longevity in C. elegans. Curr Biol 20: 2159–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichs S, Haber DA. 2007. Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell 131: 1097–1108. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doncheva NT, Assenov Y, Domingues FS, Albrecht M. 2012. Topological analysis and interactive visualization of biological networks and protein structures. Nat Protoc 7: 670–685. [DOI] [PubMed] [Google Scholar]
- Duncan DT, Prodduturi N, Zhang B. 2010. WebGestalt2: an updated and expanded version of the Web-based Gene Set Analysis Toolkit. BMC Bioinformatics 11(Suppl 4): P10. [Google Scholar]
- Ebert MS, Sharp PA. 2012. Roles for microRNAs in conferring robustness to biological processes. Cell 149: 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. 2003. MicroRNA targets in Drosophila. Genome Biol 5: R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP. 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. 2010. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330: 1775–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden TR, Melov S. 2004. Microarray analysis of gene expression with age in individual nematodes. Aging Cell 3: 111–124. [DOI] [PubMed] [Google Scholar]
- Hamilton B, Dong Y, Shindo M, Liu W, Odell I, Ruvkun G, Lee SS. 2005. A systematic RNAi screen for longevity genes in C. elegans. Genes Dev 19: 1544–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammell M, Long D, Zhang L, Lee A, Carmack CS, Han M, Ding Y, Ambros V. 2008. mirWIP: microRNA target prediction based on microRNA-containing ribonucleoprotein-enriched transcripts. Nat Methods 5: 813–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammell CM, Lubin I, Boag PR, Blackwell TK, Ambros V. 2009. nhl-2 modulates microRNA activity in Caenorhabditis elegans. Cell 136: 926–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz H, Cohen SM. 2010. MicroRNAs and gene regulatory networks: managing the impact of noise in biological systems. Genes Dev 24: 1339–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornstein E, Shomron N. 2006. Canalization of development by microRNAs. Nat Genet 38(Suppl): S20–S24. [DOI] [PubMed] [Google Scholar]
- Ibañez-Ventoso C, Yang M, Guo S, Robins H, Padgett RW, Driscoll M. 2006. Modulated microRNA expression during adult lifespan in Caenorhabditis elegans. Aging Cell 5: 235–246. [DOI] [PubMed] [Google Scholar]
- Inui M, Martello G, Piccolo S. 2010. MicroRNA control of signal transduction. Nat Rev Mol Cell Biol 11: 252–263. [DOI] [PubMed] [Google Scholar]
- Inukai S, de Lencastre A, Turner M, Slack F. 2012. Novel microRNAs differentially expressed during aging in the mouse brain. PLoS One 7: e40028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, de Lencastre A, Pincus Z, Slack FJ. 2009. Dynamic expression of small non-coding RNAs, including novel microRNAs and piRNAs/21U-RNAs, during Caenorhabditis elegans development. Genome Biol 10: R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Chen X, Inukai S, Zhao H, Slack FJ. 2011. Age-associated changes in expression of small, noncoding RNAs, including microRNAs, in C. elegans. RNA 17: 1804–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK. 2007. Common aging pathways in worms, flies, mice and humans. J Exp Biol 210: 1607–1612. [DOI] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. 2014. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42: D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, et al. 2005. Combinatorial microRNA target predictions. Nat Genet 37: 495–500. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15–20. [DOI] [PubMed] [Google Scholar]
- Li X, Cassidy JJ, Reinke CA, Fischboeck S, Carthew RW. 2009. A microRNA imparts robustness against environmental fluctuation during development. Cell 137: 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucanic M, Graham J, Scott G, Bhaumik D, Benz CC, Hubbard A, Lithgow GJ, Melov S. 2013. Age-related micro-RNA abundance in individual C. elegans. Aging (Albany NY) 5: 394–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangone M, Manoharan AP, Thierry-Mieg D, Thierry-Mieg J, Han T, Mackowiak SD, Mis E, Zegar C, Gutwein MR, Khivansara V, et al. 2010. The landscape of C. elegans 3′UTRs. Science 329: 432–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GM. 2009. Epigenetic gambling and epigenetic drift as an antagonistic pleiotropic mechanism of aging. Aging Cell 8: 761–764. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Murphy CT, Zou S, Pletcher SD, Chin CS, Jan YN, Kenyon C, Bargmann CI, Li H. 2004. Comparing genomic expression patterns across species identifies shared transcriptional profile in aging. Nat Genet 36: 197–204. [DOI] [PubMed] [Google Scholar]
- Mello C, Fire A. 1995. DNA transformation. Methods Cell Biol 48: 451–482. [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Thomas PD. 2013. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res 41: D377–D386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska EA, Alvarez-Saavedra E, Abbott AL, Lau NC, Hellman AB, McGonagle SM, Bartel DP, Ambros VR, Horvitz HR. 2007. Most Caenorhabditis elegans microRNAs are individually not essential for development or viability. PLoS Genet 3: e215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori MA, Raghavan P, Thomou T, Boucher J, Robida-Stubbs S, Macotela Y, Russell SJ, Kirkland JL, Blackwell TK, Kahn CR. 2012. Role of microRNA processing in adipose tissue in stress defense and longevity. Cell Metab 16: 336–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehammer C, Podolska A, Mackowiak SD, Kagias K, Pocock R. 2015. Specific microRNAs regulate heat stress responses in Caenorhabditis elegans. Sci Rep 5: 8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu W, Lu ZJ, Zhong M, Sarov M, Murray JI, Brdlik CM, Janette J, Chen C, Alves P, Preston E, et al. 2011. Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome Res 21: 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren Hooten N, Abdelmohsen K, Gorospe M, Ejiogu N, Zonderman AB, Evans MK. 2010. microRNA expression patterns reveal differential expression of target genes with age. PLoS One 5: e10724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osella M, Bosia C, Cora D, Caselle M. 2011. The role of incoherent microRNA-mediated feedforward loops in noise buffering. PLoS Comput Biol 7: e1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantano L, Estivill X, Marti E. 2010. SeqBuster, a bioinformatic tool for the processing and analysis of small RNAs datasets, reveals ubiquitous miRNA modifications in human embryonic cells. Nucleic Acids Res 38: e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincus Z, Smith-Vikos T, Slack FJ. 2011. MicroRNA predictors of longevity in Caenorhabditis elegans. PLoS Genet 7: e1002306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece-Hoyes JS, Deplancke B, Shingles J, Grove CA, Hope IA, Walhout AJ. 2005. A compendium of Caenorhabditis elegans regulatory transcription factors: a resource for mapping transcription regulatory networks. Genome Biol 6: R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiedel JM, Klemm SL, Zheng Y, Sahay A, Bluthgen N, Marks DS, van Oudenaarden A. 2015. Gene expression. MicroRNA control of protein expression noise. Science 348: 128–132. [DOI] [PubMed] [Google Scholar]
- Shalgi R, Lieber D, Oren M, Pilpel Y. 2007. Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput Biol 3: e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Vikos T, de Lencastre A, Inukai S, Shlomchik M, Holtrup B, Slack FJ. 2014. MicroRNAs mediate dietary-restriction-induced longevity through PHA-4/FOXA and SKN-1/Nrf transcription factors. Curr Biol 24: 2238–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somel M, Khaitovich P, Bahn S, Paabo S, Lachmann M. 2006. Gene expression becomes heterogeneous with age. Curr Biol 16: R359–R360. [DOI] [PubMed] [Google Scholar]
- Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM. 2005. Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell 123: 1133–1146. [DOI] [PubMed] [Google Scholar]
- Vallania FL, Sherman M, Goodwin Z, Mogno I, Cohen BA, Mitra RD. 2014. Origin and consequences of the relationship between protein mean and variance. PLoS One 9: e102202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez-Rifo A, Jannot G, Armisen J, Labouesse M, Bukhari SI, Rondeau EL, Miska EA, Simard MJ. 2012. Developmental characterization of the microRNA-specific C. elegans Argonautes alg-1 and alg-2. PLoS One 7: e33750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther DM, Kasturi P, Zheng M, Pinkert S, Vecchi G, Ciryam P, Morimoto RI, Dobson CM, Vendruscolo M, Mann M, et al. 2015. Widespread proteome remodeling and aggregation in aging C. elegans. Cell 161: 919–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M, Niu W, Lu ZJ, Sarov M, Murray JI, Janette J, Raha D, Sheaffer KL, Lam HY, Preston E, et al. 2010. Genome-wide identification of binding sites defines distinct functions for Caenorhabditis elegans PHA-4/FOXA in development and environmental response. PLoS Genet 6: e1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zisoulis DG, Lovci MT, Wilbert ML, Hutt KR, Liang TY, Pasquinelli AE, Yeo GW. 2010. Comprehensive discovery of endogenous Argonaute binding sites in Caenorhabditis elegans. Nat Struct Mol Biol 17: 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.