

This review by Hauer and Gasser presents an overview of the function of chromatin structure and its dynamic changes in response to genotoxic stress, focusing on both subnuclear organization and the physical mobility of DNA, and discusses the link between enhanced chromatin dynamics and histone loss.
Keywords: chromatin structure, histones, nuclear organization, DNA damage, nucleosome remodelers
Abstract
Chromatin is organized into higher-order structures that form subcompartments in interphase nuclei. Different categories of specialized enzymes act on chromatin and regulate its compaction and biophysical characteristics in response to physiological conditions. We present an overview of the function of chromatin structure and its dynamic changes in response to genotoxic stress, focusing on both subnuclear organization and the physical mobility of DNA. We review the requirements and mechanisms that cause chromatin relocation, enhanced mobility, and chromatin unfolding as a consequence of genotoxic lesions. An intriguing link has been established recently between enhanced chromatin dynamics and histone loss.
Improved live-imaging technologies and experiments that capture long-range contacts between chromosomal domains (chromosome conformation capture) have shown that the chromatin of yeast, fly, mouse, and human cell nuclei undergoes constant subdiffusive movement and plastic reorganization within interphase nuclei (for reviews, see Dekker et al. 2013; Dion and Gasser 2013). In yeast, the movement of genomic loci in vivo is influenced by both ATP levels (Marshall et al. 1997; Heun et al. 2001) and the strength of reversible contacts with fixed elements of the nucleus, such as nuclear pores and sites of chromatin anchorage along the nuclear envelope (Hediger et al. 2002; Agmon et al. 2013; Verdaasdonk et al. 2013; Strecker et al. 2016). In addition, local chromatin context and intrinsic forces between individual nucleosomes constrain chromatin fiber flexibility (Dekker 2008; Neumann et al. 2012; Hajjoul et al. 2013; Amitai et al. 2017; Hauer et al. 2017). Given that the density of nucleosome packing is regulated by ATP-dependent nucleosome remodeling complexes, it can be expected that these remodelers influence chromatin mobility as they alter nucleosome position.
In yeast, nucleosome remodelers indeed promote long-range chromatin movement and the relocation of genomic loci to specific sites of anchorage or repair (Dion et al. 2012; Neumann et al. 2012; Seeber et al. 2013a; Horigome et al. 2014; Strecker et al. 2016). Intriguingly, the subnuclear mobility of chromatin in yeast, as monitored by time-lapse microscopy, increases both at sites of induced double-strand breaks (DSBs) and genome-wide; that is, at undamaged sites in response to widespread damage. In both cases, the increase depends on both the INO80 remodeler and checkpoint kinase activation (Dion et al. 2012; Mine-Hattab and Rothstein 2012; Seeber et al. 2013a; Strecker et al. 2016). Moreover, enhanced DSB mobility was correlated with more efficient repair by homologous recombination (HR) (Dion et al. 2012; Mine-Hattab and Rothstein 2012; Neumann et al. 2012; Hauer et al. 2017). This increase in efficiency could stem from enhanced accessibility to the sites of damage and/or from a facilitated search for the homologous donor sequence, a necessary step in homology-driven repair (HDR). Ectopic homology is particularly important when the replicated sister chromatid is not available as a donor. Consistently, DSBs in mammalian genomes that exhibit increased movement correlate frequently with genomic translocations (Roukos et al. 2013). Moreover, the spatial proximity of sequences within the nuclear volume was found to influence the probability that two sequences would engage in ectopic recombination (Zhang et al. 2012; Agmon et al. 2013; Batte et al. 2017). While this suggested a function for both DNA movement and position, it remained unclear which molecular events drive changes in chromatin mobility, nor was it established how the composition of chromatin and/or its compaction respond to the DNA damage response (DDR) genome-wide.
Recently published work suggests that both reduced structural constraints and altered nucleosome occupancy change the mobility of chromatin in response to DNA damage. In other studies, the unfolding and spatial expansion of chromatin regions following DNA damage from ultraviolet (UV) light was observed (Adam et al. 2016; Strickfaden et al. 2016). Here we review the structural composition and organization of interphase chromatin and examine the changes that are known to occur during DNA damage and repair, focusing on studies that link nucleosome eviction and turnover to the repair of DNA lesions.
Nucleosomes: the building blocks of chromatin
Genomic DNA is organized into nucleosomes that are formed by wrapping 146 base pairs (bp) of DNA around an octamer of four core histones: H2A, H2B, H3, and H4 (Fig. 1). The hydrophobic histone fold domain (HFD), found in all core histones, mediates dimerization and prompts the formation of H2A–H2B and H3–H4 dimers, while exposed positive charges stabilize histone–DNA interactions (for reviews, see Malik and Henikoff 2003; Talbert et al. 2012). Whereas the nucleosomal core is compact, eight flexible lysine-rich histone tails extend from it, modulating internucleosomal contacts and providing binding sites for nonhistone proteins.
Figure 1.

Chromatin structure and function. Nucleosomes consist of octameric histone complexes and come in many different “flavors” owing to a multitude of histone variants and histone tail modifications. Both cytosolic and nuclear chaperones protect newly synthesized histones and assemble them into nucleosomes. The abundant high-mobility group proteins bind DNA and chromatin, altering nucleosome stability. In the middle panel, we show how chromatin remodeling complexes organize nucleosomes along DNA through ATP-dependent reactions. Actin-related proteins dimerize with actin within remodelers and chaperones and may mediate histone contacts. Chromatin folds into higher-order structures. Whole chromosomes assume three-dimensional organization in the nucleus, yet chromatin remains locally mobile. Chromatin interaction with the nuclear envelope and pores as well as with itself leads to subcompartmentation of the yeast nucleus.
The high-mobility group (HMG) proteins are the second most abundant class of proteins found on chromatin after the core histones (for review, see Bianchi and Agresti 2005). These small highly charged proteins have a variety of functions ranging from the establishment of proper chromatin architecture to the control of transcriptionally active and inactive chromatin regions. The three major families of HMG proteins include the HMGA proteins, which bind AT-rich DNA with an AT hook; the HMGN proteins, which bind within nucleosomes; and the HMGB proteins, which bind in the minor groove of DNA (Bianchi and Agresti 2005; Malarkey and Churchill 2012). Among these, HMGB1 has been well studied with respect to enhancing transcription (Celona et al. 2011). However, our understanding of HMG proteins in DNA repair is nascent at best.
Histone variants
Although the core histones form a highly conserved structure, nucleosome stability varies thanks to the incorporation of noncanonical histone variants and a vast array of post-translational modifications (PTMs) (Fig. 1; for review, see Campos and Reinberg 2009). Lower eukaryotes, such as the budding yeast Saccharomyces cerevisiae, have but one version of each core histone and only two histone variants: Htz1 and Hho1. The mammalian counterparts to these, called H2A.Z and H1, diverge significantly across species. Generally, there are but a few variants of H2B and H4 and many of H2A and H3, and the H1 histone is unique in being the only variant that lacks the central HFD. It functions as a linker histone, binding between nucleosomes and altering chromatin compaction and folding (Panday and Grove 2017).
H2A.Z is particularly interesting in the context of DNA damage. Apart from its role in regulating transcription (Guillemette et al. 2005; Li et al. 2005; Raisner et al. 2005; Albert et al. 2007), studies from multiple species implicate H2A.Z or Htz1 in DNA repair pathways (Kalocsay et al. 2009; Morillo-Huesca et al. 2010; Horigome et al. 2014). In addition to H2A.Z, mammals express H2A.X, an H2A variant with an additional C-terminal motif that is modified by DNA damage checkpoint (DDC) kinases on Ser139, to generate γH2A.X (Rogakou et al. 1998). In yeast, the canonical H2A contains the analogous phospho-target at Ser129. In flies, H2Aav serves as both H2A.Z and H2A.X. There are two additional H2A variants in mammals: macroH2A and H2A.Bbd (Barr body-deficient). Interestingly, macroH2A binds chromatin in a poly(ADP-ribose) (PAR)-dependent manner (Timinszky et al. 2009; Khurana et al. 2014) and accumulates at DNA DSBs as well as at sites of UV damage (Xu et al. 2012). This coincides with a dramatic reorganization of chromatin that promotes the binding of tumor suppressor and repair mediator BRCA1, which in turn promotes HDR (Khurana et al. 2014). Finally, centromere-specific H3 variants (CenpA in humans and Cse4 in budding yeast) define centromeric regions that allow proper chromosome segregation. Clearly, histone variants not only add diversity to nucleosome structure but help modulate nucleosome organization in response to stress and cell cycle stage.
Histone PTMs
In addition to the phosphorylation of H2A.X, layers of flexibility are conferred on nucleosomes by PTMs that target the N-terminal tails of histones H3 and H4 and the C-terminal tails of H2A and H2B (Campos and Reinberg 2009; Zentner and Henikoff 2013). PTMs affect chromatin structure in three ways: either through intrinsic effects on histone–histone interactions, through extrinsic effects on internucleosome contacts, or by providing binding sites for effector molecules. Intrinsic effects alter nucleosome stability by changing histone–histone or histone–DNA interactions, while extrinsic effects tend to influence longer-range contacts between nucleosomes, altering higher-order chromatin organization. Finally, chromatin-modifying proteins that recognize histone PTMs often trigger changes enzymatically. Using all three modes of action, histone variants and PTMs generate domains of transcriptionally active and inactive chromatin in response to both the environment and cell-intrinsic signals.
Among the best-studied environmentally induced modifications are those that occur in response to genotoxic stress (Smeenk and van Attikum 2013), and prominent among damage-linked histone modifications is the phosphorylation of histone variant H2A.X by the checkpoint kinases ATR and ATM (γH2A by Mec1 and Tel1 in budding yeast). This modification recruits repair factors and amplifies signaling by the DDC (Rogakou et al. 1998; Downs et al. 2000). In addition, H4 acetylation (H4K16ac) as well as histone H1 and H2B ubiquitylation coincide with the DDR. Acetylation of H4K16 is carried out by the histone acetyltransferases TIP60 and MOF (Murr et al. 2006; Sharma et al. 2010), with MOF regulating global levels of H4K16ac and TIP60 localizing to DSBs to modify H4K16 in a site-specific manner (Akhtar and Becker 2000). Importantly, cells depleted for either MOF or TIP60 were defective in DSB repair (Murr et al. 2006; Li et al. 2010; Sharma et al. 2010).
The E3 ligases that carry out ubiquitinylation also contribute to repair functions. The RNF20–40 complex monoubiquitinylates H2BK120 near DSBs, coincident with RNF8-mediated ubiquitination of histone H1 (Huen et al. 2007; Mailand et al. 2007; Thorslund et al. 2015). RNF168, another ubiquitin ligase, subsequently binds to ubi-H1 to target H2AK13 and K15 (Wang and Elledge 2007; Doil et al. 2009; Mattiroli et al. 2012). This cascade of ubiquitination events carried out by the combined action of RNF8 and RNF168 favor the recruitment of 53BP1, a protein that channels DSB repair to nonhomologous end-joining (NHEJ). In contrast, H4K16ac together with monoubiquitylated H2BK120 promotes chromatin relaxation and the recruitment of factors that mediate resection, favoring HDR (for an in-depth review, see Schwertman et al. 2016).
In addition to the PTMs that arise in response to genotoxic insults, the prelesion chromatin status can directly influence DNA repair and its outcome. For instance, H3K36 methylation, a mark for transcriptionally active chromatin, was shown to recruit the HR machinery to DSBs (Aymard et al. 2014; Pfister et al. 2014; Bleuyard et al. 2017). In contrast, methylated H4K20 (H4K20me2) provides a 53BP1-binding site that promotes NHEJ (Sanders et al. 2004; Botuyan et al. 2006; Pellegrino et al. 2017). Recent results extend this by proposing that unmethylated H4K20, which marks replicated DNA, favors HR over NHEJ (Saredi et al. 2016).
In summary, histone PTMs work alongside histone variants to define nucleosome structure and orchestrate repair. Both help partition the genome into domains that seem to have different degrees of accessibility, which in turn can affect the repair pathway used. This of course implies that there is an extensive regulation of the enzymatic machinery that controls the assembly, disassembly, and distribution of histones and nucleosomes.
Histone chaperones and nucleosome remodelers: architects of chromatin
Two large families of proteins, histone chaperones and nucleosome remodeling complexes, carry out the majority of these “architectural” tasks (Fig. 1). As soon as histones are synthesized in the cytoplasm, they are bound by dedicated histone chaperones that prevent improper nucleosome assembly, unspecific binding to DNA, and unscheduled histone degradation. Chaperones have overlapping as well as unique roles in histone delivery, buffering, and transfer (for reviews, see De Koning et al. 2007; Gurard-Levin et al. 2014). Following the chaperone-mediated assembly of nucleosomes, their variant histone composition, compaction level, and position with respect to sequence motifs can all be altered by chromatin remodelers (Fig. 1). Whereas remodelers generally do not assemble new nucleosomes from histone dimers, they nonetheless bind histones through their actin-related protein subunits (e.g., Arp4, Arp5, Arp6, Arp7, Arp8, and Arp9 in S. cerevisiae) (Fig. 1) and often work in concert with chaperones. Nucleosome remodeling complexes generally contain a large Swi2/Snf2 (switch/sucrose nonfermenting) ATPase as their catalytic subunit and use energy from ATP hydrolysis to exchange histone variants or evict, slide, and space nucleosomes along DNA. Even though their modes of action overlap to a certain extent, each remodeler carries out a dedicated function with respect to gene transcription, DNA replication, repair, and chromatin structure (for reviews, see Clapier and Cairns 2009; Papamichos-Chronakis and Peterson 2013). For the majority of Snf2 ATPases, this specificity depends on their assembly into large macromolecular complexes; each complex contains its own set of auxiliary subunits, which can include DNA helicases, histone-modifying enzymes, histone mark readers, Arps, and actin itself. Two exceptions to this, however, are the human Alc1 (amplified in liver cancer 1) and SMARCAD1 (yeast Fun30 [function unknown protein 30]) ATPases, which contribute to repair pathways on their own (for reviews, see Seeber et al. 2013b; Jeggo and Downs 2014).
Subnuclear organization and the higher-order folding of chromatin
One consequence of regular nucleosome packing, particularly in the presence of histone H1, is the folding of the 10-nm nucleosomal fiber into a solenoid helix called the 30-nm fiber (Fig. 1). Whereas the exact structure of these folded 10-nm fibers is debated, it is nonetheless important to note that chromatin domains assume different levels of higher-order packaging, which helps prevent intermingling of chromosomes in nuclear space. Particularly in higher eukaryotes, entire chromosomes were shown to occupy distinct “territories” with an ordered radial distribution that correlated with the density of genes (Gilbert et al. 2005). Budding yeast nuclei have a slightly different mode of organization, with chromosomes assuming a loose Rabl configuration in which all centromeres are clustered near the membrane-embedded spindle pole body (SPB), and telomeres are found opposite this at sites around the nuclear rim (Fig. 1; Gotta et al. 1996; Bystricky et al. 2005; Duan et al. 2010). Finally, a third element of nuclear organization concerns the distribution of repetitive sequences and silent tissue-specific genes into heterochromatin, a compact chromatin state that has H3K9 methylation and low levels of transcription and forms subnuclear clusters. Given the risk that resected repeats might misanneal to homologous but inappropriate sequences, it is not surprising that heterochromatic domains have evolved mechanisms to suppress HDR (Amaral et al. 2017). Similarly, in yeast, the nucleolar subcompartment, which contains tandem rDNA repeats, suppresses HR-mediated repair (Torres-Rosell et al. 2007). Thus, across multiple eukaryotic species, both subnuclear context and chromatin states influence repair pathways (Torres-Rosell et al. 2007; Oza et al. 2009; Chiolo et al. 2011; Agmon et al. 2013; Horigome et al. 2014; Tsouroula et al. 2016; Batte et al. 2017) just as they regulate transcription.
Chromatin movement and position during DSB repair
DDC activation and HDR
All cells, whether post-mitotic or proliferating, are challenged by thousands of DNA-damaging events every day, and damage generally occurs irrespective of the underlying sequence. Damage is induced by exogenous agents (e.g., radiation, radiomimetic cancer drugs, or toxins) as well as endogenous activities (e.g., free radicals arising from cell metabolism or replication errors) (Lindahl and Barnes 2000; Jackson and Bartek 2009), with one of the most common sources of damage being solar UV light, which forms thymidine base dimers. Other modifying agents, such as methylmethane sulfonate (MMS), add bulky adducts to DNA. If left unrepaired, such adducts pose a barrier to both DNA and RNA polymerases, causing the formation of single-stranded stretches of DNA and, occasionally, DSBs. Similar problems arise when nucleotide pools are depleted during DNA synthesis, a condition that triggers replication fork collapse and break formation. In contrast to this, ionizing radiation (IR) or radiomimetic antibiotics such as zeocin (or the related bleomycin) directly induce DNA nicks and, at low frequency, DSBs (Povirk et al. 1977). Different repair pathways have evolved to deal with these diverse lesions, yet common among many pathways is the DDC response (Fig. 2A). The DDC is initiated by the conserved damage-sensing kinases ATR and ATM. These activate downstream kinases and other enzymes, which initiate and fine-tune repair pathway choice and the delay and resumption of cell cycle progression.
Figure 2.
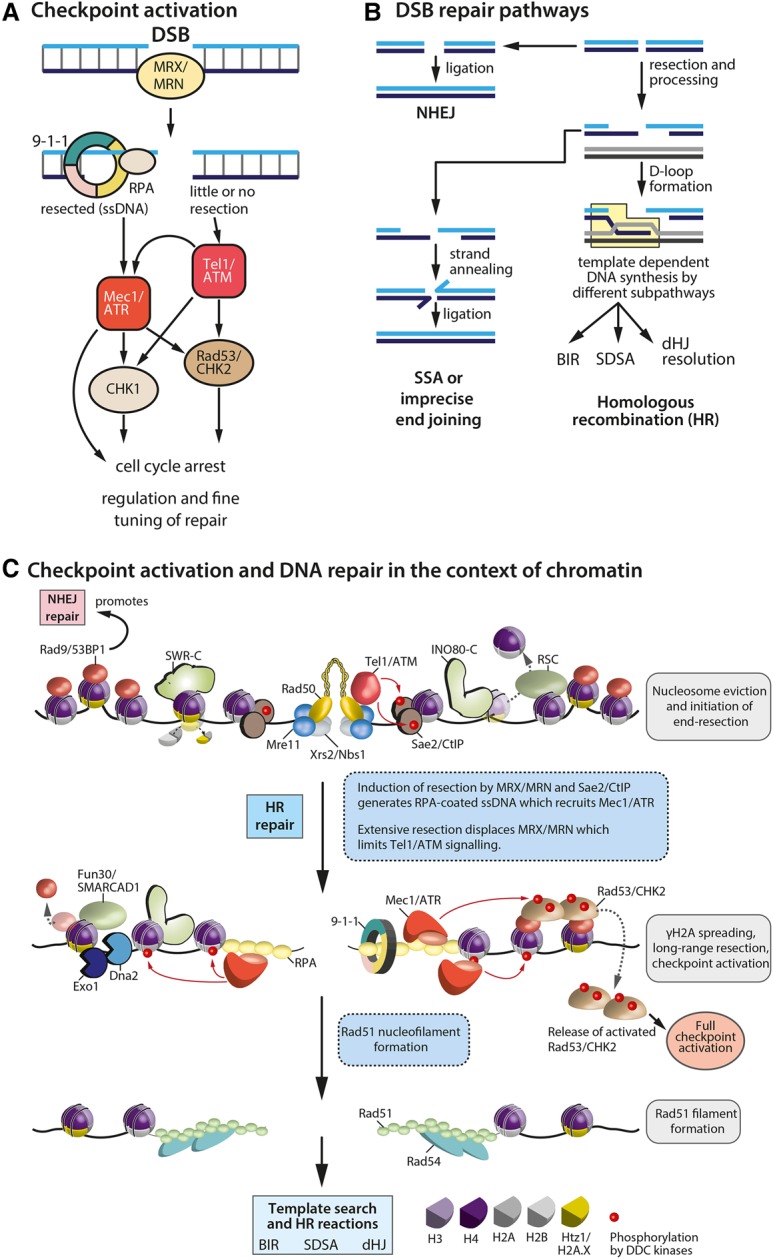
Checkpoint activation and DSB repair in the context of chromatin. (A) Proteins involved in DDC activation in response to a DSB. The checkpoint stalls the cell cycle and coordinates repair proteins acting at the site of damage. Mammalian proteins are capitalized. (B) Three of the main DSB repair pathways are shown: NHEJ, single-strand annealing (SSA), and HR (see the text for details). HR intermediates are resolved by subsequent pathways, resulting in different repair outcomes (light-blue box). (BIR) Break-induced replication; (SDSA) synthesis-dependent strand annealing; (dHJ) double-Holliday junction. (C) Effects of both the DDC and the DSB repair functions integrate on the chromatin template. The key steps and the main players during homology-directed DSB repair are listed. The first panel highlights early steps after DSB formation. The second panel illustrates DSB processing and spreading of the DDC signal. The last panel shows the Rad51 nucleofilament before homology search and after strand invasion and repair.
Two opposing pathways dominate DSB repair: NHEJ and HR (Fig. 2B). During NHEJ, free DNA ends are rapidly ligated, which in principle can be error-free. However, if there has been removal or modification of the terminal bases, as often occurs, then repair by NHEJ will be mutagenic. In contrast, DSB repair by HR copies information from an identical intact DNA template (the sister chromatid or an ectopic donor) to restore the original sequence without mutation (Heyer et al. 2010; Symington and Gautier 2011). While this is most often error-free repair, improper annealing or repeat-driven recombination can also generate large deletions, insertions, and genomic translocations. Mammalian cells prevalently use NHEJ yet have substantial levels of HR during DNA replication. Yeasts generally carry out repair during S or G2 phase and preferentially template HR-mediated repair from the sister chromatid. Like mammals, however, yeast also has a fully functional NHEJ pathway that functions throughout the cell cycle.
The first step in HR-mediated DSB repair is the recruitment of the Mre11–Rad50–Nbs1 complex (Mre1–Rad50–Xrs2 in budding yeast), which directly activates the checkpoint kinase ATM (Tel1) or triggers resection that in turn recruits and activates the alternative DDC kinase ATR/Mec1 (Fig. 2C). Transducer proteins (53BP1/Rad9 and MDC1) help transmit the damage signal to downstream effector kinases (CHK2/Rad53 and CHK1) and trigger a full checkpoint response. Whereas ATR/Mec1 stays bound to the ssDNA–RFA complex that recruits it, the response spreads throughout the entire nucleus thanks to dispersion of these weakly bound effector kinases. These stimulate the recruitment of repair proteins and downstream events, such as cell cycle arrest and transcriptional induction of functions necessary for repair. For an in-depth review of DDC activation, see Ciccia and Elledge (2010).
Early repair factors such as the RecQ helicase BLM (yeast Sgs1) can reverse fold-back structures to facilitate resection by the exonucleases CtIP/Sae2, Exo1, and Dna2, which also has a helicase domain. The requirement for helicases highlights the fact that DNA structure changes during repair. This becomes especially important during HR-mediated repair, which requires access to both the lesion and a homologous template for strand invasion (Heyer et al. 2010). Nucleosomes can obstruct end processing and repair factor access (Fig. 2C), and, to overcome this barrier, several chromatin remodeling complexes are recruited to DSBs, including INO80-C, SWR1-C/SRCAP-C, and RSC in yeast and BAF, CHD3/CHD4, and INO80 in mammals (for reviews, see Seeber et al. 2013b; Jeggo and Downs 2014). Their action is thought to increase chromatin accessibility. For instance, INO80-C binds to H2A.Z-containing nucleosomes, where it exchanges H2A.Z–H2B dimers for canonical H2A–H2B (Papamichos-Chronakis et al. 2011; Lademann et al. 2017). This has been shown recently to be important for both DNA end resection and presynaptic filament formation (Lademann et al. 2017). The reverse action is catalyzed by SWR1-C, which incorporates H2A.Z–H2B dimers into nucleosomes in a stepwise and unidirectional fashion (Luk et al. 2010). Besides INO80 and RSC (Chai et al. 2015), a third remodeler that has been implicated in DSB repair is Fun30 and its human homolog, SMARCAD1. Both the yeast and human proteins appear to promote Exo1- and Dna2-mediated long-range resection through chromatin (Chen et al. 2012; Costelloe et al. 2012; Eapen et al. 2012). In yeast, Fun30 was shown to catalyze histone dimer exchanges and nucleosome repositioning (Awad et al. 2010), while, in mammals, it promotes telomere maintenance by recombination in the absence of telomerase (Cox et al. 2016). The binding of Fun30 at DSBs depends on an interaction network that includes the damage scaffold protein TOPBP1/Dpb11 and the 9-1-1 complex (Bantele et al. 2017). The resection that it promotes creates long 3′ ssDNA overhangs flanking the break, which are in turn bound by the ssDNA-binding complex RPA, which recruits ATR/Mec1 that extends H2A.X phosphorylation (generating large stretches of γH2A.X in mammals and γH2A in yeast). Finally, following resection and repair, Fun30 was shown to have an important role in deactivating the DDC kinase cascade (Chen et al. 2012; Eapen et al. 2012).
Once resected, Rad52 mediates the replacement of RPA by Rad51, triggering the formation of a Rad51 nucleofilament, which engages in a physical search for its homologous template. This process of homology search has long been considered the one step in repair that a priori requires DNA movement by either three-dimensional diffusion or “sliding” along the chromatin (Wilson et al. 1994). To find and invade a double-stranded donor template is inherently difficult, and it is even harder if the homologous donor is spatially distant or sequestered in another chromatin domain. This may be facilitated through controlled relocalization events and events that increase the physical mobility of both damaged and undamaged DNA in response to stress (Dion and Gasser 2013).
DSB repair within the nuclear space—a matter of chromatin context
The accumulation of repair factors in “foci” has been studied for years in mammalian cells. However, the shift of DSBs to subnuclear structures for repair was initially characterized in yeast (Nagai et al. 2008) and was only recently shown to occur during DSB repair in flies and mammals, particularly for breaks occurring in a heterochromatin context. Early work in yeast focused on telomeres, which bind two integral inner nuclear membrane proteins—Mps3 (a SUN [Sad1–Unc-84-related] domain protein) and Esc1 (an acidic protein)—that suppress subtelomeric recombination (Schober et al. 2009). Similarly, the highly repetitive rDNA array is tethered at the nuclear envelope through the LEM2 homolog Heh1/Src1, which also helps repress recombination among rDNA repeats (Mekhail et al. 2008). The fact that spontaneous Rad52-containing foci, which form during canonical HR, are specifically depleted from the nuclear envelope (Bystricky et al. 2009) led to the hypothesis that binding the nuclear envelope per se might suppress recombination and favor alternative pathways of repair (Taddei and Gasser 2012; Horigome et al. 2016).
A further player in damage positioning is the nuclear pore complex (NPC). High-throughput synthetic phenotypes coupled with targeted mutant analysis showed that the NPC's Nup84 subcomplex serves as a binding site for persistent DSBs that cannot be repaired by either classic NHEJ or HR (Nagai et al. 2008; Kalocsay et al. 2009; Oza et al. 2009) and for collapsed replication forks (Nagai et al. 2008; Su et al. 2015). Subtelomeric breaks (Therizols et al. 2006) and short telomeres in the absence of telomerase (Churikov et al. 2016) also relocate to the NPC in order to be processed for alternative pathways of repair in cells lacking telomerase. Unlike fork-associated damage, DSBs were shown to bind the Nup84 complex in both G1 and S phase and to not require either the chromatin remodeler INO80-C or the recombinase activity of Rad51 (Horigome et al. 2014). On the other hand, the relocation of DSBs to Mps3 occurred uniquely in S/G2 phase and was shown to be dependent on resection and Rad51 in both budding and fission yeasts (Horigome et al. 2014; Swartz et al. 2014). Intriguingly, DSB binding to Mps3 binding required the two remodelers INO80-C and SWR1-C, while break relocation to the NPC required only SWR1-C and occurred without extensive resection (Yoshida et al. 2010; Horigome et al. 2014). Consistent with the notion that Mps3 and NPC mediate different functions in repair, it was shown that sister chromatid exchange defects were additive upon sequential loss of Mps3 and Nup84 (Horigome et al. 2014).
The Slx5/Slx8 SUMO targeted ubiquitin ligase (STUbL) is also recruited to DSBs and forms a bridge to the Nup84 subcomplex of the NPC in both yeast (Nagai et al. 2008) and flies (Ryu et al. 2015). Its loss enhances the rate of gross chromosomal rearrangements, which arise from spontaneous ectopic recombination events (Zhang et al. 2006; Nagai et al. 2008). The STUbL Slx5/Slx8 was proposed to play an active role in relocation, given that it accumulates at breaks (Cremona et al. 2012; Psakhye and Jentsch 2012; Sarangi and Zhao 2015).
Four recent studies (Ryu et al. 2015; Su et al. 2015; Churikov et al. 2016; Horigome et al. 2016) shed light on the function of SUMOylation and STUbL binding not only at induced DSBs but also at short telomeres and collapsed replication forks (for reviews, see Seeber and Gasser 2016; Amaral et al. 2017). In yeast, poly-SUMOylation mediated by the E3 ligases Siz2 and Mms21 recruited Slx5/Slx8 to persistent breaks in G1 phase, and both Slx5 and Slx8 were necessary for damage relocation to pores (Horigome et al. 2016). In contrast, during S phase, mono-SUMOylation by Mms21 was sufficient to shift a resected break to Mps3 even in the absence of Slx5/Slx8. Functionally, the interaction of DSBs with Slx5/Slx8 at the NPC favored repair by break-induced replication (BIR) and/or imprecise end-joining. In addition to DSBs, Churikov et al. (2016) showed that the shift of critically short telomeres to the yeast NPC made use of a SUMO-dependent pathway that also involved the binding of Slx5/Slx8 to poly-SUMOlyated proteins. This facilitated recombination-mediated telomere maintenance through either de-SUMOylation or proteasomal degradation (Nagai et al. 2008; Churikov et al. 2016). In a similar manner, heterochromatic DSBs in Drosophila cells were seen to move toward the edge of heterochromatin (Chiolo et al. 2011) and then undergo a second relocation event to the NPC (Ryu et al. 2015). Collectively, these results elucidated a conserved SUMOylation-dependent pathway that facilitates the association of damaged loci with either the NPC or a SUN domain-binding site in the inner nuclear membrane, each favoring distinct steps in repair.
Repair in heterochromatic domains: integrating DSB relocation with chromatin structure
Like the DSB movement to pores, the extrusion of DSBs from heterochromatin has been observed in multiple contexts. It is important to note that heterochromatic regions consist of highly repetitive sequences that are prone to nonallelic ectopic recombination. Illegitimate recombination events in heterochromatin can result in translocations, duplications, and deletions, reminiscent of the alterations found in human pathologies (Pearson et al. 2005). Moreover, NHEJ in repetitive DNA, even if imprecise, is potentially less problematic given that small insertions or deletions do less harm within tandem repeats than in genes. Given these considerations, it is clear that repair within these regions needs to be tightly regulated in order to preserve genome integrity.
The major domain of repetitive sequence in yeast is the rDNA, which is found in the nucleolus. Given the large number of repeat units, a large fraction of the rDNA is usually kept in a heterochromatic state. An early study in yeast showed that an induced DSB in the rDNA shifts into the nucleoplasm out of the nucleolus in order to be processed for repair by HR (Torres-Rosell et al. 2007). Extrusion from the nucleolus depended on the SUMOylation of Rad52 by the Smc5/6–Mms21 SUMO ligase complex. Mutations that abrogated this shift caused rDNA hyperrecombination and genome instability, although one cannot exclude that the mutants also altered other steps in repair (Torres-Rosell et al. 2007). Recently, two reports showed that this pathway is conserved in human cells (Harding et al. 2015; van Sluis and McStay 2015). In both studies, persistent DSB in the rDNA shifted away from the nucleolus and its repetitive DNA (Harding et al. 2015; van Sluis and McStay 2015). This coincided with ATM-dependent inhibition of RNA polymerase I, an important step that helps prevent collision between the repair and transcription machineries.
Pericentric and centromeric repeats in mammals and flies also form heterochromatic compartments, and the processing of DSBs within pericentric regions in Drosophila is strikingly similar to that observed in the yeast rDNA. Irradiation-induced DSBs were shown to be relocated out of heterochromatin prior to Rad51 recruitment and HDR (Chiolo et al. 2011). Moreover, pericentric DSBs were subject to SUMOylation-dependent anchoring to the NPC after being shifted to the edge of the heterochromatic domain (Ryu et al. 2015). In mouse cells, G1-phase pericentric breaks that were repaired by NHEJ remained positionally stable, whereas breaks in S/G2 were resected and relocated to the edge of the pericentric domain (Tsouroula et al. 2016). Intriguingly, breaks in the centromeric domain (which, unlike pericentric chromatin, does not bear H3K9 methylation) behaved differently; however, in both cases, repair by HR showed heterochromatic exclusion. In mice, DSB relocation was dependent on DNA resection followed by Rad51/BRCA2 binding, which stabilized DSBs to prevent repair by a more deleterious recombination pathway called single-strand annealing (SSA) (Tsouroula et al. 2016).
Given that the repair of heterochromatic DSB by HR occurs only after breaks relocate away from the repressive compartment, it was proposed that the kinetics of repair within heterochromatin might be slower than in euchromatin (Torres-Rosell et al. 2007; Ayoub et al. 2008; Chiolo et al. 2011; Jakob et al. 2011; Ryu et al. 2015; van Sluis and McStay 2015). Whereas some data suggested that HR and NHEJ in heterochromatin are temporally distinct (Harding et al. 2015), a recent study in Drosophila argued that a single induced DSB is repaired with roughly the same kinetics in euchromatic and heterochromatic domains and that both environments were able to support both pathways of repair (Janssen et al. 2016). Nonetheless, induced DSBs shift away from heterochromatin like irradiation-induced breaks (Chiolo et al. 2011; Ryu et al. 2015). Thus, in both flies and mice, there is a conserved segregation of HR-mediated repair events away from domains that are rich in sequence repeats (Tsouroula et al. 2016).
It makes sense to sequester the breaks that occur in repeats away from ectopic copies that could generate aberrant recombination events, and some of the key triggers for DSB relocation were identified. Nevertheless, it was unclear how highly compacted heterochromatin itself might change following DNA damage. One attractive hypothesis for the long-range shifts of broken DNA was that changes in flanking nucleosome organization were at the root of the rearrangement.
Heterochromatin unfolds in response to DNA damage
Heterochromatin is generally compacted and relatively inaccessible to RNA polymerases. This restrictive chromatin nature relies primarily on a dense packing of nucleosomes, repressive histone marks (H3K9me3 and H3K27me3), and the presence of other heterochromatin-binding proteins, such as HP1 and KAP-1 (Goodarzi et al. 2008). Since heterochromatin is as likely to incur breaks as euchromatin and since repair kinetics may be similar (Janssen et al. 2016), it was proposed that the accessibility of DNA within heterochromatin might change during the DDR. Several studies have indeed shown that DNA damage causes heterochromatin to unfold. In human fibroblasts or mouse NIH3T3 cells, chromatin relaxation in response to IR results from ATM-dependent KAP-1 phosphorylation and its subsequent loss from heterochromatin (Ziv et al. 2006; Goodarzi et al. 2008, 2011; Beucher et al. 2009; Woodbine et al. 2011; Lee et al. 2012). Interestingly, CHD3 (Goodarzi et al. 2011), a remodeler involved in chromatin compaction and gene repression (Denslow and Wade 2007), also was dissociated upon KAP-1 phosphorylation. Comparable events were seen after treatment with tert-butyl hydroperoxide (TBH), an agent that confers oxidative damage (Woodbine et al. 2011). Analogously, the yeast remodeler enzyme SWI/SNF was needed to facilitate Rad51- and Rad54-dependent strand invasion by alleviating heterochromatic constraints during recombinational repair of the MAT locus with HML (Sinha et al. 2009).
In Drosophila, both checkpoint kinases ATM and ATR are required for global heterochromatin expansion, which coincides with DSB relocation and the binding of HR factors such as Rad51 (Chiolo et al. 2011; Ryu et al. 2015). Whereas heterochromatin relaxation also occurs at site-specific CRISPR–Cas9-induced DSBs in mammalian cells, it was found that the compacted state in pericentric or centromeric regions was not refractory to mammalian RAD51 or KU80 binding (Tsouroula et al. 2016). Furthermore, RAD51 recruitment patterns were not altered after heterochromatin relaxation was forced by either treatment with the deacetylase inhibitor trichostatin A (TSA) or tethering of the transcriptional activator VP64 (Tsouroula et al. 2016). Thus, even though chromatin decompacts, changes in repair factor accessibility did not change significantly, and therefore it does not seem to be the critical parameter for repair.
It remained possible, however, that heterochromatin expansion and compaction would be functionally linked to damage relocation. Intriguingly, heterochromatin expansion did not reduce the heterochromatin-associated mark H3K9me3 and rather led to its increase in regions flanking the DSB (Tsouroula et al. 2016), consistent with data showing that laser damage-induced chromatin expansion was followed by a localized compaction (Burgess et al. 2014). This recompaction was found to be important for checkpoint signaling but not for repair, which suggests that specific chromatin configurations may regulate different steps of the DDR in a temporal manner. After specific and sequential steps that open chromatin for factor accessibility, a closed conformation is restored (Soria et al. 2012).
The role of chromatin compaction levels was also investigated at mammalian telomeres. Telomeres are structures with long TTAGG repeats that extend over many kilobases, ending in a 3′ ssDNA overhang. If left unprotected, the overhang looks like a resected DSB ready for recombination. Normally, the Shelterin complex (containing repeat-binding factors TRF1 and TRF2 and other proteins) binds to telomeric ends and bends them back, forming a protective “t loop.” T-loop formation blocks ATM binding and prevents DDR activation and illegitimate recombination events (Denchi and de Lange 2007; Guo et al. 2007). It was further proposed that the telomere-protective complex Shelterin might mediate chromatin compaction at telomere ends to prevent aberrant DNA damage signaling (Bandaria et al. 2016). Since DDR signaling is not strictly inhibited by chromatin compaction (Ziv et al. 2006; Goodarzi et al. 2008) and since, in some cases, chromatin recompaction can even amplify DDC signaling (Burgess et al. 2014), it was difficult to predict a priori whether chromatin compaction at telomeres stems the DDR. To probe this, Bandaria et al. (2016) used superresolution microscopy and showed that the Shelterin complex organizes human telomeric DNA into a compact globular structure in HeLa cells. Interestingly, knockdown of individual Shelterin subunits or mutations that interfered with its assembly caused a 10-fold increase in telomere volume, presumably a reflection of chromatin unfolding. This correlated directly with the accumulation of DDR signals at telomeres, leading the investigators to suggest that DNA compaction reduces DDR activation at telomeres, introducing a “telomere compaction model” for checkpoint regulation (Bandaria et al. 2016).
Two further studies challenged this idea. Superresolution stochastic optical reconstruction microscopy (STORM) in mouse cells (Timashev et al. 2017) or HeLa cells (Vancevska et al. 2017) depleted for TRF1, TRF2, or both showed that DDR markers such as γH2A.X or 53BP1 accumulate at telomeres without widespread chromatin decompaction (Timashev et al. 2017; Vancevska et al. 2017). In both studies, the knockdown of telomeric Shelterin factors affected the size of a subset of DDR-positive telomere foci, yet this was thought to reflect 53BP1-dependent clustering of dysfunctional telomeres rather than decompaction. While compacted chromatin states may still protect telomeres from end-to-end joining and unwanted recombination, these studies challenge the notion that chromatin compaction itself regulates checkpoint activation at telomeres.
The generalized response of chromatin to damage
DNA damage obviously can occur anywhere in the genome, and the results on chromatin compaction cited above seem to argue that repair factor accessibility is not the crucial factor differentiating repair in heterochromatin from euchromatin. Indeed, nucleosomal structure is a barrier to repair genome-wide. Consistently, chromatin decondensation was found to promote the repair of many different DNA lesions, including base damage, nicks, and breaks, regardless of the chromatin context. The question then is, how does damage-induced decompaction occur?
UV light causes base damage that is repaired by a process called nucleotide excision repair (NER). In this pathway, the detection of DNA lesions is stimulated by the damaged DNA-binding protein 2 (DDB2), a member of the cullin–RING ubiquitin ligase (CRL4) complex (Marteijn et al. 2014). ZRF1, another NER protein, stabilizes the CRL4 complex at damaged sites (Gracheva et al. 2016), and DICER-processed noncoding RNAs were proposed to assist repair (Francia et al. 2012; Wei et al. 2012). Fluorescence microscopy experiments in hamster cells then showed that DDB2 elicits the unfolding of large chromatin structures at UV-induced damage sites in an ATP-dependent manner (Luijsterburg et al. 2012). Interestingly, this coincided with a DDB2- and PARP1-dependent reduction in core histone density around the lesion (Luijsterburg et al. 2012). Finally, the direct targeting of DDB2, ZRF1, or DICER to chromatin by a LacI fusion protein allowed the unfolding of chromatin even without DNA damage (Adam et al. 2016; Chitale and Richly 2017). Under these conditions, the DICER and ZRF1 effects were shown to depend on PARP1, with the catalytic DICER activity being dispensable (Chitale and Richly 2017).
Chromatin decompaction at DSBs occurs independently of the repair pathway. In human U2OS cells, PARP1 was shown to recruit the CHD2 remodeler to multiphoton microirradiation-induced DSBs through a PAR-binding domain. CHD2 triggered rapid chromatin expansion and the deposition of histone variant H3.3 (Luijsterburg et al. 2016). This was further required to assemble the NHEJ complex at broken chromosomes and promote efficient DSB repair (Luijsterburg et al. 2016). Another microscopy-based study used a molecular flow assay to measure chromatin compaction around irradiation-induced breaks (Hinde et al. 2014). By analyzing the flow of EGFP molecules into chromatin before and after DSB induction, Hinde et al. (2014) argued that DNA damage induces a transient decrease in chromatin compaction, which in turn facilitates the recruitment of the NHEJ repair factor Ku70 to the lesion.
The transient decompaction of chromatin near a DSB was confirmed in yeast using improved time-lapse imaging regimes and superresolution microscopy (Amitai et al. 2017; Hauer et al. 2017). The chromatin expansion that occurred at an induced DSB was INO80-dependent. Polymer models based on mobility parameters predicted that chromatin unfolding would favor DSB extrusion from compacted domains, a prediction borne out in live-imaging experiments in yeast (Amitai et al. 2017).
In summary, both mammalian chromatin and yeast chromatin unfold and, in some cases, recompact in response to DNA damage in both heterochromatic and euchromatic domains. The processing of the DSB or the abundance of repair factors may guide different repair pathways even though the difference in accessibility between euchromatin and heterochromatin is not large. Chromatin may help protect telomeres as well, along with t loops. Interestingly, all of these findings indicate large-scale changes in chromatin structure in response to damage, yet selective break relocation from one nuclear compartment to another cannot be explained simply by chromatin expansion, and other pathways are implicated in the targeted shifts made by DSBs, such as the generation of damage-specific binding sites on nuclear structures and the alteration of microtubules and/or actin filaments (Dion and Gasser 2013; Horigome et al. 2014; Lottersberger et al. 2015; Amitai et al. 2017).
Chromatin constraints and mobility in the absence and presence of DNA breaks
Studies from several species show that the baseline movement of chromatin within an interphase nucleus occurs with a subdiffusive character (Marshall et al. 1997; Bornfleth et al. 1999; Heun et al. 2001; Vazquez et al. 2001; Albert et al. 2013; Dion and Gasser 2013; Amitai et al. 2015). This means that chromatin roams within a restricted volume that is significantly smaller than the volume of the nucleus. The subdiffusive movement indicates that internal forces, such as nucleosome–nucleosome contacts or sister cohesion (Dion et al. 2013), constrain free diffusion (Marshall et al. 1997; Heun et al. 2001; Vazquez et al. 2001; Chubb et al. 2002; Gasser et al. 2004; Bystricky et al. 2005). Moreover, the mobility of tagged loci was influenced by ATP levels (Marshall et al. 1997; Heun et al. 2001; Levi et al. 2005) or DNA interaction with fixed elements at the nuclear envelope, such as pores, the SPB (Verdaasdonk et al. 2013; Horigome et al. 2014; Strecker et al. 2016; Lawrimore et al. 2017), or the inner nuclear membrane protein Esc1 (Gasser et al. 2004). Indeed, the continuity of the chromatin fiber itself was shown to restrict movement, given that an excised circular chromatin locus of 16 kb diffused much more freely than the identical chromosomal locus (Neumann et al. 2012). Experiments that compared chromatin flexibility and the degree of movement at different loci along the genome enforced this notion (Verdaasdonk et al. 2013; Dickerson et al. 2016). Finally, altering local chromatin organization was shown to impact movement, as illustrated by the targeting of a chromatin remodeler to a fluorescently tagged locus (Neumann et al. 2012). This could also be provoked by a forced loss of nucleosomes from DNA through mutation or histone shutoff (Bouck and Bloom 2007; Verdaasdonk et al. 2013; Hauer et al. 2017). Collectively, these results argued that in response to DNA damage, chromatin remodeling and histone eviction may indeed increase chromatin mobility, which might have multiple implications for both compartment shifting and homology search.
The phenomenon of increased chromatin mobility in response to DNA breaks—both locally at the induced DSBs (Fig. 3) and genome-wide—is now robustly documented (Dion et al. 2012, 2013; Mine-Hattab and Rothstein 2012; Hauer et al. 2017; Herbert et al. 2017). Increased mobility requires damage that activates the DDC kinase Mec1 in yeast, while, at dysfunctional telomeres in mammals, increased telomere movement occurred in a 53BP1-dependent and ATM kinase-dependent manner (Lottersberger et al. 2015). The increase in telomere movement correlated with elevated rates of telomere–telomere fusions (Dimitrova et al. 2008) and was also dependent on SUN domain-containing proteins that bridge from the nucleoplasm through the double nuclear envelope to the cytoskeleton, forming the so-called LINC (linker of nucleoskeleton and cytoskeleton) complex (Lottersberger et al. 2015). The ablation of cytoskeleton-bound kinesins also compromised mobility and reduced the rates of end-to-end telomere fusion. Thus, one source of movement acts through chromatin-binding components of the nuclear envelope, and kinesins appears to enhance chromatin dynamics indirectly by moving the entire nucleus and in turn impacting repair (Lottersberger et al. 2015). Furthermore, 53BP1-independent movement and the clustering of critically short telomeres for recombination-dependent end maintenance through the ALT (alternative lengthening of telomeres) pathway have been observed (Cho et al. 2014; O'Sullivan et al. 2014; Cox et al. 2016).
Figure 3.

Concepts of local and global chromatin mobility in response to DNA damage. (A) A summary of local DSB mobility events. Formation of a DSB activates the DDC kinase Mec1/ATR, which phosphorylates downstream effector proteins (in yeast, Rad9 and Rad53) as well as chromatin remodeling complexes (INO80-C). If the DSB is repaired by HR, local repair proteins process the lesion, leading to the formation of the Rad51 nucleofilament, which will engage in homology search and recombinational repair. Chromatin locally unfolds at the break site, and histones and other proteins are modified by PARylation, SUMOylation, and ubiquitination. These promote the extrusion of damage from heterochromatic domains and increase access for repair factors. DSBs become more mobile, which is thought to facilitate homology search throughout the nuclear volume and promote relocation events to the nuclear periphery. Both the DDC and chromatin remodelers are needed to enhance DSB mobility. (B) Model for global chromatin mobility. Multiple random DSBs and single-strand breaks cause a global increase in chromatin mobility, which in yeast requires Mec1, Rad53, and INO80-C. Both the checkpoint and INO80-C trigger proteasome-dependent degradation of core histones in response to damage, which decompacts chromatin, enhances global mobility, and facilitates repair. The impact of forced histone reduction is highlighted in blue. (C) Multiple mechanisms contribute to chromatin mobility following DNA damage in yeast. Chromatin expands in response to damage-driven histone degradation. Cytoplasmic actin filaments can enhance nuclear rotation. Potentially, a loss of attachment through events such as centromere release might further increase chromatin mobility.
At internal DSBs, mobility is thought to enhance the search for donor sequences required by HDR (Rudin and Haber 1988; Wilson et al. 1994; Weiner et al. 2009; Neumann et al. 2012), which is particularly important if the sister chromatid is also broken or is absent, necessitating a search for an ectopic template for HDR. Mobility could also help disassociate illegitimate pairing events during HR, reattach a loose end for NHEJ (Lottersberger et al. 2015), or help DSBs shift away from the bulk of heterochromatic repeats (Torres-Rosell et al. 2007; Chiolo et al. 2011; Harding et al. 2015; Ryu et al. 2015; van Sluis and McStay 2015; Tsouroula et al. 2016). However, it should not be forgotten that DSBs that exhibit increased movement are not always positive, as they can be a source of genomic translocations (Roukos et al. 2013).
Histone loss and the cytoskeleton affect chromatin mobility
Two mechanisms have been shown to drive the increased chromatin mobility observed at site-specific DSBs, at uncapped mammalian telomeres, and genome-wide. First, as mentioned above, the movement of dysfunctional mammalian telomeres was shown to be driven at least partly by microtubule-bound kinesins. The damage-induced increase in mobility was reversibly inhibited by treating mouse cells with the microtubule poison taxol or nocodazole or by depleting two LINC complex components (SUN1 and SUN2) or the membrane-spanning KASH domain Nesprin proteins (Lottersberger et al. 2015). Also needed was the DDC checkpoint activator 53BP1 (Dimitrova et al. 2008; Lottersberger et al. 2015). Similarly, a recent study in mammalian cells used high-throughput chromosome conformation capture (capture Hi-C) to investigate the clustering of induced breaks. Again, DSB clustering occurred in 53BP1-independent actively transcribed regions in a 53BP1-independent manner that required both DDR proteins and cytoskeleton organizers. MRN, Formin 2 (FMN2, a nuclear actin organizer), and the LINC complex were all involved (Aymard et al. 2017), suggesting that forces applied by microtubule motors linked to the nuclear envelope are transduced to internal chromatin domains.
In yeast, microtubule depolymerization by nocodazole did not reduce chromatin movement but rather increased it in both the presence and absence of damage (Marshall et al. 1997; Amitai et al. 2017). On the other hand, depolymerization of the actin cytoskeleton reduced but did not eliminate damage-induced chromatin mobility (Amitai et al. 2017). This discrepancy probably reflects the different roles of cytoskeletal filaments in yeast and humans: In yeast, microtubule depolymerization releases centromere anchoring even in interphase nuclei, augmenting movement, while loss of actin interferes with subcellular organelle position, including the nucleus. In another study, the depletion of the KASH-like protein Csm4, a putative LINC complex component, increased telomere movement, possibly by triggering telomere detachment, while, again, depolymerization of the actin cytoskeleton by Latrunculin A (LatA) reduced movement (Spichal et al. 2016). Despite this global reduction of basal chromatin movement upon perturbation of the actin cytoskeleton, there is nonetheless a persistent increase in movement following the induction of DNA damage in the presence of LatA (Amitai et al. 2017). Using high-resolution imaging of the dynamics, it was shown that LatA suppresses actin-driven nuclear oscillations but does not interfere with changes occurring on the level of chromatin (Amitai et al. 2017). We do not exclude, however, that LatA also impacts actin-containing remodelers, such as INO80-C or Tip60. Since INO80-C activity was necessary for damage-associated mobility (Dion et al. 2012; Seeber et al. 2013a), LatA, upon extended incubation, could interfere with remodeler function.
A recent study confirmed that both local DSB mobility and global damage-induced movement of chromatin not only stem from external forces but also arise from a DDC-triggered INO80-C-dependent alteration of chromatin structure (Fig. 3B). Importantly, this latter correlated with a significant reduction in nucleosomes on DNA, reflecting the proteolytic degradation of 30%–40% of core histones in yeast (Fig. 3C; Hauer et al. 2017). A direct causal link between nucleosome packing, chromatin unfolding, and enhanced movement was shown by artificially reducing histone levels by either transcriptionally repressing the histone H3/H4 genes or ablating the HMGB1 homologs of budding yeast, Nhp6a and Nhp6b (Hauer et al. 2017). Earlier work had shown that loss of the HMG protein Nhp6a/b (HMGB1 in mammals) is sufficient to reduce histone levels even without exposing cells to damage (Celona et al. 2011). Down-regulating histones or deleting the HMGB1 homologs alone led to enhanced chromatin mobility, domain unfolding, and increased fiber flexibility (Hauer et al. 2017). This directly links chromatin composition to physical DNA movement and places nucleosome stability at the heart of damage-related mobility. In another recent study, chromatin unfolding and expansion of domains in response to UV-induced damage were documented in mammalian cells (Adam et al. 2016). This was partially dependent on the DDB2 subunit of the CRL4 ubiquitin ligase complex but stemmed mainly from histone eviction and redistribution rather than degradation (Adam et al. 2016).
Intriguingly, a previous study had triggered the transcriptional shutdown of histone H3 alone in yeast and had monitored a loss—not a gain—of locus mobility (Verdaasdonk et al. 2013). This difference may be explained by the very long times used for H3 repression in this study, which allowed cells to accumulate in G2 phase. In G2 phase, sister–sister cohesion adds additional constraints to chromatin mobility (Dion et al. 2013). Alternative explanations, of course, are also possible.
A study by the Durocher group (Strecker et al. 2016) recently has postulated a release of chromosomal tethers around the centromere as the source of alterations in both local and global chromatin mobility. The investigators identified Cep3, an inner kinetochore protein in yeast, as a target of the DDC kinase Rad53 and showed that damage-induced phosphorylation of Cep3 triggers a release of centromeres from their SPB tether. This was proposed to enhance chromatin movement in response to damage, an effect that appeared to be abrogated in a phospho-acceptor site mutant, cep3 (S575A) (Strecker et al. 2016). Strikingly, the cep3-S575A mutation had no effect on repair by HR, which was interpreted as showing that chromatin mobility does not facilitate the homology search (Strecker et al. 2016). However, again, cell cycle-related changes were not analyzed, and several laboratories reported that they do not score centromere release in response to damage (Herbert et al. 2017; Lawrimore et al. 2017).
The fate of nucleosomes during DNA damage and repair in yeast
Pioneering studies from the Verreault and Gunjan laboratories (Gunjan and Verreault 2003), again in budding yeast, have shown that changing histone levels can impact DNA damage sensitivity. Early work identified a Rad53-dependent surveillance mechanism that regulates histone protein levels in the context of DNA repair (Gunjan and Verreault 2003). In this study, histone H3 was overexpressed from the galactose promoter, leading to an accumulation of excess histones. In cells depleted for the DDR mediator Rad53 (rad53Δ), this resulted in slow growth, DNA damage sensitivity, and chromosome loss phenotypes (Gunjan and Verreault 2003; Liang et al. 2012). Surprisingly, rad53Δ sensitivity to genotoxic agents was significantly suppressed by reducing histone H3/H4 gene dosage. A similar benefit to survival was observed following induction of a site-specific DSB: The reduction of core histone levels through deletion of genes encoding H3 and H4 improved survival, possibly by enhancing HR-mediated repair (Liang et al. 2012). Interestingly, and in contrast to Hauer et al. (2017), hht2-hhf2Δ in the absence of damage seemed to reduce primarily the free pool of histones rather than chromatin-bound histones (Gunjan and Verreault 2003; Liang et al. 2012). Indeed, Rad53 preferentially phosphorylates nonincorporated histones, targeting them for degradation (Gunjan and Verreault 2003; Singh et al. 2009). The investigators suggested that an excess of histones might bind repair proteins and sequester them away from the damage, reducing cell survival in the face of genotoxic stress (Gunjan and Verreault 2003). Conversely, it would follow that histone depletion promotes survival after damage by increasing the amount of free repair proteins. This may act additively with the enhanced accessibility of damage to the repair machinery that stems from a reduced nucleosome density and an increased mobility of chromatin during the DDR.
Extending damage-induced histone loss and remodeler action beyond yeast
The question arises of how widespread this phenomenon of histone loss in response to stress or other genomic conditions might be. It is useful to note that a drop in histone levels has also been shown to occur during replicative aging of human fibroblasts (Oberdoerffer 2010; O'Sullivan et al. 2010; Pal and Tyler 2016). Indeed, late passage cells show decreased levels of both histone transcription and histone proteins. Interestingly, this effect seemed to stem from a chronic checkpoint response triggered by telomere shortening in the course of aging (O'Sullivan et al. 2010). Similar degrees of age-related histone level reductions were seen in yeast (Feser et al. 2010), and longevity could be increased by bringing histone amounts back to their native level (Feser et al. 2010). These physiological fluxes in histone levels have parallels to a process in which histone genes are transiently repressed during the recovery of mammalian cells from apoptosis (Tang et al. 2017). The process has been termed “anastasis” and raises the hypothesis that transient histone level reductions could be beneficial for growth recovery, although the mechanisms were not yet established (Tang et al. 2017). Naturally, there may also be indirect effects of histone level reduction that influence cell growth (Gunjan et al. 2005; Eriksson et al. 2012)
The notion that histones must be removed and replaced during the repair of DNA damage has been elaborated in a prime–repair–restore model (Soria et al. 2012). This model proposes that a lesion must first be made accessible to the repair machinery (“primed”). Second, the chromatin context of the lesion must be dealt with during repair. Finally, the chromatin/nucleosomal context must be restored once repair is completed. The disruption and restoration of chromatin during the DDR impact both cell survival and epigenome maintenance (Soria et al. 2012; Adam et al. 2015; Gerhold et al. 2015), and the process is naturally not restricted to DSBs and repair by HR. It is important to note that newly deposited histones may lack histone PTMs or bear others that specifically mark the domain as a site of repair and that this in itself may be of benefit to the cell. Depending on the chromatin, it may be that the original chromatin state is restored in a process that requires both chaperones and remodelers in a later stage of the cell cycle, allowing for a transient chromatin-based memory of damage.
The role of remodelers in nucleosome dynamics at IR-induced DSBs and at sites of UV damage has been demonstrated in mammalian cells as well (Luijsterburg et al. 2012, 2016). CHD2-dependent remodeling of chromatin around breaks in human U2OS cells relied on PARylation, which caused chromatin expansion and deposition of the histone variant H3.3 (Luijsterburg et al. 2016). Finally, fluorescence recovery after photobleaching (FRAP) microscopy revealed a DDB2- and PARP1-dependent loss of core histones from UV-damaged chromatin in hamster cells (Luijsterburg et al. 2012). Apart from the importance of PARylation in DDR signaling, ATM and NBS1 recruitment to I-PpoI-induced DSBs was shown to cause localized disruption of nucleosomes in human MCF7 cells, and chromatin immunoprecipitation experiments showed a drop in H2B levels at breaks and a selective loss of H2A and H2B, but not of H3 or H4, around I-PpoI sites (Berkovich et al. 2007; Goldstein et al. 2013). Similar results were obtained with FRAP experiments at localized UV damage sites in human cell lines (Dinant et al. 2013). In this case, selective H2A/H2B loss at the damaged sites was scored and was shown to require Spt16, a subunit of the histone chaperone FACT that enables transcriptional restart after repair (Dinant et al. 2013). The down-regulation of nucleolin, a nucleolar protein with histone chaperone activity, also abrogated nucleosome disruption, repair factor recruitment, and DSB repair (Goldstein et al. 2013).
Mammalian nucleosomes are not only lost locally in response to UV, IR, or radiomimetic damage but also altered genome-wide. Chromatin fractionation and salt extraction studies showed that histones were destabilized and released from chromatin upon irradiation in human 293T cells (Xu et al. 2010). Quantitative measures argue that the core histones H2B, H3, and H4 as well as the histone variant H2A.X are more readily extractable from chromatin after damage, thanks to an active process that required the ATPase activity of the p400 ATPase in the SWI/SNF complex and histone acetylation by the Tip60 acetyltransferase (Xu et al. 2010). Also implicated in such responses is the human CUL4–DDB–ROC1 ubiquitin ligase, which modifies histones in vitro and ubiquitinates H3 and H4 in response to global UV damage in vivo (Wang et al. 2006). Ubiquitination also weakens histone–histone interactions to enhance nucleosome eviction, although it is unclear whether subsequent proteasomal degradation occurred (Wang et al. 2006), as was shown in yeast (Hauer et al. 2017). While we can expect there to be species-specific aspects of histone mobilization and turnover, it is becoming clear that such events are central to genome maintenance across eukaryotic species.
Conclusions
We draw two general conclusions. First, chromatin undergoes dramatic changes in response to DNA damage not only locally but genome-wide. Second, we note that despite the prevalent view that histones and nucleosomes are stable units of chromatin structure, their turnover and degradation appear to be at the heart of conserved pathways that help cells deal with genotoxic stress. Limited histone degradation can drive chromatin mobility, increase access, and perhaps even enhance genomic stability over the lifetime of cells and organisms. Indeed, controlled histone proteolysis has been shown to have roles in a variety of cellular processes, such as developmental transitions, spermatogenesis, the immune response, and neuronal plasticity (Maze et al. 2015). As pointed out in a recent review (Dhaenens et al. 2015), it is time to investigate more intensely how histone levels and histone proteolysis regulate processes beyond the DDR, such as aging. Future studies may show that nucleosome dynamics and rates of histone turnover not only control repair and genome stability but also distinguish differentiated chromatin states that regulate transcription (Taneja and Grewal 2017).
Acknowledgments
We apologize to those scientists who made valuable contributions to the field over the years but were not cited here due to space constraints. We thank all members of the Gasser laboratory for discussion, and A. Seeber and A. Cheblal for critical reading of the manuscript. M.H.H. was supported by a PhD fellowship of the Boehringer Ingelheim Fonds. S.M.G. thanks the Human Frontiers Science Program, the Swiss National Science Foundation, the Swiss Cancer League, and the Novartis Research Foundation for support.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.307702.117.
References
- Adam S, Dabin J, Polo SE. 2015. Chromatin plasticity in response to DNA damage: the shape of things to come. DNA Repair 32: 120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam S, Dabin J, Chevallier O, Leroy O, Baldeyron C, Corpet A, Lomonte P, Renaud O, Almouzni G, Polo SE. 2016. Real-time tracking of parental histones reveals their contribution to chromatin integrity following DNA damage. Mol Cell 64: 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agmon N, Liefshitz B, Zimmer C, Fabre E, Kupiec M. 2013. Effect of nuclear architecture on the efficiency of double-strand break repair. Nat Cell Biol 15: 694–699. [DOI] [PubMed] [Google Scholar]
- Akhtar A, Becker PB. 2000. Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol Cell 5: 367–375. [DOI] [PubMed] [Google Scholar]
- Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF. 2007. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 446: 572–576. [DOI] [PubMed] [Google Scholar]
- Albert B, Mathon J, Shukla A, Saad H, Normand C, Leger-Silvestre I, Villa D, Kamgoue A, Mozziconacci J, Wong H, et al. 2013. Systematic characterization of the conformation and dynamics of budding yeast chromosome XII. J Cell Biol 202: 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral N, Ryu T, Li X, Chiolo I. 2017. Nuclear dynamics of heterochromatin repair. Trends Genet 33: 86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amitai A, Toulouze M, Dubrana K, Holcman D. 2015. Analysis of single locus trajectories for extracting in vivo chromatin tethering interactions. PLoS Comput Biol 11: e1004433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amitai A, Seeber A, Gasser SM, Holcman D. 2017. Visualization of chromatin decompaction and break site extrusion as predicted by statistical polymer modeling of single-locus trajectories. Cell Rep 18: 1200–1214. [DOI] [PubMed] [Google Scholar]
- Awad S, Ryan D, Prochasson P, Owen-Hughes T, Hassan AH. 2010. The Snf2 homolog Fun30 acts as a homodimeric ATP-dependent chromatin-remodeling enzyme. J Biol Chem 285: 9477–9484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, Iacovoni JS, Daburon V, Miller KM, Jackson SP, et al. 2014. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol 21: 366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymard F, Aguirrebengoa M, Guillou E, Javierre BM, Bugler B, Arnould C, Rocher V, Iacovoni JS, Biernacka A, Skrzypczak M, et al. 2017. Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nat Struct Mol Biol 24: 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. 2008. HP1-β mobilization promotes chromatin changes that initiate the DNA damage response. Nature 453: 682–686. [DOI] [PubMed] [Google Scholar]
- Bandaria JN, Qin P, Berk V, Chu S, Yildiz A. 2016. Shelterin protects chromosome ends by compacting telomeric chromatin. Cell 164: 735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantele SC, Ferreira P, Gritenaite D, Boos D, Pfander B. 2017. Targeting of the Fun30 nucleosome remodeller by the Dpb11 scaffold facilitates cell cycle-regulated DNA end resection. Elife 6: e21687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batte A, Brocas C, Bordelet H, Hocher A, Ruault M, Adjiri A, Taddei A, Dubrana K. 2017. Recombination at subtelomeres is regulated by physical distance, double-strand break resection and chromatin status. EMBO J 36: 2609–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovich E, Monnat RJ Jr, Kastan MB. 2007. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol 9: 683–690. [DOI] [PubMed] [Google Scholar]
- Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, Goodarzi AA, Krempler A, Jeggo PA, Lobrich M. 2009. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J 28: 3413–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi ME, Agresti A. 2005. HMG proteins: dynamic players in gene regulation and differentiation. Curr Opin Genet Dev 15: 496–506. [DOI] [PubMed] [Google Scholar]
- Bleuyard JY, Fournier M, Nakato R, Couturier AM, Katou Y, Ralf C, Hester SS, Dominguez D, Rhodes D, Humphrey TC, et al. 2017. MRG15-mediated tethering of PALB2 to unperturbed chromatin protects active genes from genotoxic stress. Proc Natl Acad Sci 114: 7671–7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornfleth H, Edelmann P, Zink D, Cremer T, Cremer C. 1999. Quantitative motion analysis of subchromosomal foci in living cells using four-dimensional microscopy. Biophys J 77: 2871–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. 2006. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127: 1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouck DC, Bloom K. 2007. Pericentric chromatin is an elastic component of the mitotic spindle. Curr Biol 17: 741–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RC, Burman B, Kruhlak MJ, Misteli T. 2014. Activation of DNA damage response signaling by condensed chromatin. Cell Rep 9: 1703–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystricky K, Laroche T, van Houwe G, Blaszczyk M, Gasser SM. 2005. Chromosome looping in yeast: telomere pairing and coordinated movement reflect anchoring efficiency and territorial organization. J Cell Biol 168: 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystricky K, Van Attikum H, Montiel MD, Dion V, Gehlen L, Gasser SM. 2009. Regulation of nuclear positioning and dynamics of the silent mating type loci by the yeast Ku70/Ku80 complex. Mol Cell Biol 29: 835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos EI, Reinberg D. 2009. Histones: annotating chromatin. Annu Rev Genet 43: 559–599. [DOI] [PubMed] [Google Scholar]
- Celona B, Weiner A, Di Felice F, Mancuso FM, Cesarini E, Rossi RL, Gregory L, Baban D, Rossetti G, Grianti P, et al. 2011. Substantial histone reduction modulates genomewide nucleosomal occupancy and global transcriptional output. PLoS Biol 9: e1001086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai B, Huang J, Cairns BR, Laurent BC. 2005. Distinct roles for the RSC and Swi/Snf ATP-dependent chromatin remodelers in DNA double-strand break repair. Genes Dev 19: 1656–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Cui D, Papusha A, Zhang X, Chu CD, Tang J, Chen K, Pan X, Ira G. 2012. The Fun30 nucleosome remodeller promotes resection of DNA double-strand break ends. Nature 489: 576–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiolo I, Minoda A, Colmenares SU, Polyzos A, Costes SV, Karpen GH. 2011. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 144: 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitale S, Richly H. 2017. DICER and ZRF1 contribute to chromatin decondensation during nucleotide excision repair. Nucleic Acids Res 45: 5901–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho NW, Dilley RL, Lampson MA, Greenberg RA. 2014. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 159: 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubb JR, Boyle S, Perry P, Bickmore WA. 2002. Chromatin motion is constrained by association with nuclear compartments in human cells. Curr Biol 12: 439–445. [DOI] [PubMed] [Google Scholar]
- Churikov D, Charifi F, Eckert-Boulet N, Silva S, Simon MN, Lisby M, Geli V. 2016. SUMO-dependent relocalization of eroded telomeres to nuclear pore complexes controls telomere recombination. Cell Rep 15: 1242–1253. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. 2009. The biology of chromatin remodeling complexes. Annu Rev Biochem 78: 273–304. [DOI] [PubMed] [Google Scholar]
- Costelloe T, Louge R, Tomimatsu N, Mukherjee B, Martini E, Khadaroo B, Dubois K, Wiegant WW, Thierry A, Burma S, et al. 2012. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 489: 581–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox KE, Marechal A, Flynn RL. 2016. SMARCAL1 resolves replication stress at ALT telomeres. Cell Reports 14: 2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona CA, Sarangi P, Yang Y, Hang LE, Rahman S, Zhao X. 2012. Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the mec1 checkpoint. Mol Cell 45: 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J. 2008. Mapping in vivo chromatin interactions in yeast suggests an extended chromatin fiber with regional variation in compaction. J Biol Chem 283: 34532–34540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Marti-Renom MA, Mirny LA. 2013. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Gen 14: 390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denchi EL, de Lange T. 2007. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448: 1068–1071. [DOI] [PubMed] [Google Scholar]
- De Koning L, Corpet A, Haber JE, Almouzni G. 2007. Histone chaperones: an escort network regulating histone traffic. Nat Struct Mol Biol 14: 997–1007. [DOI] [PubMed] [Google Scholar]
- Denchi EL, de Lange T. 2007. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448: 1068–1071. [DOI] [PubMed] [Google Scholar]
- Denslow SA, Wade PA. 2007. The human Mi-2/NuRD complex and gene regulation. Oncogene 26: 5433–5438. [DOI] [PubMed] [Google Scholar]
- Dhaenens M, Glibert P, Meert P, Vossaert L, Deforce D. 2015. Histone proteolysis: a proposal for categorization into ‘clipping’ and ‘degradation’. Bioessays 37: 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson D, Gierlinski M, Singh V, Kitamura E, Ball G, Tanaka TU, Owen-Hughes T. 2016. High resolution imaging reveals heterogeneity in chromatin states between cells that is not inherited through cell division. BMC Cell Biol 17: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, Chen YCM, Spector DL, de Lange T. 2008. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 456: 524–U551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinant C, Ampatziadis-Michailidis G, Lans H, Tresini M, Lagarou A, Grosbart M, Theil AF, van Cappellen WA, Kimura H, Bartek J, et al. 2013. Enhanced chromatin dynamics by FACT promotes transcriptional restart after UV-induced DNA damage. Mol Cell 51: 469–479. [DOI] [PubMed] [Google Scholar]
- Dion V, Gasser SM. 2013. Chromatin movement in the maintenance of genome stability. Cell 152: 1355–1364. [DOI] [PubMed] [Google Scholar]
- Dion V, Kalck V, Horigome C, Towbin BD, Gasser SM. 2012. Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat Cell Biol 14: 502–509. [DOI] [PubMed] [Google Scholar]
- Dion V, Kalck V, Seeber A, Schleker T, Gasser SM. 2013. Cohesin and the nucleolus constrain the mobility of spontaneous repair foci. EMBO Rep 14: 984–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, et al. 2009. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 136: 435–446. [DOI] [PubMed] [Google Scholar]
- Downs JA, Lowndes NF, Jackson SP. 2000. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 408: 1001–1004. [DOI] [PubMed] [Google Scholar]
- Duan Z, Andronescu M, Schutz K, McIlwain S, Kim YJ, Lee C, Shendure J, Fields S, Blau CA, Noble WS. 2010. A three-dimensional model of the yeast genome. Nature 465: 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eapen VV, Sugawara N, Tsabar M, Wu WH, Haber JE. 2012. The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol Cell Biol 32: 4727–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson PR, Ganguli D, Nagarajavel V, Clark DJ. 2012. Regulation of histone gene expression in budding yeast. Genetics 191: 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, Tyler JK. 2010. Elevated histone expression promotes life span extension. Mol Cell 39: 724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francia S, Michelini F, Saxena A, Tang D, de Hoon M, Anelli V, Mione M, Carninci P, d'Adda di Fagagna F. 2012. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 488: 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser SM, Hediger F, Taddei A, Neumann FR, Gartenberg MR. 2004. The function of telomere clustering in yeast: the Circe effect. Cold Spring Harb Symp Quant Biol 69: 327–337. [DOI] [PubMed] [Google Scholar]
- Gerhold CB, Hauer MH, Gasser SM. 2015. INO80-C and SWR-C: guardians of the genome. J Mol Biol 427: 637–651. [DOI] [PubMed] [Google Scholar]
- Gilbert N, Gilchrist S, Bickmore WA. 2005. Chromatin organization in the mammalian nucleus. Int Rev Cytol 242: 283–336. [DOI] [PubMed] [Google Scholar]
- Goldstein M, Derheimer FA, Tait-Mulder J, Kastan MB. 2013. Nucleolin mediates nucleosome disruption critical for DNA double-strand break repair. Proc Natl Acad Sci 110: 16874–16879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, Jeggo PA. 2008. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell 31: 167–177. [DOI] [PubMed] [Google Scholar]
- Goodarzi AA, Kurka T, Jeggo PA. 2011. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nat Struct Mol Biol 18: 831–839. [DOI] [PubMed] [Google Scholar]
- Gotta M, Laroche T, Formenton A, Maillet L, Scherthan H, Gasser SM. 1996. The clustering of telomeres and colocalization with Rap1, Sir3, and Sir4 proteins in wild-type Saccharomyces cerevisiae. J Cell Biol 134: 1349–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracheva E, Chitale S, Wilhelm T, Rapp A, Byrne J, Stadler J, Medina R, Cardoso MC, Richly H. 2016. ZRF1 mediates remodeling of E3 ligases at DNA lesion sites during nucleotide excision repair. J Cell Biol 213: 185–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemette B, Bataille AR, Gevry N, Adam M, Blanchette M, Robert F, Gaudreau L. 2005. Variant histone H2A.Z is globally localized to the promoters of inactive yeast genes and regulates nucleosome positioning. PLoS Biol 3: e384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunjan A, Verreault A. 2003. A Rad53 kinase-dependent surveillance mechanism that regulates histone protein levels in S. cerevisiae. Cell 115: 537–549. [DOI] [PubMed] [Google Scholar]
- Gunjan A, Paik J, Verreault A. 2005. Regulation of histone synthesis and nucleosome assembly. Biochimie 87: 625–635. [DOI] [PubMed] [Google Scholar]
- Guo X, Deng Y, Lin Y, Cosme-Blanco W, Chan S, He H, Yuan G, Brown EJ, Chang S. 2007. Dysfunctional telomeres activate an ATM–ATR-dependent DNA damage response to suppress tumorigenesis. EMBO J 26: 4709–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurard-Levin ZA, Quivy JP, Almouzni G. 2014. Histone chaperones: assisting histone traffic and nucleosome dynamics. Annu Rev Biochem 83: 487–517. [DOI] [PubMed] [Google Scholar]
- Hajjoul H, Mathon J, Ranchon H, Goiffon I, Mozziconacci J, Albert B, Carrivain P, Victor JM, Gadal O, Bystricky K, et al. 2013. High-throughput chromatin motion tracking in living yeast reveals the flexibility of the fiber throughout the genome. Genome Res 23: 1829–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Boiarsky JA, Greenberg RA. 2015. ATM dependent silencing links nucleolar chromatin reorganization to DNA damage recognition. Cell Rep 13: 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauer MH, Seeber A, Singh V, Thierry R, Sack R, Amitai A, Kryzhanovska M, Eglinger J, Holcman D, Owen-Hughes T, et al. 2017. Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat Struct Mol Biol 24: 99–107. [DOI] [PubMed] [Google Scholar]
- Hediger F, Neumann FR, Van Houwe G, Dubrana K, Gasser SM. 2002. Live imaging of telomeres: yKu and Sir proteins define redundant telomere-anchoring pathways in yeast. Curr Biol 12: 2076–2089. [DOI] [PubMed] [Google Scholar]
- Herbert S, Brion A, Arbona JM, Lelek M, Veillet A, Lelandais B, Parmar J, Fernandez FG, Almayrac E, Khalil Y, et al. 2017. Chromatin stiffening underlies enhanced locus mobility after DNA damage in budding yeast. EMBO J 36: 2595–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heun P, Laroche T, Shimada K, Furrer P, Gasser SM. 2001. Chromosome dynamics in the yeast interphase nucleus. Science 294: 2181–2186. [DOI] [PubMed] [Google Scholar]
- Heyer WD, Ehmsen KT, Liu J. 2010. Regulation of homologous recombination in eukaryotes. Annu Rev Genet 44: 113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinde E, Kong X, Yokomori K, Gratton E. 2014. Chromatin dynamics during DNA repair revealed by pair correlation analysis of molecular flow in the nucleus. Biophys J 107: 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horigome C, Oma Y, Konishi T, Schmid R, Marcomini I, Hauer MH, Dion V, Harata M, Gasser SM. 2014. SWR1 and INO80 chromatin remodelers contribute to DNA double-strand break perinuclear anchorage site choice. Mol Cell 55: 626–639. [DOI] [PubMed] [Google Scholar]
- Horigome C, Bustard DE, Marcomini I, Delgoshaie N, Tsai-Pflugfelder M, Cobb JA, Gasser SM. 2016. PolySUMOylation by Siz2 and Mms21 triggers relocation of DNA breaks to nuclear pores through the Slx5/Slx8 STUbL. Genes Dev 30: 931–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. 2007. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 131: 901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. 2009. The DNA-damage response in human biology and disease. Nature 461: 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob B, Splinter J, Conrad S, Voss KO, Zink D, Durante M, Lobrich M, Taucher-Scholz G. 2011. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res 39: 6489–6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, Breuer GA, Brinkman EK, van der Meulen AI, Borden SV, van Steensel B, Bindra RS, LaRocque JR, Karpen GH. 2016. A single double-strand break system reveals repair dynamics and mechanisms in heterochromatin and euchromatin. Genes Dev 30: 1645–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeggo PA, Downs JA. 2014. Roles of chromatin remodellers in DNA double strand break repair. Exp Cell Res 329: 68–77. [DOI] [PubMed] [Google Scholar]
- Kalocsay M, Hiller NJ, Jentsch S. 2009. Chromosome-wide Rad51 spreading and SUMO–H2A.Z-dependent chromosome fixation in response to a persistent DNA double-strand break. Mol Cell 33: 335–343. [DOI] [PubMed] [Google Scholar]
- Khurana S, Kruhlak MJ, Kim J, Tran AD, Liu J, Nyswaner K, Shi L, Jailwala P, Sung MH, Hakim O, et al. 2014. A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Rep 8: 1049–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lademann CA, Renkawitz J, Pfander B, Jentsch S. 2017. The INO80 complex removes H2A.Z to promote presynaptic filament formation during homologous recombination. Cell Rep 19: 1294–1303. [DOI] [PubMed] [Google Scholar]
- Lawrimore J, Barry TM, Barry RM, York AC, Cook DM, Akialis K, Tyler J, Vasquez P, Yeh E, Bloom K. 2017. Microtubule dynamics drive enhanced chromatin motion and mobilize telomeres in response to DNA damage. Mol Biol Cell 28: 1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Goodarzi AA, Adelmant GO, Pan Y, Jeggo PA, Marto JA, Chowdhury D. 2012. Phosphoproteomic analysis reveals that PP4 dephosphorylates KAP-1 impacting the DNA damage response. EMBO J 31: 2403–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi V, Ruan Q, Plutz M, Belmont AS, Gratton E. 2005. Chromatin dynamics in interphase cells revealed by tracking in a two-photon excitation microscope. Biophys J 89: 4275–4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Eirin-Lopez JM, Ausio J. 2005. H2AX: tailoring histone H2A for chromatin-dependent genomic integrity. Biochem Cell Biol 83: 505–515. [DOI] [PubMed] [Google Scholar]
- Li X, Corsa CA, Pan PW, Wu L, Ferguson D, Yu X, Min J, Dou Y. 2010. MOF and H4 K16 acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1. Mol Cell Biol 30: 5335–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, Burkhart SL, Singh RK, Kabbaj MH, Gunjan A. 2012. Histone dosage regulates DNA damage sensitivity in a checkpoint-independent manner by the homologous recombination pathway. Nucleic Acids Res 40: 9604–9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T, Barnes DE. 2000. Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol 65: 127–133. [DOI] [PubMed] [Google Scholar]
- Lottersberger F, Karssemeijer RA, Dimitrova N, de Lange T. 2015. 53BP1 and the LINC complex promote microtubule-dependent DSB mobility and DNA repair. Cell 163: 880–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg MS, Lindh M, Acs K, Vrouwe MG, Pines A, van Attikum H, Mullenders LH, Dantuma NP. 2012. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J Cell Biol 197: 267–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg MS, de Krijger I, Wiegant WW, Shah RG, Smeenk G, de Groot AJ, Pines A, Vertegaal AC, Jacobs JJ, Shah GM, et al. 2016. PARP1 links CHD2-mediated chromatin expansion and H3.3 deposition to DNA repair by non-homologous end-joining. Mol Cell 61: 547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk E, Ranjan A, Fitzgerald PC, Mizuguchi G, Huang Y, Wei D, Wu C. 2010. Stepwise histone replacement by SWR1 requires dual activation with histone H2A.Z and canonical nucleosome. Cell 143: 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. 2007. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 131: 887–900. [DOI] [PubMed] [Google Scholar]
- Malarkey CS, Churchill ME. 2012. The high mobility group box: the ultimate utility player of a cell. Trends Biochem Sci 37: 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik HS, Henikoff S. 2003. Phylogenomics of the nucleosome. Nat Struct Biol 10: 882–891. [DOI] [PubMed] [Google Scholar]
- Marshall WF, Straight A, Marko JF, Swedlow J, Dernburg A, Belmont A, Murray AW, Agard DA, Sedat JW. 1997. Interphase chromosomes undergo constrained diffusional motion in living cells. Curr Biol 7: 930–939. [DOI] [PubMed] [Google Scholar]
- Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. 2014. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 15: 465–481. [DOI] [PubMed] [Google Scholar]
- Mattiroli F, Vissers JH, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, Sixma TK. 2012. RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell 150: 1182–1195. [DOI] [PubMed] [Google Scholar]
- Maze I, Wenderski W, Noh KM, Bagot RC, Tzavaras N, Purushothaman I, Elsasser SJ, Guo Y, Ionete C, Hurd YL, et al. 2015. Critical role of histone turnover in neuronal transcription and plasticity. Neuron 87: 77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekhail K, Seebacher J, Gygi SP, Moazed D. 2008. Role for perinuclear chromosome tethering in maintenance of genome stability. Nature 456: 667–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mine-Hattab J, Rothstein R. 2012. Increased chromosome mobility facilitates homology search during recombination. Nat Cell Biol 14: 510–517. [DOI] [PubMed] [Google Scholar]
- Morillo-Huesca M, Clemente-Ruiz M, Andujar E, Prado F. 2010. The SWR1 histone replacement complex causes genetic instability and genome-wide transcription misregulation in the absence of H2A.Z. PLoS One 5: e12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, Herceg Z. 2006. Histone acetylation by Trrap–Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol 8: 91–99. [DOI] [PubMed] [Google Scholar]
- Nagai S, Dubrana K, Tsai-Pflugfelder M, Davidson MB, Roberts TM, Brown GW, Varela E, Hediger F, Gasser SM, Krogan NJ. 2008. Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science 322: 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann FR, Dion V, Gehlen LR, Tsai-Pflugfelder M, Schmid R, Taddei A, Gasser SM. 2012. Targeted INO80 enhances subnuclear chromatin movement and ectopic homologous recombination. Genes Dev 26: 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdoerffer P. 2010. An age of fewer histones. Nat Cell Biol 12: 1029–1031. [DOI] [PubMed] [Google Scholar]
- O'Sullivan RJ, Kubicek S, Schreiber SL, Karlseder J. 2010. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol 17: 1218–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan RJ, Arnoult N, Lackner DH, Oganesian L, Haggblom C, Corpet A, Almouzni G, Karlseder J. 2014. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat Struct Mol Biol 21: 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oza P, Jaspersen SL, Miele A, Dekker J, Peterson CL. 2009. Mechanisms that regulate localization of a DNA double-strand break to the nuclear periphery. Genes Dev 23: 912–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Tyler JK. 2016. Epigenetics and aging. Sci Adv 2: e1600584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panday A, Grove A. 2017. Yeast HMO1: linker histone reinvented. Microbiol Mol Biol Rev 81: e00037–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papamichos-Chronakis M, Peterson CL. 2013. Chromatin and the genome integrity network. Nat Rev Genet 14: 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papamichos-Chronakis M, Watanabe S, Rando OJ, Peterson CL. 2011. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell 144: 200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson CE, Nichol Edamura K, Cleary JD. 2005. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet 6: 729–742. [DOI] [PubMed] [Google Scholar]
- Pellegrino S, Michelena J, Teloni F, Imhof R, Altmeyer M. 2017. Replication-coupled dilution of H4K20me2 guides 53BP1 to pre-replicative chromatin. Cell Rep 19: 1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister SX, Ahrabi S, Zalmas LP, Sarkar S, Aymard F, Bachrati CZ, Helleday T, Legube G, La Thangue NB, Porter AC, et al. 2014. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep 7: 2006–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povirk LF, Wubter W, Kohnlein W, Hutchinson F. 1977. DNA double-strand breaks and alkali-labile bonds produced by bleomycin. Nucleic Acids Res 4: 3573–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psakhye I, Jentsch S. 2012. Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 151: 807–820. [DOI] [PubMed] [Google Scholar]
- Raisner RM, Hartley PD, Meneghini MD, Bao MZ, Liu CL, Schreiber SL, Rando OJ, Madhani HD. 2005. Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell 123: 233–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273: 5858–5868. [DOI] [PubMed] [Google Scholar]
- Roukos V, Voss TC, Schmidt CK, Lee S, Wangsa D, Misteli T. 2013. Spatial dynamics of chromosome translocations in living cells. Science 341: 660–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin N, Haber JE. 1988. Efficient repair of HO-induced chromosomal breaks in Saccharomyces cerevisiae by recombination between flanking homologous sequences. Mol Cell Biol 8: 3918–3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu T, Spatola B, Delabaere L, Bowlin K, Hopp H, Kunitake R, Karpen GH, Chiolo I. 2015. Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat Cell Biol 17: 1401–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, Kouzarides T. 2004. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 119: 603–614. [DOI] [PubMed] [Google Scholar]
- Sarangi P, Zhao X. 2015. SUMO-mediated regulation of DNA damage repair and responses. Trends Biochem Sci 40: 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saredi G, Huang H, Hammond CM, Alabert C, Bekker-Jensen S, Forne I, Reveron-Gomez N, Foster BM, Mlejnkova L, Bartke T, et al. 2016. H4K20me0 marks post-replicative chromatin and recruits the TONSL–MMS22L DNA repair complex. Nature 534: 714–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober H, Ferreira H, Kalck V, Gehlen LR, Gasser SM. 2009. Yeast telomerase and the SUN domain protein Mps3 anchor telomeres and repress subtelomeric recombination. Genes Dev 23: 928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwertman P, Bekker-Jensen S, Mailand N. 2016. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat Rev Mol Cell Biol 17: 379–394. [DOI] [PubMed] [Google Scholar]
- Seeber A, Gasser SM. 2016. Chromatin organization and dynamics in double-strand break repair. Curr Opin Genet Dev 43: 9–16. [DOI] [PubMed] [Google Scholar]
- Seeber A, Dion V, Gasser SM. 2013a. Checkpoint kinases and the INO80 nucleosome remodeling complex enhance global chromatin mobility in response to DNA damage. Genes Dev 27: 1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeber A, Hauer MH, Gasser SM. 2013b. Nucleosome remodelers in double-strand break repair. Curr Opin Genet Dev 23: 174–184. [DOI] [PubMed] [Google Scholar]
- Sharma GG, So S, Gupta A, Kumar R, Cayrou C, Avvakumov N, Bhadra U, Pandita RK, Porteus MH, Chen DJ, et al. 2010. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol Cell Biol 30: 3582–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, Kabbaj MH, Paik J, Gunjan A. 2009. Histone levels are regulated by phosphorylation and ubiquitylation-dependent proteolysis. Nat Cell Biol 11: 925–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha M, Watanabe S, Johnson A, Moazed D, Peterson CL. 2009. Recombinational repair within heterochromatin requires ATP-dependent chromatin remodeling. Cell 138: 1109–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeenk G, van Attikum H. 2013. The chromatin response to DNA breaks: leaving a mark on genome integrity. Annu Rev Biochem 82: 55–80. [DOI] [PubMed] [Google Scholar]
- Soria G, Polo SE, Almouzni G. 2012. Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell 46: 722–734. [DOI] [PubMed] [Google Scholar]
- Spichal M, Brion A, Herbert S, Cournac A, Marbouty M, Zimmer C, Koszul R, Fabre E. 2016. Evidence for a dual role of actin in regulating chromosome organization and dynamics in yeast. J Cell Sci 129: 681–692. [DOI] [PubMed] [Google Scholar]
- Strecker J, Gupta GD, Zhang W, Bashkurov M, Landry MC, Pelletier L, Durocher D. 2016. DNA damage signalling targets the kinetochore to promote chromatin mobility. Nat Cell Biol 18: 281–290. [DOI] [PubMed] [Google Scholar]
- Strickfaden H, McDonald D, Kruhlak MJ, Haince JF, Th'ng JP, Rouleau M, Ishibashi T, Corry GN, Ausio J, Underhill DA, et al. 2016. Poly(ADP-ribosyl)ation-dependent transient chromatin decondensation and histone displacement following laser microirradiation. J Biol Chem 291: 1789–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su XA, Dion V, Gasser SM, Freudenreich CH. 2015. Regulation of recombination at yeast nuclear pores controls repair and triplet repeat stability. Gene Dev 29: 1006–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz RK, Rodriguez EC, King MC. 2014. A role for nuclear envelope-bridging complexes in homology-directed repair. Mol Biol Cell 25: 2461–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Gautier J. 2011. Double-strand break end resection and repair pathway choice. Annu Rev Genet 45: 247–271. [DOI] [PubMed] [Google Scholar]
- Taddei A, Gasser SM. 2012. Structure and function in the budding yeast nucleus. Genetics 192: 107–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert PB, Ahmad K, Almouzni G, Ausio J, Berger F, Bhalla PL, Bonner WM, Cande WZ, Chadwick BP, Chan SW, et al. 2012. A unified phylogeny-based nomenclature for histone variants. Epigenetics Chromatin 5: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja N, Grewal SIS. 2017. Shushing histone turnover: it's FUN protecting epigenome–genome. Cell Cycle 16: 1731–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang HM, Talbot CC Jr, Fung MC, Tang HL. 2017. Molecular signature of anastasis for reversal of apoptosis. F1000Res 6: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therizols P, Fairhead C, Cabal GG, Genovesio A, Olivo-Marin JC, Dujon B, Fabre E. 2006. Telomere tethering at the nuclear periphery is essential for efficient DNA double strand break repair in subtelomeric region. J Cell Biol 172: 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorslund T, Ripplinger A, Hoffmann S, Wild T, Uckelmann M, Villumsen B, Narita T, Sixma TK, Choudhary C, Bekker-Jensen S, et al. 2015. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 527: 389–393. [DOI] [PubMed] [Google Scholar]
- Timashev LA, Babcock H, Zhuang X, de Lange T. 2017. The DDR at telomeres lacking intact Shelterin does not require substantial chromatin decompaction. Genes Dev 31: 578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timinszky G, Till S, Hassa PO, Hothorn M, Kustatscher G, Nijmeijer B, Colombelli J, Altmeyer M, Stelzer EH, Scheffzek K, et al. 2009. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat Struct Mol Biol 16: 923–929. [DOI] [PubMed] [Google Scholar]
- Torres-Rosell J, Sunjevaric I, De Piccoli G, Sacher M, Eckert-Boulet N, Reid R, Jentsch S, Rothstein R, Aragon L, Lisby M. 2007. The Smc5-Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat Cell Biol 9: 923–931. [DOI] [PubMed] [Google Scholar]
- Tsouroula K, Furst A, Rogier M, Heyer V, Maglott-Roth A, Ferrand A, Reina-San-Martin B, Soutoglou E. 2016. Temporal and spatial uncoupling of DNA double strand break repair pathways within mammalian heterochromatin. Mol Cell 63: 293–305. [DOI] [PubMed] [Google Scholar]
- Vancevska A, Douglass KM, Pfeiffer V, Manley S, Lingner J. 2017. The telomeric DNA damage response occurs in the absence of chromatin decompaction. Genes Dev 31: 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Sluis M, McStay B. 2015. A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes Dev 29: 1151–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez J, Belmont AS, Sedat JW. 2001. Multiple regimes of constrained chromosome motion are regulated in the interphase Drosophila nucleus. Curr Biol 11: 1227–1239. [DOI] [PubMed] [Google Scholar]
- Verdaasdonk JS, Vasquez PA, Barry RM, Barry T, Goodwin S, Forest MG, Bloom K. 2013. Centromere tethering confines chromosome domains. Mol Cell 52: 819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Elledge SJ. 2007. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci 104: 20759–20763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zhai L, Xu J, Joo HY, Jackson S, Erdjument-Bromage H, Tempst P, Xiong Y, Zhang Y. 2006. Histone H3 and H4 ubiquitylation by the CUL4–DDB–ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol Cell 22: 383–394. [DOI] [PubMed] [Google Scholar]
- Wei W, Ba Z, Gao M, Wu Y, Ma Y, Amiard S, White CI, Rendtlew Danielsen JM, Yang YG, Qi Y. 2012. A role for small RNAs in DNA double-strand break repair. Cell 149: 101–112. [DOI] [PubMed] [Google Scholar]
- Weiner A, Zauberman N, Minsky A. 2009. Recombinational DNA repair in a cellular context: a search for the homology search. Nat Rev Microbiol 7: 748–755. [DOI] [PubMed] [Google Scholar]
- Wilson JH, Leung WY, Bosco G, Dieu D, Haber JE. 1994. The frequency of gene targeting in yeast depends on the number of target copies. Proc Natul Acad Sci 91: 177–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbine L, Brunton H, Goodarzi AA, Shibata A, Jeggo PA. 2011. Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and Artemis for their repair. Nucleic Acids Res 39: 6986–6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Sun Y, Jiang X, Ayrapetov MK, Moskwa P, Yang S, Weinstock DM, Price BD. 2010. The p400 ATPase regulates nucleosome stability and chromatin ubiquitination during DNA repair. J Cell Biol 191: 31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Xu Y, Gursoy-Yuzugullu O, Price BD. 2012. The histone variant macroH2A1.1 is recruited to DSBs through a mechanism involving PARP1. FEBS Lett 586: 3920–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Shimada K, Oma Y, Kalck V, Akimura K, Taddei A, Iwahashi H, Kugou K, Ohta K, Gasser SM, et al. 2010. Actin-related protein Arp6 influences H2A.Z-dependent and -independent gene expression and links ribosomal protein genes to nuclear pores. PLoS Genet 6: e1000910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Henikoff S. 2013. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol 20: 259–266. [DOI] [PubMed] [Google Scholar]
- Zhang C, Roberts TM, Yang J, Desai R, Brown GW. 2006. Suppression of genomic instability by SLX5 and SLX8 in Saccharomyces cerevisiae. DNA Repair 5: 336–346. [DOI] [PubMed] [Google Scholar]
- Zhang Y, McCord RP, Ho YJ, Lajoie BR, Hildebrand DG, Simon AC, Becker MS, Alt FW, Dekker J. 2012. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell 148: 908–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. 2006. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol 8: 870–876. [DOI] [PubMed] [Google Scholar]