

This Outlook by Sump and Brickner highlights the work of Franks et al., which provides new mechanistic insight into the molecular basis by which Nup98 promotes gene activation in normal hematopoietic cells and how that process is altered by translocations to cause excess expression of developmental genes in leukemia.
Keywords: Nup98, Wdr82, Set1A, histone 3 Lys4 trimethylation, transcription, acute myeloid leukemia
Abstract
Nuclear pore proteins (Nups) interact with chromosomes to regulate gene expression and chromatin structure. A new study by Franks and colleagues (pp. 2222–2234) provides new mechanistic insight into the molecular basis by which Nup98 promotes gene activation in normal hematopoietic cells and how that process is altered by translocations to cause excess expression of developmental genes in leukemia.
The nuclear pore complex (NPC) is a large regulatory channel found in the nuclear envelope (NE), made up of ∼30 different nuclear pore proteins (Nups) (Wente and Rout 2010). These Nups localize both at the NPC and as soluble proteins within the nucleoplasm. In addition to their well-understood function in regulating the trafficking of proteins and RNA between the cytoplasm and the nucleus, Nups physically interact with chromatin, and this interaction impacts gene expression (Ptak et al. 2014). Translocations involving Nup98 cause acute myeloid leukemia (AML); translocations that fuse the N terminus of Nup98 with the C terminus of one of several partners cause aberrant gene expression, leading to transformation (Xu and Powers 2009; Franks and Hetzer 2013). However, because it has been unclear precisely how Nup98 mediates gene activation in normal cells, the molecular effects of these Nup98 translocations have remained unknown.
Previous work in flies and mammals has shown that Nup98 promotes recruitment of the MLL/Trx complex (which catalyzes the methylation of histone H3 on Lys4 [H3K4]) to developmentally regulated genes (Light et al. 2013; Xu et al. 2016). The Nup98 translocations that cause AML, including Nup98-HOXA9, Nup98-HOXD13, Nup98-Top1, and Nup98-Nsd1, share the N terminus of Nup98, suggesting that this portion of Nup98 is responsible for the changes in gene expression that drive AML (Oka et al. 2016; Xu et al. 2016).
Using both mouse hematopoietic progenitor cells (HPCs) and immortalized human cells as a model, Franks et al. (2017) explored both how Nup98 promotes gene activation in normal cells and (using Nup98-Nsd1) how Nup98 fusion proteins promote aberrant gene activation (Franks 2017). Chromatin immunoprecipitation combined with high-throughput sequencing of Nup98 in wild-type mouse HPCs revealed that Nup98 binding correlated with trimethylation of H3K4 even more so than transcription factors such as HOXB4 that are associated with active promoters within the nucleoplasm in HPCs. This aligns nicely with previous results showing an association between the FG-rich segments of Nup98 and the transcriptional coactivator CBP/p300 (Kasper et al. 1999). The investigators found that Nup98 physically interacts with Wdr82. Using a C-terminal-truncated form of Nup98 (that cannot interact with the NPC and is localized in the nucleoplasm) for coimmunoprecipitation/mass spectrometry identified the protein Wdr82 as a binding partner for nucleoplasmic Nup98. This protein is a member of the Wdr82–Set1A/B–COMPASS (complex of proteins associated with Set1) (WSC) complex, suggesting that Nup98 might recruit this complex to particular chromosomal sites to promote trimethylation of H3K4. Consistent with this idea, a large number of the Nup98-bound promoters also recruited the WSC complex. Furthermore, knockdown of Nup98 led to loss of Set1A recruitment to promoter regions and reduced H3K4 trimethylation at the promoters to which they both normally bind. This change correlates with changes in the expression of these promoters. This suggests that Nup98 is critically involved in recruitment of the WSC complex to active genes, promoting stronger expression (Fig. 1).
Figure 1.
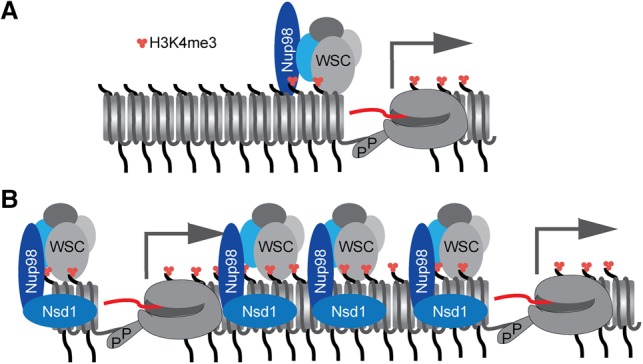
Model for Nup98 and Nup98-Nsd1 regulation of chromatin modification and gene expression. (A) In wild-type mouse HPCs, Nup98 facilitates the recruitment of the WSC complex to active promoters, which catalyzes H3K4 trimethylation, promoting RNA polymerase II-dependent transcription. (B) In cells expressing the Nup98-Nsd1 fusion protein, the WSC complex is recruited by Nup98-Nsd1 to ectopic sites, leading to inappropriate methylation of H3K4. This promotes inappropriate expression of genes such as HoxA9 and Meis1, leading to AML.
Because the N-terminal portion of Nup98 binds to WSC, this suggests that the domain may mediate recruitment of WSC to particular chromosomal loci. If so, then translocations to the N terminus of Nup98 would recruit both the fusion partner protein and WSC to these sites, potentially altering chromatin modifications and transcription. Indeed, Nup98-Nsd1 association with the HOXA locus leads to increased H3K4 methylation and expression. Moreover, the binding of the Nup98 fusion protein and the pattern of H3K4 methylation is much broader in cells expressing Nup98-Nsd1, and this correlates with increased expression of genes in the HOXA locus such as HOXA7, HOXA9, and HOXA10. Thus, the mechanism of recruitment of the fusion protein is somewhat different from that of endogenous Nup98: It reflects the recruitment of endogenous Nup98 but is either more efficient or more stable or can spread (perhaps because the fusion partners provide multivalency). This change in the recruitment of Nup98 leads to broader recruitment of WSC, altering local gene expression and driving AML.
Previous work had suggested critical roles for the interaction of Nup98 fusion proteins with the nuclear exportin Crm1 (Oka et al. 2016) and the H3K4 methyltransferase MLL1 (Xu et al. 2016) to recruit Nup98 to chromosomal sites, promoting AML. The study by Franks et al. (2017) proposes an additional/alternative explanation for the downstream effects of recruitment of Nup98 fusion proteins to chromatin, raising a number of interesting questions that will be addressed in the future. First, how is Nup98 recruited to particular chromosomal loci in both normal cells and AML? This likely plays a critical role in determining the severity of AML. The two most frequently up-regulated developmental genes in AML are HOXA9 and Meis1; in mouse models, up-regulation of HOXA9 alone leads to leukemia in 4 mo, while up-regulation of both HOXA9 and Meis1 leads to leukemia in 2 mo (Xu and Powers 2009). While Nup98-Nsd1 causes up-regulation of both of these genes, other Nup98 fusion proteins, such as Nup98-HOXD13, up-regulate HOXA9 but not Meis1. Work in flies and yeast has suggested that certain transcription factors can recruit Nup98 (Nup100 in yeast) to particular chromosomal sites (Light et al. 2013). If this is generally true, transcription factors may regulate recruitment of Nup98-WSC to particular promoters in normal cells and, in AML, to additional sites. Indeed, Nup98 binding is correlated with a number of transcription factors in stem cells (Liang et al. 2013). If this correlation is causative, perhaps transcription factors also recruit Nup98 to active genes in addition to the CRM1/MLL1-dependent mechanisms.
Second, does binding of Nup98 to the WSC occur prior to or after transcription? In flies and mammals, Nup98 binds to transcriptionally active loci, and NUP98 knockdown has broad, complex effects on transcription (Franks and Hetzer 2013). Thus, Nup98 may promote transcription, which could increase H3K4 methylation. However, in yeast and HeLa cells, Nup98/Nup100 is required for H3K4 methylation of repressed promoters to poise them for future expression, which suggests that Nup98 binding can be independent of transcription (Light et al. 2013). Resolving the role of transcription in the recruitment of Nup98 will clarify our thinking about the role of Nup98 in regulating gene expression.
Finally, what distinguishes Nup98-dependent and Nup98-independent mechanisms of Set1A/B–COMPASS recruitment, and how might this change during differentiation or in diseased states? Although Nup98 physically interacts with Wdr82, it does not stably interact with the full WSC, and there are many sites that bind Set1A/B–COMPASS but not Nup98. Curiously, Nup98 knockdown has broad effects on H3K4me3, including at sites to which it does not obviously bind. Understanding these mechanisms of WSC recruitment will provide important insights into both the basic regulation of H3K4 methylation and the molecular mechanisms that drive Nup98-dependent AML.
Acknowledgments
We acknowledge members of the Brickner laboratory for helpful discussions, and support from National Institutes of Health R01 GM118712.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.310359.117.
References
- Franks TM, Hetzer MW. 2013. The role of Nup98 in transcription regulation in healthy and diseased cells. Trends Cell Biol 23: 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks TM, McCloskey A, Shokirev M, Benner C, Rathore A, Hetzer MW. 2017. Nup98 recruits the Wdr82–Set1A/COMPASS complex to promoters to regulate H3K4 trimethylation in hematopoietic progenitor cells. Genes Dev (this issue) doi: 10.1101/gad.306753.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper LH, Brindle PK, Schnabel CA, Pritchard CE, Cleary ML, van Deursen JM. 1999. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol Cell Biol 19: 764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Franks TM, Marchetto MC, Gage FH, Hetzer MW. 2013. Dynamic association of NUP98 with the human genome. PLoS Genet 9: e1003308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light WH, Freaney J, Sood V, Thompson A, D'Urso A, Horvath CM, Brickner JH. 2013. A conserved role for human Nup98 in altering chromatin structure and promoting epigenetic transcriptional memory. PLoS Biol 11: e1001524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka M, Mura S, Yamada K, Sangel P, Hirata S, Maehara K, Kawakami K, Tachibana T, Ohkawa Y, Kimura H, et al. 2016. Chromatin-prebound Crm1 recruits Nup98-HoxA9 fusion to induce aberrant expression of Hox cluster genes. Elife 5: e09540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak C, Aitchison JD, Wozniak RW. 2014. The multifunctional nuclear pore complex: a platform for controlling gene expression. Curr Opin Cell Biol 28: 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wente SR, Rout MP. 2010. The nuclear pore complex and nuclear transport. Cold Spring Harb Perspect Biol 2: a000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Powers MA. 2009. Nuclear pore proteins and cancer. Semin Cell Dev Biol 20: 620–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Valerio DG, Eisold ME, Sinha A, Koche RP, Hu W, Chen CW, Chu SH, Brien GL, Park CY, et al. 2016. NUP98 fusion proteins interact with the NSL and MLL1 complexes to drive leukemogenesis. Cancer Cell 30: 863–878. [DOI] [PMC free article] [PubMed] [Google Scholar]