

SUMMARY
Whole-exome sequencing (WES) and de novo variant detection have proven a powerful approach to gene discovery in complex neurodevelopmental disorders. We have completed WES of 325 Tourette disorder trios from the Tourette International Collaborative Genetics cohort and a replication sample of 186 trios from the Tourette Syndrome Association International Consortium on Genetics (511 total). We observe strong and consistent evidence for the contribution of de novo likely gene-disrupting (LGD) variants (rate ratio [RR] 2.32, p = 0.002). Additionally, de novo damaging variants (LGD and probably damaging missense) are overrepresented in probands (RR 1.37, p = 0.003). We identify four likely risk genes with multiple de novo damaging variants in unrelated probands: WWC1 (WW and C2 domain containing 1), CELSR3 (Cadherin EGF LAG seven-pass G-type receptor 3), NIPBL (Nipped-B-like), and FN1 (fibronectin 1). Overall, we estimate that de novo damaging variants in approximately 400 genes contribute risk in 12% of clinical cases.
INTRODUCTION
Tourette disorder (TD) is an often-disabling developmental neuropsychiatric syndrome characterized by persistent motor and vocal tics. Onset is typically in early childhood, and estimates of the worldwide prevalence are between 0.3% and 1% (Centers for Disease Control and Prevention, 2009; Robertson, 2008; Scharf et al., 2015). The vast majority of children and adults who present for medical attention have other impairing co-occurring psychiatric disorders, including obsessive-compulsive disorder (OCD) (do Rosário and Miguel Filho, 1997; Ghanizadeh and Mosallaei, 2009; Hounie et al., 2006), attention-deficit/hyperactivity disorder (ADHD) (Burd et al., 2005; Leckman, 2003; Roessner et al., 2007), and mood and anxiety disorders (Cavanna et al., 2009; Hirschtritt et al., 2015). Rates of OCD-like conditions, such as trichotillomania and pathologic skin picking (Lochner et al., 2005), are likewise elevated.
Current treatments for tics and TD have limited efficacy and pharmacotherapies may carry significant long-term adverse effects. A fundamental obstacle to identifying novel therapeutic targets is a limited understanding of underlying pathophysiology. There is widespread agreement that genetics plays a significant role in TD etiology based on twin and family studies (Browne et al., 2015; Mataix-Cols et al., 2015; Pauls et al., 1981, 1991; Price et al., 1985). To date, non-parametric linkage analyses (The Tourette Syndrome Association International Consortium for Genetics, 1999, 2007) and a genome-wide association study (Scharf et al., 2013) have not yet led to reproducible, statistically significant findings. Studies of rare pedigrees have identified putative risk genes expressed in the developing striatum and mediating neurite outgrowth (Abelson et al., 2005; Stillman et al., 2009) or involved in histaminergic neurotransmission (Ercan-Sencicek et al., 2010), signal transduction and cell adhesion (Lawson-Yuen et al., 2008; Verkerk et al., 2003), or serotonin transport (Moya et al., 2013). None of these findings can yet be considered definitive.
Our group and others have reported on copy number variations (CNVs) in TD (Fernandez et al., 2012; McGrath et al., 2014; Nag et al., 2013; Sundaram et al., 2010), confirming a role for rare structural variants and showing a trend toward enrichment of de novo events. These findings also provide additional support for the involvement of histaminergic neurotransmission, as well as dopaminergic neurotransmission, in the pathogenesis of TD (Ercan-Sencicek et al., 2010; Fernandez et al., 2012) and suggest a potential overlap with CNVs contributing to other neurodevelopmental syndromes (Malhotra and Sebat, 2012).
Studies of de novo sequence variation using whole-exome sequencing (WES) have proven to be a powerful approach to systematic gene discovery in genetically complex neurodevelopmental disorders (NDDs) apart from TD (Bilgüvar et al., 2010; de Ligt et al., 2012; Deciphering Developmental Disorders Study, 2015, 2017; Epi4K Consortium, 2016; Allen et al., 2013; EuroEPINOMICS-RES Consortium et al., 2014; Rauch et al., 2012), particularly autism spectrum disorders (ASDs) (De Rubeis et al., 2014; Dong et al., 2014; Iossifov et al., 2014; Iossifov et al., 2012; Neale et al., 2012; O’Roak et al., 2011, 2012; Sanders et al., 2012, 2015; Willsey et al., 2013). In light of these findings and our previous results suggesting a role for de novo CNVs, we performed WES in 325 (311 after quality control) TD parent-child trios from the Tourette International Collaborative Genetics group (TIC Genetics; http://tic-genetics.org) to identify de novo single-nucleotide variants (SNVs) and insertion-deletion variants (indels). We observe significant evidence for the contribution of de novo likely gene-disrupting (LGD) variants (insertion of premature stop codon, frameshift, or canonical splice-site variant) to TD. We then replicate these findings in WES data from 186 parent-child trios (173 after quality control) from the Tourette Syndrome Association International Consortium for Genetics (TSAICG; https://www.findtsgene.org/). We also observe evidence for the contribution of de novo damaging variants (LGD and probably damaging missense). Overall, we estimate that 12% of clinical cases will carry a de novo damaging variant (LGD and probably damaging missense) mediating TD risk and that approximately 400 genes are vulnerable to these variants. Finally, a combined analysis identifies one high-confidence TD (hcTD) risk gene (false discovery rate [FDR] < 0.1), WWC1 (WW and C2 domain containing 1), and three additional probable TD (pTD) risk genes (FDR < 0.3) CELSR3 (Cadherin EGF LAG seven-pass G-type receptor 3), NIPBL (Nipped-B-like), and FN1 (fibronectin 1). See Figure 1 for an overview of this study.
Figure 1. Study Overview.
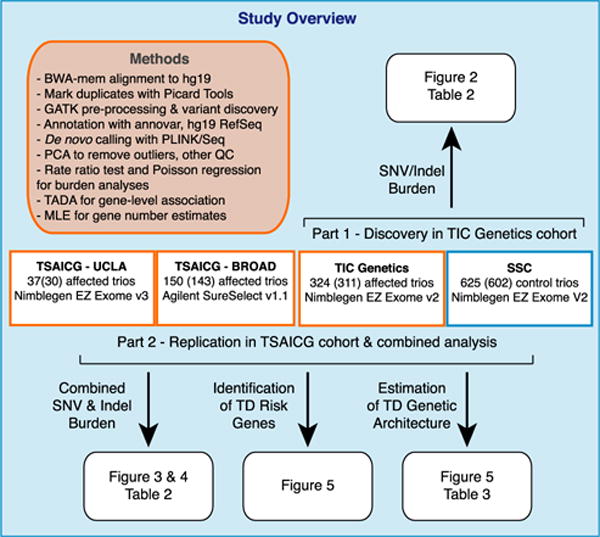
Using WES, we assessed the burden of de novo variants in Tourette disorder (TD) in the Tourette International Collaborative Genetics group (TIC Genetics; http://tic-genetics.org) and the Tourette Syndrome Association International Collaboration for Genetics (TSAICG; https://www.findtsgene.org/) cohorts. We performed an initial analysis of de novo single-nucleotide variant (SNV) and insertion-deletion variants (indel) in the TIC Genetics cohort (n = 325, 311 in parentheses passed quality control [QC]). This was followed by replication in the TSAICG cohort (n = 186, 173 passed QC: 143 of 149 samples sequenced at the Broad Institute and 30 of 37 samples sequenced at UCLA) and then a combined analysis (n = 484 trios). We obtained control trios, consisting of unaffected parents and unaffected sibling controls, from the Simons Simplex Collection (SSC; n = 625, 602 passed QC). In this figure, affected cohorts are outlined in a red box and control trios in blue. After assessing the contribution of de novo variants to TD risk, we assessed the number of TD genes that contribute to TD risk via damaging de novo variants (likely gene disrupting, a.k.a. LGD, and probably damaging missense, a.k.a. missense 3 or Mis3). We then utilized the TADA algorithm (He et al., 2013) to identify TD risk genes based on per-gene burden of de novo variants. Finally, we predicted the gene discovery yield as additional TD trios are sequenced. See Table S1 for detailed sample- and cohort-level information, Table S2 for a list of annotated de novo variants, and Table S4 for TADA gene association p and q values.
RESULTS
De Novo LGD Variants Are Associated with TD Risk in the TIC Genetics Cohort
We first conducted WES of 325 TIC Genetics trios. Dietrich et al. (2015) have previously described ascertainment and phenotyping of this cohort (Dietrich et al., 2015). We utilized the SeqCap EZ Human Exome v.2.0 library kit (Roche NimbleGen) to capture exomes from whole-blood-derived DNA and then sequenced with Illumina HiSeq 2000 technology. In all analyses, we compared TD trios passing quality control to control trios from the Simons Simplex Collection (SSC) (Fischbach and Lord, 2010a). The SSC consists of simplex families: two unaffected parents with a single child affected with autism spectrum disorder (ASD). Approximately 80% of these families also include one or more unaffected siblings. Therefore, our control trios consist of two unaffected parents and an unaffected sibling. We randomly selected 625 control trios from among the 96.9% (n = 2,438 of 2,517) of SSC families that had been captured with the same library used for the TIC Genetics cohort and sequenced using Illumina technology. 311 TD trios (311/325, 95.7%) and 602 SSC trios (602/625, 96.3%) passed quality control (Table 1; Table S1). We summarize sequencing metrics in Table 1 and detail all sample- and cohort-level data in Table S1. The distribution of de novo coding variants per individual in the TIC Genetics cohort and in the SSC siblings follow an expected Poisson distribution (Figure S1), as has been observed in other disorders (e.g., Fromer et al., 2014; Homsy et al., 2015; Neale et al., 2012; Sanders et al., 2012; Xu et al., 2012).
Table 1.
Cohort-Level Information, Quality Control, and Sequencing Metrics
| Cohort | TIC Genetics | TSAICG-Broad | TSAICG-UCLA | SSC Siblings |
|---|---|---|---|---|
| Samples sequenced (trios) | 975 (325) | 447 (149) | 111 (37) | 2,124 (708) |
| Samples passing QC (trios)a | 933 (311) | 429 (143) | 90 (30) | 1,806 (602) |
| Male:Female (sex ratio)b | 244:67 (3.64) | 115:28 (4.11) | 29:1 (29.0) | 275:327 (0.84) |
| Paternal agec | 32.83 (±0.61) | 33.29 (±0.81) | 31.25 (±2.20) | 32.99 (±0.47) |
| Exome capture platform | Nimblegen EZ Exome V2 |
Agilent SureSelect v1.1 | Nimblegen EZ Exome V3 |
Nimblegen EZ Exome V2 |
| Size of capture region | 44,001,748 bp | 32,760,120 bp | 63,564,965 bp | 44,001,748 bp |
| RefSeq hg19 coding region coveredd | 32,586,393 bp | 31,844,591 bp | 33,644,238 bp | 32,586,393 bp |
| % Refseq hg19 coding region covered | 96.33% | 94.13% | 99.45% | 96.33% |
| Mean callable exome (million bp) | 27.52 (±0.66) | 26.84 (±1.11) | 27.09 (±3.63) | 25.80 (±1.06) |
| Total callable exome (million bp) | 8,560.01 | 3,838.13 | 812.71 | 15,529.00 |
| Total callable exome + other (million bp) | 17,108.37 | 6,549.29 | 2,089.62 | 28,584.79 |
| Mean total reads per sample (million) | 99.03 (±1.42) | 86.45 (±2.68) | 105.17 (±3.71) | 111.33 (±1.89) |
| Mean read length | 76 (±0) | 75.98 (±0.004) | 100 (±0) | 77.64 (±0.71) |
| Passing unique aligned reads (million) | 93.64 (±1.33) | 73.40 (±2.04) | 86.17 (±3.04) | 102.43 (±1.64) |
| % passing, unique reads aligned | 99.76% (±0.01%) | 98.70% (±0.03%) | 99.68% (±0.03%) | 98.95% (±0.12%) |
| Number of bases in target (million)e | 2,748.94 (±39.20) | 2,505.88 (±66.45) | 1,964.89 (±67.91) | 2,256.09 (±41.76) |
| % Duplicate reads | 5.17% (±0.07%) | 13.19% (±0.35%) | 17.52% (±1.39%) | 6.46% (±0.20%) |
| Mean coverage in targete | 80.79 (±1.15) | 73.65 (±1.95) | 57.75 (±2.00) | 66.31 (±1.23) |
| Median coverage in targete | 67.72 (±1.00) | 62.41 (±1.68) | 46.19 (±1.60) | 55.73 (±1.05) |
| % target at 2×e | 94.89% (±0.05%) | 92.53% (±0.03%) | 96.49% (±0.07%) | 94.76% (±0.07%) |
| % target at 10×e | 90.20% (±0.13%) | 87.72% (±0.14%) | 92.38% (±0.25%) | 88.34% (±0.13%) |
| % target at 20×e | 83.76% (±0.22%) | 81.57% (±0.31%) | 84.72% (±0.95%) | 79.51% (±0.30%) |
| % target at 40×e | 68.77% (±0.40%) | 66.41% (±0.81%) | 57.97% (±1.97%) | 59.37% (±0.63%) |
| Fold 80 vase penalty | 3.27 (±0.03) | 3.29 (±0.01) | 2.37 (±0.02) | 3.65 (±0.37) |
| Het SNP 1uality | 10.97 (±0.04) | 9.27 (±0.05) | 12.68 (±0.10) | 11.08 (±0.04) |
| Base pair error rate | 0.0034 (±0.0001) | 0.0043 (±0.0001) | 0.0043 (±0.0001) | 0.0044 (±0.0001) |
| Novel transition/transversion ratio | 2.01 (±0.02) | 2.30 (±0.02) | 2.23 (±0.05) | 2.13 (±0.01) |
| Novel insertion/deletion ratio | 0.51 (±0.01) | 0.31 (±0.02) | 0.52 (±0.03) | 0.42 (±0.01) |
We sequenced two Tourette disorder (TD) cohorts in this study: TIC Genetics and TSAICG. Two different locations sequenced the TSAICG—the Broad Institute and UCLA. We compared these cohorts to control trios from the Simons Simplex Collection (SSC). Three different library kits captured exomes with different target sizes and, therefore, varying coverage of RegSeq hg19 coding regions. Mean and median coverage differed across the cohorts. To control for these factors, we determined the number of “callable” bp, or the number of bp in each family that have ≥20× coverage in all family members. We summed these lengths across all families within a given cohort to determine the “total callable” bp. We then intersected these coordinates with RefSeq hg19 coding exons to determine the “total callable exome,” or the number of bp within RefSeq coding exons that had sufficient coverage for de novo calling. Picard Tools (https://broadinstitute.github.io/picard/) generated capture, sequencing, alignment, and variant level quality metrics, and GATK DepthOfCoverage generated coverage metrics for the exome intervals. We remove samples with excess de novo variants (>5). A panel of informative genotypes confirmed familiality, and trios were removed if expected family relationships did not confirm or if there were unexpected relationships within or across families. If any family member failed QC, we removed all family members from the analysis. Principal-component analysis (PCA) revealed outliers, which we removed from analysis (Figure S4). Sequencing metrics summarized in this table are from samples passing QC only. We provide quality control information and detailed sequencing metrics for all subjects in Table S1.
Statistics were estimated from passing samples only
Calculated from children only. Mutation rates were not significantly different between males and females in the TIC Genetics (p = 0.4, two-sided rate ratio test, STAR Methods), combined TSAICG (p = 0.9), or SSC siblings cohorts (p = 0.3). See also Table S1
Calculated from parents only
Size determined from intersection of exome capture array intervals plus 100 bp interval padding (added during GATK processing) with RefSeq hg19 coding intervals
Target refers to entire Refseq hg19 coding regions (33,828,798 bp). Where applicable, sequencing metrics include ± 95% confidence intervals
We conducted Sanger-sequencing-based validation for all de novo variants predicted in TD probands and observed confirmation rates of 97.2% in the TIC Genetics trios (97.8% for SNVs and 60% for indels). We did not do so in SSC controls, and consequently, for all burden analyses, we compared unconfirmed de novo variants identified using identical methods from both cohorts. For analysis of recurrent mutations in probands, we relied solely on confirmed variants. See Table S2 for detailed annotations of each predicted de novo variant, including validation status.
We compared the rate of de novo mutation per base pair (bp), restricting these analyses to base pairs covered at ≥20× in all members of a trio (our minimum criteria for de novo calling; Sanders et al., 2012). We calculated the rate per base pair as the total number of variants observed within this target region. More specifically, for the overall (coding plus non-coding) mutation rate (e.g., Table 2), this encompassed the exome capture array intervals plus the 100 bp of interval padding added during GATK processing (denoted as “total callable” in Table 1). For coding mutation rate (e.g., Table 2; Figures 2A, 2B, 3A, and 3B), this encompassed the intersection of these intervals with the coding portion of the exome based on RefSeq hg19 gene definitions (“total callable exome” in Table 1). This strategy normalizes for differences in capture array design and coverage distribution across the exome.
Table 2.
Distribution of De Novo SNVs and Indels in TD Cases and Controls
| Variant Type | Total # Variants | Mean Rate per bp (×10−8) (±95% CI) | Theoretical Rate per Individual (±95% CI) | Rate Ratio (±95% CI) | p Value | |||
|---|---|---|---|---|---|---|---|---|
| Cohort | TIC Genetics | |||||||
| TIC Gen | Control | TIC Gen | Control | TIC Gen | Control | |||
| (N = 311) | (N = 602) | (N = 311) | (N = 602) | (N = 311) | (N = 602) | |||
| All | 525 | 862 | 1.53 (1.41–1.65) | 1.50 (1.40–1.60) | 1.04 (0.96–1.12) | 1.01 (0.95–1.08) | 1.02 (0.93–1.12) | p = 0.4 |
| Coding | 301a | 484b | 1.75 (1.56–1.95) | 1.56 (1.42–1.70) | 1.19 (1.06–1.32) | 1.05 (0.96–1.15) | 1.13 (1.00–1.28) | p = 0.05 |
| Synonymous | 67 | 134 | 0.39 (0.30–0.49) | 0.43 (0.36–0.50) | 0.27 (0.20–0.33) | 0.29 (0.24–0.34) | 0.91 (0.70–1.17) | p = 0.8 |
| Nonsynonymous | 233 | 343 | 1.36 (1.18–1.53) | 1.10 (0.99–1.22) | 0.92 (0.8–1.03) | 0.75 (0.67–0.83) | 1.23 (1.07–1.42) | p = 0.008 |
| Missense (Mis) | 207 | 321 | 1.21 (1.04–1.37) | 1.03 (0.92–1.15) | 0.82 (0.70–0.93) | 0.70 (0.62–0.78) | 1.17 (1.01–1.36) | p = 0.04 |
| Missense 3 (Mis3) | 111 | 158 | 0.65 (0.53–0.77) | 0.51 (0.43–0.59) | 0.44 (0.36–0.52) | 0.34 (0.29–0.40) | 1.27 (1.03–1.57) | p = 0.03 |
| Likely Gene Disrupting (LGD) | 26 | 22 | 0.15 (0.092–0.21) | 0.071 (0.042–0.10) | 0.10 (0.062–0.14) | 0.048 (0.028–0.068) | 2.14 (1.28–3.61) | p = 0.006 |
| Damaging (LGD + Mis3) | 137 | 180 | 0.80 (0.67–0.93) | 0.58 (0.49–0.67) | 0.54 (0.45–0.63) | 0.39 (0.33–0.45) | 1.38 (1.14–1.67) | p = 0.003 |
| LGD SNV | 16 | 19 | 0.092 (0.048–0.14) | 0.061 (0.034–0.088) | 0.063 (0.033–0.093) | 0.041 (0.023–0.060) | 1.53 (0.82–2.82) | p = 0.1 |
| LGD FS Indel | 10 | 3 | 0.058 (0.022–0.093) | 0.0095 (−0.0013–0.020) | 0.039 (0.015–0.063) | 0.0064 (−0.00086–0.014) | 6.05 (1.85–25.65) | p = 0.003 |
| In-Frame Indel | 1 | 4 | 0.0058 (−0.0056–0.017) | 0.012 (0.00022–0.024) | 0.0039 (−0.0038–0.012) | 0.0081 (0.00015–0.016) | 0.45 (0.02–3.48) | p = 0.9 |
| Cohort | TSAICG | |||||||
| TSAICG | Control | TSAICG | Control | TSAICG | Control | |||
| (N = 173) | (N = 602) | (N = 173) | (N = 602) | (N = 173) | (N = 602) | |||
| All | 258 | 862 | 1.49 (1.31–1.67) | 1.50 (1.40–1.60) | 1.01 (0.89–1.13) | 1.01 (0.95–1.08) | 0.99 (0.88–1.11) | p = 0.6 |
| Coding | 153 | 484b | 1.64 (1.39–1.9) | 1.56 (1.42–1.70) | 1.11 (0.94–1.29) | 1.05 (0.96–1.15) | 1.06 (0.90–1.23) | p = 0.3 |
| Synonymous | 44 | 134 | 0.47 (0.34–0.61) | 0.431 (0.36–0.50) | 0.32 (0.23–0.41) | 0.292 (0.24–0.34) | 1.10 (0.81–1.47) | p = 0.3 |
| Nonsynonymous | 109 | 343 | 1.17 (0.95–1.39) | 1.10 (0.99–1.22) | 0.79 (0.64–0.94) | 0.75 (0.67–0.83) | 1.06 (0.88–1.28) | p = 0.3 |
| Missense (Mis) | 96 | 321 | 1.03 (0.82–1.24) | 1.03 (0.92–1.15) | 0.70 (0.56–0.84) | 0.70 (0.62–0.78) | 1.00 (0.82–1.21) | p = 0.5 |
| Missense 3 (Mis3) | 49 | 158 | 0.53 (0.36–0.70) | 0.51 (0.43–0.59) | 0.36 (0.25–0.47) | 0.34 (0.29–0.40) | 1.04 (0.78–1.37) | p = 0.4 |
| Likely Gene Disrupting (LGD) | 13 | 22 | 0.14 (0.059–0.21) | 0.071 (0.042–0.10) | 0.092 (0.040–0.14) | 0.048 (0.028–0.068) | 1.97 (1.03–3.68) | p = 0.04 |
| Damaging (LGD + Mis3) | 62 | 180 | 0.67 (0.49–0.84) | 0.58 (0.49–0.67) | 0.45 (0.33–0.57) | 0.39 (0.33–0.45) | 1.15 (0.89–1.48) | p = 0.2 |
| LGD SNV | 8 | 19 | 0.083 (0.026–0.14) | 0.061 (0.034–0.088) | 0.056 (0.018–0.094) | 0.041 (0.023–0.060) | 1.41 (0.62–2.98) | p = 0.3 |
| LGD FS Indel | 5 | 3 | 0.053 (0.0068–0.10) | 0.0095 (−0.0013–0.020) | 0.036 (0.0046–0.068) | 0.0064 (−0.00086–0.014) | 5.56 (1.36–26.71) | p = 0.02 |
| In-Frame Indel | 2 | 4 | 0.021 (−0.0081–0.050) | 0.012 (0.00022–0.024) | 0.014 (−0.0055–0.034) | 0.0081 (0.00015–0.016) | 1.67 (0.22–8.97) | p = 0.4 |
| Cohort | Combined | |||||||
| TD | Control | TD | Control | TD | Control | |||
| (N = 484) | (N = 602) | (N = 484) | (N = 602) | (N = 484) | (N = 602) | |||
| All | 783 | 862 | 1.52 (1.42–1.62) | 1.51 (1.40–1.60) | 1.03 (0.96–1.09) | 1.02 (0.95–1.08) | 1.01 (0.93–1.10) | p = 0.4 |
| Coding | 454c | 484b | 1.72 (1.56–1.87) | 1.56 (1.42–1.70) | 1.16 (1.06–1.26) | 1.05 (0.96–1.15) | 1.10 (0.99–1.23) | p = 0.07 |
| Synonymous | 111 | 134 | 0.42 (0.35–0.50) | 0.43 (0.36–0.50) | 0.29 (0.23–0.34) | 0.29 (0.24–0.34) | 0.97 (0.78–1.21) | p = 0.6 |
| Nonsynonymous | 342 | 343 | 1.29 (1.15–1.43) | 1.10 (0.99–1.22) | 0.87 (0.78–0.96) | 0.75 (0.67–0.83) | 1.17 (1.03–1.33) | p = 0.02 |
| Missense (Mis) | 303 | 321 | 1.14 (1.01–1.28) | 1.03 (0.92–1.15) | 0.77 (0.69–0.86) | 0.70 (0.62–0.78) | 1.11 (0.97–1.27) | p = 0.1 |
| Missense 3 (Mis3) | 160 | 158 | 0.61 (0.51–0.70) | 0.51 (0.43–0.59) | 0.41 (0.34–0.48) | 0.34 (0.29–0.40) | 1.19 (0.98–1.44) | p = 0.07 |
| Likely Gene Disrupting (LGD) | 39 | 22 | 0.15 (0.099–0.19) | 0.071 (0.042–0.10) | 0.10 (0.067–0.13) | 0.048 (0.028–0.068) | 2.08 (1.31–3.37) | p = 0.004 |
| Damaging (LGD + Mis3) | 199 | 180 | 0.75 (0.65–0.85) | 0.58 (0.49–0.67) | 0.51 (0.44–0.58) | 0.39 (0.33–0.45) | 1.30 (1.09–1.55) | p = 0.006 |
| LGD SNV | 24 | 19 | 0.089 (0.054–0.12) | 0.061 (0.040–0.088) | 0.060 (0.037–0.084) | 0.041 (0.023–0.060) | 1.48 (0.86–2.59) | p = 0.1 |
| LGD FS Indel | 15 | 3 | 0.056 (0.028–0.084) | 0.0097 (−0.0013–0.020) | 0.038 (0.019–0.057) | 0.0065 (−0.000859–0.0136) | 5.88 (1.95–23.82) | p = 0.001 |
| In-Frame Indel | 3 | 4 | 0.011 (−0.0015–0.024) | 0.013 (0.00022–0.024) | 0.0075 (−0.0010–0.016) | 0.0087 (0.00015–0.016) | 0.88 (0.17–4.04) | p = 0.7 |
After de novo variant identification, we estimated the per base mutation rates for each class of variant by dividing the number of variants by the total number of callable base pairs (either within coding regions [“callable exome”] or within all regions [“total callable”]; Table 1). We further divided this rate by two in order to account for the diploid genome. We also estimated the theoretical number of variants per individual (exome) based on the mutation rates per bp and the size of the total possible target (for all de novo variants; varied by capture array, see Table 1) or RefSeq hg19 coding intervals (coding de novo variants; 33,828,798 bp). We determined rate ratios by dividing the observed rate in TD probands by the observed rate in the SSC controls and estimated p values with a one-sided rate ratio test. See also Table S3.
One (1) coding variant (301 total) is annotated as “unknown” effect by Annovar and therefore is not present in the synonymous (67) or nonsynonymous (233) counts (total = 300)
Seven (7) coding variants (484 total) are annotated as “unknown” effect by Annovar and therefore are not present in the synonymous (134) or nonsynonymous (343) counts (total = 477)
One (1) coding variant (454 total) is annotated as “unknown” effect by Annovar and therefore is not present in the synonymous (111) or nonsynonymous (342) counts (total = 453)
Figure 2. De Novo Variants Are Associated with Risk in the TIC Genetics Cohort.
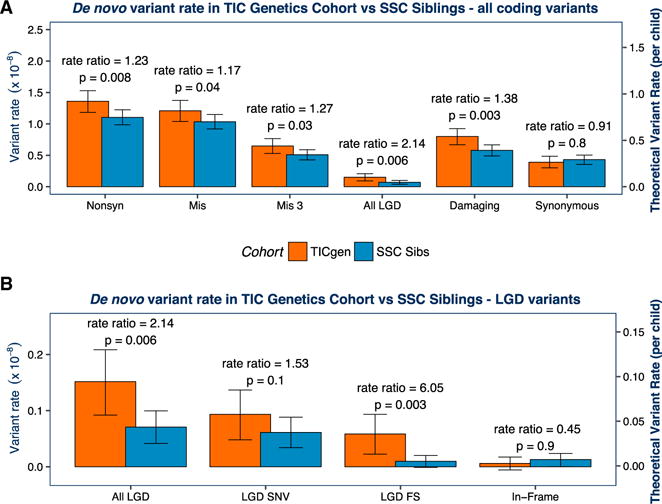
We first compared the rate of de novo mutation per base pair(bp) in the TIC Genetics and SSC cohorts. We determined the “total callable exome” for each TD proband or SSC sibling (Table 1; Table S1). We then calculated the mutation rate per bp for each individual based on the observed number of de novo variants and the size of the callable exome. The mean of these rates is plotted by cohort in (A) and (B) (see left y axis; see also Table 2). To estimate rate ratios and p values, we compared the number of mutations observed per the number of callable bp assessed using a one-sided rate ratio test. We estimated the theoretical rate of coding de novo variants per individual by multiplying the variant rate by the size of the “coding” exome (RefSeq hg19 coding exons; 33,828,798 bp). We display this as the right y axis in (A) and (B). We compare the main classes of variants in (A). All classes of de novo non-synonymous variants show a significantly elevated rate ratio in TD probands (red) versus SSC siblings (blue). As expected, de novo synonymous variants are not significantly overrepresented in TD probands (p = 0.8). We compare subclasses of LGD variants in (B). Frameshift (FS) indels trend toward a higher rate ratio (RR) than LGD SNVs (RR 6.0, p = 0.003 versus RR 1.5,p = 0.1). In-frame indels, which are not expected to have marked biological impact, are not significantly overrepresented in TD probands (p = 0.9).Aone- sided binomial exact test to assess the significance of the observed burden differences in TD cases versus controls produced consistent results (Figure S2). Mis3, missense variants predicted to be damaging by PolyPhen (Missense 3 or Mis3; PolyPhen2 [HDIV] score ≥ 0.957).
Figure 3. Association of De Novo Variants with TD Is Confirmed in the TSAICG Cohort.
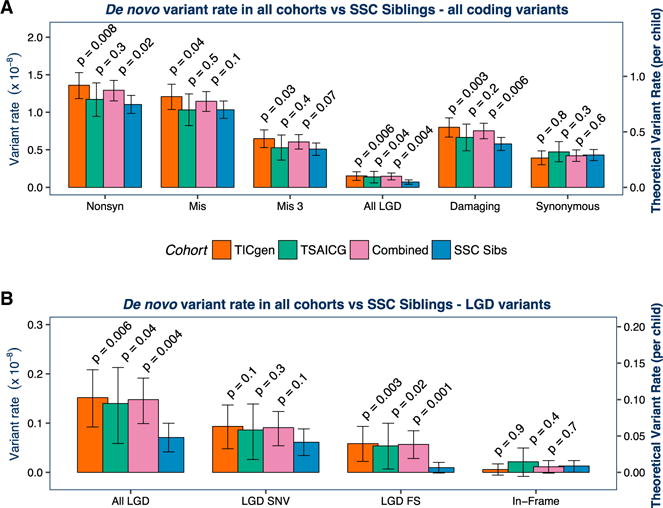
We next repeated the analyses in a non-overlapping cohort, ascertained and characterized by the TSAICG. De novo mutation rate per bp and theoretical mutation rate per child were calculated as in Figure 2. The TIC Genetics cohort is in red, TSAICG in green, the “Combined” TD cohort of TIC Genetics and TSAICG in purple, and the SSC control trios in blue. We compared the rate of de novo variants within the total callable exome with a one-sided rate ratio test (see Figure 2; Table 1). As in the TIC Genetics cohort, de novo LGD variants are elevated in TSAICG TD probands (p = 0.04) (A). De novo damaging variants as a group (LGD + Mis3) showed a trend toward enrichment in probands (p = 0.2). Again, FS indels occur at a substantially elevated rate (p = 0.02) (B). Neither synonymous de novo variants (p = 0.3; A) nor de novo in-frame indels (p = 0.4; B) showed any differences between TD and controls. Finally, we combined the TIC Genetics and TSAICG cohorts to obtain an overall estimate for de novo variant burden in TD (purple bars in A and B). De novo LGD variants are strongly associated with TD risk, occurring 2-fold more frequently in TD probands (RR 2.1, 95% CI 1.3–3.4, p = 0.004). De novo damaging variants (LGD + Mis3) are also associated (RR 1.3, 95% CI 1.1–1.5, p = 0.006). The distribution of de novo coding variants per individual in the TIC Genetics and TSAICG cohorts, as well as in the SSC siblings, follows an expected Poisson distribution (FigureS1). Mis3, missense variants predicted to be damaging by PolyPhen (Missense 3 or Mis3; PolyPhen2 [HDIV] score ≥ 0.957).
We calculated the mutation rate per base pair for each individual. For Figures 2 and 3, we plotted the mean of these rates by cohort, along with the 95% confidence intervals (see also Table 2). We utilized a one-sided rate ratio test, comparing the number of variants per the number of callable base pairs assessed to estimate rate ratios and p values (Table S3).
Based on consistent observations in other NDDs and our TD CNV analyses, we hypothesized that de novo LGD variants would be significantly overrepresented in TD cases versus controls. We confirmed this expectation, with de novo LGD variants showing a significantly elevated rate ratio in TD probands (rate ratio [RR] 2.14, 95% CI 1.28–3.61, p = 0.006; Figure 2A; Table 2). De novo missense (Mis) variants, particularly those predicted to be damaging by PolyPhen2 (Missense 3 or Mis3; PolyPhen2 [HDIV] score ≥ 0.957; Adzhubei et al., 2010, 2013), are also enriched in probands (RR 1.27, 95% CI 1.03–1.57, p = 0.03; Figure 2A). As a group, therefore, damaging de novo variants (Mis3 and LGD variants) occur at a significantly higher rate in coding regions in TD probands versus SSC controls (RR 1.38, 95% CI 1.14–1.67, p = 0.003). No differences were seen in the rate of de novo synonymous variants (RR 0.91, 95% CI 0.70–1.17, p = 0.8) nor in the rate of in-frame indels (RR 0.45, 95% CI 0.019–3.48, p = 0.9). A one-sided binomial exact test, which is typically used in WES studies to assess the significance of observed burden differences in cases versus controls (e.g., Iossifov et al., 2014; Sanders et al., 2012; Willsey et al., 2013), produced consistent results (Figure S2). Indeed, these results more strongly support the association of de novo variants with TD. However, as the distribution of callable base pairs per sample varied across the cohorts due to differences in experimental design (e.g., library capture protocol or sequencing coverage; Table 1; Figure S3), we felt the rate ratio test would provide a more accurate estimate of the significance of true variant burden. This approach compares the number of variants while also controlling for the per sample differences in the number of base pairs with sufficient coverage and quality for de novo detection.
We also estimated the theoretical number of variants per individual (exome) based on the total size of RefSeq hg19 coding intervals (33,828,798 bp); this is shown as a second axis on the same plots (Figures 2A and 2B). We reasoned this would provide a second and potentially more accurate comparison metric versus the rate of observed de novo variants per individual because the callable exome differed by cohort (Figure S3). The theoretical rate also has the advantage of providing an estimate of the total number of expected de novo variants under 100% coverage, as opposed to the number of observed variants per individual, and is therefore a useful metric for comparing across sequencing studies.
Analysis of the TSAICG Cohort Replicates Association of De Novo Likely Gene-Disrupting Variants
We next evaluated 186 TD trios ascertained through the Tourette Syndrome Association International Consortium for Genetics (TSAICG). Scharf et al. (2013) have previously described the ascertainment and phenotyping of this cohort. We compared the 173/186 (93.0%) trios passing our quality control metrics (Table S1) to the same set of 602 SSC control trios (Table 2; Figures 3A and 3B). Within the TSAICG cohort, we attempted validation on only a subset of de novo variants based on their validation likelihood (De Rubeis et al., 2014). Within the variants from the TSAICG cohort prioritized for validation, 94.3% of de novo variants confirmed, with 96.4% of SNVs and 60% of indels confirmed (Table S2). Again, all burden analyses were based on unconfirmed de novo variants in both TD and control cohorts. The distribution of de novo coding variants per individual in the TSAICG also follows an expected Poisson distribution (Figure S1).
This analysis replicates the association of de novo LGD variants with TD (RR 1.97, 95% CI 1.03–3.68, p = 0.04, one-sided rate ratio test; Figure 3A; Table 2). Again, neither synonymous de novo variants (RR 1.10, 95% CI 0.81–1.47, p = 0.3) nor de novo in-frame indels (RR 1.67, 95% CI 0.22–8.97, p = 0.4) showed any differences between TD and controls.
There is a male:female sex bias in both the TIC Genetics (3.64; Table 1) and TSAICG (4.97) cohorts but not in the SSC sibling trios (0.84); however, mutation rates were not significantly different between males and females in the TIC Genetics (p = 0.4, two-sided rate ratio test; Table S1), TSAICG (p = 0.9), or SSC siblings (p = 0.3) cohorts. Therefore, despite the differences in sex ratio, the direction of effect suggests that, if anything, there is a slightly higher rate of de novo variants in females, and therefore, a male-biased TD cohort and a non-male-biased control cohort should result in conservative burden estimates.
Managing Batch Effects across Multiple Cohorts and Array Types
We hypothesized that batch effects might confound the combined analyses due to the use of three different exome capture arrays and sequencing at different centers (Table 1). Indeed, the three cohorts have different coverage distributions (Figure S3) and cluster separately in principal-component analysis (PCA) based on sequencing quality metrics (Figure S4). Likewise, we observed that “naïve” estimates of de novo variant rates were highly divergent across cohorts (Figure S5). However, we did not observe a significant difference in the “normalized” de novo variant rates between TIC Genetics, TSAICG, and the SSC control trios, suggesting that we adequately controlled for these confounds in our analyses.
Nonetheless, to ensure that the observed increases in de novo burden were not due to additional batch effects, we also performed a Poisson regression (Figure 4) to control for other factors potentially influencing de novo variant rate and detection. In iterative univariate multiple regression analyses, we observed that paternal age, sequencing coverage (percent of exome at 2× coverage), sequencing coverage uniformity (fold 80 base penalty), heterozygous SNP quality, and the number of de novo synonymous variants provided the best model for de novo coding variants. We used the size of the callable coding exome as an offset (Table 1; Table S1; Figure S3). The correlation between paternal age and de novo variant rate has been previously observed (e.g., Iossifov et al., 2012, 2014; Kong et al., 2012a; Neale et al., 2012; O’Roak et al., 2012; Sanders et al., 2012). Sex was not a significant predictor (Table S1). After controlling for these additional covariates in the Poisson multiple regression, de novo LGD variants still remained significantly associated with TD risk (Figure 4; RR 2.20, 95% CI 1.19–4.08, p = 0.01; RR 2.23, 95% CI 1.04–4.82, p = 0.04 in TIC Genetics and TSAICG, respectively). Additionally, de novo damaging variants (LGD + Mis3) showed enrichment in the TIC Genetics cohort (RR 1.38, 95% CI 1.08–1.76, p = 0.009) and a trend toward enrichment in the TSAICG cohort (RR 1.37, 95% CI 0.98–1.92, p = 0.07). Mis3 variants alone were no longer significantly associated in either cohort, although we still observed evidence of modest effects in the TIC Genetics (RR 1.27, 95% CI 0.97–1.65, p = 0.08) cohort.
Figure 4. Poisson Regression to Control for Paternal Age and Sequencing Coverage Confirms Association of De Novo LGD Variants.
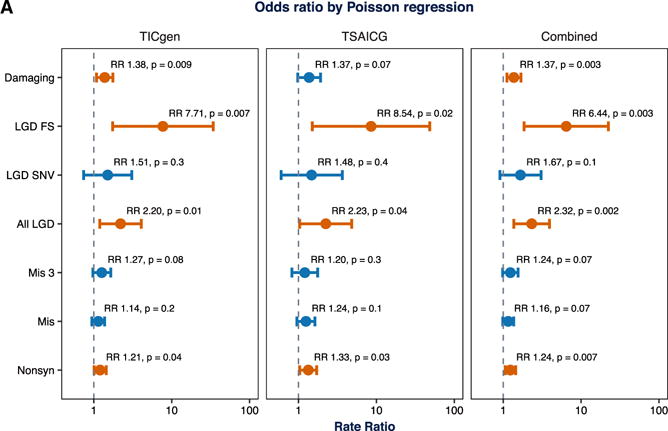
To ensure that the observed differences in burden were not due to additional batch effects (Figures S3–S5), we performed a Poisson regression to control for other factors influencing de novo variant rate and detection. We first confirmed that the distribution of de novo coding variants per individual in the TIC Genetics and TSAICG cohorts, as well as in the SSC siblings, follow an expected Poisson distribution (Figure S1). Next, after several model building steps, we selected paternal age, sequencing coverage (percent of exome at 2× coverage), sequencing coverage uniformity (fold 80 base penalty), heterozygous SNP quality, and the number of de novo synonymous variants as covariates, along with affected status, in the regression analysis (Figure S3). The size of the callable coding exome served as the offset, and the number of de novo variants in a particular class was the response variable. After controlling for these covariates, de novo LGD variants remained associated with TD risk in both cohorts, and in the combined cohort, we estimate the rate ratio as 2.32 (95% CI 1.37–3.93, p = 0.002). Additionally, de novo damaging variants (LGD + Mis3) showed enrichment in the TIC Genetics cohort, a trend toward enrichment in the TSAICG cohort, and are significantly enriched overall with a rate ratio of 1.37 (95% CI 11.11–1.69, p = 0.003). Using this approach to analysis, Mis3 variants alone are not significantly associated in either cohort but show a trend toward enrichment in the combined data (rate ratio 1.24,95% CI 0.98–1.55, p = 0.07). Other approaches to correct for batch effects consistently supported an increased burden of de novo LGD and damaging variants in TD probands (see Figures S2 and S6 for details). Mis3, missense variants predicted to be damaging by PolyPhen (Missense 3 or Mis3; PolyPhen2 [HDIV] score ≥ 0.957).
Combined Analysis Estimates a Rate Ratio of 2.32 for De Novo LGD Variants
Having observed that putatively deleterious de novo variants are overrepresented in both TIC Genetics and TSAICG probands separately, and that the overall rate of de novo mutations was not significantly different by cohort (Figure S5), we combined the TIC Genetics and TSAICG cohorts (484 TD trios) to obtain an overall estimate for de novo variant burden in TD (Figure 4). We observed a significant excess of de novo LGD variants (RR 2.32, 95% CI 1.37–3.93, p = 0.002, Poisson regression) and de novo damaging (LGD + Mis3) variants (RR 1.37, 95% CI 1.11–1.69, p = 0.003). Mis3 variants alone again showed a trend toward enrichment in the combined data (RR 1.24, 95% CI 0.98–1.55, p = 0.07). We observed similar results with the binomial exact and rate ratio tests (Figures 3A and 3B; Table 2; Table S2), as well as a Fisher exact test normalizing for the rate of de novo synonymous variants (Figure S6).
De Novo Damaging Variants Contribute to TD Risk in Approximately 12% of Cases
As previously noted, we can estimate the theoretical de novo variant rate per individual (exome) by multiplying the observed rate per base pair by the total size of all RefSeq hg19 coding regions. By subtracting the theoretical rate, per exome, of de novo variants in controls from the theoretical rate in probands, we can then estimate the percentage of probands in whom a de novo variant is contributing to TD risk (Iossifov et al., 2014; Sanders et al., 2015). Based on this calculation, we estimate that 5.0% (95% CI 1.3%–8.7%) of cases have a de novo LGD variant and 11.6% (95% CI 2.4%–20.8%) of cases have a de novo damaging variant contributing to TD risk (Table 3). Similarly, 6.9% (95% CI 4.9%–8.9%) of ASD cases have a de novo LGD variant mediating ASD risk (Sanders et al., 2015).
Table 3.
Contribution of De Novo SNVs to TD Risk
| Variant Type | Theoretical rate per child (±95% CI)a | % of cases with mutation mediating risk (±95% CI) | % of mutations carrying risk (±95% CI) | |
|---|---|---|---|---|
| Combined TD Cohort | ||||
| TD | Control | |||
| (N = 484) | (N = 602) | |||
| Likely Gene Disrupting (LGD) | 0.098 (0.067 – 0.13) | 0.048 (0.028 – 0.067) | 5.0% (1.3%–8.7%) | 51.3% (13.7%–89.0%) |
| Damaging (LGD + Mis3) | 0.51 (0.44 – 0.58) | 0.39 (0.33 – 0.45) | 11.6% (2.4%–20.8%) | 22.9% (4.8%–41.0%) |
To estimate the percentage of probands in whom a de novo variant is contributing to TD risk, we subtracted the theoretical rate, per exome, of de novo variants in controls from the theoretical rate in probands (Iossifov et al., 2014; Sanders et al., 2015). We predict that 5.0% (95% CI 1.3%–8.7%) of cases have a de novo LGD variant and 11.6% (95% CI 2.4%–20.8%) of cases have a de novo damaging variant contributing TD risk. To estimate the fraction of observed proband de novo variants that contribute to TD risk, we divided the difference in theoretical rate by the theoretical rate in probands (Iossifov et al., 2014; Sanders et al., 2015). Based on this approach, we predict that 51.3% (95% CI 13.7%–89.0%) of de novo LGD and 22.9% (95% CI 4.8%–41.0%) of de novo damaging variants carry TD risk.
Theoretical rate per child was calculated per individual. Mean theoretical rate and 95% CI was then calculated per cohort based on individual rates (see STAR Methods and Table 2 for more details)
We can also estimate the fraction of observed proband de novo variants that contribute to TD risk (Iossifov et al., 2014; Sanders et al., 2015) by dividing the difference in theoretical rate by the theoretical rate in probands. Using this approach, we estimate that 51.3% (95% CI 13.7%–89.0%) of de novo LGD and 22.9% (95% CI 4.8%–41.0%) of de novo damaging variants carry TD risk (Table 3). Again, the estimate for de novo LGD variants in TD is similar to that for ASD (45.9%, 95% CI 31.8%–55.5%) (Sanders et al., 2015).
Maximum Likelihood Estimation Predicts that Approximately 400 Genes Contribute TD Risk
We next utilized a maximum likelihood estimation (MLE) procedure to estimate the number of genes contributing risk to TD, based on vulnerability to de novo damaging variants, as has been done recently in congenital heart disease (Homsy et al., 2015). We observed 192 confirmed damaging de novo variants in 484 TD probands. Therefore, for every possible number of risk genes, from 1 to 2,500, we simulated 192 variants. 50,000 permutations were conducted: in each permutation, we randomly selected risk genes and then, based on the fraction of damaging variants estimated to carry risk, randomly assigned a percentage of variants to the risk genes and the rest of the variants to the non-risk genes. We weighted the probability of variation by gene size and GC content (He et al., 2013). We then determined the combined number of risk and non-risk genes harboring multiple de novo variants and recorded when the number of genes with two variants and the number of genes with three or more variants in the simulated data matched the number observed in our study (4 and 1, respectively). Based on the frequency of these occurrences versus gene number, we determined the MLE of the number of TD risk genes to be 420 genes (Figure 5A). Alternatively, based on methods we have used previously to evaluate target size in ASD (Sanders et al., 2011, 2012), we estimate 447 genes (95% CI 136.7–932.7).
Figure 5. Recurrent De Novo Damaging Variants Identify Four Likely TD Risk Genes.
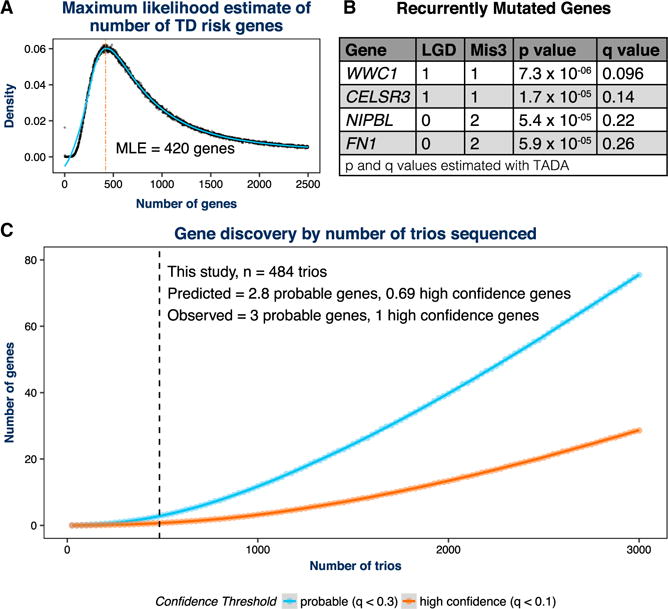
(A) Given the number of confirmed damaging de novo variants observed in 484 TD probands (192) and an empirical estimate of the fraction of these carrying risk, we used a maximum likelihood estimation (MLE) procedure to estimate the total number of “target” genes. After 50,000 permutations, we estimate that 420 genes contribute to TD risk based on vulnerability to de novo damaging variants. We identified five genes with recurrent de novo LGD or Mis3 variants confirmed using PCR and Sanger sequencing (Table S2).
(B) We estimated the per-gene p values and q values for recurrence with TADA using the de novo only algorithm (He et al., 2013). Based on previously established q value (false discovery rate) thresholds (see De Rubeis et al., 2014; He et al., 2013; Sanders et al., 2015), one of these genes, WWC1, is a high-confidence TD (hcTD) risk gene (q < 0.1), and three of these genes are probable TD (pTD) risk genes (q < 0.3; shown in A). The fifth gene, TTN, did not meet this threshold (q = 0.76), as expected given its large size.
(C) The estimate of 420 genes derived from (A) was utilized to predict the likely future gene discovery yield as additional TD trios are whole-exome sequenced. For each of 10,000 permutations, we ran simulated variants through the TADA de novo algorithm to assess per-gene q values. We then recorded the number of pTD genes (q < 0.3) and hcTD genes (q < 0.1) observed at each cohort size and plotted the smoothed trend line using local polynomial regression fitting. The regression model also predicted the number of genes identified at a given number of trios. The predicted number of TD genes for the cohort presented in this study (484 trios) tracked very closely with our empirical results: we predict 2.8 pTD genes (we observed 3) and 0.69 hcTD genes (we observed 1). Mis3, missense variants predicted to be damaging by PolyPhen (Missense 3 or Mis3; PolyPhen2 [HDIV] score ≥ 0.957).
Recurrent De Novo Variants Identify Four Candidate Genes
We next asked whether de novo variants cluster within specific genes. Here we considered only de novo damaging variants that confirmed using Sanger sequencing (Table S2). We chose to focus on de novo damaging variants because both LGD and Mis3 variants showed evidence of TD association. We identified five genes with multiple (two or more) de novo LGD or Mis3 variants. None of these had two de novo LGD variants. Based on our MLE of 420 risk genes, we estimated the per-gene p and q values for these observations with TADA, using the de novo only algorithm (He et al., 2013). Based on previously established q value thresholds (FDR thresholds) (De Rubeis et al., 2014; He et al., 2013; Sanders et al., 2015), one of these genes is a high-confidence TD (hcTD) gene (q < 0.1)— WWC1 (WW and C2 domain containing 1)—and three of these genes are probable TD (pTD) risk genes (q < 0.3)— CELSR3 (Cadherin EGF LAG seven-pass G-type receptor 3), NIPBL (Nipped-B-like), and FN1 (fibronectin 1) (Figure 5B).
Prediction of the Number of Risk Genes Identified by Cohort Size
We also utilized our MLE of the number of genes involved in TD risk to predict the likely future gene discovery yield from WES. We fixed the gene number at 420 and varied the cohort size. Therefore, we calculated the number of variants in each iteration based on the cohort size and the observed variant rate per proband. In each iteration, we randomly selected 420 TD risk genes and then assigned a fraction of the permuted variants to these TD risk genes and the leftover fraction to the remaining non-TD risk genes. This allocation was determined based on the fraction of variants estimated to carry risk. We performed 10,000 permutations at each cohort size, separately randomly generating LGD and Mis3 variants, using their observed rates and per-gene likelihoods. These data were then combined, and each permutation was run through the TADA de novo algorithm to assess the per gene q values. We then recorded the number of pTD genes (q < 0.3) and hcTD genes (q < 0.1) observed at each cohort size (Figure 5C). Based on the smoothed curves, the predicted number of probable genes for the cohort presented in this study (484 trios) tracked very closely with our empirical results: we predict 2.8 pTD genes (we observed 3) and 0.69 hcTD genes (we observed 1). Moreover, we can further predict that, at 1,000 trios, we will identify approximately 11.8 pTD genes and 3.2 hcTD genes and, at 2,000 trios, will identify 39.8 pTD genes and 13.4 hcTD genes.
DISCUSSION
Exome sequencing of TD trios establishes the increased rate of de novo LGD variants in cases versus controls. We observe this excess burden in two independently ascertained cohorts: TIC Genetics and TSAICG. We also observe evidence for enrichment of de novo Mis3 variants in TD probands, though statistical significance is not reached in all tests. Sequencing of additional trios is certain to clarify this result. As has been well established in exome studies of other NDDs, these results provide a highly reliable avenue for gene discovery based on the recurrence of damaging de novo mutations. In the current dataset, one gene, WWC1, meets the threshold for high-confidence association and three genes meet the threshold for probable association.
The four likely TD genes span a range of biological pathways and functional ontologies and are all clearly brain expressed (Kang et al., 2011; Kapushesky et al., 2012; Petryszak et al., 2014, 2016). Indeed, these genes provide interesting avenues for additional investigations: WWC1, also known as KIBRA (kidney and brain expressed protein), is a cytoplasmic phosphoprotein that shows evidence of interaction with multiple proteins and pathways (Kremerskothen et al., 2003; Rebhan et al., 1997; Zhang et al., 2014). For instance, it may be a transcriptional co-activator of estrogen receptor 1 (ESR1), regulate the collagen-stimulated activation of ERK-MAPK cascade, and regulate the Hippo/SWH signaling pathway (Zhang et al., 2014). It has been demonstrated to have roles in cell polarity, migration, and trafficking, as well as learning and memory (Schneider et al., 2010). It is also likely regulated by PRKCZ (protein kinase C zeta), a kinase known to play a role in synaptic plasticity and memory formation (Büther et al., 2004).
CELSR3 belongs to the flamingo subfamily of non-classic cadherins, which are defined by non-interaction with catenins and seven transmembrane domains (Feng et al., 2012). The protein encoded by this gene may be involved in the regulation of contact-dependent neurite outgrowth (Chai et al., 2015). In mice, Celsr3 appears to be critical for axon pathfinding in the central nervous system, with cortico-cortical and cortico-subcortical connections defective in mutant mice (Tissir et al., 2005; Zhou et al., 2008). Moreover, the role of Celsr3 in steering motor axons innervating the dorsal hindlimb and in the anterior-posterior patterning of monoaminergic neurons has also recently been demonstrated (Chai et al., 2014; Fenstermaker et al., 2010).
NIPBL, also known as Delangin, appears to have two critical functions: (1) it is essential for loading the cohesin complex onto sister chromatids during meiosis I and DNA double-stranded break repair (Peters et al., 2008) and (2) it may influence gene expression during development (Zuin et al., 2014). Variants in NIPBL are associated with Cornelia de Lange syndrome (CdLS), a developmental disorder characterized by slow growth, moderate to severe intellectual disability, and abnormalities of bones in the arms, hands, and fingers (Brachmann, 1916; De Lange, 1933). Many affected individuals also have behavior problems, including compulsive repetition, anxiety, OCD, and ADHD (Mulder et al., 2017; Oliver et al., 2008). Given that approximately 60% of CdLS cases have a heterozygous NIPBL variant (Mannini et al., 2013), we were surprised to observe variants in this gene in our subjects. However, we only observed Mis3 variants, perhaps suggesting that these variants have less severe consequences. Indeed, cdLS severity is highly correlated to the expression levels of NIPBL (Kaur et al., 2016), and the de novo Mis3 variants that we observed were in exons 29 and 47 whereas exon 10 has the greatest proportion of pathogenic CdLS variants (Mannini et al., 2013). In addition, both patients have some phenotypic aspects that are consistent with CdLS: (1) the TIC Genetics proband has failure to thrive in childhood with adult short stature (final height in the fifth percentile for males), generalized anxiety, and irritable bowel, and (2) the TSAICG proband has developmental delay, intellectual disability, and mild hearing loss, although birthweight, height, and weight were within normal limits.
FN1 codes for two types of fibronectin-1 protein: a soluble plasma protein, mainly produced by the liver and involved in blood clotting and wound healing; and an insoluble protein released to the extracellular space, helping with formation of fibrils and the extracellular matrix (Frantz et al., 2010; Pankov and Yamada, 2002; Rebhan et al., 1997). Both types of proteins are involved in cell adhesion, spreading, migration, and differentiation (Frantz et al., 2010; Pankov and Yamada, 2002). It is therefore possible that this gene will be similarly involved in neurite outgrowth or a similar process during brain development. Indeed, homozygous knockout mice display neural tube defects and shortened anterior-posterior axes (George et al., 1993). Variants in FN1 also appear to be involved in glomerulopathy with fibronectin deposits (Castelletti et al., 2008). Unlike the probands with NIPBL variants, affected individuals do not appear to have overlapping phenotypic characteristics, although this disorder has a late onset (Castelletti et al., 2008).
Given the current results, a comparison to recent studies of ASD may be instructive: to date 2,517 simplex SSC ASD families have been reported, and both de novo LGD and Mis3 variants have been associated with ASD risk, with mutation rates and effect sizes consistent with those observed here (e.g., rate ratio of 2.08 versus 1.74 for de novo LGD variants in TD versus ASD) (Iossifov et al., 2014). Of note, the ascertainment strategies used in both TD cohorts did not restrict to apparently simplex families, as was done in the SSC. Given the evidence for an increased burden of de novo variation in simplex versus multiplex families in ASD (e.g., Leppa et al., 2016), it would be reasonable to hypothesize that the current analysis may underestimate the rate ratios for de novo variants in simplex TD families.
The widespread success in gene discovery leveraging de novo variation in ASD (De Rubeis et al., 2014; Dong et al., 2014; Iossifov et al., 2012, 2014; Neale et al., 2012; O’Roak et al., 2011, 2012; Sanders et al., 2012, 2015; Willsey et al., 2013) strongly argues for additional WES in TD. The current gene discovery by cohort size curves predict that increasing our study size to 2,517 trios would lead to the identification of ~21 hcTD genes, which is a similar order of magnitude to the 27 hcASD genes identified from 2,517 trios in Iossifov et al. (2014). Moreover, the integration of de novo CNV data should further increase the yield of risk genes (Sanders et al., 2015). The discovery of a large number of TD-associated genes will provide a critical substrate for model systems and systems-biological studies aimed at understanding the spatial, temporal, and cell-level dynamics of TD pathology (Parikshak et al., 2013; Willsey et al., 2013; Willsey and State, 2015; Xu et al., 2014) and, importantly, for the development of novel, more effective therapeutic targets.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| TIC Genetics trios (n = 325) | Tourette International Collaborative Genetics Study | https://tic-genetics.org/ |
| TSAICG trios (n = 273) | Tourette Syndrome Association International Consortium for Genetics | https://www.findtsgene.org |
| Deposited Data | ||
| Whole exome sequencing data from TIC Genetics (n = 325) | This paper. SRA: SUB2101648 | SRA url |
| Whole exome sequencing data from TSAICG (n = 286) | This paper. SRA: XXX. | SRA url |
| Whole exome sequencing data from SSC control trios (n = 625) | Iossifov et al., 2014 | SRA url |
| Software and Algorithms | ||
| Genome Analysis Tool Kit (GATK) | DePristo et al., 2011; McKenna et al., 2010; Van der Auwera et al., 2013 | https://software.broadinstitute.org/gatk/best-practices/ |
| BWA-mem | Li and Durbin, 2009 | http://bio-bwa.sourceforge.net/ |
| Picard Tools | Broad Institute | https://broadinstitute.github.io/picard/ |
| Annovar | Wang et al., 2010 | http://annovar.openbioinformatics.org/en/latest/ |
| PLINK/SEQ | Fromer et al., 2014 | https://atgu.mgh.harvard.edu/plinkseq/ |
| Primer Design | This paper | http://primerdesign.willseylab.com/ |
| DeNovoFinder | De Rubeis et al., 2014 | |
| Perl & Shell script code for data processing & analysis | This paper | https://bitbucket.org/willseylab/tourette_phase1 |
| R code for data analysis | This paper | https://bitbucket.org/willseylab/tourette_phase1 |
| TADA | He et al., 2013 | http://wpicr.wpic.pitt.edu/WPICCompGen/TADA/TADA_homepage.htm |
| Other | ||
| 1000 Genomes GRCh37 hg19 genome build | N/A | http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/human_g1k_v37.fasta.gz |
| RefSeq hg19 gene annotation | N/A | http://genome.ucsc.edu/cgi-bin/hgTables?command=start |
| Intervals file for NimbleGen SeqCap EZ Exome v2 | Roche NimbleGen | https://bitbucket.org/willseylab/tourette_phase1 |
| Intervals file for NimbleGen SeqCap EZ Exome v3 | Agilent Technologies | https://bitbucket.org/willseylab/tourette_phase1 |
| Intervals file for Agilent SureSelect v1.1 | Roche NimbleGen | https://bitbucket.org/willseylab/tourette_phase1 |
| Coding regions only from RefSeq hg19 gene annotation | This paper | https://bitbucket.org/willseylab/tourette_phase1 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Matthew W State (matthew.state@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Sample Selection
Two independent collaborative groups recruited 511 parent-child trios subjects for DNA sequencing: the Tourette International Collaborative Genetics group (325 trios; TIC Genetics; http://tic-genetics.org) and the Tourette Syndrome Association International Consortium for Genetics (186 trios; TSAICG; https://www.findtsgene.org/). Dietrich et al. (2015) and Scharf et al. (2013) have previously described recruitment criteria in detail for each group. Briefly, each of the parent-child trios recruited by TIC Genetics or TSAICG consisted of an affected child (proband) meeting criteria for TD or a chronic tic disorder based on the Diagnostic and Statistical Manual of Mental Disorders-Fourth edition, Text Revision (DSM-IV-TR) (American Psychiatric Association, 2000). Phenotypic data available for each cohort is described in more detail in Tables 1 and S1, but briefly: both the TIC Genetics and TSAICG cohorts have phenotype data for sex, parental age, co-morbid OCD and/or ADHD in the proband, and most have data on whether there is any history of tic disorders in the first degree relatives. The TIC Genetics cohort generally has data on tic disorders in second-degree relatives as well. Among the 511 total trios sequenced here, parental clinical data was available for 487 families. Of these, 442 families (90.8%) had no evidence of a tic disorder in either parent. Sibling and second-degree relative histories were available for 278 families. Of these, 206 families (74.1%) had no evidence of a tic disorder in parents or siblings, and 163 families (58.6%) had no evidence of a tic disorder in parents, siblings, or a second degree relative (Table S1).
All adult participants and parents of children provided written informed consent along with written or oral assent of their participating child. The Institutional Review Board of each participating site approved the study.
METHOD DETAILS
Whole Exome Sequencing
Exome Capture and Sequencing
We performed whole-exome capture and sequencing of DNA from 511 affected children and their parents (1,533 samples total). We derived all DNA samples from primary blood cells. Of the 511 trios, we sequenced 325 (TIC Genetics) at the Yale Center for Genomic Analysis (YCGA), using the NimbleGen SeqCap EZ Exome v2 capture library (Roche NimbleGen, Madison, WI, USA) and the Illumina HiSeq 2000 sequencing platform (74 bp paired-end reads; Illumina, San Diego, CA); 149 (TSAICG) at the Broad Institute, using the Agilent SureSelect v1.1 capture library (Agilent Technologies, Santa Clara, CA, USA) and Illumina HiSeq 2500 sequencing platform; and 37 (TSAICG) at UCLA using the NimbleGen SeqCap EZ Exome v3 capture library (Roche NimbleGen, Madison, WI, USA) and the Illumina HiSeq 2500 sequencing platform.
Control Data
We obtained WES data from unaffected parent-child trios (n = 625) from the Simons Simplex Collection (SSC) (De Rubeis et al., 2014; Dong et al., 2014; Iossifov et al., 2012, 2014; Neale et al., 2012; O’Roak et al., 2011, 2012; Sanders et al., 2012, 2015; Willsey et al., 2013). These children and their parents have no evidence of autism spectrum or other neurodevelopmental disorders (Fischbach and Lord, 2010b). Like the TIC Genetics data, these SSC data were generated from primary blood-derived DNA and sequenced on the Illumina HiSeq 2000 sequencing platform after capture with the NimbleGen SeqCap EZ Exome v2 array.
Variant Detection
We utilized GATK best practices (https://software.broadinstitute.org/gatk/best-practices/) for pre-processing and variant discovery (DePristo et al., 2011; McKenna et al., 2010; Van der Auwera et al., 2013). We processed each cohort separately, including the TSAICG – Broad, and TSAICG – UCLA sub-cohorts. BWA-mem (Li and Durbin, 2009) aligned raw reads to the 1000 Genomes GRCh37 hg19 genome build, Picard Tools (https://broadinstitute.github.io/picard/) marked duplicates, and GATK conducted base quality score recalibration. We conducted variant calling per sample with HaplotypeCaller in GVCF mode. Subsequent joint genotyping conducted across each cohort produced a multi-sample VCF callset for each cohort. Where appropriate, we utilized a list of capture targets corresponding to each cohort’s respective library capture kit, with an interval padding of 100. We applied variant quality score recalibration (VQSR) to each VCF to refine the callset. We utilized passing variants only in downstream analyses. Example commands for variant calling are located in the project bitbucket repository at https://bitbucket.org/willseylab/tourette_phase1. Annovar (Wang et al., 2010) annotated variants according to RefSeq hg19 gene definitions.
De Novo Variant Detection
We called de novo variants using a combination of PLINK/SEQ (Fromer et al., 2014) and in-house scripts (see https://bitbucket.org/willseylab/tourette_phase1 for example commands). Empirically validated filters identified high confidence de novo SNVs and indels (see Sanders et al., 2012). Briefly, we called a de novo variant if:
The child was heterozygous for a variant with alternate allele frequency (AB) between 0.3 and 0.7 in the child and ≤ 0.05 in the parents (i.e., not present)
Minimum sequencing depth ≥ 20 in all family members at the variant position
Allelic depth for the alternate allele (AD) ≥ 8
Observed allele frequency in the respective cohort ≤ 0.001
Minimum map quality ≥ 30
Minimum phred-scaled genotype likelihood ≥ 20
De Novo Variant Validation
We attempted validation of all de novo coding variants predicted in TD subjects. We PCR-amplified whole-blood derived DNA and then Sanger sequenced the amplicon. We assessed all family members to ensure that (1) the variant was present in the child, and (2) absent in both parents. We predicted 301 de novo coding variants in the 311 TIC Genetics trios; however, we were unable to attempt confirmations on 51 of these variants due to difficulties with primer design, PCR amplification, and/or Sanger sequence quality. In the 250 variants with confirmation data, 243 confirmed as true de novos (97.2%). 240/245 of the de novo SNVs confirmed (97.8% confirmation rate), and 3/5 of the de novo indels confirmed (60%).
In total, we identified 153 de novo coding variants in the TSAICG data. For each identified de novo variant, a Python script developed by Samocha and colleagues (DeNovoFinder) (De Rubeis et al., 2014) estimated both the relative probability of a true de novo event versus an inherited variant, and the likelihood of validation given a variety of quality control metrics. This script estimates the relative probability of true de novo (p_dn) based on the genotype likelihood of all trio members (PL, normalized Phred-scaled likelihoods), the allele frequency, and the average mutation rate per genome. We assigned one of three levels of validation likelihood (high, medium, and low) to each de novo variant as a result of combination of the relative probability of true de novo (p_dn), allele balance and read depth of all trio members, and allele frequency. Samocha and colleagues’ work shows that the de novo SNV and indel variants with high likelihood of validation have validation rate 97.3% and 92.3%, respectively. Therefore, we carried out validation on all low de novo variants and a subset of medium and high indel de novo variants by Sanger Sequencing or by Sequenom SNP genotyping. Due to previously demonstrated high validation rate, we randomly chose only 87% of the high SNV variants for validation. For the TSAICG cohort, we were unable to attempt validation on 35 variants, and we did not attempt validation on 30 based on the validation prediction described above. In the remaining 88, 83 confirmed (94.3%); 80/83 were de novo SNVs (96.4%) and 3/5 were de novo indels (60%). We provide A list of all predicted de novo variants and their confirmation status in Table S2.
Quality Control
We created a panel of informative genotypes to confirm family relationships, and we omitted trios if expected family relationships did not confirm, or if there were unexpected relationships within or across families (Supplemental Experimental Procedures). Additionally, we removed samples with excess de novo variants (> 5).
Picard Tools (https://broadinstitute.github.io/picard/) generated capture, sequencing, alignment, and variant level quality metrics; and GATK DepthOfCoverage generated coverage metrics for the exome intervals (Table 1; Table S1). Principal components analysis (PCA) of these data focused on the first four principal components (PCs) to identify outliers (Figure S4). We considered samples greater than three standard deviations (SD) from the mean in any of the first four principal components as outliers and removed them from analysis. See next section for more details on the PCA.
311 TIC Genetics trios (311/325, 95.7%), 173 TSAICG trios (173/186, 93.0%), and 602 SSC trios (602/625, 96.3%) passed quality control (Table S1). We provide sequencing metrics for all subjects following alignment in Table S1.
Principal-Components Analysis
Although processed concurrently and with the same pipeline, the 484 TD trios from the TIC Genetics and TSAICG cohorts, as well as the 602 SSC control trios, were sequenced at different times using different capture platforms, sequencing machines, and genomic core facilities (Figure 1). Therefore, we performed principal components analysis (PCA) to check for potential batch effects (Figure S4). We collected sequencing quality metrics using the Picard tools CollectHsMetrics, CollectAlignmentSummaryMetrics, and Collect-VariantCallingMetrics. We also estimated the number of callable base pairs within each trio as the number of base pairs at ≥ 20× coverage in all family members (we refer to this as joint coverage at 20×). These metrics, as well as paternal and maternal age, where available, informed the PCA (Table S1). The PCA revealed clear batch effects based on sequencing facility, particularly with respect to the TSAICG UCLA and Broad subsets, and within the SSC control trios (Figure S4). We focused on the first four principal components (PCs), which explain 61.6% of the variance in the quality metrics. We considered samples greater than three standard deviations (SD) from the mean in any of the first four principal components outliers, and consequently, we removed from the analysis the entire family containing that sample (n = 23 of 1219 families or 1.89% of all families; Table S1; Figure S4).
Burden Analyses
Comparison to Poisson Distribution
To compare the observed distribution of de novo coding variants per individual to the corresponding expected Poisson distribution we determined the frequency of the counts per individual for each cohort (TIC Genetics, TSAICG, and SSC Siblings) and then plotted this as a histogram. We next plotted a Poisson distribution using the dpois R function with lambda (λ) equal to the mean of the counts per individual. l was determined per cohort (see #2 below). All three cohorts appear to follow the expected Poisson distribution. However, to confirm this, we conducted a Chi Square goodness-of-fit test between the observed and expected distributions with the following steps (example R code in italics):
Determine the number of individuals with 0, 1, 2, 3, 4, 5, or more de novo coding variants. Note that because during quality control we trimmed individuals with > 5 de novo variants the number of individuals with > 5 de novo coding variants is 0 for each cohort.counts = count(< df with number of de novos per individual >, numPassingCoding)$n
Create a vector of the number of de novo coding variants within each, separate individual and calculate the mean of this vector.x = rep(0:5, times = counts)distMean = mean(x)
Estimate the probabilities of 0, 1, 2, 3, 4, or 5 de novo coding variants in a given individual with the dpois R function, with l = the mean calculated in 2. probs = dpois(0:5, lambda = distMean)
Estimate the probability of > 5 de novo coding variants in a given individual by determining the complement of (3). In other words, 1 – the sum of probabilities estimated in (3).comp = 1-sum(probs)
Using a Chi-Square test in R (chisq.test), determine the p value for the observed distribution being different that the expected Poisson distribution, based on λ = the mean calculated in 1. We estimated p values through Monte Carlo simulation.pvalue < - chisq.test(x = c(counts, 0), p = c(probs, comp), simulate.p.value = TRUE)$p.value
In all three cohorts, the distribution of observed de novo coding variants per individual was not significantly different from the expected Poisson distribution (TIC Genetics, p = 0.96; TSAICG, p = 0.74; SSC Siblings, p = 0.77; Figure S1), suggesting the observed distributions can be modeled by the Poisson distribution.
Definition of Coding Portion of Exome
We defined the coding portion of the RefSeq hg19 by restricting to coding exons only (i.e., excluding UTRs, etc) in the RefSeq hg19 gene definitions (downloaded from the UCSC Genome Browser, “Table Browser” tool), and then merging all overlapping or book-ended intervals with bedtools sort and then bedtools merge (Quinlan and Hall, 2010). We then calculated the combined size by summing the intervals with awk. The resulting “coding exome” is 33,828,798 bp. The final bedfile, along with commands to create it and to calculate the total size are available on bitbucket (https://bitbucket.org/willseylab/tourette_phase1).
Estimation of Mutation Rate per BP
Within each family, we estimated the rate of de novo mutations per base pair (bp). To accurately calculate the rate, we first precisely defined the interval within which de novo variant calling was possible. We then calculated the rate per base pair as the total number of variants within this target region. We did this both for all de novo variants (coding + non-coding) and only for de novo coding variants.
We started with the exome capture array intervals plus the 100 bp of interval padding added during GATK processing but further restricted to the portion covered at ≥ 20× in all family members (with minimum base quality ≥ 10 and minimum map quality ≥ 20). The coverage threshold matches our threshold for de novo calling and the base and map quality thresholds correspond to the minimum considered by GATK during variant calling. We denote this interval “total callable” (see Table 1). We further defined the “total callable exome” (Table 1) as the intersection of this interval with the coding portion of the exome, according to RefSeq hg19 gene definitions (see previous section).
For the overall de novo mutation rate (i.e., coding plus non-coding variants), we considered all variants identified within the total callable (exome + other) interval. For coding mutation rate (e.g., Table 2; Figures 2A, 2B, 3A, and 3B), we applied the same approach with the total callable exome. This strategy normalizes for differences in capture array design and coverage distribution across the exome and precisely estimates coding mutation rate.
To plot mutation rates per cohort (e.g., Figures 2 and 3), we determined the mutation rate per individual (number of variants divided by total callable exome), and then determined the mean mutation rate per cohort. We provided one vector of data, corresponding to the individual mutation rates, to the t.test function in R to estimate the 95% confidence interval.
Alternatively, we can estimate an overall rate per bp as the number of de novo variants of a particular class observed cohort-wide divided by the number of callable bp assessed cohort-wide. These mutation rates were very similar to the mean rates estimated per bp (Table S3).
Estimation of Mutation Rate per Child
We estimated the theoretical rate of coding de novo variants per child by multiplying the per bp mutation rate by the size of the RefSeq hg19 coding exome (33,828,798 bp; see “Definition of Coding Portion of Exome”). We then determined the mean mutation rate per cohort. Again, we estimated the 95% CI in R using the t.test function with one vector of data corresponding to the individual mutation rates. We obtained an identical estimate by simply multiplying the mean mutation rate per bp by the size of the coding exome.
Rate Ratio Test
We compared de novo mutation rates per base pair in R using a one-sided rate ratio test. Essentially, the rate ratio test compares the number of variants observed between two cohorts while using the number of base pairs assessed as a denominator. Therefore, for this test we utilized the total number of variants observed across each cohort, and the total callable bp assessed across each cohort. We used this general format in R:
Where we set x, the number of events, to a vector of length two, containing the number of proband variants and the number of sibling variants; T, the event counts, to a vector of length two, containing the number of callable bp assessed in probands and in siblings. This function estimates the rate ratio, along with 95% CI.
Chi-square Analysis of Deviance
We utilized a chi-square analysis of deviance test to determine whether the mutation rates per bp differed between the TIC Genetics, the TSAICG, and the SSC Sibling trios. We first conducted a poisson regression with the glm function in R and then utilized the anova function to test the resulting model.
The Poisson regression utilized the number of passing de novos of a particular class as the response variable, with cohort as the predictor, and callable bp as the offset.
We then passed the model to the anova function and extracted the p value for cohort:
Binomial Exact Test
The binomial exact test has been used in many whole exome sequencing studies to assess the burden of de novo variants in cases versus controls (e.g., Iossifov et al., 2012, 2014; Sanders et al., 2012). We therefore performed this test as a comparison to the rate ratio test conducted in the main text. We utilized the R function binom.test, and conducted a one-sided test with ‘hypothesis = “greater”’. We set the test up to compare the number of observed de novo variants of a particular class in probands versus siblings, accounting for the number of samples in each cohort by estimating the “probability of success” based on the proportion of samples that were TD probands versus controls. Hence, we used this general format in R:
Where we set x, the number of successes, to the number of proband variants; n, the number of trials, to the total number of proband and sibling variants; and p, the hypothesized probability of success, to the fraction of individuals that are probands (numberProbands=numberProbands + numberSiblings).
Poisson Regression
We also performed a Poisson regression to control for factors influencing de novo mutation rate and detection, such as paternal age and sequencing coverage (Iossifov et al., 2014; Kong et al., 2012b; O’Roak et al., 2012; Sanders et al., 2012), respectively. We used the Akaike information criterion (AIC), implemented in R, to assess the relative quality of different Poisson models for predicting the number of de novo coding variants. During model selection, we assessed potential covariates versus the response variable of coding de novo mutation rate in SSC control trios, and without including affected status as a covariate. We chose to look in the SSC trios only, because we observed that most batch effects observed across the cohorts were strongly correlated with phenotype status. However, repeating these steps across all of the cohorts resulted in the same final model (not shown). We determined that paternal age, sequencing coverage (percent of exome at 2× coverage), sequencing coverage uniformity (fold 80 base penalty), and heterozygous SNP quality provided the best model. Additionally, however, we reasoned that the number of de novo synonymous mutations per individual could potentially control for additional batch effects affecting the rate of de novo variant detection. Indeed, when we included the number of de novo synonymous variants, along with the aforementioned covariates, in a Poisson regression to predict the number of de novo nonsynonymous mutations (we chose nonsynonymous because coding mutations include the synonymous mutations), we observed a stronger model (better AIC) than excluding de novo synonymous variants. We used the size of the callable coding exome as an offset because each base pair represents an opportunity for a de novo variant. Therefore, the final model to estimate the rate ratios, confidence intervals, and p values for association was:
Because of the inclusion of the number of de novo synonymous mutations in the model, we did not estimate the rate ratios for de novo synonymous mutations, as was done with the other methods. We conducted regression analyses in R using the glm function.
Additionally, we utilized the Bayesian information criterion (BIC) for model selection, implemented in R using the bic.glm function from the BMA package. Based on the BIC, the best model included percent of bases ≥ 2× and maternal age only. However, we chose to utilize all of the covariates identified with the AIC because we felt it was important to include additional covariates that may be more relevant when considering other classes of de novo mutation. Nonetheless, Poisson regression with the simplified model above results in consistent results (not shown).
Fisher Exact Test
As a third, independent method to estimate burden of de novo variants in TD probands versus SSC Siblings, we performed a Fisher exact test that “normalizes” by the number of de novo synonymous mutations (Sanders et al., 2012). We chose this method because we hypothesized that the number of de novo synonymous mutations per individual should be unrelated to phenotypic status, and therefore, could potentially control for batch effects affecting the rate of de novo variant detection. For this analysis, we constructed a 2×2 contingency table from the counts of de novo mutations of a particular class in probands and SSC sibling controls, and the counts of de novo synonymous mutations in probands and SSC sibling controls. A one-sided Fisher exact test then estimated the odds ratio and p value for each class of mutation. For example, for de novo LGD variants in the combined cohort the contingency table is:
|
| ||
| TD Probands | SSC Siblings | |
|
| ||
| De novo LGD | 39 | 22 |
|
| ||
| De novo synonymous | 111 | 134 |
|
| ||
We then conducted the Fisher exact test in R as:
This estimates an odds ratio of 2.1 with p = 0.0068 for LGD variants.
Percent of Variants Contributing Risk
As mentioned previously (see “Estimate of Mutation Rate per Child”), we estimated the theoretical de novo coding mutation rate per child based on the observed mutation rate per bp and the number of bp in the RefSeq hg19 coding exome (Table 3). We then estimated two parameters: (1) the percent of cases that have a de novo variant contributing TD risk, and (2) the fraction of observed proband de novo variants that contribute to TD risk.
The percent of cases that havea de novo variant contributing TDrisk. In order to calculate this difference, while also estimating 95% CI’s, we leveraged the t.test function in R to determine the mean difference between the rate in TD probands and SSC controls. We input two vectors of data, one with mutation rate. This function also outputs 95% CI’s for this difference. See Table 3 for results.
The fraction of observed proband de novo variants that contribute TD risk. We estimated this parameter based on the results in (1): we divided the difference in theoretical rate (percent of cases that have a de novo variant contributing risk) calculated above by the theoretical rate in probands. To estimate the 95% CI’s, we utilized the upper and lower bounds of the 95% CI calculated for (1) in the same formula.
TADA
The enrichment of de novo damaging (LGD + Mis3) variants in TD, as well as the observation of 5 genes with multiple de novo damaging variants raises the possibility that this class of variant targets a set of genes that mediates TD risk. We tested this hypothesis with the transmitted and de novo association (TADA) test, a Bayesian model that can effectively combine data from de novo variants, inherited variants in families, and standing variants in the population (via case-control cohorts) to assess the association of specific genes with TD risk. In this study, we elected not to include rare inherited exome variants because we have not yet associated this class of variant with TD risk, as expected given their small effect size. Instead, we used a specialized version of TADA that analyzes only the de novo variants from exome sequencing data, called TADA-Denovo (He et al., 2013).
The TADA-Denovo test considers two types of variants, de novo LGD and de novo severe missense (those predicted by PolyPhen2-HDIV to be “probably damaging” to protein function, abbreviated as “Mis3”; Adzhubei et al., 2010, 2013). The main input is the number of de novo LGD and number of de novo Mis3 variants per gene. Additionally, we utilized the included mutation rates (mu) for all human genes, which are based on Sanders et al. (2012). The test analyzes each of these event types separately and then combines the evidence in a Bayesian fashion, weighting each type of variant differently.
To compute the Bayes factors and p values, TADA-Denovo requires the following parameters:
ntrio: the number parent-child trios passing QC (484)
mu: per gene mutation probabilities for each class of de novo variant assessed; in our case, de novo LGD and de novo Mis3 variants. The per gene probability of any mutation, from Sanders et al. (2012), was modified to reflect the chance of each class of mutation by the following steps:
- The fraction of de novo variants that are LGD was estimated as:
- The fraction of de novo variants that are Mis3 was estimated as:
- Next, the per gene overall de novo mutation probability was multiplied by each of these fractions to estimate the per gene probabilities of each class of mutation:
gamma.mean.dn (γ): the average relative risk (γ) is related to the fold-enrichment (λ, relative to random expectation) and the fraction of causal genes (π) by the following equation: π (γ − 1) = λ − 1.
- We calculated the fraction of causal genes (π) as:
- Fold-enrichment (λ) for LGD as:
- Fold-enrichment (λ) for Mis3 as:
- Solving for the relative risk (γ):
Using these parameters, TADA-Denovo calculates the Bayes factors of all input genes. Next, it computes the p value for each gene by generating random mutational data, based on each gene’s specified mutation rate, to obtain a null distribution of Bayes factors. We used 1,000 samplings of de novo variants in each gene to determine null distributions. Finally, TADA-Denovo calculates a false discovery rate (FDR) q-value for each gene using a Bayesian “direct posterior approach.” A low q-value represents strong evidence for TD association.
Estimation of Number of TS Genes
Maximum Likelihood Estimation
In the 484 TD trios passing quality control, we observed 199 damaging de novo variants. However, seven of these variants did not pass validation, leaving 192 damaging variants. Within these, four genes had two damaging de novo variants, and one gene had ≥ 3 damaging variants. Therefore, to estimate the number of genes contributing risk to TD, we leveraged a maximum likelihood estimation (MLE) procedure to identify the number of genes (we tested between 1:2500) that best fits these observations (Homsy et al., 2015).
For each possible number of risk genes, from 1 to 2,500, we simulated 192 variants. We repeated this 50,000 times. In each permutation, we randomly selected risk genes and randomly assigned a percentage of variants to the risk genes and the rest of the variants to the non-risk genes. We based these percentages on the fraction of damaging variants estimated to carry risk (27.3%; see below). We utilized the per gene probabilities of mutation from TADA, which are weighted by gene size and GC content (He et al., 2013; Sanders et al., 2012). We then counted the number of both risk and non-risk genes that harbored multiple variants and recorded when the number of genes with two variants and the number of genes with three or more variants in the simulated data matched the number observed in our study (4 and 1, respectively). We then calculated the frequency of concordance between the permuted data and the observed data. Finally, we determined the MLE by plotting the smoothed trend line of frequency versus number of risk genes using local polynomial regression fitting (loess in R) and used the predict function in R to estimate the MLE of the number of risk genes (420 genes).
To estimate the fraction of damaging mutations carrying risk (E), we calculated M1 and M2, the observed rates of de novo damaging variants per TD proband and per SSC sibling control, respectively. More specifically, M1 = (199/484) and M2 = (180/602). We then estimated E as:
For this calculation, we calculated E from unconfirmed counts (199 proband mutations, 180 SSC sibling mutations), as confirmation of the sibling de novo mutations was not attempted and we reasoned that both populations should have a similar rate of false positives.
We performed a similar estimate in the main text: we divided the difference in theoretical rate between probands and SSC sibling controls by the theoretical rate in probands. Using this approach, we estimated that 51.3% (95% CI 13.7 – 89.0%) of de novo LGD and 22.9% (95% CI 4.8 – 41.0%) of de novo damaging variants contribute risk to TD. However, we chose to estimate E using the first method to match work done by (Homsy et al., 2015).
‘Unseen Species’
As has been done previously in ASD (Sanders et al., 2012), we estimated the number of risk genes (C) based on the framework developed for the ‘unseen’ species problem. This estimate requires four parameters: (1) number of risk associated variants (d), (2) total number of observed risk genes (c), (3) number of genes mutated once (c1), and (4) probability that newly added variant hits a previously mutated gene (u).
d was estimated as the number of damaging variants observed (199) minus the expected number of damaging variants, where the expected number of variants was estimated from the SSC control trios. More specifically, we scaled the observed number of damaging variants in the 602 control trios (180) to the expected number in 484 trios (180 *(484/602) = 145). Therefore, we estimated d as 199−145 = 54.
We estimated c, the number of observed risk genes, as d minus the number of recurrent variants (11) plus the number of genes with recurrent variants (5). d is the total number of risk associated variants, so we subtracted the number of de novo variants present in recurrently mutated genes to account for the multiple variants in the same gene. We then added back the number of genes with recurrent variants to obtain the final estimate of c (essentially, we removed the extra variants within genes with multiple de novo variants). Therefore, c was equal to 54 − 11 + 5 = 48.
We estimated c1 as c minus the number of recurrent genes: c1 = 48 − 5 = 43
We estimated u as 1 − (c1/d) = 1 − (43/54) = 0:2037
Finally, combining all of these parameters, we estimated the total number of risk genes (C) to be approximately 447:
To calculate the 95% CI for this estimate, we repeated the analysis, substituting in the upper and lower 95% CI’s for the expected number of damaging variants. More specifically, recall that we scaled the observed number of damaging variants in the 602 control trios (180) down to the expected number in 484 trios (180 * (484/602) = 145). Thus, we similarly estimated the upper limit of the expected number of damaging variants by determining the upper CI of the number of de novo damaging variants per child in the SSC (0.3443 variants per child) using the t.test function in R, and multiplying this estimate by 602 trios and rounding up. For example, we determined the upper estimate of the number of expected damaging variants as:
Therefore, we estimated d as 199−167 = 32.
We similarly determined the lower estimate of the number of expected damagaing variants as:
Therefore, we estimated d as 199−123 = 76
Utilizing the same formula as above, we then estimated the 95% CI as 136.7–932.7.
Estimation of Gene Discovery by Cohort Size
After estimating the number of genes involved in TD risk, we utilized this number to predict the gene discovery yield, as additional TD trios are whole-exome sequenced. We fixed the gene number at 420, and varied the cohort size. Therefore, we calculated the number of variants in each iteration based on the observed mutation rate in probands (based on 199 de novo damaging mutations – 7 that failed confirmation = 192 variants). As was done in the MLE, we randomly selected TD risk genes, and then assigned a fraction of these variants to TD risk genes and the remaining fraction to non-TD risk genes (see below for estimation of fractions). We performed 10,000 permutations at each cohort size, and randomly generated LGD and Mis3 variants separately, using their respective rates and per gene likelihoods. We then combined these data and each permutation was run through the TADA de novo algorithm with the same parameters used above for the observed data to assess the per gene q-values. We then recorded the number of probable genes (q < 0.3) and high confidence genes (q < 0.1) that were observed at each cohort size and plotted the smoothed trend line using local polynomial regression fitting (loess in R). The regression model also predicted the number of genes identified at a given number of trios.
To estimate the fraction of LGD and Mis3 variants carrying risk (ELGD and EMis3), we calculated M1 and M2, the observed rates of de novo LGD or Mis3 variants per TD proband and per SSC sibling control, respectively. More specifically, for LGD variants, M1 = (39/484) and M2 = (22/602), and therefore, we estimated E as: E = ((M1−M2)/M1) = ((39/484) − (22/602)/(39/484)) = 0.546. For Mis3 variants, M1 = (303/484) and M2 = (158/602), and therefore, we estimated E as: E = ((M1−M2)/M1) = ((303/484) − (158/602)/(303/484) = 0.206.
As was previously done, we calculated E from unconfirmed counts because we did not attempt confirmation of the sibling de novo mutations and we reasoned that both populations should have a similar rate of false positives. Moreover, in order to keep these calculations consistent with the maximum likelihood estimate and TADA, we did not utilize the theoretical rate per individual, as we did in the main text.
QUANTIFICATION AND STATISTICAL ANALYSIS
We conducted all statistical analyses in R (v ≥ 3.31). We have made the R scripts used in these analyses available on bitbucket at https://bitbucket.org/willseylab/tourette_phase1. Where appropriate, we present data as mean ± the 95% confidence interval (CI). We estimate mean and 95% CI with the t.test function. We describe the value of n in the main text and/or in Tables 1, 2, and 3, and n stands for number of samples (trios), number of base pairs, or number of variants as indicated. We conducted the primary burden analyses with a rate ratio test, using the poisson.test function, and comparing, across two cohorts, the number of de novo variants per the number of callable bp assessed. When comparing TD probands versus SSC controls, we utilized a one-sided test (alternative = “greater”), given the prior evidence for the role of de novo mutations in TD and other neurodevelopmental disorders. However, we compared rates between TD cohorts with a two-sided test because we did not expect these rates to differ. In secondary burden analyses, one-sided binomial exact tests (binom.test) and Fisher’s exact tests (fisher.test), as well as a Poisson regression (see “Poisson Regression”) also assessed significance.
We did not correct p values for multiple comparisons because our primary hypotheses focused on de novo LGD variants, followed by secondary characterization of other variant classes. We considered a p value < 0.05 statistically significant and we list individual p values in the main text, Figures 2, 3, 4, and 5, and Tables 1, 2, and 3.
As described above in the STAR Methods, we estimated p- and q-values for individual association with TD risk with the algorithm, TADA, which is described in detail in He et al. (2013).
DATA AND SOFTWARE AVAILABILITY
Data
We have deposited aligned whole exome sequencing data (.bam files) in the Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra/) under accession IDs SUB2101648 (Tic Genetics data) and YYY (TSAICG data).
Software
Perl, Shell, and R code used to process these data and complete statistical analyses are available on bitbucket at https://bitbucket.org/willseylab/tourette_phase1. Our in-house primer design software that generated primer sets for variant confirmations is located at http://primerdesign.willseylab.com/.
ADDITIONAL RESOURCES
Description: url. TIC Genetics website. https://tic-genetics.org/
Description: url. TSAICG website. https://www.findtsgene.org
Description: url. Bitbucket repository. https://bitbucket.org/willseylab/tourette_phase1
Supplementary Material
Highlights.
Exome sequencing links damaging de novo sequence variants with Tourette disorder
De novo variants in 420 genes contribute risk in 12% of clinical cases
Recurrent de novo variants identify one high-confidence TD risk gene: WWC1
Gene discovery will exponentially increase as additional cohorts are sequenced
Acknowledgments
We wish to thank the families who have participated in and contributed to this study. We also thank the NIMH Repository and Genomics Resource (U24MH068457 to J.A.T.) at RUCDR Infinite Biologics for transforming cell lines and providing DNA samples and the Simons Foundation for providing genotyping and sequencing data used as controls in this study. This study was supported by grants from the National Institute of Mental Health (R01MH092290 to L.W. Brown; R01MH092291 to S. Kuperman; R01MH092292 to B.J. Coffey; R01MH092293 to G.A.H. and J.A.T.; R01MH092513 to S.H. Zinner; R01MH092516 to D.E. Grice; R01MH092520 to D.L. Gilbert; R01MH092289 to M.W.S.; K08MH099424 to T.V.F.; K23MH085057 to J.M.S.), the National Institute of Neurological Disorders and Stroke (U01NS40024-09S1 and K02 NS085048 to J.M.S.), the Harvard Clinical and Translational Science Center (UL TR001102to J.M.S.), the Tourette Association of America (to C.A.M. and J.M.S.), the New Jersey Center for Tourette Syndrome and Associated Disorders (NJCTS; to G.A.H. and J.A.T.), and the Overlook International Fund (to M.W.S.). We are also grateful to the NJCTS for facilitating the inception and organization of the TIC Genetics study. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was additionally supported by grants from Spain (to P. Mir): the Instituto de Salud Carlos III (PI10/01674, PI13/01461), the Con-sejería de Economía, Innovación, Ciencia y Empresa de la Junta de Andalucía (CVI-02526, CTS-7685), the Consejería de Salud y Bienestar Social de la Junta de Andalucía (PI-0741/2010, PI-0437-2012, and PI-0471-2013), the Sociedad Andaluza de Neurología, the Fundación Alicia Koplowitz, the Fundación Mutua Madrileña, and the Jaques and Gloria Gossweiler Foundation; grants from Germany (to A. Münchau): Deutsche Forschungsgemeinschaft (DFG: MU 1692/3-1, MU 1692/4-1, and project C5 of the SFB 936). This research was supported by the NIHR Great Ormond Street Hospital Biomedical Research Centre (GOSH BRC). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
This research was also supported in part by an Informatics Starter Grant from the PhRMA Foundation (to Y. Bromberg). We thank all of the individuals involved in recruitment and assessment of the subjects reported in this study: Denmark: Nikoline Frost and Heidi B. Biernat (Copenhagen); Germany: Stephanie Enghardt, Yvonne Friedrich, Christiane Michel (Dresden), Jenny Schmalfeld (Lübeck), Hanife Kling, and Ariane Saccarello (Ulm); Spain: María T. Cáceres, Fátima Carrillo, Marta Correa, Pilar Gómez-Garre, and Laura Vargas (Sevilla); the Netherlands: Vivian op de Beek (Amsterdam), Marieke Mes-schendorp (Groningen), Nicole Driessen, Nadine Schalk (Nijmegen), Noor Tromp (Alkmaar), Els van den Ban (Utrecht), Jolanda Blom, Rudi Bruggemans, and MariAnne Overdijk (Barendrecht); UK: Anup Kharod (London GOSH); USA: Sarah Jacobson (Cincinnati), Angie Cookman (Iowa City), Zoey Shaw (Mount Sinai/NKI), Julia Brillante, Daniela B. Colognori, Joseph Conerty, Alycia Davis, Carolyn Spiro, Donna Tischfield (Rutgers), Shannon Granillo, and JD Sandhu (Seattle Children’s). Finally, we thank Adife Gulhan Ercan-Sencicek (Yale), Xin He (University of Chicago), Kathryn Roeder (CMU), Bernie Devlin (University of Pittsburgh), Helen Willsey (UCSF), the Willsey lab (UCSF), and all who may not have been mentioned.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, A.J.W., T.V.F., S.J.S., B.M.N., C.A.M., J.A.T., J.M.S., M.W.S., and G.A.H.; Methodology, A.J.W., T.V.F., D.Y., A.Y.H., K.E.S., B.M.N., J.M.S., and M.W.S.; Software, A.J.W., J.D.M., and L.S.; Validation, A.J.W., T.V.F., D.Y., A.Y.H., P.R., G.C., and J.M.S.; Formal Analysis, A.J.W., T.V.F., J.D.M., and S.D.; Investigation, A.J.W., T.V.F., J.D.M., A.Y.H., P.R., L.S., and J.M.S.; Resources, TIC Genetics, TSAICG, C.A.M., J.A.T., J.M.S., M.W.S., and G.A.H.; Data Curation, A.J.W., T.V.F., D.Y., J.D.M., and J.M.S.; Writing – Original Draft, A.J.W., T.V.F., and M.W.S.; Writing – Review & Editing, A.J.W., T.V.F., D.Y., A.Y.H., TIC Genetics, TSAICG, G.C., C.A.M., J.A.T., J.M.S., M.W.S., and G.A.H.; Visualization, A.J.W.; Supervision, C.A.M., J.A.T., J.M.S., M.W.S., and G.A.H.; Project Administration, J.A.T., J.M.S., M.W.S., and G.A.H.; Funding Acquisition, C.A.M., J.A.T., J.M.S., M.W.S., and G.A.H.
CONSORTIA
The members of the Tourette International Collaborative Genetics (TIC Genetics) consortium are: Mohamed Abdulkadir, Julia Bohnenpoll, Yana Bromberg, Lawrence W. Brown, Keun-Ah Cheon, Barbara J. Coffey, Li Deng, Andrea Dietrich, Shan Dong, Lonneke Elzerman, Thomas V. Fernandez, Odette Fründt, Blanca Garcia-Delgar, Erika Gedvilaite, Donald L. Gilbert, Dorothy E. Grice, Julie Hagstrøm, Tammy Hedderly, Gary A. Heiman, Isobel Heyman, Pieter J. Hoekstra, Hyun Ju Hong, Chaim Huyser, Laura Ibanez-Gomez, Young Key Kim, Young-Shin Kim, Robert A. King, Yun-Joo Koh, Sodahm Kook, Samuel Kuperman, Andreas Lamerz, Bennett Leventhal, Andrea G. Ludolph, Claudia Lühr da Silva, Marcos Madruga-Garrido, Jeffrey D. Mandell, Athanasios Maras, Pablo Mir, Astrid Morer, Alexander Münchau, Tara L. Murphy, Cara Nasello, Thaïra J. C. Openneer, Kerstin J. Plessen, Petra Richer, Veit Roessner, Stephan Sanders, Eun-Young Shin, Deborah A. Sival, Louw Smith, Dong-Ho Song, Jungeun Song, Matthew W. State, Anne Marie Stolte, Nawei Sun, Jay A. Tischfield, Jennifer Tübing, Frank Visscher, Michael F. Walker, Sina Wanderer, Shuoguo Wang, A. Jeremy Willsey, Martin Woods, Jinchuan Xing, Yeting Zhang, Anbo Zhou, and Samuel H. Zinner. The members of the Tourette Syndrome Association International Consortium for Genetics (TSAICG) are: Cathy L. Barr, James R. Batterson, Cheston Berlin, Ruth D. Bruun, Cathy L. Budman, Danielle C. Cath, Sylvain Chouinard, Giovanni Coppola, Nancy J. Cox, Sabrina Darrow, Lea K. Davis, Yves Dion, Nelson B. Freimer, Marco A. Grados, Matthew E. Hirschtritt, Alden Y. Huang, Cornelia Illmann, Robert A. King, Roger Kurlan, James F. Leckman, Gholson J. Lyon, Irene A. Malaty, Carol A. Mathews, William M. MaMahon, Benjamin M. Neale, Michael S. Okun, Lisa Osiecki, David L. Pauls, Danielle Posthuma, Vasily Ramensky, Mary M. Robertson, Guy A. Rouleau, Paul Sandor, Jeremiah M. Scharf, Harvey S. Singer, Jan Smit, Jae-Hoon Sul, and Dongmei Yu.
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures, four tables, and consortia member names and can be found with this article online at http://dx.doi.org/10.1016/j.neuron.2017.04.024.
A video abstract is available at http://dx.doi.org/10.1016/j.neuron.2017.04.024#mmc6.
References
- Abelson JF, Kwan KY, O’Roak BJ, Baek DY, Stillman AA, Morgan TM, Mathews CA, Pauls DL, Rasin MR, Gunel M, et al. Sequence variants in SLITRK1 are associated with Tourette’s syndrome. Science. 2005;310:317–320. doi: 10.1126/science.1116502. [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013 doi: 10.1002/0471142905.hg0720s76. Chapter 7, Unit 7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, et al. Epi4K Consortium; Epilepsy Phenome/Genome Project De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. DSM-IV-TR. American Psychiatric Association; 2000. [Google Scholar]
- Bilgüvar K, Oztürk AK, Louvi A, Kwan KY, Choi M, Tatli B, Yalnizoǧlu D, Tüysüz B, Caǧlayan AO, Gökben S, et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature. 2010;467:207–210. doi: 10.1038/nature09327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann W. Ein fall von symmetrischer monodaktylie durch Ulnadefekt, mit symmetrischer flughautbildung in den ellenbeugen, sowie anderen abnormitaten (zwerghaftogkeit, halsrippen, behaarung) Jarb Kinder Phys Erzie. 1916;84:225–235. [Google Scholar]
- Browne HA, Hansen SN, Buxbaum JD, Gair SL, Nissen JB, Nikolajsen KH, Schendel DE, Reichenberg A, Parner ET, Grice DE. Familial clustering of tic disorders and obsessive-compulsive disorder. JAMA Psychiatry. 2015;72:359–366. doi: 10.1001/jamapsychiatry.2014.2656. [DOI] [PubMed] [Google Scholar]
- Burd L, Freeman RD, Klug MG, Kerbeshian J. Tourette Syndrome and learning disabilities. BMC Pediatr. 2005;5:34. doi: 10.1186/1471-2431-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büther K, Plaas C, Barnekow A, Kremerskothen J. KIBRA is a novel substrate for protein kinase Czeta. Biochem Biophys Res Commun. 2004;317:703–707. doi: 10.1016/j.bbrc.2004.03.107. [DOI] [PubMed] [Google Scholar]
- Castelletti F, Donadelli R, Banterla F, Hildebrandt F, Zipfel PF, Bresin E, Otto E, Skerka C, Renieri A, Todeschini M, et al. Mutations in FN1 cause glomerulopathy with fibronectin deposits. Proc Natl Acad Sci USA. 2008;105:2538–2543. doi: 10.1073/pnas.0707730105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanna AE, Servo S, Monaco F, Robertson MM. The behavioral spectrum of Gilles de la Tourette syndrome. J Neuropsychiatry Clin Neurosci. 2009;21:13–23. doi: 10.1176/jnp.2009.21.1.13. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (CDC) Prevalence of diagnosed Tourette syndrome in persons aged 6–17 years - United States, 2007. MMWR Morb Mortal Wkly Rep. 2009;58:581–585. [PubMed] [Google Scholar]
- Chai G, Zhou L, Manto M, Helmbacher F, Clotman F, Goffinet AM, Tissir F. Celsr3 is required in motor neurons to steer their axons in the hindlimb. Nat Neurosci. 2014;17:1171–1179. doi: 10.1038/nn.3784. [DOI] [PubMed] [Google Scholar]
- Chai G, Goffinet AM, Tissir F. Celsr3 and Fzd3 in axon guidance. Int J Biochem Cell Biol. 2015;64:11–14. doi: 10.1016/j.biocel.2015.03.013. [DOI] [PubMed] [Google Scholar]
- De Lange C. Sur un type nouveau de dégénération (typus Amstelodamensis) Arch Med Enfants. 1933;36:713–719. [Google Scholar]
- de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. DDD Study; Homozygosity Mapping Collaborative for Autism; UK10K Consortium Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519:223–228. doi: 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–438. doi: 10.1038/nature21062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich A, Fernandez TV, King RA, State MW, Tischfield JA, Hoekstra PJ, Heiman GA, Group, T.I.C.G.C.; TIC Genetics Collaborative Group The Tourette International Collaborative Genetics (TIC Genetics) study, finding the genes causing Tourette syndrome: objectives and methods. Eur Child Adolesc Psychiatry. 2015;24:141–151. doi: 10.1007/s00787-014-0543-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- do Rosário MC, Miguel Filho EC. Obsessive-compulsive disorder and Tourette syndrome: is there a relationship? Sao Paulo Med J. 1997;115:1410–1411. doi: 10.1590/s1516-31801997000200008. [DOI] [PubMed] [Google Scholar]
- Dong S, Walker MF, Carriero NJ, DiCola M, Willsey AJ, Ye AY, Waqar Z, Gonzalez LE, Overton JD, Frahm S, et al. De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Rep. 2014;9:16–23. doi: 10.1016/j.celrep.2014.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi4K Consortium. De novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am J Hum Genet. 2016;99:287–298. doi: 10.1016/j.ajhg.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan-Sencicek AG, Stillman AA, Ghosh AK, Bilguvar K, O’Roak BJ, Mason CE, Abbott T, Gupta A, King RA, Pauls DL, et al. L-histidine decarboxylase and Tourette’s syndrome. N Engl J Med. 2010;362:1901–1908. doi: 10.1056/NEJMoa0907006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EuroEPINOMICS-RES Consortium; Epilepsy Phenome/Genome Project; Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet. 2014;95:360–370. doi: 10.1016/j.ajhg.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Han Q, Zhou L. Planar cell polarity genes, Celsr1–3, in neural development. Neurosci Bull. 2012;28:309–315. doi: 10.1007/s12264-012-1232-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenstermaker AG, Prasad AA, Bechara A, Adolfs Y, Tissir F, Goffinet A, Zou Y, Pasterkamp RJ. Wnt/planar cell polarity signaling controls the anterior-posterior organization of monoaminergic axons in the brainstem. J Neurosci. 2010;30:16053–16064. doi: 10.1523/JNEUROSCI.4508-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez TV, Sanders SJ, Yurkiewicz IR, Ercan-Sencicek AG, Kim YS, Fishman DO, Raubeson MJ, Song Y, Yasuno K, Ho WSC, et al. Rare copy number variants in tourette syndrome disrupt genes in histaminergic pathways and overlap with autism. Biol Psychiatry. 2012;71:392–402. doi: 10.1016/j.biopsych.2011.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach GD, Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010a;68:192–195. doi: 10.1016/j.neuron.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Fischbach GD, Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010b;68:192–195. doi: 10.1016/j.neuron.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123:4195–4200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119:1079–1091. doi: 10.1242/dev.119.4.1079. [DOI] [PubMed] [Google Scholar]
- Ghanizadeh A, Mosallaei S. Psychiatric disorders and behavioral problems in children and adolescents with Tourette syndrome. Brain Dev. 2009;31:15–19. doi: 10.1016/j.braindev.2008.03.010. [DOI] [PubMed] [Google Scholar]
- He X, Sanders SJ, Liu L, De Rubeis S, Lim ET, Sutcliffe JS, Schellenberg GD, Gibbs RA, Daly MJ, Buxbaum JD, et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 2013;9:e1003671. doi: 10.1371/journal.pgen.1003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschtritt ME, Lee PC, Pauls DL, Dion Y, Grados MA, Illmann C, King RA, Sandor P, McMahon WM, Lyon GJ, et al. Tourette Syndrome Association International Consortium for Genetics Lifetime prevalence, age of risk, and genetic relationships of comorbid psychiatric disorders in Tourette syndrome. JAMA Psychiatry. 2015;72:325–333. doi: 10.1001/jamapsychiatry.2014.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350:1262–1266. doi: 10.1126/science.aac9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hounie AG, do Rosario-Campos MC, Diniz JB, Shavitt RG, Ferrão YA, Lopes AC, Mercadante MT, Busatto GF, Miguel EC. Obsessive-compulsive disorder in Tourette syndrome. Adv Neurol. 2006;99:22–38. [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, Sousa AM, Pletikos M, Meyer KA, Sedmak G, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapushesky M, Adamusiak T, Burdett T, Culhane A, Farne A, Filippov A, Holloway E, Klebanov A, Kryvych N, Kurbatova N, et al. Gene Expression Atlas update—a value-added database of microarray and sequencing-based functional genomics experiments. Nucleic Acids Res. 2012;40:D1077–D1081. doi: 10.1093/nar/gkr913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur M, Mehta D, Noon SE, Deardorff MA, Zhang Z, Krantz ID. NIPBL expression levels in CdLS probands as a predictor of mutation type and phenotypic severity. Am J Med Genet C Semin Med Genet. 2016;172:163–170. doi: 10.1002/ajmg.c.31495. [DOI] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012a;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012b;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremerskothen J, Plaas C, Büther K, Finger I, Veltel S, Matanis T, Liedtke T, Barnekow A. Characterization of KIBRA, a novel WW domain-containing protein. Biochem Biophys Res Commun. 2003;300:862–867. doi: 10.1016/s0006-291x(02)02945-5. [DOI] [PubMed] [Google Scholar]
- Lawson-Yuen A, Saldivar JS, Sommer S, Picker J. Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur J Hum Genet. 2008;16:614–618. doi: 10.1038/sj.ejhg.5202006. [DOI] [PubMed] [Google Scholar]
- Leckman JF. Phenomenology of tics and natural history of tic disorders. Brain Dev. 2003;25(Suppl 1):S24–S28. doi: 10.1016/s0387-7604(03)90004-0. [DOI] [PubMed] [Google Scholar]
- Leppa VM, Kravitz SN, Martin CL, Andrieux J, Le Caignec C, Martin-Coignard D, DyBuncio C, Sanders SJ, Lowe JK, Cantor RM, Geschwind DH. Rare inherited and de novo CNVs reveal complex contributions to ASD risk in multiplex families. Am J Hum Genet. 2016;99:540–554. doi: 10.1016/j.ajhg.2016.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochner C, Hemmings SM, Kinnear CJ, Niehaus DJ, Nel DG, Corfield VA, Moolman-Smook JC, Seedat S, Stein DJ. Cluster analysis of obsessive-compulsive spectrum disorders in patients with obsessive-compulsive disorder: clinical and genetic correlates. Compr Psychiatry. 2005;46:14–19. doi: 10.1016/j.comppsych.2004.07.020. [DOI] [PubMed] [Google Scholar]
- Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223–1241. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannini L, Cucco F, Quarantotti V, Krantz ID, Musio A. Mutation spectrum and genotype-phenotype correlation in Cornelia de Lange syndrome. Hum Mutat. 2013;34:1589–1596. doi: 10.1002/humu.22430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mataix-Cols D, Isomura K, Pérez-Vigil A, Chang Z, Rück C, Larsson KJ, Leckman JF, Serlachius E, Larsson H, Lichtenstein P. Familial risks of Tourette syndrome and chronic tic disorders. A population-based cohort study JAMA Psychiatry. 2015;72:787–793. doi: 10.1001/jamapsychiatry.2015.0627. [DOI] [PubMed] [Google Scholar]
- McGrath LM, Yu D, Marshall C, Davis LK, Thiruvahindrapuram B, Li B, Cappi C, Gerber G, Wolf A, Schroeder FA, et al. Copy number variation in obsessive-compulsive disorder and tourette syndrome: a cross-disorder study. J Am Acad Child Adolesc Psychiatry. 2014;53:910–919. doi: 10.1016/j.jaac.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya PR, Wendland JR, Rubenstein LM, Timpano KR, Heiman GA, Tischfield JA, King RA, Andrews AM, Ramamoorthy S, McMahon FJ, Murphy DL. Common and rare alleles of the serotonin transporter gene, SLC6A4, associated with Tourette’s disorder. Mov Disord. 2013;28:1263–1270. doi: 10.1002/mds.25460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder PA, Huisman SA, Hennekam RC, Oliver C, van Balkom ID, Piening S. Behaviour in Cornelia de Lange syndrome: a systematic review. Dev Med Child Neurol. 2017;59:361–366. doi: 10.1111/dmcn.13361. [DOI] [PubMed] [Google Scholar]
- Nag A, Bochukova EG, Kremeyer B, Campbell DD, Muller H, Valencia-Duarte AV, Cardona J, Rivas IC, Mesa SC, Cuartas M, et al. Tourette Syndrome Association International Consortium for Genetics CNV analysis in Tourette syndrome implicates large genomic rearrangements in COL8A1 and NRXN1. PLoS ONE. 2013;8:e59061. doi: 10.1371/journal.pone.0059061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver C, Arron K, Sloneem J, Hall S. Behavioural phenotype of Cornelia de Lange syndrome: case-control study. Br J Psychiatry. 2008;193:466–470. doi: 10.1192/bjp.bp.107.044370. [DOI] [PubMed] [Google Scholar]
- Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115:3861–3863. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, Geschwind DH. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–1021. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauls DL, Cohen DJ, Heimbuch R, Detlor J, Kidd KK. Familial pattern and transmission of Gilles de la Tourette syndrome and multiple tics. Arch Gen Psychiatry. 1981;38:1091–1093. doi: 10.1001/archpsyc.1981.01780350025002. [DOI] [PubMed] [Google Scholar]
- Pauls DL, Raymond CL, Stevenson JM, Leckman JF. A family study of Gilles de la Tourette syndrome. Am J Hum Genet. 1991;48:154–163. [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Tedeschi A, Schmitz J. The cohesin complex and its roles in chromosome biology. Genes Dev. 2008;22:3089–3114. doi: 10.1101/gad.1724308. [DOI] [PubMed] [Google Scholar]
- Petryszak R, Burdett T, Fiorelli B, Fonseca NA, Gonzalez-Porta M, Hastings E, Huber W, Jupp S, Keays M, Kryvych N, et al. Expression Atlas update—a database of gene and transcript expression from microarray- and sequencing-based functional genomics experiments. Nucleic Acids Res. 2014;42:D926–D932. doi: 10.1093/nar/gkt1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryszak R, Keays M, Tang YA, Fonseca NA, Barrera E, Burdett T, Fullgrabe A, Füentes AM, Jupp S, Koskinen S, et al. Expression Atlas update—an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 2016;44(D1):D746–D752. doi: 10.1093/nar/gkv1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price RA, Kidd KK, Cohen DJ, Pauls DL, Leckman JF. A twin study of Tourette syndrome. Arch Gen Psychiatry. 1985;42:815–820. doi: 10.1001/archpsyc.1985.01790310077011. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D. GeneCards: integrating information about genes, proteins and diseases. Trends Genet. 1997;13:163. doi: 10.1016/s0168-9525(97)01103-7. [DOI] [PubMed] [Google Scholar]
- Robertson MM. The prevalence and epidemiology of Gilles de la Tourette syndrome. Part 1: the epidemiological and prevalence studies. J Psychosom Res. 2008;65:461–472. doi: 10.1016/j.jpsychores.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Roessner V, Becker A, Banaschewski T, Freeman RD, Rothenberger A, Tourette Syndrome International Database Consortium Developmental psychopathology of children and adolescents with Tourette syndrome–impact of ADHD. Eur Child Adolesc Psychiatry. 2007;16(Suppl 1):24–35. doi: 10.1007/s00787-007-1004-6. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. Autism Sequencing Consortium Insights into Autism Spectrum Disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87:1215–1233. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf JM, Yu D, Mathews CA, Neale BM, Stewart SE, Fagerness JA, Evans P, Gamazon E, Edlund CK, Service SK, et al. North American Brain Expression Consortium; UK Human Brain Expression Database Genome-wide association study of Tourette’s syndrome. Mol Psychiatry. 2013;18:721–728. doi: 10.1038/mp.2012.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf JM, Miller LL, Gauvin CA, Alabiso J, Mathews CA, Ben-Shlomo Y. Population prevalence of Tourette syndrome: a systematic review and meta-analysis. Mov Disord. 2015;30:221–228. doi: 10.1002/mds.26089. [DOI] [PubMed] [Google Scholar]
- Schneider A, Huentelman MJ, Kremerskothen J, Duning K, Spoelgen R, Nikolich K. KIBRA: a new gateway to learning and memory? Front Aging Neurosci. 2010;2:4. doi: 10.3389/neuro.24.004.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillman AA, Krsnik Z, Sun J, Rasin MR, State MW, Sestan N, Louvi A. Developmentally regulated and evolutionarily conserved expression of SLITRK1 in brain circuits implicated in Tourette syndrome. J Comp Neurol. 2009;513:21–37. doi: 10.1002/cne.21919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram SK, Huq AM, Wilson BJ, Chugani HT. Tourette syndrome is associated with recurrent exonic copy number variants. Neurology. 2010;74:1583–1590. doi: 10.1212/WNL.0b013e3181e0f147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Tourette Syndrome Association International Consortium for Genetics. A complete genome screen in sib pairs affected by Gilles de la Tourette syndrome. Am J Hum Genet. 1999;65:1428–1436. doi: 10.1086/302613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Bar I, Jossin Y, De Backer O, Goffinet AM. Protocadherin Celsr3 is crucial in axonal tract development. Nat Neurosci. 2005;8:451–457. doi: 10.1038/nn1428. [DOI] [PubMed] [Google Scholar]
- The Tourette Syndrome Association International Consortium for Genetics. Genome scan for Tourette disorder in affected-sibling-pair and multi-generational families. Am J Hum Genet. 2007;80:265–272. doi: 10.1086/511052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk AJ, Mathews CA, Joosse M, Eussen BH, Heutink P, Oostra BA, Tourette Syndrome Association International Consortium for Genetics CNTNAP2 is disrupted in a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. Genomics. 2003;82:1–9. doi: 10.1016/s0888-7543(03)00097-1. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, State MW. Autism spectrum disorders: from genes to neurobiology. Curr Opin Neurobiol. 2015;30:92–99. doi: 10.1016/j.conb.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Ionita-Laza I, Roos JL, Boone B, Woodrick S, Sun Y, Levy S, Gogos JA, Karayiorgou M. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat Genet. 2012;44:1365–1369. doi: 10.1038/ng.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Wells AB, O’Brien DR, Nehorai A, Dougherty JD. Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J Neurosci. 2014;34:1420–1431. doi: 10.1523/JNEUROSCI.4488-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yang S, Wennmann DO, Chen Y, Kremerskothen J, Dong J. KIBRA: in the brain and beyond. Cell Signal. 2014;26:1392–1399. doi: 10.1016/j.cellsig.2014.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Bar I, Achouri Y, Campbell K, De Backer O, Hebert JM, Jones K, Kessaris N, de Rouvroit CL, O’Leary D, et al. Early forebrain wiring: genetic dissection using conditional Celsr3 mutant mice. Science. 2008;320:946–949. doi: 10.1126/science.1155244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuin J, Franke V, van Ijcken WF, van der Sloot A, Krantz ID, van der Reijden MI, Nakato R, Lenhard B, Wendt KS. A cohesin-independent role for NIPBL at promoters provides insights in CdLS. PLoS Genet. 2014;10:e1004153. doi: 10.1371/journal.pgen.1004153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.