

Geter et al. show that tamoxifen resistance involves selective mRNA translational reprogramming to an anti-estrogen state by Runx2 and other mRNAs. Tamoxifen-resistant translational reprogramming is shown to be mediated by increased expression of eIF4E and its increased availability by hyperactive mTOR and to require phosphorylation of eIF4E at Ser209 by increased MNK activity.
Keywords: translational control, tamoxifen, eIF4E, translation initiation, cap-dependent translation, ER+ breast cancer
Abstract
The majority of breast cancers expresses the estrogen receptor (ER+) and is treated with anti-estrogen therapies, particularly tamoxifen in premenopausal women. However, tamoxifen resistance is responsible for a large proportion of breast cancer deaths. Using small molecule inhibitors, phospho-mimetic proteins, tamoxifen-sensitive and tamoxifen-resistant breast cancer cells, a tamoxifen-resistant patient-derived xenograft model, patient tumor tissues, and genome-wide transcription and translation studies, we show that tamoxifen resistance involves selective mRNA translational reprogramming to an anti-estrogen state by Runx2 and other mRNAs. Tamoxifen-resistant translational reprogramming is shown to be mediated by increased expression of eIF4E and its increased availability by hyperactive mTOR and to require phosphorylation of eIF4E at Ser209 by increased MNK activity. Resensitization to tamoxifen is restored only by reducing eIF4E expression or mTOR activity and also blocking MNK1 phosphorylation of eIF4E. mRNAs specifically translationally up-regulated with tamoxifen resistance include Runx2, which inhibits ER signaling and estrogen responses and promotes breast cancer metastasis. Silencing Runx2 significantly restores tamoxifen sensitivity. Tamoxifen-resistant but not tamoxifen-sensitive patient ER+ breast cancer specimens also demonstrate strongly increased MNK phosphorylation of eIF4E. eIF4E levels, availability, and phosphorylation therefore promote tamoxifen resistance in ER+ breast cancer through selective mRNA translational reprogramming
Estrogen receptor-positive (ER+) breast cancers comprise the majority (70%–80%) of breast cancers and the majority of breast cancer deaths resulting from metastatic disease (Fisher et al. 2004; Early Breast Cancer Trialists’ Collaborative Group 2005). Anti-estrogen therapy with tamoxifen remains a cornerstone of therapy for ER+ premenopausal breast cancer, but resistance occurs in a third of patients and often progresses to metastasis and death (Musgrove and Sutherland 2009; Droog et al. 2013). ER drives survival and proliferation pathways in breast cancer (Fullwood et al. 2009), functions as a nuclear hormone receptor responsible for integrating signals relayed by estrogen, and plays a critical role in breast cell transformation and carcinogenesis (Sommer and Fuqua 2001). Of the two main isoforms, ERα is implicated primarily in the onset of breast cancer (Sommer and Fuqua 2001). ERα binds transcriptional coactivators and regulators (e.g., NCOA1, NCOA2, and NCOA3) that specify differential transcriptional activity (Sommer and Fuqua 2001; Oxelmark et al. 2006; Simpson et al. 2010). Tamoxifen is an ER-antagonizing small molecule that blocks ER transcriptional activity (Osborne et al. 2000) and inhibits ER+ breast cancer cell proliferation and survival (Osborne et al. 2000).
Most tamoxifen resistance in breast cancer does not involve loss of ERα receptor expression, although mutations are common (Garcia-Quiroz et al. 2014). Tamoxifen resistance often involves hyperactivation of epidermal growth factor receptors (EGFRs) through a variety of mechanisms, including PI3K mutation and activation and MAPK–ERK activation (Campbell et al. 2001; Clark et al. 2002; Miller et al. 2010), resulting in uncoupled signaling from tamoxifen blockade (deGraffenried et al. 2004; Miller et al. 2011; Osborne and Schiff 2011). Both EGFR and ERα signaling can hyperactivate the MAPK–ERK and mTOR pathways, which are known effectors of tamoxifen resistance (Schiff et al. 2004; Massarweh et al. 2008; Miller et al. 2010, 2011; Sabnis and Brodie 2011; Baselga et al. 2012; Bostner et al. 2013; Beelen et al. 2014a,b; Karthik et al. 2015). mTOR consist of two complexes: mTORC1 and mTORC2. mTORC1 regulates protein synthesis, lipid synthesis, and ribosome biogenesis (Dancey 2010; Laplante and Sabatini 2012) and includes the proteins mTOR, Raptor, and GβL, among others (Sabatini 2006). mTORC1 phosphorylates (inactivates) the negative regulator of cap-dependent mRNA translation known as the eIF4E-binding protein (4E-BP1). mTORC2 includes the proteins mTOR, Rictor, and GβL, among others; regulates cytoskeleton organization in response to growth signals; and promotes cell survival and proliferation through activation of AKT (Zoncu et al. 2011). Increased signaling through these pathways is often caused by up-regulated expression of EGFR and IGF-IR proteins, which is also seen in the majority of tamoxifen-resistant ER+ breast cancers and tamoxifen-resistant cell lines (Treeck et al. 2006; Cottu et al. 2014).
Investigation of mTOR-directed endocrine resistance mechanisms have focused primarily on pathway cross-talk and activating upstream mutations. Phosphorylation by S6 kinase 1 (S6K1), a target of mTORC1, establishes endocrine-independent activation of ERα (Yamnik and Holz 2010). Similarly, EGFR activation by dimerization through activating mutations or ligands stimulates both mTORC1/2 and MAPK–ERK pathways and is also associated with tamoxifen and endocrine therapy resistance (Nicholson et al. 2004). Moreover, increased AKT signaling is associated with resistance to anti-hormonal therapy, and, accordingly, inhibition of mTOR partially restores sensitivity (deGraffenried et al. 2004; Beeram et al. 2007). Furthermore, inhibiting both mTORC1/2 complexes blocks upstream AKT activation and increases resensitization to anti-endocrine agents (Leung et al. 2010; Jordan et al. 2014). However, little is known regarding the molecular basis for tamoxifen resistance downstream from mTOR, apart from pathway cross-talk. Downstream effectors of mTOR activity in tamoxifen and endocrine resistance remain unknown. mTOR and ERK signaling pathways converge on the control of mRNA translation (Silvera et al. 2010). Translation of mRNA begins with recognition of the 5′ inverted methyl7-GTP “cap” structure by the translation initiation complex consisting of cap-binding protein eIF4E, RNA helicase eIF4A, and scaffolding protein eIF4G, which recruits the 40S ribosomal subunit and other initiation factors. Many of the translation initiation factors are regulated by mTORC1 activity. In particular, mTOR hyperphosphorylates the 4E-BPs (4E-BP1 is the major form in epithelial cells), preventing them from sequestering eIF4E by competing with the scaffolding protein eIF4G, thereby promoting translation. mRNA translation is also regulated by the MAPK/ERK pathway in response to growth factors, cytokines, and oncogenic signaling (Topisirovic and Sonenberg 2011b). ERK acts on mRNA translation by activating the eIF4G-associated kinase MNK1. MNK1 phosphorylates eIF4E at S209, which is associated with increased transformation potential, although a mechanism is lacking (Wendel et al. 2007; Silvera et al. 2010; Wheater et al. 2010; Konicek et al. 2011; Topisirovic and Sonenberg 2011b; Wolfe et al. 2014). The nature of the increased requirement for eIF4E with malignancy is thought to involve selective mRNA translation, but the mechanism is complex and remains only partially understood (Waskiewicz et al. 1999; Topisirovic and Sonenberg 2011a; Bhat et al. 2015). Up-regulation of the abundance and/or activity of eIF4E, eIF4A, and/or eIF4G occurs widely in breast and other cancers and selectively up-regulates translation of certain mRNAs involved in survival, proliferation, and metastasis (Avdulov et al. 2004; Braunstein et al. 2007; Kim et al. 2009; Silvera et al. 2010; Badura et al. 2012; Decarlo et al. 2015). In fact, increased abundance of eIF4E has been shown to be important in resistance to a variety of PI3K–AKT–mTOR inhibitors (Avdulov et al. 2004; Kim et al. 2009; Silvera et al. 2009a, 2010; Bitterman and Polunovsky 2010; Hsieh et al. 2010; Ilic et al. 2010; Burris 2013; Bhat et al. 2015; Fagan et al. 2017).
Certain mRNAs possess long or structured 5′ untranslated regions (UTRs) that serve to more highly regulate their translation and often encode transforming and survival proteins important for cancer development and progression (Koromilas et al. 1992; Svitkin et al. 2005; Badura et al. 2012). These mRNAs typically display a greater requirement for eIF4E, often a result of increased secondary structure close to the cap (Koromilas et al. 1992; Badura et al. 2012; Hsieh et al. 2012). Certain sequence motifs are also thought to increase the requirement for eIF4E interaction, although direct binding by eIF4E has not been shown (Hsieh et al. 2012). Here we show that an essential mechanism of tamoxifen resistance involves genome-wide translational reprogramming to select for the translation of mRNAs that specifically provide anti-estrogen and ER activities and requires increased expression and availability of eIF4E and its increased phosphorylation by MNK1. Only blockade of both mTORC1 and MNK1 re-establishes tamoxifen sensitivity and blocks selective translation of the small group of mRNAs that provide tamoxifen resistance. We propose a mechanism by which increased levels and phosphorylation of eIF4E promote selective translation of certain mRNAs.
Results
Increased eIF4E abundance, eIF4E S209 phosphorylation, and mTORC1 and MNK activity in tamoxifen-resistant breast cancers and cell lines
We characterized established MCF7 tamoxifen-responsive LCC1 cells (referred to here as TamS cells) and isotype-matched tamoxifen-resistant LCC9 cells (referred to here as TamR cells) used widely as a clinically relevant model of tamoxifen therapy resistance (e.g., Brunner et al. 1997; Clarke et al. 2003; Howell et al. 2004). Tamoxifen-sensitive cells demonstrate impaired growth and fail to transition through G1 to S phase during treatment (Brunner et al. 1997), consistent with the primary inhibitory effect of tamoxifen. TamR cells are resistant to inhibition and maintain normal proliferation and survival to clinically relevant doses of tamoxifen (Supplemental Fig. S1A–C). An ER+ tamoxifen-resistant patient-derived xenograft (PDX) known as BR7 was also insensitive to tamoxifen, as shown by normal cell cycle distribution despite tamoxifen treatment (Supplemental Fig. S1D). TamR and BR7 cells were insensitive to tamoxifen-induced repression of canonical ER signaling as well, in contrast to TamS cells (Supplemental Fig. S1E,F). mTORC1 activity, measured by phosphorylation of 4E-BP1 (S65) and ribosomal protein S6, was elevated in TamR compared with TamS cells (Fig. 1A), as were eIF4E levels (Fig. 1B) and eIF4E phosphorylation (Fig. 1C, normalized for eIF4E levels). eIF4E levels and S209 phosphorylation were also increased in BR7 (PDX) cells compared with TamS cells (Fig. 1D). Therefore, increased mTORC1 and MNK pathway activity is associated with endocrine resistance in cell lines and in a PDX model of endocrine-resistant disease. Increased levels of eIF4E do not typically increase overall protein synthesis very strongly, which was also observed here (Fig. 1E). However, increased levels of eIF4E can selectively increase the translation of specific mRNAs in different physiological conditions (Avdulov et al. 2004; Holcik and Pestova 2007; Pelletier et al. 2015; Truitt et al. 2015). Importantly, there were no significant differences in the levels of other key translation factors in TamR compared with TamS cells, apart from the increased expression of eIF4E (Supplemental Fig. S2A).
Figure 1.
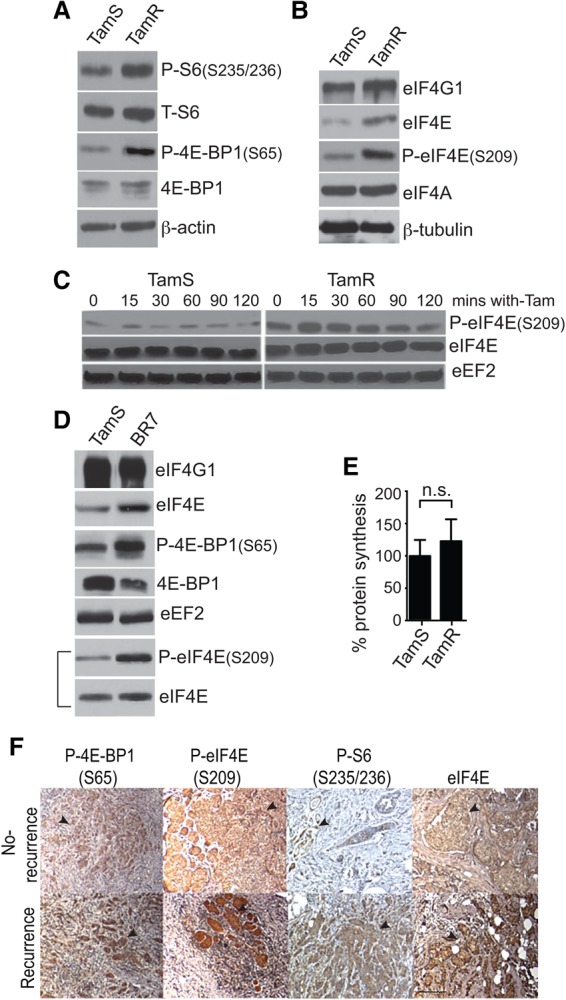
mTORC1 and MAPK pathway hyperactivation in tamoxfien resistance in ER+ breast cancer. (A) Immunoblot of TamS and TamR cells lysed in NP-40 buffer during exponential growth and probed for mTORC1 target proteins. β-Actin was used as a loading control. (B) Immunoblot of representative TamS and TamR cells lysed in NP-40 buffer and probed for the eIF4F complex proteins. eIF4A was also used as a loading control. (C) Immunoblot analysis of protein lysates from TamS and TamR cells following 4-hydroxytamoxifen (4-OHT) treatment in serum-free medium. Cells were treated using serum-free medium, with the 0 time point indicating untreated control samples using ImageJ software. eEF2 was used as a loading control. The bracketed eIF4E-P and eIF4E blots were normalized to loading equal levels of eIF4E. All other blots used equal protein amounts. The image is representative of three independent experiments. (D) NP40 cytoplasmic protein extracts were subjected to immunoblot as shown. Representative results comparing TamS with BR7 PDX cells are shown. eIF4E-P and eIF4E blots were normalized to loading equal levels of eIF4E. All other blots used equal protein amounts. (E) The overall protein synthesis activity of TamS and TamR cells was measured by [35S]-methionine metabolic labeling normalized to TamR cells. A representative of three independent experiments is shown. (n.s.) Not significant. (F) Immunohistochemical staining of representative recurrent and nonrecurrent tumor specimens for P-4E-BP1 (S65), P-S6 (S235/236), P-eIF4E (S209), and total eIF4E. Bar, 20 µm. The immunoblots shown are representative of three independent experiments.
We investigated biopsy specimens from ER+ invasive intraductal breast cancer patients who progressed on treatment (de novo resistance) or recurred within 5 yr of tamoxifen treatment, a standard for resistance, compared with nonrecurrent treated tumors at 10 yr. Despite the small sample size (due to difficulty in obtaining well-validated resistant and sensitive tumor specimens), tamoxifen-resistant tumors showed significantly increased eIF4E S209 phosphorylation (Mnk1 activation) compared with tamoxifen-sensitive tumors (Supplemental Table S1, P = 0.05), as did tamoxifen- or aromatase-resistant tumors (P = 0.016) (Supplemental Table S2). Given the fact that mTORC1 is already highly active and that eIF4E is already overexpressed as a driver of breast cancer, it is not surprising that there was only a trend toward increased mTORC1 activity (P-4E-BP1) and slightly increased eIF4E levels with tamoxifen or aromatase resistance that did not reach statistical significance. The lower saturation level of immunohistochemistry compared with immunoblot may also contribute to the smaller detectable increase in eIF4E levels, although it was apparent in many of the specimens (Fig. 1F).
Reduced overexpression of eIF4E and its S209 phosphorylation are required to restore tamoxifen sensitivity to resistant cells
The role of eIF4E-selective mRNA translation in endocrine therapy resistance was tested by stably transducing TamS and TamR cells with doxycycline (Dox)-inducible shRNAs targeting the 3′ UTR of eIF4E. Quantitative RT–PCR (qRT–PCR) and immunoblot analysis showed an average fourfold reduction of eIF4E mRNA and protein levels (Fig. 2A,B). Interestingly, whereas levels of eIF4E silencing were similar in both cell lines, it resulted in a larger (50% greater) reduction in overall protein synthesis only in TamR cells, indicating a moderate addiction to elevated levels of eIF4E with the acquisition of tamoxifen resistance (Fig. 2C).
Figure 2.
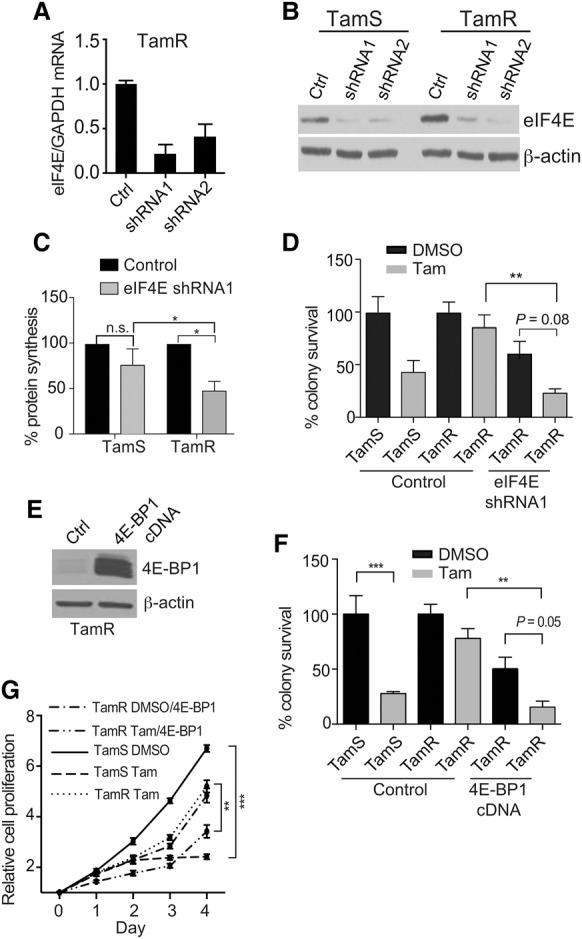
Blocking eIF4F complex formation by targeting eIF4E partially restores tamoxifen sensitivity. (A) mRNA expression of eIF4E in TamR cells following 72 h of 1 µg/mL Dox induction of eIF4E shRNAs. Equal amounts of RNA were quantified by quantitative real-time PCR (RT-qPCR) and normalized to GAPDH using the −ΔΔCt method. (B) Immunoblot of equal amounts of protein from NP-40-extracted TamS and TamR sh-control or sh-eIF4E cells 72 h after Dox addition. β-Actin was used as a loading control. Representative immunoblots are shown. (C) Overall protein synthesis activity of TamS and TamR cells with or without eIF4E silencing by [35S]-methionine metabolic labeling. Three independent studies were averaged. (*) P < 0.05 by two-way ANOVA; (n.s.) not significant. (D) Colony survival growth assay was performed by low-density seeding (1000 cells) of stably transduced TamS and TamR cells treated with vehicle (DMSO) or 1 µM 4-OHT 24 h after plating. Dox (1 µg/mL) was administered 24 h after plating and removed at 72 h. Colonies were scored after 10 d, counting only ≥50 cells per colony. Results from three independent experiments were normalized to DMSO control. (**) P < 0.01. Comparisons were by two-way ANOVA. (E) Representative immunoblot of equal amounts of protein lysate from 4E-BP1-overexpressing cells. Dox (1 µg/mL) was added 72 h prior to lysis in NP-40 buffer. β-Actin was used as a loading control. (F) Colony survival assays were performed as in D after plating with Dox-induced 4E-BP1 expression. (**) P < 0.01; (***) P < 0.001 by two-way ANOVA. (G) Cell proliferation was assayed with Dox-induced overexpression of 4E-BP1, as shown. Cell proliferation was assayed by MTT and treated with vehicle (DMSO) or 1 µM 4-OHT. Dox was added at day 0 to induce 4E-BP1 expression. Results from three independent experiments were normalized to day 0. (**) P < 0.01; (***) P < 0.001 by t-test.
We therefore asked whether the increased expression and/or phosphorylation of eIF4E is essential for selective mRNA translation and tamoxifen resistance. Silencing eIF4E in the presence of 4-hydroxytamoxifen (4-OHT), the active metabolite of tamoxifen, reduced clonogenic cell survival and cell growth of TamR cells by fourfold compared with the nonsilencing control and threefold to fourfold compared with silenced but untreated TamR cells (Fig. 2D; Supplemental Fig. S2B,C). Similarly inducible overexpression of 4E-BP1 in TamR-resistant cells (Fig. 2E) reduced eIF4E cap-binding complexes (Supplemental Fig. S2D) and resulted in a fourfold reduction in cell proliferation and survival of tamoxifen-treated TamR cells compared with controls (Fig. 2F,G). Moreover, silencing eIF4E in the BR7 PDX model restored tamoxifen sensitivity, as shown by delayed cell cycling in response to treatment (Supplemental Fig. S2E). Everolimus (RAD001) is an inhibitor of the mTORC1 signaling pathway and is currently approved for the treatment of hormone receptor-positive endocrine therapy-resistant breast cancers. To confirm the involvement of mTORC1 in endocrine resistance, TamR cells were treated with RAD001, which partially resensitized them to tamoxifen, as shown by the restoration of inhibition of cell proliferation and decreased survival (Supplemental Fig. S2F,G). Both increased mTORC1 activity and eIF4E availability are therefore required for tamoxifen resistance. Collectively, these data suggest a critical role for the eIF4E/4E-BP1 balance in regulating tamoxifen resistance and responsiveness by mTORC1 activity.
A requirement for eIF4E S209 phosphorylation (MNK1-mediated) has been implicated in tumorigenesis and metastasis (Bianchini et al. 2008; Wheater et al. 2010; Robichaud et al. 2015), and we observed increased eIF4E S209 phosphorylation in tamoxifen-resistant breast tumor tissues and cell lines. We therefore examined the role of eIF4E phosphorylation in tamoxifen resistance. A serine-to-alanine HA-tagged eIF4E protein (S209A) or a serine-to-aspartic acid protein (S209D) was expressed in TamS and TamR cells (Supplemental Fig. S3A). Endogenous eIF4E was silenced to eliminate its contribution, and cells were assayed for proliferation in the presence or absence of 4-OHT (Fig. 3A,B). Notably, TamR cells were blocked in proliferation by expression of the nonphosphorylated S209A eIF4E mutant only in the presence of tamoxifen (implicating an essential role for MNK1-mediated eIF4E phosphorylation at S209) and overexpression of eIF4E in tamoxifen resistance. However, expression of the S209D phosphomimetic eIF4E protein in TamS cells did not confer tamoxifen-resistant proliferation. These data suggest two possibilities: that acquisition of tamoxifen resistance is multigenic and not solely the result of eIF4E overexpression and phosphorylation (whereas resistance can be reversed by impairing either because both are important) and/or that the phospho-mimetic eIF4E variant protein cannot fully recapitulate the effects of eIF4E phosphorylation, consistent with a previous report (Topisirovic et al. 2004).
Figure 3.

eIF4E S209 phosphorylation promotes tamoxifen resistance. (A) TamS cells were transfected with either empty vector or an eIF4E S209D-expressing construct 48 h prior to proliferation assay. Endogenous eIF4E was silenced by shRNA. Cell proliferation was assayed by MTT assay with cells treated with DMSO vehicle or 1 µM 4-OHT. Results are from three independent experiments. (B) TamR cells were transfected with empty vector or eIF4E S209A-expressing vector 48 h prior to proliferation assay. Endogenous eIF4E was silenced by shRNA. Cell proliferation was assayed by MTT assay using conditions described in the legend for Figure 2. Results from three independent experiments are shown. (C) Immunoblot of TamS and TamR cells treated with escalating doses of CGP57380 (GCP) for 2 h and lysed in NP-40 buffer; equal protein amounts were probed for P-eIF4E, total eIF4E, and β-actin (loading control). (D) Cell proliferation was assayed as above. Cells were treated with DMSO, 1 µM 4-OHT, or 4-OHT and MNK1 inhibitor CGP. Results of three independent experiments are shown. (**) P < 0.01 by t-test. (E) Colony survival assays were performed as described in the legend for Figure 2. Cells were treated with DMSO, 1 µM 4-OHT, 10 µM CGP, or combination therapy 24 h after plating. Drugs were restored every 72 h. Data from three independent experiments were normalized to DMSO control. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001 by two-way ANOVA; (n.s.) not significant. (F) TamR colony survival assays were performed as described in the legend for Figure 2 and treated as in E plus 20 mM RAD001. Data from three independent experiments were normalized to DMSO control. (**) P < 0.01 by t-test.
We next investigated the effect of inhibition of MNK1 on tamoxifen sensitivity by the small molecule MNK1 inhibitor CGP57380. Dose escalation studies on both TamS and TamR cells established a concentration of 10 µM for complete inhibition of eIF4E phosphorylation by CGP57380 (identical in dose to previously reported) (Fig. 3C), where it has no inhibitory activity on other families of related kinases (p38, JNK1, and RSKs) (Knauf et al. 2001; Rowlett et al. 2008). This was confirmed in TamR cells by examining ATF2 phosphorylation at Thr69/71 (a target of RSK, JNK1, and p38 MAPK) and eIF4B S422 (a target of RSKs)—the next most sensitive kinases of CGP57380 inhibition. There was no change in phosphorylation of either protein with treatment (Supplemental Fig. S3B). Combined treatment with tamoxifen and CGP57380 significantly resensitized TamR cells to tamoxifen, as shown by a >60% reduction in proliferation and clonogenic survival compared with untreated controls (Fig. 3D,E). As described previously, cotreatment with mTORC1 inhibitor RAD001 produced a further additive reduction in cell survival (Fig. 3F; Supplemental Fig. S3C), with only a 10% reduction in global protein synthesis despite complete ablation of eIF4E phosphorylation (Supplemental Fig. S3D). Polysome profiling of tamoxifen-resistant cells following CGP57380 treatment showed no significant differences (Supplemental Fig. S3E), suggesting a role for eIF4E phosphorylation in selectively reprogramming the translation of a small subset of mRNAs involved in tamoxifen resistance.
Both overexpression of eIF4E and its phosphorylation are required to promote tamoxifen resistance in normally sensitive ER+ breast cancer cells
Since merely expressing a phospho-mimetic eIF4E is insufficient to confer tamoxifen resistance to normally sensitive cells, we determined whether both overexpression of eIF4E and increased eIF4E S209 phosphorylation were required. TamS cells were stably transfected with a Dox-inducible HA-tagged eIF4E cDNA that tripled eIF4E levels (Fig. 4A). Tamoxifen-sensitive TamS cells were unable to proliferate in the presence of tamoxifen regardless of eIF4E overexpression (Fig. 4B). However, since eIF4E availability and phosphorylation are limited by 4E-BP1, we hyperactivated mTORC1 by disrupting the repressing TSC1/TSC2 complex through shRNA silencing of Tsc2 (Fig. 4C). Silencing Tsc2 strongly increases mTORC1 signaling (Sato et al. 2012), demonstrated here by increased phosphorylation of 4E-BP1 and ribosomal protein S6. Importantly, Tsc2 silencing conferred tamoxifen resistance to normally sensitive ER+ breast cancer cells (Fig. 4D). Cosilencing Tsc2 and overexpressing eIF4E slightly reduced tamoxifen resistance for unknown reasons but might be related to homeostatic regulation of eIF4E levels. We noted somewhat lower levels of eIF4E and 4E-BP1 phosphorylation in Tsc2 silenced eIF4E-overexpressing cells, consistent with this possibility. The importance of eIF4E S209 phosphorylation using a phospho-dead protein could not be tested due to the inability to sufficiently silence endogenous eIF4E in cells that were already drug-selected twice. Nevertheless, eIF4E and its phosphorylation, increased mTORC1 activity, and increased levels of available eIF4E and its phosphorylation can confer tamoxifen resistance. We note somewhat less eIF4E and 4E-BP1 phosphorylation in Tsc2 silenced cells with eIF4E overexpression, supportive of this possibility (Fig. 4C). There was no change in basal ER signaling under these conditions, as shown by induction of ER biomarker mRNAs (Fig. 4E).
Figure 4.
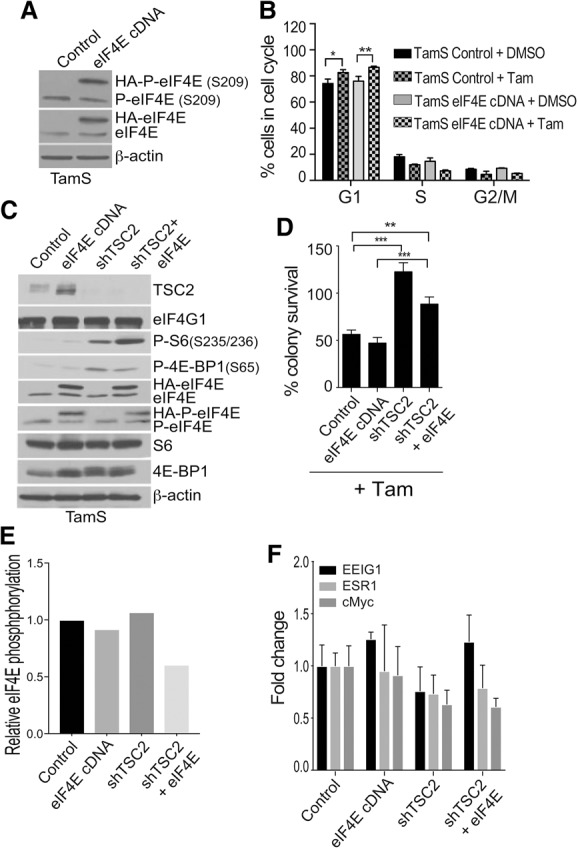
Hyperactivation of mTORC1 and eIF4E overexpression reprogram the cancer genome to mimic tamoxifen resistance. (A) Representative immunoblot analysis of equal amounts of protein lysate from eIF4E-overexpressing cells. Dox (2 µg/mL) was added 72 h prior to lysis in NP-40 buffer. Equal protein amounts were immunoblotted as shown. β-Actin was used as a loading control. (B) Cell cycle analysis of TamS control and TamS cells overexpressing eIF4E and treated with DMSO (vehicle) or 1µM 4-OHT for 72 h in 1% CS-FBS. Dox (2 µg/mL) was added for 72 h. Cells were subjected to exhaustive RNase A and stained with propidium iodide (PI). Flow cytometry data were collected using a FACScalibur and analyzed with FloJo software. The average of three studies is shown. (C) Immunoblot analysis of mTORC1 pathway proteins in TamS control, eIF4E-overexpressing, TSC2 silenced, or TSC2 silenced and eIF4E-overexpressing cells. Cells were treated with 2 µg/mL Dox for 72 h and lysed in NP-40 buffer. Equal protein amounts were immunoblotted. β-Actin was used as a loading control. Representative results are shown. (D) Colony survival assays from three studies were performed as described in the legend for Figure 2. TamS sh-control, eIF4E-overexpressing, shTSC2, and shTSC2 and eIF4E-overexpressing cells were treated with either DMSO or 1 µM 4-OHT. (**) P < 0.01; (***) P < 0.0001 by two-way ANOVA. (E) Quantitation of eIF4E S209 phosphorylation in the cells treated in C. (F) Markers of ER signaling in TamS cells were quantified by RT-qPCR of mRNAs with cDNA overexpression of eIF4E, shRNA silencing of TSC2, or both. Results are the average of three independent studies.
mRNAs are altered in abundance and translation in tamoxifen-resistant compared with tamoxifen-sensitive breast cancer cells
Research on tamoxifen-resistant disease has not yet been focused on differential mRNA translation. Here we sought to identify mRNAs that are selectively altered in translation in tamoxifen-resistant cells. We conducted a genome-wide translatome and transcriptome analysis using RNA sequencing (RNA-seq) of TamR and TamS cells. Three sets of conditions were analyzed to fully represent the genome-wide changes in mRNA abundance and translation: (1) expression levels for total mRNA (transcription), (2) changes in translation (heavy polysome fraction) regardless of mRNA abundance or translational regulation, and (3) translation-specific changes (ratio of heavy polysome mRNA/total mRNA) (Fig. 5A,B; Supplemental Table S3). Analyses used a cutoff of log2 1.0 (twofold) for total mRNA and log2 0.6 (1.5-fold) for heavy polysome association; the latter was set lower because smaller changes in protein expression can have significant physiological effects. Significance was set at P < 0.05 for both mRNA and polysome analysis. Gene ontology (GO) analyses of significantly altered genes in both transcription and translation revealed an enrichment of developmental, cell survival, and differentiation pathways in endocrine therapy-resistant cells (Fig. 5C–G). We note specific enrichment in up-regulated Hox and DNA recombination genes, with a concomitant repression of estrogen and Tgfβ genes (Supplemental Table S4). Moreover, Hox genes encode transcription factors that specify stem cell fate determination and are also important in oncogenesis (Shah and Sukumar 2010). Both the ER and TGF-β pathways play a pivotal role in tumor suppression (Bachman and Park 2005; Berger et al. 2013).
Figure 5.

Selective translation of mRNAs important in cell proliferation, survival, and genomic reprogramming in tamoxifen-resistant compared with tamoxifen-sensitive breast cancer cells. (A,B) Genome-wide transcription and translation mRNA profiling of TamS compared with TamR cells with 1 µM 4-OHT for 48 h. Results are from two independent studies. Total mRNA and purified fractions containing four or more bound ribosomes (heavy) were sequenced using Illumina HiSeq 2500 single read. Volcano plots represent differences in transcription and translation. Blue dots identify mRNAs significantly changed in abundance. Transcription parameters were P ≤ 0.05 and −1.0 ≤ log2 ≥ 1.0, translation parameters were P ≤ 0.05 and −0.6 ≤ log2 ≥ 0.6. Red dots identify mRNAs not significantly changed in abundance. Statistical analysis was performed using the limma R package. (C,D) The top molecular functions of mRNAs significantly altered in total abundance and translation from heavy (well-translated) fractions (four or more ribosomes), respectively. (E,F) The top biological functions of mRNAs significantly altered in total abundance and translation from heavy (well-translated) fractions (four or more ribosomes), respectively. (G) Relative pathway summation of transcriptional and translational changes in mRNAs in TamR cells relative to TamS cells.
Identification of mRNAs highly dependent on overexpression of P-eIF4E for translation in tamoxifen-resistant breast cancer cells
We next identified mRNAs associated with tamoxifen resistance that were selectively altered in translation resulting from increased expression, availability, and phosphorylation of eIF4E. We used genome-wide transcriptomic and translatomic analyses in TamR cells with and without modest eIF4E reduction to identify this data set by silencing eIF4E to levels similar to TamS cells (Fig. 6A). Total mRNA and polysomal mRNA profiling showed only a very slight overall reduction in mRNA and polysome content in tamoxifen-resistant cells after eIF4E silencing (Fig. 6A–C; Supplemental Fig. S4A–C). Surprisingly, the number of mRNAs that changed significantly only at the translation-specific level was small (with most of them down-regulated) but included a small number that were translationally up-regulated as well (Fig. 6C; Supplemental Fig. S4A; Supplemental Table S6). Select genes from RNA-seq analysis were validated by qPCR of total and heavy polysome fractions (Supplemental Fig. S4B,C). GO analyses and Ingenuity Pathway Analyses (IPAs) revealed similar biological and molecular functions for transcriptionally and translationally increased mRNAs associated with tamoxifen resistance (Fig. 6D–F; Supplemental Fig. S4E–G).
Figure 6.

Tamoxifen resistance is associated with eIF4E overexpression and selective mRNA translation. (A) Polysome profiles of TamR cells without and with eIF4E silencing. Representative results are shown. (Inset) Immunoblot showing eIF4E levels and control β-actin. (B,C) Genome-wide transcription and translation mRNA profiling of TamR cells with or without eF4E silencing plus 1 µM 4-OHT for 48 h. Results are from two independent studies. Total mRNA and purified fractions containing four or more bound ribosomes (heavy) were sequenced and analyzed using the same parameters as described in the legend for Figure 5. (D,E) The top molecular functions of mRNAs significantly altered in total abundance (D) and in translation (E) from heavy (well-translated) fractions (four or more ribosomes), respectively. (F) The top biological functions of mRNAs significantly altered in translation from heavy polyribosomes.
We assessed the biological functions of mRNAs identified as highly eIF4E-dependent in tamoxifen-resistant cells. Supplemental Table S5 lists mRNAs that meet these stringent criteria. Of these, Runx2 was particularly notable because it encodes a protein with a number of activities that could play a role in tamoxifen resistance, as it is an important inhibitor of estrogen signaling and stimulates oncogenic pathways. RUNX2 is a transcription factor involved in regulating cell determination (Young et al. 2007; Blyth et al. 2010) and the TGF-β and Wnt/β-catenin pathways (which are also involved in cancer development, progression, and metastasis) (Young et al. 2007; Yang et al. 2015) and opposes ER signaling, leading to more aggressive ER+ breast cancer (Tandon et al. 2014). Interestingly, recent studies have also shown that RUNX2 plays a crucial role in regulating mammary stem cell regeneration (Ferrari et al. 2015). Surprisingly, we found Runx2 to be the only Runx gene within the family to be transcriptionally or translationally up-regulated in both TamR and PDX tamoxifen-resistant cell lines (Fig. 7A,B; Supplemental Table S4). Total Runx2 mRNA levels were unchanged with eIF4E reduction in TamR cells, but heavy polysome association was reduced threefold, which corresponds to a fourfold to fivefold reduction in RUNX2 protein levels (Fig. 7C,D). We also determined whether Runx2 mRNA requires eIF4E S209 phosphorylation by Mnk1. Cells were untreated or treated with CGP57380, and Runx2 mRNA and protein levels were determined. There was no statistically significant difference in Runx2 mRNA levels with drug treatment, whereas RUNX2 protein levels were reduced more than threefold (Fig. 7E). Therefore, increased levels or availability of eIF4E and increased eIF4E S209 phosphorylation by Mnk1 promote selectively increased translation of Runx2 mRNA.
Figure 7.

Silencing Runx2 mRNA partially restores tamoxifen sensitivity to resistant cells. (A) Relative levels of Runx2 mRNA and levels in heavy polysomes in TamR compared with TamS cells. (B) Levels of Runx2 mRNA and protein in BR7 compared with TamS cells. mRNA levels were determined by RT-qPCR, as above. The average of three studies is shown. The immunoblot is representative of three independent studies. (C) Relative levels of Runx2 mRNA in total and heavy polysomes in TamR cells in Dox-inducible sh-control nonsilencing (NS) and sh-eIF4E silencing for 72 h. Equal amounts of RNA were quantified by RT-qPCR and normalized to GAPDH using the −ΔΔCt method. An average of three studies is shown. (D) Representative immunoblot analysis of equal amounts of protein lysate from TamR cells silenced with nonsilencing (NS) control or sh-eIF4E for 72 h. Equal protein amounts were immunoblotted as shown. eEF2 was used as a loading control. (E) TamS and TamR cells were treated with CGP57380 for 6 h, and equal protein amounts of lysates were examined by immunoblot as shown. (F) Relative levels of Runx2 mRNA in TamR cells silenced with nonsilencing (NS) control or sh-Runx2 were analyzed as above. (G) Cell proliferation was assayed as described in the legend for Figure 2. Cells were treated with DMSO and 1 µM 4-OHT and silenced for Runx2 or nonsilencing (NS). The results of three independent experiments are shown. (H) Colony survival assays from three studies were performed as described in the legend for Figure 2 using TamR cells with sh-nonsilencing (sh-NS) control or sh-Runx2 in the presence of 1 µM 4-OHT. (I) STRING analysis of the top 20 protein interactors of the Runx2–ERα complex. Light-blue and purple lines represent validated interactions, and green, red, and dark-blue lines represent predicted interactions based on past literature. (J) Levels of identified mRNAs in TamR cells treated with DMSO, 1 µM 4-OHT, or 4-OHT and silenced for Runx2 or nonsilencing (NS) after 72 h of treatment. (**)P < 0.01; (***)P < 0.001 by t-test.
Computational analysis of the Runx2 5′ UTR is consistent with a greater eIF4E dependency for translation, showing significant secondary structure and a high GC content within 30 nucleotides of the cap as well as a ΔG of approximately −40 kcal/mol (Supplemental Fig. S4H). To this end, we asked whether mRNAs that are translationally down-regulated upon eIF4E reduction have features in their 5′ UTRs that dictate a strong dependence on eIF4E levels. We conducted a genome-wide analysis of the 5′ UTR of mRNAs translationally down-regulated with eIF4E silencing to the level of tamoxifen-sensitive cells; however, we did not observe any statistical differences in GC content or length when compared with all cellular mRNAs (Supplemental Fig. S5A,B). This is consistent with previously published results regarding 5′ UTR analysis of translationally altered mRNAs upon eIF4E down-regulation (Truitt et al. 2015). To investigate the importance of RUNX2 in tamoxifen resistance in ER+ cells, we used shRNA to reduce Runx2 mRNA levels approximately threefold, to levels found in tamoxifen-sensitive TamS cells and TamR cells silenced for eIF4E (Fig. 7F). The threefold reduction in RUNX2 in TamR cells resulted in a strong impairment in proliferation in the presence of tamoxifen (Fig. 7H) as well as a significant reduction in clonogenic cell survival of normally drug-resistant cells (Fig. 7I).
RUNX2 establishes a molecular program that opposes ERα signaling in both the normal and transformed settings (Chimge and Frenkel 2013; McDonald et al. 2014). In support of these studies, using computational STRING analysis, we identified interactions between RUNX2 and ERα, including established tamoxifen resistance genes (Fig. 7I). Furthermore, ERα–RUNX2 interaction analysis is consistent with RUNX2 stimulation of both the mTORC1 and MAPK translational control pathways (Fig. 7I) to promote drug resistance. In fact, it has been reported that breast tumors expressing high levels of RUNX2 generally express low levels of ERα and vice versa. To this end, we performed an extensive bioinformatics search of the TCGA (The Cancer Genome Atlas) breast cancer database as well as analysis of breast cancer cell lines and found an almost perfect inverse correlation between ERα (ESR1) and Runx2 mRNA expression in 594 patients diagnosed with ER+ breast cancer (Supplemental Fig. S5C,D). To understand the significance of the RUNX2–ERα axis in relation to tamoxifen responsiveness, we investigated whether silencing Runx2 in TamR cells reverses the RUNX2 blockade of canonical ERα signaling as a mechanism to re-establish drug sensitivity. qRT–PCR analysis of ER target genes indicated that reduction of RUNX2 did not restore ERα signaling in resistant cells (Fig. 7J), indicating that RUNX2 expression may permanently overwrite classical ERα signaling, leading to genomic changes that establish permanent anti-estrogen resistance.
Discussion
The majority of ER+ breast cancer patients treated with tamoxifen will relapse with resistant disease even decades after curative care (Ali and Coombes 2002). Evidence has shown that a cross-talk exists between the ER and the PI3K/Akt/mTOR and MAPK signaling pathways in promoting tamoxifen resistance (Sommer and Fuqua 2001; Fan et al. 2014). Our findings indicate that tamoxifen resistance also involves the mRNA translational regulation of these pathways and is manifested by increased eIF4E levels, availability, and phosphorylation, resulting in selective mRNA translational reprogramming that establishes an anti-ER and anti-estrogen signaling state. Furthermore, these results have broader implications in understanding resistance to other endocrine therapies; notably, resistance to aromatase inhibitors. While aromatase inhibitors are also clinically used for the treatment of metastatic ER+ breast cancers, resistance occurs and is thought to arise through mechanisms similar to tamoxifen; namely, mTORC1 inhibition in combination with aromatase inhibitors leads to an overall increase in patient survival similar to results obtained in this and other studies regarding anti-estrogen resistance.
Alterations in eIF4E-dependent translation can promote selective translation of mRNAs that reprogram cancer cells for survival, invasion, metastasis, and possibly drug resistance (Silvera et al. 2009b; Hsieh et al. 2012; Boussemart et al. 2014). We previously showed a causal role for selective mRNA translation in therapy resistance in breast and other cancers (Braunstein et al. 2009; Ramírez-Valle et al. 2010; Badura et al. 2012; Korets et al. 2014). Here we disclose a mechanism by which increased expression, availability, and phosphorylation of eIF4E form a regulatory nexus important in anti-estrogen resistance in ER+ breast cancer. During therapeutic treatment, when cell surface EGF and IGF receptors are activated, they transactivate downstream MAPK/ERK/MNK and PI3K/Akt/mTOR pathways and ultimately converge on eIF4E, increasing its activity, phosphorylation, and availability. This leads to eIF4E-mediated selective translation of key mRNAs, such as Runx2. Our genome-wide transcription/translation analysis of tamoxifen-resistant cells revealed that several key pathways are down-regulated upon eIF4E silencing, with a majority of the down-regulated pathways with eIF4E silencing involved in cellular organization and motility, genetic recombination, and developmental processes (Ramírez-Valle et al. 2008; Cao et al. 2016). Most of these mRNAs are involved in DNA–protein interactions and the regulation of transcription factor binding. From these findings, we identified RUNX2 at the intersection of these molecular functions and demonstrated a strong translational down-regulation with eIF4E silencing in tamoxifen-resistant cells.
RUNX2 belongs to the family of RUNX transcription factors (RUNX1,2,3), which are involved in lineage-specific cell fate determination that is recapitulated in cellular transformation and tumorigenesis. RUNX proteins regulate gene expression by functioning as molecular scaffolds to recruit chromatin remodeling enzymes (e.g., SWI/SNF and CTCF) and modulate promoter accessibility (Young et al. 2007; Wu et al. 2014). Studies involving the role of RUNX2 in breast cancer have demonstrated the importance of overexpression of RUNX2 in regulating tumor growth, epithelial–mesenchymal transition, and metastasis (Pratap et al. 2009; Chimge et al. 2011; Karlin et al. 2014). Furthermore, RUNX2 has been shown to regulate the expression of genes involved in WNT/β-catenin and TGF-β signaling—two key pathways known to be dysregulated in many cancers, particularly breast cancer (Chimge and Frenkel 2013; Ferrari et al. 2015). Importantly, WNT and TGF-β signaling has been shown to promote cancer progression to a more poorly differentiated state (Barcellos-Hoff and Akhurst 2009; Ferrari et al. 2015). Notably, other studies describing RUNX2 transcriptional activity have shown that it regulates ER signaling by direct and indirect interactions (McDonald et al. 2014; Jeselsohn et al. 2017). Thus, our studies demonstrate that established genes and signaling pathways that confer tamoxifen resistance (and possibly other forms of endocrine therapy resistance) do so by acting on eIF4E abundance and phosphorylation to selectively translationally reprogram the breast cancer cell for estrogen- and ER-opposing activities.
Materials and methods
Chemicals and inhibitors
Final concentrations of chemicals and inhibitors used were 0.02% DMSO, 1 µM 4-OHT (Millipore), 20 nM RAD001 (Selleck Chemicals), and 10 µM CGP57380 (Sigma).
Cell lines and cell culture
MCF7 and BR7 cells were maintained in improved MEM (IMEM) with L-glutamine without phenol red (Cellgro), 5% fetal bovine serum (FBS) (Gibco), 0.4% gentamicin sulfate (Lonza), 0.5 µg/mL fungizone (Gibco), and 5 µg/mL plasmocin at 37°C in a 5% CO2 tissue culture incubator. 4-OHT (1 µM) was added to TamR cells every 72 h. HEK293FT cells were maintained in DMEM with L-glutamine (Corning), 10% FBS, 1% penicillin–streptomycin (Life Technologies), 1 mM sodium pyruvate (Thermo Scientific), and 1% MEM nonessential amino acids (Thermo Scientific). Cells were routinely checked for mycoplasma contamination.
Patient cohorts and tissues
Archival tumor tissue specimens were obtained with prior Institutional Review Board (IRB) approval for patients ≥18 yr of age with ER+ (≥5% ER+ staining) invasive ductal breast cancer (IDC) stage II/III treated with adjuvant tamoxifen and/or aromatase inhibitor (Supplemental Tables S1, S2). Patients who recurred within 5 yr were considered resistant. A pathology database of all available treated tumor specimens was queried to identify cases between 2002 and 2011 that had a clinical description of <5-yr recurrence or no recurrence at 10 yr.
Anchorage-dependent colony formation assays
Cells were trypsinized, filtered, and counted using an automated cell counter (Bio-Rad). Cells (1 × 103) were seeded in triplicate in six-well culture dishes using IMEM supplemented with 5% charcoal-stripped FBS and 0.4% gentamicin sulfate and allowed to adhere overnight. The medium was changed, and the indicated treatments were carried out. The medium and treatments were changed every 72 h for 10–12 d. Colonies were washed, fixed, and stained with 0.5% Crystal Violet in 6% glutaraldehyde. Colonies containing ≥50 cells were scored.
Cell cycle analysis
Cells were trypsinzed, filtered, and counted using an automated cell counter (Bio-Rad). Cells (7 × 105) were seeded on 10-cm culture plates in IMEM (Corning) with 5% FBS (Gibco) and 0.4% gentamicin sulfate (Lonza) and allowed to adhere overnight. The medium was changed to IMEM (Corning) with 1% charcoal-stripped FBS (HyClone) and 0.4% gentamicin sulfate for 48 h. Cells were treated with the appropriate drug for 72 h and with fresh drug after 48 h. Cells were trypsinized and fixed in 70% ethanol overnight at 4°C. Cells were washed with PBS and treated with 0.5 mg/mL RNase A for 30 min at 37°C. Cells were washed again with PBS and stained with 50 µg/mL propidium iodide (PI) or Hoechst 33342 for 45 min at room temperature and protected from light. Data were collected using a FACScalibur or LSRII UV and analyzed with FlowJo 10.0.
Polysome-associated mRNA isolation
Isolation of ribosome-bound mRNA by polysome separation was performed as described previously with minor modifications (Silvera et al. 2017). Briefly, MCF7 cells were seeded 48 h prior to treatment, and cells were treated with 100 µg/mL cycloheximide for 10 min at 37°C, trypsinized, and collected in ice-cold PBS containing protease inhibitor cocktail and EDTA-free (Roche Diagnostics). All subsequent steps contained 100 µg/mL cycloheximide. Cells were resuspended in low-salt buffer (LSB; 20 mM Tris at pH 7.4–7.5, 10 mM NaCl, 3 mM MgCl2, ribonuclease inhibitor [Thermo Scientific]) and incubated for 3–5 min on ice. Detergent buffer (LSB with 1.2% Triton X-100, 0.2 M sucrose) was then added, and cells were lysed with 15–20 strokes in a sterilized Dounce homogenizer at 4°C. Lysates were cleared by microfuge centrifugation at maximum speed for 5 min, and supernatant was combined with 100 µL of heparin buffer (LSB with 10 mg/mL heparin, 1.5 M NaCl) and then layered on a 15%–50% sucrose gradient in LSB using equal OD260 units of samples. Gradients were centrifuged at 36,000 rpm for 2 h in a SW40Ti rotor (Beckman Coulter), and polysome profiles were done at UV absorbance 254 nm by continuous flow cell monitoring and collected using an Isco UA-6 absorbance detector (Teledyne ISCO) and with a Foxy R1 fraction collector (Teledyne ISCO) at 1.5 mL/min.
RNA-seq and analysis
RNA was extracted and purified from pooled polysome fractions using the RNeasy minikit (Qiagen) as per the manufacturer's instructions. RNA quality was measured by a Bioanalyzer (Agilent Technologies). Fractions containing two to three bound ribosomes were considered poorly translated (light fractions), and those containing four or more bound ribosomes were considered well translated (heavy fraction). RNA-seq was carried out by the New York University School of Medicine Genome Technology Core using the Illumina HiSeq 2500 single read. To quantify translational efficiency, the difference in log2 intensity between matched polysomal mRNA and total mRNA was determined. To examine differences in transcription and translation, total mRNA and polysome mRNA were quantile-normalized separately. Statistical analysis was performed using the limma R package (Ritchie et al. 2015). Gene enrichment analysis was performed using IPA software, and GO analysis was performed using the DAVID online tool.
Quantitative real-time PCR (RT-qPCR) and analysis
RNA was extracted using Trizol as per the manufacturer's instructions. One microgram of RNA was used for reverse transcription reaction using Promega GoScript, as per the manufacturer's instructions. qRT–PCR was completed using iTaq Universal 2× SYBR Green qPCR master mix (Bio-Rad) and an Applied Biosystems 7500 Fast RT–PCR machine as per the manufacturers’ instructions. Fold change was calculated using the 2−ΔΔCt method. A list of the primers used is available on request.
Cap chromatography
In brief, cells were lysed in NP-40 buffer (50 mM HEPES at pH7.0, 150 mM NaCl, 2 mM EDTA, 25 mM NaF, 25 mM β-glycerophosphate, 2 mM Na3VO4, 1% IGEPAL, Complete miniprotease inhibitor cocktail tablet ± EDTA [Roche]), and lysates were cleared by microcentrifugation at 13,000 rpm for 10 min at 4°C. Lysate protein concentration was determined by BCA assay, equal amounts were incubated with m7GTP Sepharose beads for 1 h at 4°C, and beads were collected by centrifugation, washed three times with lysis buffer, resolved by 10% or 12% SDS-PAGE, and transferred to a PVDF transfer membrane (Millipore). The membrane was blocked in 5% BSA in TBS-T at 4°C. Primary antibodies were incubated overnight at 4°C. Secondary ECL antibodies (GE Healthcare) were incubated for 1 h at room temperature in 5% reconstituted dried milk in TBS-T. Protein was imaged using the chemiluminescence method and Genemate autoradiography film.
Cell proliferation assay
Cell proliferation was measured using the CellTiter 96 nonradioactive cell proliferation assay kit (Promega) according to the manufacturer's instructions. MCF7 cells were plated at 1500 cells per well in triplicate in 96-well culture plates. Cells were allowed to attach overnight. On day 0, 15 µL of dye solution containing tetrazolium was added to each well and incubated for 4 h at 37°C. One-hundred microliters of Stop Six was added to each well to solubilize the formazan products using the overnight method in a humidified chamber. Absorbance was measured at 570 nm. Four more time points were collected on days 1–4. Time points were normalized to day 0.
Statistical analysis
Unpaired t-test and two-way or one-way ANOVA were used for biological studies when applicable to determine statistical significance. Biomarkers were of ordinal measurements and used Fisher's exact test. Data were analyzed using GraphPad Prism 6.0e. Significant values were considered P < 0.05 (*), P < 0.01 (**), or P < 0.001 (***).
Supplementary Material
Acknowledgments
We thank the New York University Flow Cytometry and Cell Sorting Core and the New York University Center for Biospecimen and Research and Development. We thank Jerson M.M. Castro and Emilio A. Galvan for helpful assistance in certain studies. We thank Dr. Hua Zhong (New York University) for statistical analyses of immunohistochemistry results. This work was supported by the Training in Pharmacological Sciences (5T32 GM066704 to P.A.G.), UL1 TR00038 from the National Center for Advancing Translational Sciences (NCATS) for core services support, the Howard Hughes Medical Institute Gilliam Fellowship (to P.A.G.), the Breast Cancer Research Foundation (BCRF-16-143), the National Institutes of Health (R01CA207893), and the New York State Department of Health (DOH01-ROWLEY-2015-00035 to R.J.S.). P.A.G. and R.J.S. developed the study and wrote the manuscript. P.A.G., A.G., A.A., and S.B. performed the experiments. P.A.G. and A.W.E. performed the computational and bioinformatic analysis for the RNA-seq data. J.B. developed the BR7 PDX tamoxifen-resistant cell line. R.A. conducted immunohistochemical analyses. S.G. scored the immunohistochemistry results. All authors contributed to the interpretation of the results.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.305631.117.
References
- Ali S, Coombes RC. 2002. Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer 2: 101–112. [DOI] [PubMed] [Google Scholar]
- Avdulov S, Li S, Michalek V, Burrichter D, Peterson M, Perlman DM, Manivel JC, Sonenberg N, Yee D, Bitterman PB, et al. 2004. Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell 5: 553–563. [DOI] [PubMed] [Google Scholar]
- Bachman KE, Park BH. 2005. Duel nature of TGF-β signaling: tumor suppressor vs. tumor promoter. Curr Opin Oncol 17: 49–54. [DOI] [PubMed] [Google Scholar]
- Badura M, Braunstein S, Zavadil J, Schneider RJ. 2012. DNA damage and eIF4G1 in breast cancer cells reprogram translation for survival and DNA repair mRNAs. Proc Natl Acad Sci 109: 18767–18772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcellos-Hoff MH, Akhurst RJ. 2009. Transforming growth factor-β in breast cancer: too much, too late. Breast Cancer Res 11: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Campone M, Piccart M, Burris HA III, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, et al. 2012. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 366: 520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beelen K, Opdam M, Severson TM, Koornstra RH, Vincent AD, Wesseling J, Muris JJ, Berns EM, Vermorken JB, van Diest PJ, et al. 2014a. Phosphorylated p-70S6K predicts tamoxifen resistance in postmenopausal breast cancer patients randomized between adjuvant tamoxifen versus no systemic treatment. Breast Cancer Res 16: R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beelen K, Opdam M, Severson TM, Koornstra RH, Vincent AD, Wesseling J, Muris JJ, Berns EM, Vermorken JB, van Diest PJ, et al. 2014b. PIK3CA mutations, phosphatase and tensin homolog, human epidermal growth factor receptor 2, and insulin-like growth factor 1 receptor and adjuvant tamoxifen resistance in postmenopausal breast cancer patients. Breast Cancer Res 16: R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeram M, Tan QT, Tekmal RR, Russell D, Middleton A, DeGraffenried LA. 2007. Akt-induced endocrine therapy resistance is reversed by inhibition of mTOR signaling. Ann Oncol 18: 1323–1328. [DOI] [PubMed] [Google Scholar]
- Berger C, Qian Y, Chen X. 2013. The p53–estrogen receptor loop in cancer. Curr Mol Med 13: 1229–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I. 2015. Targeting the translation machinery in cancer. Nat Rev Drug Discov 14: 261–278. [DOI] [PubMed] [Google Scholar]
- Bianchini A, Loiarro M, Bielli P, Busa R, Paronetto MP, Loreni F, Geremia R, Sette C. 2008. Phosphorylation of eIF4E by MNKs supports protein synthesis, cell cycle progression and proliferation in prostate cancer cells. Carcinogenesis 29: 2279–2288. [DOI] [PubMed] [Google Scholar]
- Bitterman PB, Polunovsky VA. 2010. Translational control of cancer: implications for targeted therapy. In mTOR pathway and mTOR inhibitors in cancer therapy (ed. Polunovsky VA, Houghton P), pp. 237–255. Springer, New York. [Google Scholar]
- Blyth K, Vaillant F, Jenkins A, McDonald L, Pringle MA, Huser C, Stein T, Neil J, Cameron ER. 2010. Runx2 in normal tissues and cancer cells: a developing story. Blood Cells Mol Dis 45: 117–123. [DOI] [PubMed] [Google Scholar]
- Bostner J, Karlsson E, Pandiyan MJ, Westman H, Skoog L, Fornander T, Nordenskjöld B, Stål O. 2013. Activation of Akt, mTOR, and the estrogen receptor as a signature to predict tamoxifen treatment benefit. Breast Cancer Res Treat 137: 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussemart L, Malka-Mahieu H, Girault I, Allard D, Hemmingsson O, Tomasic G, Thomas M, Basmadjian C, Ribeiro N, Thuaud F, et al. 2014. eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature 513: 105–109. [DOI] [PubMed] [Google Scholar]
- Braunstein S, Karpisheva K, Pola C, Goldberg J, Hochman T, Yee H, Cangiarella J, Arju R, Formenti SC, Schneider RJ. 2007. A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Mol Cell 28: 501–512. [DOI] [PubMed] [Google Scholar]
- Braunstein S, Badura ML, Xi Q, Formenti SC, Schneider RJ. 2009. Regulation of protein synthesis by ionizing radiation. Mol Cell Biol 29: 5645–5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner N, Boysen B, Jirus S, Skaar TC, Holst-Hansen C, Lippman J, Frandsen T, Spang-Thomsen M, Fuqua SA, Clarke R. 1997. MCF7/LCC9: an antiestrogen-resistant MCF-7 variant in which acquired resistance to the steroidal antiestrogen ICI 182,780 confers an early cross-resistance to the nonsteroidal antiestrogen tamoxifen. Cancer Res 57: 3486–3493. [PubMed] [Google Scholar]
- Burris H. 2013. Overcoming acquired resistance to anticancer therapy: focus on the PI3K/Akt/mTOR pathway. Cancer Chemother Pharmacol 71: 829–842. [DOI] [PubMed] [Google Scholar]
- Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. 2001. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor α: a new model for anti-estrogen resistance. J Biol Chem 276: 9817–9824. [DOI] [PubMed] [Google Scholar]
- Cao Y, Wei M, Li B, Liu Y, Lu Y, Tang Z, Lu T, Yin Y, Qin Z, Xu Z. 2016. Functional role of eukaryotic translation initiation factor 4γ1 (EIF4G1) in NSCLC. Oncotarget 7: 24242–24251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimge NO, Frenkel B. 2013. The RUNX family in breast cancer: relationships with estrogen signaling. Oncogene 32: 2121–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimge NO, Baniwal SK, Little GH, Chen YB, Kahn M, Tripathy D, Borok Z, Frenkel B. 2011. Regulation of breast cancer metastasis by Runx2 and estrogen signaling: the role of SNAI2. Breast Cancer Res 13: R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AS, West K, Streicher S, Dennis PA. 2002. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol Cancer Ther 1: 707–717. [PubMed] [Google Scholar]
- Clarke R, Liu MC, Bouker KB, Gu Z, Lee RY, Zhu Y, Skaar TC, Gomez B, O'Brien K, Wang Y, et al. 2003. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene 22: 7316–7339. [DOI] [PubMed] [Google Scholar]
- Cottu P, Bieche I, Assayag F, El Botty R, Chateau-Joubert S, Thuleau A, Bagarre T, Albaud B, Rapinat A, Gentien D, et al. 2014. Acquired resistance to endocrine treatments is associated with tumor-specific molecular changes in patient-derived luminal breast cancer xenografts. Clin Cancer Res 20: 4314–4325. [DOI] [PubMed] [Google Scholar]
- Dancey J. 2010. mTOR signaling and drug development in cancer. Nat Rev Clin Oncol 7: 209–219. [DOI] [PubMed] [Google Scholar]
- Decarlo L, Mestel C, Barcellos-Hoff MH, Schneider RJ. 2015. eIF4E is a feed-forward translational coactivator of TGFβ early pro-transforming events in breast epithelial cells. Mol Cell Biol 35: 2597–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deGraffenried LA, Friedrichs WE, Russell DH, Donzis EJ, Middleton AK, Silva JM, Roth RA, Hidalgo M. 2004. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt activity. Clin Cancer Res 10: 8059–8067. [DOI] [PubMed] [Google Scholar]
- Droog M, Beelen K, Linn S, Zwart W. 2013. Tamoxifen resistance: from bench to bedside. Eur J Pharmacol 717: 47–57. [DOI] [PubMed] [Google Scholar]
- Early Breast Cancer Trialists’ Collaborative Group. 2005. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 365: 1687–1717. [DOI] [PubMed] [Google Scholar]
- Fagan DH, Fettig LM, Avdulov S, Beckwith H, Peterson MS, Ho YY, Wang F, Polunovsky VA, Yee D. 2017. Acquired tamoxifen resistance in MCF-7 breast cancer cells requires hyperactivation of eIF4F-mediated translation. Horm Cancer 8: 219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan P, Agboke FA, Cunliffe HE, Ramos P, Jordan VC. 2014. A molecular model for the mechanism of acquired tamoxifen resistance in breast cancer. Eur J Cancer 50: 2866–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari N, Riggio AI, Mason S, McDonald L, King A, Higgins T, Rosewell I, Neil JC, Smalley MJ, Sansom OJ, et al. 2015. Runx2 contributes to the regenerative potential of the mammary epithelium. Sci Rep 5: 15658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher B, Jeong JH, Bryant J, Anderson S, Dignam J, Fisher ER, Wolmark N. 2004. Treatment of lymph-node-negative, oestrogen-receptor-positive breast cancer: long-term findings from National Surgical Adjuvant Breast and Bowel Project randomised clinical trials. Lancet 364: 858–868. [DOI] [PubMed] [Google Scholar]
- Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, et al. 2009. An oestrogen-receptor-α-bound human chromatin interactome. Nature 462: 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Quiroz J, Rivas-Suarez M, Garcia-Becerra R, Barrera D, Martinez-Reza I, Ordaz-Rosado D, Santos-Martinez N, Villanueva O, Santos-Cuevas CL, Avila E, et al. 2014. Calcitriol reduces thrombospondin-1 and increases vascular endothelial growth factor in breast cancer cells: implications for tumor angiogenesis. J Steroid Biochem Mol Biol 144: 215–222. [DOI] [PubMed] [Google Scholar]
- Holcik M, Pestova TV. 2007. Translation mechanism and regulation: old players, new concepts. Meeting on translational control and non-coding RNA. EMBO Rep 8: 639–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell SJ, Johnston SR, Howell A. 2004. The use of selective estrogen receptor modulators and selective estrogen receptor down-regulators in breast cancer. Best Pract Res Clin Endocrinol Metab 18: 47–66. [DOI] [PubMed] [Google Scholar]
- Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR, Meyuhas O, Shokat KM, Ruggero D. 2010. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell 17: 249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, et al. 2012. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485: 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic N, Utermark T, Widlund HR, Roberts TM. 2010. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci 108: E699–E708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeselsohn R, Cornwell M, Pun M, Buchwalter G, Nguyen M, Bango C, Huang Y, Kuang Y, Paweletz C, Fu X, et al. 2017. Embryonic transcription factor SOX9 drives breast cancer endocrine resistance. Proc Natl Acad Sci 114: E4482–E4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan NJ, Dutkowski CM, Barrow D, Mottram HJ, Hutcheson IR, Nicholson RI, Guichard SM, Gee JM. 2014. Impact of dual mTORC1/2 mTOR kinase inhibitor AZD8055 on acquired endocrine resistance in breast cancer in vitro. Breast Cancer Res 16: R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin KL, Mondal G, Hartman JK, Tyagi S, Kurley SJ, Bland CS, Hsu TY, Renwick A, Fang JE, Migliaccio I, et al. 2014. The oncogenic STP axis promotes triple-negative breast cancer via degradation of the REST tumor suppressor. Cell Rep 9: 1318–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthik GM, Ma R, Lovrot J, Kis LL, Lindh C, Blomquist L, Fredriksson I, Bergh J, Hartman J. 2015. mTOR inhibitors counteract tamoxifen-induced activation of breast cancer stem cells. Cancer Lett 367: 76–87. [DOI] [PubMed] [Google Scholar]
- Kim YY, Von Weymarn L, Larsson O, Fan D, Underwood JM, Peterson MS, Hecht SS, Polunovsky VA, Bitterman PB. 2009. Eukaryotic initiation factor 4E binding protein family of proteins: sentinels at a translational control checkpoint in lung tumor defense. Cancer Res 69: 8455–8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauf U, Tschopp C, Gram H. 2001. Negative regulation of protein translation by mitogen-activated protein kinase-interacting kinases 1 and 2. Mol Cell Biol 21: 5500–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konicek BW, Stephens JR, McNulty AM, Robichaud N, Peery RB, Dumstorf CA, Dowless MS, Iversen PW, Parsons S, Ellis KE, et al. 2011. Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res 71: 1849–1857. [DOI] [PubMed] [Google Scholar]
- Korets SB, Musa F, Curtin J, Blank SV, Schneider RJ. 2014. Dual mTORC1/2 inhibition in a preclinical xenograft tumor model of endometrial cancer. Gynecol Oncol 132: 468–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koromilas AE, Lazaris-Karatzas A, Sonenberg N. 1992. mRNAs containing extensive secondary structure in their 5′ non-coding region translate efficiently in cells overexpressing initiation factor eIF-4E. EMBO J 11: 4153–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. 2012. mTOR signaling in growth control and disease. Cell 149: 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung E, Kannan N, Krissansen GW, Findlay MP, Baguley BC. 2010. MCF-7 breast cancer cells selected for tamoxifen resistance acquire new phenotypes differing in DNA content, phospho-HER2 and PAX2 expression, and rapamycin sensitivity. Cancer Biol Ther 9: 717–724. [DOI] [PubMed] [Google Scholar]
- Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R. 2008. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res 68: 826–833. [DOI] [PubMed] [Google Scholar]
- McDonald L, Ferrari N, Terry A, Bell M, Mohammed ZM, Orange C, Jenkins A, Muller WJ, Gusterson BA, Neil JC, et al. 2014. RUNX2 correlates with subtype-specific breast cancer in a human tissue microarray, and ectopic expression of Runx2 perturbs differentiation in the mouse mammary gland. Dis Model Mech 7: 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TW, Hennessy BT, Gonzalez-Angulo AM, Fox EM, Mills GB, Chen H, Higham C, Garcia-Echeverria C, Shyr Y, Arteaga CL. 2010. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest 120: 2406–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TW, Balko JM, Arteaga CL. 2011. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J Clin Oncol 29: 4452–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musgrove E, Sutherland R. 2009. Biological determinants of endocrine resistance in breast cancer. Nat Rev 9: 631–643. [DOI] [PubMed] [Google Scholar]
- Nicholson RI, Staka C, Boyns F, Hutcheson IR, Gee JM. 2004. Growth factor-driven mechanisms associated with resistance to estrogen deprivation in breast cancer: new opportunities for therapy. Endocr Relat Cancer 11: 623–641. [DOI] [PubMed] [Google Scholar]
- Osborne CK, Schiff R. 2011. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med 62: 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne CK, Zhao H, Fuqua SA. 2000. Selective estrogen receptor modulators: structure, function, and clinical use. J Clin Oncol 18: 3172–3186. [DOI] [PubMed] [Google Scholar]
- Oxelmark E, Roth JM, Brooks PC, Braunstein SE, Schneider RJ, Garabedian MJ. 2006. The cochaperone p23 differentially regulates estrogen receptor target genes and promotes tumor cell adhesion and invasion. Mol Cell Biol 26: 5205–5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, Graff J, Ruggero D, Sonenberg N. 2015. Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res 75: 250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratap J, Imbalzano KM, Underwood JM, Cohet N, Gokul K, Akech J, van Wijnen AJ, Stein JL, Imbalzano AN, Nickerson JA, et al. 2009. Ectopic runx2 expression in mammary epithelial cells disrupts formation of normal acini structure: implications for breast cancer progression. Cancer Res 69: 6807–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez-Valle F, Braunstein S, Zavadil J, Formenti SC, Schneider RJ. 2008. eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J Cell Biol 181: 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez-Valle F, Badura ML, Braunstein S, Narasimhan M, Schneider RJ. 2010. Mitotic Raptor promotes mTORC1 activity, G(2)/M cell cycle progression, and internal ribosome entry site-mediated mRNA translation. Mol Cell Biol 30: 3151–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaud N, del Rincon SV, Huor B, Alain T, Petruccelli LA, Hearnden J, Goncalves C, Grotegut S, Spruck CH, Furic L, et al. 2015. Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP-3. Oncogene 34: 2032–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowlett RM, Chrestensen CA, Nyce M, Harp MG, Pelo JW, Cominelli F, Ernst PB, Pizarro TT, Sturgill TW, Worthington MT. 2008. MNK kinases regulate multiple TLR pathways and innate proinflammatory cytokines in macrophages. Am J Physiol Gastrointest Liver Physiol 294: G452–G459. [DOI] [PubMed] [Google Scholar]
- Sabatini DM. 2006. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer 6: 729–734. [DOI] [PubMed] [Google Scholar]
- Sabnis G, Brodie A. 2011. Adaptive changes results in activation of alternate signaling pathways and resistance to aromatase inhibitor resistance. Mol Cell Endocrinol 340: 142–147. [DOI] [PubMed] [Google Scholar]
- Sato A, Kasai S, Kobayashi T, Takamatsu Y, Hino O, Ikeda K, Mizuguchi M. 2012. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat Commun 3: 1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff R, Massarweh SA, Shou J, Bharwani L, Mohsin SK, Osborne CK. 2004. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin Cancer Res 10: 331S–336S. [DOI] [PubMed] [Google Scholar]
- Shah N, Sukumar S. 2010. The Hox genes and their roles in oncogenesis. Nat Rev Cancer 10: 361–371. [DOI] [PubMed] [Google Scholar]
- Silvera D, Arju R, Darvishian F, Levine P, Goldberg JG, Hockman T, Formenti SC, Schneider RJ. 2009a. Inflammatory breast cancer pathogenesis mediated by translation initiation factor eIF4G overexpression and unorthodox protein synthesis. Nature Cell Biol 11: 903–910. [DOI] [PubMed] [Google Scholar]
- Silvera D, Arju R, Darvishian F, Levine PH, Zolfaghari L, Goldberg J, Hochman T, Formenti SC, Schneider RJ. 2009b. Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nat Cell Biol 11: 903–908. [DOI] [PubMed] [Google Scholar]
- Silvera D, Formenti SC, Schneider RJ. 2010. Translational control in cancer. Nat Rev Cancer 10: 254–266. [DOI] [PubMed] [Google Scholar]
- Silvera D, Ernlund A, Arju R, Connolly E, Volta V, Wang J, Schneider RJ. 2017. mTORC1 and -2 coordinate transcriptional and translational reprogramming in resistance to DNA damage and replicative stress in breast cancer cells. Mol Cell Biol 37: e00577–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson NE, Lambert WM, Watkins R, Giashuddin S, Huang SJ, Oxelmark E, Arju R, Hochman T, Goldberg JD, Schneider RJ, et al. 2010. High levels of Hsp90 cochaperone p23 promote tumor progression and poor prognosis in breast cancer by increasing lymph node metastases and drug resistance. Cancer Res 70: 8446–8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer S, Fuqua SA. 2001. Estrogen receptor and breast cancer. Semin Cancer Biol 11: 339–352. [DOI] [PubMed] [Google Scholar]
- Svitkin YV, Herdy B, Costa-Mattioli M, Gingras AC, Raught B, Sonenberg N. 2005. Eukaryotic translation initiation factor 4E availability controls the switch between cap-dependent and internal ribosomal entry site-mediated translation. Mol Cell Biol 25: 10556–10565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon M, Chen Z, Pratap J. 2014. Runx2 activates PI3K/Akt signaling via mTORC2 regulation in invasive breast cancer cells. Breast Cancer Res 16: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topisirovic I, Sonenberg N. 2011a. mRNA translation and energy metabolism in cancer: the role of the MAPK and mTORC1 pathways. Cold Spring Harb Symp Quant Biol 76: 355–367. [DOI] [PubMed] [Google Scholar]
- Topisirovic I, Sonenberg N. 2011b. Translational control by the eukaryotic ribosome. Cell 145: 333–334. [DOI] [PubMed] [Google Scholar]
- Topisirovic I, Ruiz-Gutierrez M, Borden KL. 2004. Phosphorylation of the eukaryotic translation initiation factor eIF4E contributes to its transformation and mRNA transport activities. Cancer Res 64: 8639–8642. [DOI] [PubMed] [Google Scholar]
- Treeck O, Wackwitz B, Haus U, Ortmann O. 2006. Effects of a combined treatment with mTOR inhibitor RAD001 and tamoxifen in vitro on growth and apoptosis of human cancer cells. Gynecol Oncol 102: 292–299. [DOI] [PubMed] [Google Scholar]
- Truitt Morgan L, Conn Crystal S, Shi Z, Pang X, Tokuyasu T, Coady Alison M, Seo Y, Barna M, Ruggero D. 2015. Differential requirements for eIF4E dose in normal development and cancer. Cell 162: 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waskiewicz AJ, Johnson JC, Penn B, Mahalingam M, Kimball SR, Cooper JA. 1999. Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol Cell Biol 19: 1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel HG, Silva RL, Malina A, Mills JR, Zhu H, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Teruya-Feldstein J, Pelletier J, et al. 2007. Dissecting eIF4E action in tumorigenesis. Genes Dev 21: 3232–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheater MJ, Johnson PW, Blaydes JP. 2010. The role of MNK proteins and eIF4E phosphorylation in breast cancer cell proliferation and survival. Cancer Biol Ther 10: 728–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe AL, Singh K, Zhong Y, Drewe P, Rajasekhar VK, Sanghvi VR, Mavrakis KJ, Jiang M, Roderick JE, Van der Meulen J, et al. 2014. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature 513: 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Whitfield TW, Gordon JAR, Dobson JR, Tai PWL, van Wijnen AJ, Stein JL, Stein GS, Lian JB. 2014. Genomic occupancy of Runx2 with global expression profiling identifies a novel dimension to control of osteoblastogenesis. Genome Biol 15: R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamnik RL, Holz MK. 2010. mTOR/S6K1 and MAPK/RSK signaling pathways coordinately regulate estrogen receptor α serine 167 phosphorylation. FEBS Lett 584: 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Quaresma AJC, Nickerson JA, Green KM, Shaffer SA, Imbalzano AN, Martin-Buley LA, Lian JB, Stein JL, van Wijnen AJ, et al. 2015. Subnuclear domain proteins in cancer cells support the functions of RUNX2 in the DNA damage response. J Cell Sci 128: 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young DW, Hassan MQ, Yang X-Q, Galindo M, Javed A, Zaidi SK, Furcinitti P, Lapointe D, Montecino M, Lian JB, et al. 2007. Mitotic retention of gene expression patterns by the cell fate-determining transcription factor Runx2. Proc Natl Acad Sci 104: 3189–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM. 2011. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12: 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.