

Abstract
Pancreatic ductal adenocarcinomas (PDAC) are highly metastatic with poor prognosis, mainly due to delayed detection. We hypothesized that intercellular communication is critical for metastatic progression. Here, we show that PDAC- derived exosomes induce liver pre-metastatic niche formation in naïve mice and consequently increase liver metastatic burden. Uptake of PDAC-derived exosomes by Kupffer cells caused transforming growth factor β secretion and upregulation of fibronectin production by hepatic stellate cells. This fibrotic microenvironment enhanced recruitment of bone marrow-derived macrophages. We found that macrophage migration inhibitory factor (MIF) was highly expressed in PDAC-derived exosomes, and its blockade prevented liver pre-metastatic niche formation and metastasis. Compared to patients whose pancreatic tumors did not progress, MIF was markedly higher in exosomes from stage I PDAC patients who later developed liver metastasis. These findings suggest that exosomal MIF primes the liver for metastasis and may be a prognostic marker for the development of PDAC liver metastasis.
One of the most lethal cancers, pancreatic cancer has a 5-year survival rate of about 6% and a median survival rate of about 6 months1,2, with pancreatic ductal adenocarcinoma (known as PDAC) being the most common type that accounts for more than 90% of cases3. The poor prognosis of PDAC is due to a combination of factors, including difficulties in detecting early stage disease, its high metastatic potential, and resistance to conventional therapies. Therefore, a better understanding of the initial events in PDAC development is needed in order to improve early detection and disease intervention.
Exosomes, membrane vesicles of endocytic origin ranging in size from 30 to 150nm4–6, are emerging as key players in intercellular communication between cancer cells and their microenvironment through horizontal transfer of information via their cargo, which includes proteins, DNAs, mRNAs and microRNAs7–13. Recently, the formation of pre-metastatic niches, a sequence of events which prepares future metastatic sites for the influx of tumor cells and which supports engraftment and survival of these incoming metastatic cells9,14,15, has been shown to depend on tumor-derived exosomes16,17.
Here, we present for the first time a detailed analysis of the sequential steps involved in liver pre-metastatic niche formation in the context of pancreatic cancer metastasis. We demonstrate that exosomes derived from malignant pancreatic lesions play a key role in liver pre-metastatic niche initiation. Selective uptake of exosomes by Kupffer cells (KCs) in the liver causes activation of fibrotic pathways, and the establishment of a pro-inflammatory milieu that ultimately supports metastasis. Specifically, we show that exosomal macrophage migration inhibitory factor (MIF) induces the release of transforming growth factor β (TGFβ) by KCs, which, in turn, promotes fibronectin (FN) production by hepatic stellate cells (hStCs). FN deposits subsequently promote the arrest of bone marrow-derived macrophages and neutrophils in the liver, completing the formation of the pre-metastatic niche. MIF knockdown prevents all sequential steps in liver pre-metastatic niche formation and, as a result, blocks exosome-induced PDAC metastasis. Importantly, we demonstrate that MIF is elevated in plasma exosomes isolated from a mouse model of pancreatic cancer (PKCY mice) bearing either pancreatic intraepithelial neoplasia (PanIN) or PDAC lesions. Moreover, MIF is also highly expressed in plasma exosomes isolated from PDAC patients whose disease progressed post diagnosis relative to patients with no evidence of disease five years post diagnosis and to healthy control subjects. These observations suggest that exosomal MIF may be a marker, as well as a functional component of PDAC liver metastasis. In summary, our study describes a previously unknown pro-metastatic circuit through which PDAC-derived exosomes can induce the formation of liver pre-metastatic niches that foster the development of metastatic disease.
Results
Pancreatic ductal adenocarcinoma-derived exosomes preferentially fuse with Kupffer cells and enhance metastatic burden in liver
To determine if PDAC-derived exosomes play a role in liver metastasis, we used an experimental model of intra-splenic injection of PAN02 murine PDAC cells18,19, which typically generates metastases restricted to the liver. We first analyzed the structure of PAN02-derived exosomes by electron microscopy, which revealed a typical exosome structure and size of approximately 100nm (Supplementary Fig. 1a). Naïve, wild-type mice were injected retro-orbitally every other day for three weeks with 5μg of PAN02-derived exosomes in a process that we previously defined as “education”16. After exosome education, we injected mCherry+ PAN02 cells intra-splenically (into the portal circulation) and analyzed the incidence of liver metastasis. We found that exosome education increased macrometastatic burden 21 days after injection, as demonstrated by quantification of mCherry+ cells (Fig. 1a, left and middle panels) and by measurement of liver weight (Fig. 1a, right panel). We verified that a control cohort educated with exosomes isolated from conditioned media of primary cultures of normal pancreas tissue from tumor-free mice (NP) did not cause an increase in metastatic burden (Fig. 1b). Furthermore, education with exosomes derived from two additional murine PDAC cell lines isolated from tumors developed in the KPC20 and PKCY21 mouse models (hereon referred to as R6560B and PKCY, respectively) resulted in increased liver metastatic burden, as assessed 3 weeks after injection of PAN02 cells (Supplementary Fig. 1b). These data suggest that PDAC-derived exosomes may support early tumor cell engraftment and metastasis by conditioning metastatic organs with favorable microenvironments (i.e., pre-metastatic niches).
Figure 1. Pancreatic ductal adenocarcinoma-derived exosomes target and activate Kupffer cells, induce liver fibrosis pathways, and increase liver metastasis.

(a) Evaluation of liver metastasis by liver weight (grams) in mice injected intra- splenically with PAN02 cells (TU, for tumor bearing) following pre-education with phosphate-buffered saline (PBS) or PAN02 exosomes (Exo). Percentage of mCherry+ PAN02 cells in the liver was measured by immunofluorescence (left panels; n = 4 mice were pooled from two independent experiments) and livers were weighed (right panel; n = 9 (CTL) and n = 8 (TU and Exo+TU) mice were pooled from three independent experiments) at day 21 post-PAN02 cell injection. Representative liver images are shown. **P < 0.01 by two-tailed t-test (middle panel), ***P < 0.05 by ANOVA (right panel). Scale bars, immunofluorescence: 200μm, whole organ images: 1cm. (b) Evaluation of liver metastasis by liver weight (grams) in mice pre-educated with PBS, exosomes from PAN02 cells, or normal pancreas (NP) prior to intra-splenic injection of PAN02 cells; n = 4 (TU) and n = 5 (PAN02exo + TU and NPexo + TU) mice from one experiment. **P < 0.01 by ANOVA. Scale bar, 1cm. (c) Fluorescence microscopy analysis of PKH67-labeled exosome incorporation (green) by F4/80+ Kupffer cells (red). Exosomes were isolated from NP, PAN02, and PKCY cells. Scale bars, 50μm. (d) Representative flow cytometric profiles of CD11b and F4/80 expression in liver cells treated with unlabeled exosomes or with PKH67-labeled NP or PDAC-derived exosomes (PAN02 and PKCY). Quantification of PKH67-positive liver cells (upper panel) and of PKH67- positive liver cells expressing CD11b and F4/80 (lower panel). For NP, n = 4 mice from one experiment; n = 7 (PAN02) and n = 8 (PKCY) mice pooled from two experiments. ***P < 0.01, **P < 0.01, *P < 0.05 by ANOVA. (e) Analysis of canonical pathways enriched in genes upregulated by Kupffer cells following in vitro education with PAN02 or NP exosomes. The list is comprised of genes related to liver fibrosis. Data were obtained from one experiment assessing three biologically independent samples. Statistical source data are presented in Supplementary Table 1. All data are represented as mean±s.e.m.
We next evaluated the cells that uptake tumor exosomes in the liver following retro-orbital injection of labeled PDAC-derived exosomes in naïve mice. For this purpose we used fluorescently labeled exosomes isolated from several PDAC models, including PAN02, PKCY and R6560B, as well as human BxPC-3 and HPAF-II PDAC cells. By analyzing the frequency of exosome positive cells in a million cells from each organ, we found that in all cases, although they first circulate through the lung due to the retro-orbital injection methodology, PDAC-derived exosomes were more frequently represented in the liver than in the lung (Supplementary Fig. 1c). In addition, we found that over 80% of liver cells that uptake exosomes were F4/80+ or F4/80+CD11b+, a phenotype consistent with KCs (Fig. 1c,d and Supplementary Fig. 1d). In contrast, control exosomes isolated from healthy normal mouse pancreas were not efficiently incorporated by KCs or other tissues analyzed (Fig. 1c,d and Supplementary Fig. 1c). Importantly, PDAC exosomes failed to fuse with other cells in the liver microenvironment, such as αSMA+ cells, S100A4+ fibroblasts, CD31+ endothelial cells, or EpCAM+ epithelial cells (Supplementary Fig. 1e). Therefore, our data suggest that KCs are the predominant cell type that uptakes PDAC-derived exosomes and likely responsible for promoting the initial steps of liver pre-metastatic niche formation.
To further define mechanisms involved in liver pre-metastatic niche formation by KCs, we educated human KCs with PAN02 or BxPC-3 exosomes in vitro and performed gene expression analysis by mRNA sequencing. Our data demonstrate that, of more than 300 canonical pathways, the liver fibrosis pathways – particularly those associated with the upregulation of genes encoding soluble factors such as CTGF, EDN, IGF, PDGF, and TGFβ (Fig. 1e and Supplementary Fig. 1f) – were the most highly expressed and over-represented (Supplementary Tables 1 and 2).
Fibronectin upregulation by hepatic stellate cells is important for liver pre- metastatic niche formation by pancreatic ductal adenocarcinoma-derived exosomes
To determine how PDAC-derived exosomes elicit a fibrotic liver microenvironment we examined, by immunofluorescence, the changes in expression of different extracellular matrix (ECM) proteins following PAN02 exosome education. We observed a downregulation of vitronectin and tenascin C, a stable but minimal expression level of collagen type I, and a marked increase in fibronectin (FN) expression (Fig. 2a). Next we investigated the source of FN upregulation by PDAC- derived exosomes by co-staining sections of pre-metastatic livers with antibodies against FN and CD31 or αSMA. We found a predominant population of αSMA+FN+ cells in FN-enriched areas of educated livers, but a lack of FN and CD31 co-staining (Supplementary Fig. 2a), suggesting that activated hStCs produced most of the FN (Fig. 2b). In contrast to education with NP-derived exosomes, education of naïve mice with PKCY or R6560B cell-derived exosomes also increased αSMA and FN expression in the liver (Supplementary Fig. 2b). These results demonstrate an induction of FN production by hStCs and support the hypothesis that PDAC-derived exosomes can establish a fibrotic liver pre-metastatic niche.
Figure 2. Role of pancreatic ductal adenocarcinoma-derived exosome education in extracellular matrix component expression and liver pre-metastatic niche formation.
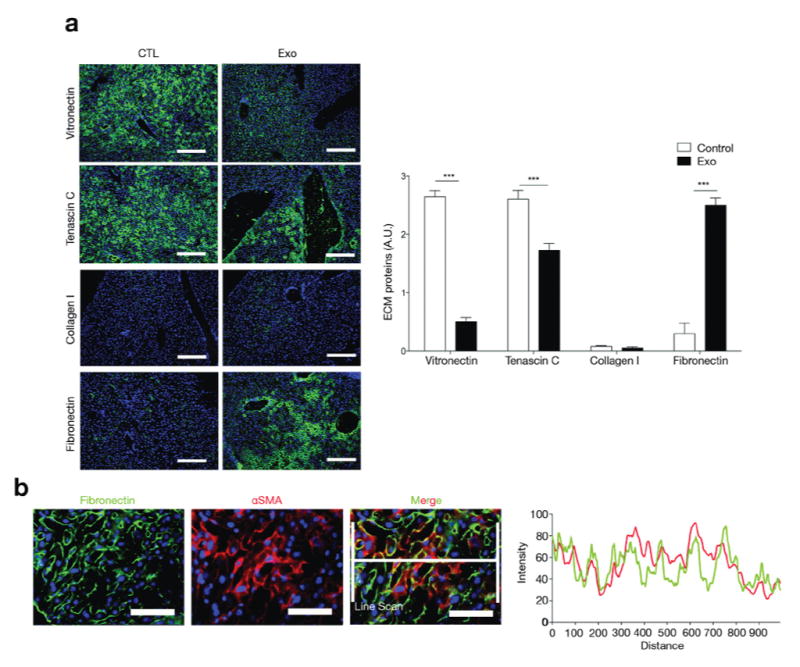
(a) Immunofluorescence quantification in arbitrary units (A.U.) of vitronectin, tenascin C, collagen I and fibronectin (FN) expression in liver after education with PAN02 exosomes (Exo) or PBS (CTL); n = 4 mice pooled from two experiments. Statistical source data are presented in Supplementary Table 4. The data are represented as mean±s.e.m. ***P < 0.001 by two-tailed t-test. Scale bars, 200μm. (b) Fluorescence microscopy analysis of FN expression in αSMA+ hStCs in livers of mice educated with PAN02 exosomes. Line scan for histogram calculation is shown. Scale bars, 50μm.
Pancreatic ductal adenocarcinoma-derived exosomes induce bone marrow- derived cell migration to liver
To evaluate if PDAC-derived exosomes play a role in preparing the liver microenvironment for the arrival of metastatic cells by eliciting macrophage recruitment, wild-type mice were educated with exosomes isolated from PAN02, PKCY, or R6560B cells. Interestingly, PDAC exosomes, but not NP exosomes, were sufficient to increase the frequency of macrophage marker F4/80-expressing cells in the liver (Fig. 3a and Supplementary Fig. 2c). A detailed time course analysis during the three-week PAN02 exosome education showed a significant increase in FN levels as early as the second week of exosome education. By the third week of education, we observed a significant and sustained increase in the levels of F4/80+ cells in the liver compared to livers that had not been educated (Fig. 3a). Moreover, to determine if the increase in macrophage frequency was the result of bone marrow (BM)-derived macrophage recruitment, we transplanted GFP-expressing BM cells into irradiated wild-type C57Bl/6 mice, followed by PAN02 exosome education and F4/80 immunostaining. We observed an overall increase in GFP+ cells in the liver after exosome education, with approximately 67% of these cells being F4/80+ (Fig. 3b, upper panels). Importantly, the GFP-F4/80+ population, likely composed of liver resident KCs, remained unchanged after PAN02 exosome education (Fig. 3b, upper-right panel). Furthermore, we found that a smaller subset, approximately 30% of the GFP+ cells, was positive for the myeloid differentiation marker Gr-1, and this population also increased in the liver following PAN02 exosome education (Fig. 3b, lower panels). Our results indicate that PAN02 exosome education promotes recruitment of BM-derived cells to the liver, including F4/80+ macrophages and, to a lesser extent, Gr-1+ neutrophils.
Figure 3. Pancreatic ductal adenocarcinoma-derived exosomes induce fibronectin expression and migration of bone marrow-derived cells to the liver.
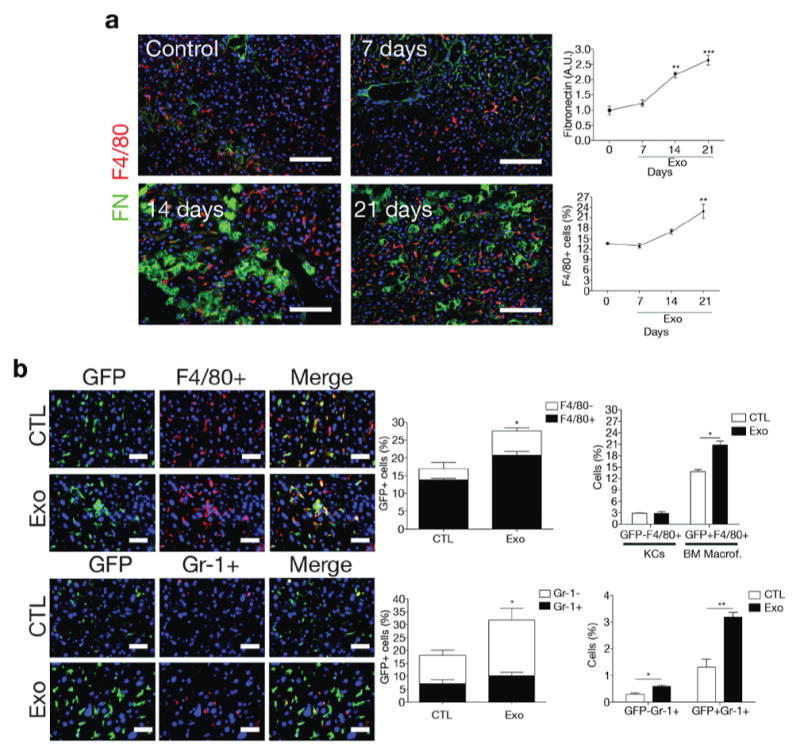
(a) Immunofluorescence quantification of FN expression in arbitrary units (A.U.) and F4/80+ cell frequency in the livers of mice during the course of PAN02 exosome education; n = 4 mice from one experiment. **P < 0.01, ***P < 0.001 by ANOVA relative to control (day 0). Scale bars, 100μm. (b) Immunofluorescence quantification of the frequency of bone marrow (BM)-derived F4/80+ (upper) and Gr1+ (lower) cells in the liver following transplantation of GFP+ BM from mice educated with PBS (CTL) or PAN02 exosomes (Exo); n = 4 (F4/80) and n = 3 (Gr1) mice from one experiment. **P < 0.01, *P < 0.05 by two-tailed t-test. Scale bars, 50μm. All statistical source data are presented in Supplementary Table 4. All data are represented as mean±s.e.m.
Macrophage recruitment to the liver pre-metastatic niche follows TGFβ signaling-induced fibronectin upregulation
Upon KC education with PDAC-derived exosomes in vitro, TGFβ was among the most highly upregulated soluble factors related to liver fibrosis and hStCs activation (60–148% increase) produced by KCs (Fig. 1e and Supplementary Fig. 1f). TGFβ mediates fibrogenesis by inducing hStC activation and myofibroblast differentiation, leading to ECM protein production22–26. We therefore hypothesized that KC-derived TGFβ might be responsible for inducing FN production by hStCs27,28. Consistent with this hypothesis, treatment with a TGF-beta type I receptor inhibitor (A83-01)29 during the three-week course of PAN02 exosome education reduced the frequency of αSMA+ hStCs, FN deposition, and F4/80+ macrophage migration to the liver (Fig. 4 and Supplementary Fig. 3). Thus, FN deposition and macrophage recruitment are both TGFβ-dependent steps that occur during liver pre-metastatic niche formation.
Figure 4. TGFβ signaling induces fibronectin upregulation and macrophage recruitment to the liver pre-metastatic niche.
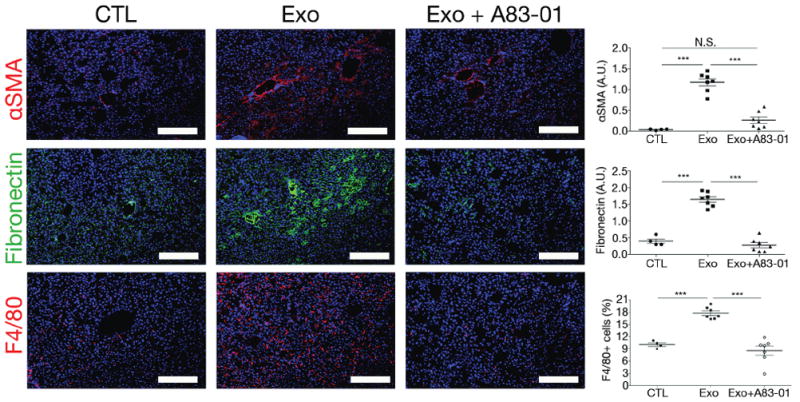
Immunofluorescence quantification of FN and αSMA expression in arbitrary units (A.U.) and F4/80+ cells in livers of mice educated with PBS (CTL), PAN02 exosomes alone (Exo), or in combination with the TGFβ receptor inhibitor A83-01 (Exo+A83-01); n = 4 (CTL) and n = 7 (Exo and Exo+A83-01) mice pooled from two experiments. The data are represented as mean±s.e.m. ***P < 0.001, N.S. stands for not significant by ANOVA. Scale bars, 200μm.
To further define the sequential events involved in liver pre-metastatic niche development, we used a conditional FN knockout murine model in which tamoxifen- driven Cre recombinase expression depletes FN during PAN02 exosome education. We verified that FN deposition in the liver was reduced to control Rosa-CreER−;Fnfl/fl levels upon tamoxifen treatment of Rosa-CreER+;Fnfl/fl, while αSMA expression was not affected (Fig. 5a). Interestingly, FN depletion led to a reduction in the frequency of F4/80+ macrophages in the liver, demonstrating that FN deposition is required to increase macrophage frequency in the pre-metastatic liver (Fig. 5a). To test if FN depletion plays a functional role in PDAC liver metastasis, we intra-splenically injected PAN02 cells into tamoxifen-treated Rosa-CreER+;Fnfl/fl and control (Rosa- CreER−;Fnfl/fl) mice educated with PAN02 exosomes. Consistent with our hypothesis that FN plays a critical role in liver pre-metastatic niche formation, FN depletion during the three-week PAN02 exosome education step reverted the pro-metastatic phenotype to resemble livers harvested from PBS-educated PAN02 tumor bearing mice (Fig. 5b). Of note, reduction of FN levels in non-educated mice using the same system did not affect baseline liver metastasis (Fig. 5c). Our results confirm that FN is required for PDAC exosome mediated liver pre-metastatic niche formation.
Figure 5. Fibronectin and macrophages play major roles in PAN02 exosome- mediated liver pre-metastatic niche formation.
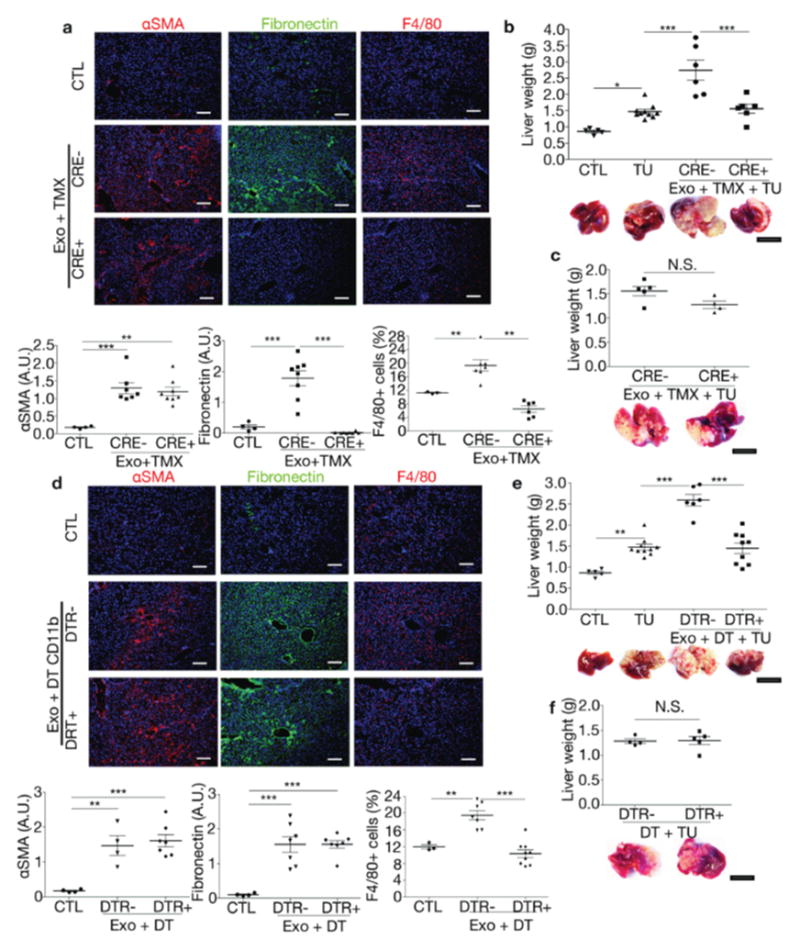
(a) Immunofluorescence quantification of FN and αSMA expression and F4/80+ cell frequency in livers of FN-conditional knockout mice. Cre−/ −Fnfl/fl (CRE-) and Cre+/−Fnfl/fl (CRE+) mice were tamoxifen-treated (TMX) and educated with PAN02 exosomes (Exo). Control PBS-educated livers (CTL): n = 3 (CTL F4/80), n = 4 (CTL αSMA, FN), n = 6 (CRE+ F4/80), n = 7 (CRE− αSMA, F4/80), and n = 8 (CRE− FN; CRE+ αSMA, FN) mice from two experiments. ***P < 0.001, **P < 0.01 by ANOVA. (b) Evaluation of liver metastasis by liver weight (grams) in tumor-free mice (CTL), PBS-educated mice injected intra-splenically with PAN02 cells (TU), and tumor-bearing CRE− and CRE+ mice TMX-treated during Exo education; n = 4 (CTL), n = 6 (CRE− and CRE+), and n = 10 (TU) mice from three experiments. ***P < 0.001, *P < 0.05 by ANOVA. (c) Evaluation of liver metastasis in TMX-treated CRE− versus CRE+ mice injected intra-splenically with PAN02 cells (TU); n = 4 (CRE+) and n = 5 (CRE-) mice from one experiment. (d) Quantification of FN and αSMA expression and F4/80+ cell frequency in mice transiently depleted of CD11b+ cells using a diphtheria toxin (DT)- inducible system during PAN02 exosomes (Exo) education. Control PBS-educated livers (CTL): n = 3 (CTL F4/80), n = 4 (CTL αSMA, FN; DTR− αSMA), n = 7 (DTR− FN, F4/80; DTR+ αSMA, FN), and n = 9 (DTR+ F4/80) mice from two experiments. ***P < 0.001, **P < 0.01 by ANOVA. (e) Evaluation of liver metastasis in tumor-free mice (CTL), PBS-educated mice injected intra-splenically with PAN02 cells (TU), and tumor-bearing CD11b-DTR− or -DTR+ mice DT-treated during Exo education; n = 5 (CTL), n = 6 (DTR-), n = 9 (DTR+), and n = 10 (TU) mice pooled from three experiments. ***P < 0.001, **P < 0.01 by ANOVA. (f) Evaluation of liver metastasis in mice transiently depleted of CD11b+ cells and injected intra-splenically with PAN02 cells; n = 4 (DTR-) and n = 5 (DTR+) mice from one experiment. All data are represented as mean±s.e.m. N.S. for not significant by two-tailed t-test. Scale bars, immunofluorescence: 200μm, whole organ: 1cm.
Macrophage ablation blocks the pro-metastatic effect of pancreatic ductal adenocarcinoma-derived exosomes in the liver
Bone-marrow macrophages are recruited to the liver during the early stages of exosome-mediated liver pre-metastatic niche formation. Therefore, we first investigated whether macrophages present in the liver pre-metastatic niche are physically associated with liver metastatic lesions. To this end, we tracked disseminated PAN02 cells in the liver of mice educated for three weeks with PAN02 exosomes, 24 hours after splenic injection. We found that metastatic cells localized adjacent to F4/80+ macrophages (Supplementary Fig. 4). Next, we sought to determine if macrophages are required for liver pre-metastatic niche formation. To distinguish between direct effects of FN deposition and FN-mediated effects on liver F4/80+ cell frequency during PAN02 exosome-mediated liver pre-metastatic niche formation and liver metastasis, we used a diphtheria toxin (DT)-inducible system that transiently depletes CD11b+ cells (including macrophages and neutrophils) expressing the DT receptor (DTR)30. DT treatment during the second week of PAN02 exosome education significantly reduced the frequency of F4/80+ macrophages without affecting αSMA+ hStC or FN levels in the liver (Fig. 5d and Supplementary Fig. 3). We then examined the effects of macrophage depletion on metastatic progression by injecting PAN02 cells into exosome-educated mice depleted of CD11b+ cells. Macrophage depletion was sufficient to revert the effects of PAN02 exosome education on liver metastasis (Fig. 5e). Of note, reduction of macrophage levels in non-educated mice using the same approach did not alter baseline liver metastasis (Fig. 5f). Taken together, these results suggest that macrophages are required for liver pre-metastatic niche-dependent metastasis formation.
MIF depletion in pancreatic ductal adenocarcinoma-derived exosomes inhibits liver pre-metastatic niche initiation
To gain insight into the mechanisms through which PDAC-derived exosomes promote KC activation and subsequent liver pre-metastatic niche formation, we mined pancreatic exosome mass spectrometry data for potential inflammatory mediators. Among the candidates with previously described roles in either or both macrophage activation and inflammatory modulation, we found that macrophage migration inhibitory factor (MIF) was highly expressed in PDAC-derived exosomes (Supplementary Table 3). MIF knockdown in PAN02 cells did not affect exosome size, quantity of protein per exosome, or the binding of these exosomes to liver CD11b+F4/80+ cells (Supplementary Fig. 5a–c). However, the effects of MIF knockdown on liver pre-metastatic niche formation were striking, as evidenced by a pronounced reduction of TGFβ expression in F4/80+ cells and of αSMA in hStCs (Fig. 6a and Supplementary Fig. 3). Moreover, MIF knockdown decreased both FN deposition and F4/80+ macrophage frequency (Fig. 6a and Supplementary Fig. 3). Additionally, we observed a reduction in tumor cell retention after 24 hours in mice educated with PAN02 MIF knockdown exosomes, compared to control PAN02 exosomes (Supplementary Fig. 5d). These results suggest that MIF orchestrates the sequential events in PDAC-derived exosome-induced liver pre-metastatic niche formation.
Figure 6. MIF-expressing PAN02 exosomes induce liver pre-metastatic niche formation.
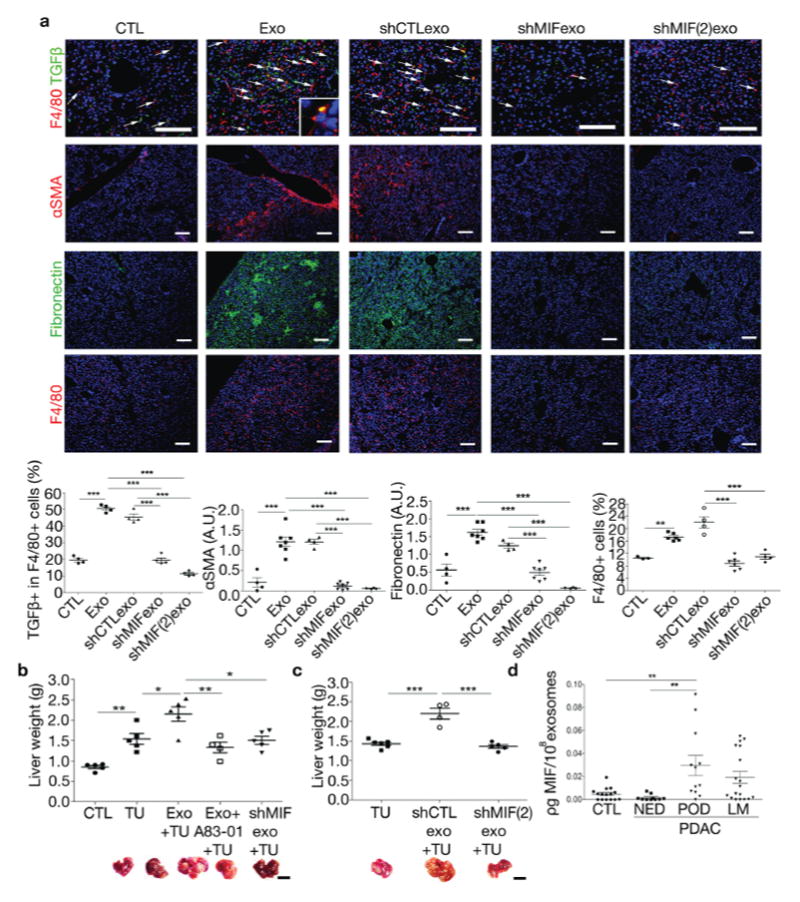
(a) Representative images and quantification of pre-metastatic niche markers in livers educated with PAN02 exosomes (Exo), PAN02shCTL exosomes (shCTLexo), PAN02shMIF exosomes (shMIFexo and shMIF(2)exo) or PBS control (CTL). Immunofluorescence analysis shows frequency of TGFβ-expressing F4/80+ cells, αSMA and FN expression as well as F4/80+ cell frequency. Inset shows TGFβ+/F4/80+ cell; n = 3 (CTL F4/80), n = 4 (CTL TGFβ, αSMA, FN; Exo TGFβ; all shCTL; shMIF TGFβ; all shMIF(2), n = 6 (Exo F4/80; shMIF F4/80), n = 7 (Exo αSMA, FN; shMIF αSMA, FN) mice pooled from two experiments. ***P < 0.001, **P < 0.001 by ANOVA. Scale bars, 100μm. (b) Evaluation of liver metastasis by liver weight (grams) in tumor-free mice (CTL), mice injected intra-splenically with PAN02 cells either pre-educated with PBS (TU), with PAN02 exosomes (Exo+TU), with PAN02 exosomes in combination with A83-01 (Exo+A83-01+TU), or with PAN02shMIF exosomes (shMIFexo+TU); n = 4 (Exo+A83-01+TU), n = 5 (CTL, TU, Exo+TU, and shMIFexo+TU) mice pooled from two experiments. **P < 0.01, *P < 0.05 by ANOVA. Scale bar, 1cm. (c) Evaluation of liver metastasis in mice injected intra-splenically with PAN02 cells either pre-educated with PBS (TU), with exosomes isolated from PAN02 cells infected with control shRNA (shCTLexo+TU) or shMIF(2) (shMIF(2)exo+TU) lentiviral vectors; n = 4 (shCTLexo+TU), n = 5 (shMIF(2)exo+TU) and n = 6 (TU) mice from one experiment. ***P < 0.01 by ANOVA. Scale bar, 1cm. (d) Enzyme-linked immune assay (ELISA) reveals increased levels of MIF (picogram per 108 exosomes) in exosomes isolated from patients with pancreatic ductal adenocarcinoma (PDAC) with progression of disease post-diagnosis (POD) compared to PDAC patients with no evidence of disease 5 years post-diagnosis (NED) and to healthy controls (CTL), but not PDAC patients with liver metastasis (LM); n = 10 (NED), n = 12 (POD), n = 15 (CTL) and n = 18 (LM) patients. All patient samples were analyzed once as part of three independent ELISA assays. **P < 0.01 by ANOVA. All data are represented as mean±s.e.m
To further evaluate the functional role of exosomal MIF in liver metastatic progression, we compared the consequences of intra-splenic injection of PAN02 cells into non-educated mice versus pre-educated mice that were treated with either PAN02 or PAN02 MIF knockdown exosomes for three weeks. In addition to preventing KC education and the sequential steps in liver pre-metastatic niche induction by PAN02 exosomes (Fig. 6a), and analogous to TGFβ1R signaling inhibition by A83-01 (Fig. 4), MIF knockdown also reduced the ability of PAN02 exosomes to promote liver metastasis (Fig. 6b,c). Control lentiviral vectors did not alter the effects of PAN02 exosomes on liver pre-metastatic niche formation (Fig. 6a,c). Collectively our data demonstrates that exosomal MIF is mediating the increase in metastatic burden observed upon PDAC-derived exosome education.
Next, we sought to determine the clinical relevance of these findings. To this end, we explored whether MIF levels varied between exosomes isolated from plasma samples of control healthy subjects and PDAC patients with liver metastasis, with either no evidence of disease five years post diagnosis (NED) or progression of disease post diagnosis (POD). We found that POD patients expressed significantly higher exosomal MIF levels compared to NED patients and healthy control subjects, suggesting that exosomal MIF could be used as a biomarker of PDAC prognosis. A trend toward lower MIF levels was apparent in exosomes isolated from PDAC patients with liver metastasis versus those with POD, although the difference between patient groups was not statistically significant (Fig. 6d). Taken together, our findings indicate that MIF-expressing exosomes are essential for liver pre-metastatic niche initiation and efficient liver metastasis.
Liver pre-metastatic niche formation precedes the establishment of pancreatic ductal adenocarcinoma lesions
To further characterize changes in the liver niche during metastatic progression, we analyzed the livers of PKCY mice that develop PDAC with reproducible kinetics, starting with “early” PanIN (Pancreatic Intraepithelial Neoplasia) lesions at 4–6 weeks, more advanced PanINs at 8–11 weeks, and invasive cancers at 16–20 weeks that metastasizes to the liver21. Livers isolated from mice that do not develop pancreatic lesions (Cre transgenic mice lacking activating p53 Kras mutations, from hereon referred to as CY mice) were used as controls. We found an increase in αSMA+ cells (6-fold), FN accumulation (7-fold), and an infiltration of F4/80+ cells (2-fold) in PKCY mice at the late PanIN stage (Fig. 7a). Next, we evaluated TGFβ expression levels during pancreatic cancer progression in PKCY mice. Importantly, we found that TGFβ was upregulated in KCs during the early PanIN stage (2-fold), suggesting that PDAC-derived exosomes play a role during the pre-tumoral stages of liver pre- metastatic niche formation (Fig. 7a). Finally, to determine whether exosomal MIF protein levels correlate with the progression of pancreatic lesions, we evaluated exosomes isolated from the plasma of PKCY mice at different stages of pancreatic lesion progression. We found increased levels of MIF in exosomes from mice with PDAC when compared to exosomes from healthy control mice (Fig. 7b). Interestingly, the increase in exosomal MIF levels was already detectable in exosomes isolated from mice with PanIN lesions (early or late stage) (Fig. 7b). Collectively, these results demonstrate that changes in MIF levels in PDAC-derived exosomes reflect liver pre-metastatic niche formation during pre-malignant stages of PDAC metastatic progression.
Figure 7. Liver pre-metastatic niche formation and increased exosomal MIF levels precede pancreatic ductal adenocarcinoma lesion development in PKCY mice.
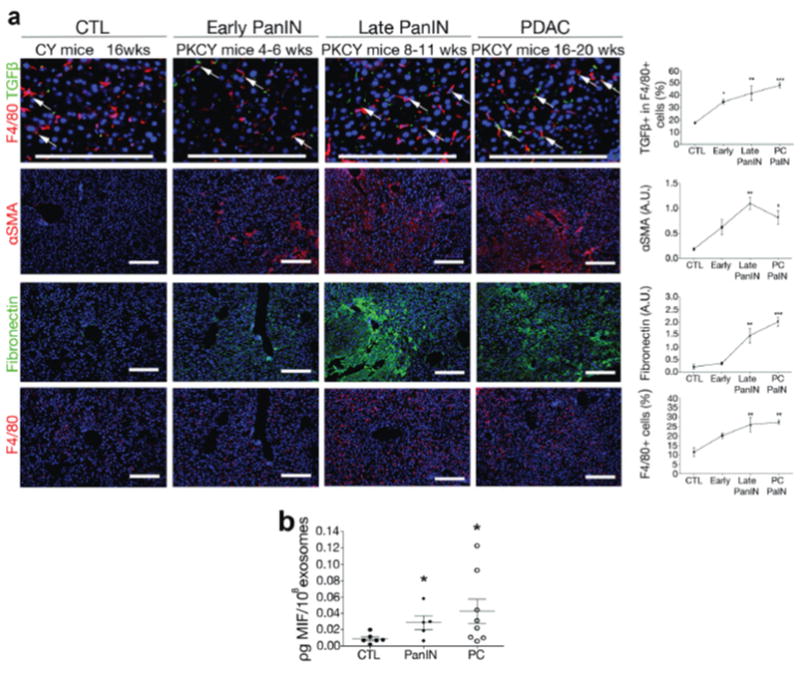
(a) Immunofluorescence quantification of αSMA and FN expression in arbitrary units (A.U.) and F4/80+ cell frequency in livers of control CY mice at 16 weeks (wks) (CTL), PKCY mice at early and late PanIN phases (4–6 and 8–11 wks, respectively), and PDAC livers (16–20 wks). Upregulation of αSMA, FN, and increased frequency of F4/80+ cells can be observed starting at the PanIN stages; n = 4 (all CTL; all early PanIN; all late PanIN; PDAC TGFβ and αSMA) and n = 5 (PDAC FN and F4/80) mice pooled from three experiments. Statistical source data can be found in Supplementary Table 4. ***P < 0.001, **P < 0.01, *P < 0.05 by ANOVA when compared to CTL. Scale bars, 200μm. (b) Levels of MIF in exosomes isolated from plasma of CY and PKCY mice, at PanIN (both early and late) and PDAC stages measured by ELISA; n = 5 (PanIN), n = 6 (CTL), and n = 8 (PDAC) mice plasma samples pooled from two experiments. *P < 0.05 by ANOVA when compared to CTL. All data are represented as mean±s.e.m.
Discussion
Circulating exosomes can be used as biological markers for pancreatic cancers4,31. However, the cellular and molecular mechanisms through which PDAC-derived exosomes affect metastasis have yet to be determined. Here, we delineate for the first time the sequential steps of liver pre-metastatic niche formation by PDAC- derived exosomes. PDAC-derived exosomes induce TGFβ signaling in KCs, leading to activation of hStCs and ECM remodeling. In turn, FN accumulation promotes an influx of BM-derived macrophages (and potentially neutrophils) to the liver, providing a favorable niche for liver metastasis (Fig. 8). Importantly, we demonstrate that this stepwise progression also occurs in a spontaneous murine model of pancreatic cancer, the PKCY mouse model, in which key early liver pre-metastatic niche features (such as hStC activation, FN accumulation, and macrophage recruitment to the liver) are already evident during the PanIN stages.
Figure 8. Model for the sequential steps in liver pre-metastatic niche formation induced by pancreatic ductal adenocarcinoma-derived exosomes.
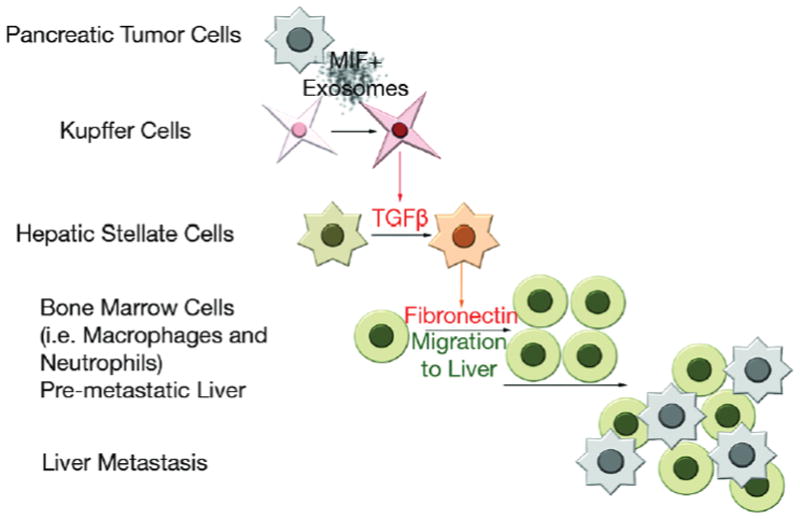
Education with MIF+ PDAC-derived exosomes, which bind predominantly to Kupffer cells in the liver, induces TGFβ production by these cells. TGFβ activates hStCs, which in turn upregulate FN. Bone marrow-derived cells (i.e. macrophages) bind to FN-enriched hepatic sites, ultimately leading to liver pre-metastatic niche formation.
Unbiased mass spectrometry analysis of PDAC-derived exosome protein cargo led us to identify MIF as a candidate mediator of liver education. Although one study has described MIF as an anti-fibrotic factor in a chemically induced liver fibrosis model32, MIF is a well-known mediator of liver inflammation and fibrosis, of BM cell recruitment to the liver and liver metastasis, and furthermore MIF tissue and plasma levels correlate with PDAC aggressiveness33–37. Here, we propose a novel role for exosomal MIF in PDAC liver metastasis and describe a mechanism through which MIF orchestrates this process.
Our functional experiments in murine models of pancreatic ductal adenocarcinoma demonstrate that exosomal MIF upregulation is an early event during cancer progression and can be detected in plasma-derived exosomes isolated from mice with pre-tumoral pancreatic lesions. Importantly, high exosomal MIF levels were present in plasma from patients with stage I PDAC, prior to liver metastasis, indicating its potential prognostic value. In contrast, exosomal MIF levels had a bimodal distribution in patients with established liver metastatic disease. The presence of low exosomal MIF levels in a subset of PDAC patients with liver metastasis indicates that MIF-dependent mechanisms may be more important during earlier stages of liver metastasis. Taken together, these observations justify future long-term retrospective and prospective studies to determine whether exosomal MIF levels could serve as an early biomarker for liver pre-metastatic niche formation and a prognostic factor for metastatic risk in patients with pre-tumoral lesions, such as pancreatitis, PanINs, intra-ductal papillary mucinous neoplasms (IPMNs), and pre- metastatic PDACs. Moreover, these results also raise the possibility that high MIF levels may be an indicator of risk for liver pre-metastatic niche formation and liver metastasis in other gastrointestinal tract tumors, such as colorectal and gastric cancer. MIF inhibition by either small molecules or neutralizing antibodies has been shown to prevent macrophage activation, TGFβ expression, and tissue fibrosis38–41. Further studies are required to determine the effects of systemic MIF inhibition on liver pre-metastatic niche formation and its therapeutic potential in pancreatic cancer.
High levels of TGFβ in patients with pancreatic cancers is associated with poor prognosis42. Anti-TGFβ compounds have shown efficacy in preclinical and clinical studies43,44, suppressing liver metastasis by acting on both incoming tumor cells and the liver microenvironment45. However, systemic TGFβ inhibition carries a substantial risk for patients as this pathway is implicated in multiple homeostatic processes and may also have tumor-suppressor functions depending on the tumor stage and stromal activation status46–48. Although our data suggest that targeting TGFβ signaling may be beneficial for preventing liver pre-metastatic niche formation, given the context-dependent roles of TGFβ, its potential anti-tumorigenic effects need to be excluded before TGFβ inhibitors are considered for metastasis prevention in PDAC patients in the clinic.
Several studies have correlated KC49–51 and ECM changes52–54 with circulating tumor cell arrest and liver metastasis. In addition, myeloid cell recruitment to the liver was also shown to precede liver metastasis55–58. Here, we demonstrate for the first time that liver pre-metastatic niche formation during PDAC metastasis depends on tumor exosome-derived MIF. Moreover, we show that this process is driven by TGFβ-signaling, FN deposition, and recruitment of BM-derived macrophages to future liver metastatic niches. By individually targeting MIF, FN, and macrophages, we successfully reverted the effects of PDAC-derived exosome education at various steps during liver pre-metastatic niche formation. Strategies aimed at targeting these particular molecules (TGFβ, MIF, FN) and cell types (BM-derived cells) have been previously explored50,59–62. We propose that our detailed dissection of the sequential events of PDAC metastasis initiation provides an understanding of how tumor secreted factors, specifically exosomes, orchestrate this process and our findings open new avenues for early detection, prevention, and therapeutic intervention.
Online Methods
Cells
Primary cultures of murine pancreatic ductal adenocarcinomas (PKCY) were obtained by dissociating cells from tumors of a 20 week-old Pdx1-Cre transgenic mouse69 in which a RosaYFP allele is expressed concurrently with pancreas-specific mutations in p53 and Kras, referred as PKCY mice21. The murine pancreatic tumor cell line with high metastatic potential to the liver, R6560B, was isolated from tumors of a Pdx1-Cre transgenic mouse69 with pancreas-specific mutations in p53 and Kras20 and was generously provided by Dr. David L. Bajor (Vonderheide lab, University of Pennsylvania). The C57Bl/6 murine pancreatic adenocarcinoma cell line PAN02 (also identified as Panc 02)18 was purchased from the DTP, DCTD Tumor Repository, NIH. MIF knockdown PAN02 cells were generated by using MIF shRNA lentiviral particle infection, generating the PAN02 variants shMIF (sc-37138-V, Santa Cruz) and shMIF(2) (iV042354, abmGood). As a control, PAN02 cells were infected with control shRNA lentiviral particles-A (sc-108080, Santa Cruz). In all cases, infected cells were selected with puromycin according to the manufacturer’s instructions. For several experiments, exosomes were isolated from human PDAC cell lines BxPC-3 and HPAF-II (ATCC). For in vitro education of human Kupffer cells (Life Technologies) with healthy normal pancreas-derived exosomes (NP), PAN02, or BxPC-3 exosomes, cells were maintained in culture for 14 days, with media containing 0 or 5μg/ml of exosomes, replenished every other day. Cells were cultured in RPMI supplemented with 10% exosome-depleted FBS (FBS, Gibco) and penicillin-streptomycin, and maintained in a humid incubator with 5% CO2 at 37°C. FBS was depleted of bovine exosomes by ultracentrifugation at 100,000g for 70 minutes (min). In some experiments, normal pancreas exosomes were obtained by culturing 25 pancreata isolated from healthy 4–6 week-old mice in 3ml of FBS-free RPMI for 12 hours.
Exosome isolation, characterization, and analyses
Isolation of exosomes for mass spectrometry and all other experiments was done by ultracentrifugation. Exosome preparation was verified by electron microscopy. Supernatant fractions collected from 72-hour cell cultures or plasma samples were pelleted by centrifugation at 500g for 10 min. The supernatant was centrifuged at 12,000g for 20 min. Exosomes were then harvested by centrifugation at 100,000g for 70 min. The exosome pellet was resuspended in 20ml of phosphate-buffered saline (PBS) and collected by ultracentrifugation at 100,000g for 70 min (Beckman Ti70). Exosome size and particle number were analyzed using the LM10 or DS500 nanoparticle characterization system (NanoSight) equipped with a blue laser (405nm). MIF levels in exosomes were measured by ELISA (ab100594, Abcam for human samples; ABIN415583, Antibodies Online for mouse samples), using 2μg of exosomes per 100μl of sample diluent, in duplicate reactions, according to manufacturer’s instructions.
Proteomics
Mass spectrometry analyses of exosomes were performed using 10μg of exosomal protein. Samples were denatured using 8M urea, reduced using 10mM DTT, and alkylated using 100mM iodoacetamide. This was followed by proteolytic digestion with endoproteinase LysC (Wako Chemicals) overnight at room temperature, and subsequent digestion with trypsin (Promega) for 5 hours at 37°C. The digestion was quenched with formic acid and the resulting peptide mixtures were desalted using in- house made C18 Empore (3M) StAGE tips. Samples were dried and solubilized in the sample loading buffer containing 2% acetonitrile and 2% formic acid. Approximately 3–5μg of each sample was analyzed by reversed phase nano-LC- MS/MS (Ultimate 3000 coupled to QExactive, Thermo Scientific). Following loading on the C18 trap column (5μm beads, Thermo Scientific) at a flow rate of 3μl/min, peptides were separated using a 75μm inner diameter C18 column (3μm beads Nikkyo Technos Co., Ltd. Japan) at a flow rate of 200nl/min, with a gradient increasing from 5% Buffer B (0.1% formc acid in acetonitrile)/95% Buffer A (0.1% formic acid) to 40% Buffer B/60% Buffer A, over 140 min. All LC-MS/MS experiments were performed in data dependent mode. Precursor mass spectra were recorded in a 300–1400 m/z mass range at 70,000 resolution, and 17,500 resolution for fragment ions (lowest mass: m/z 100). Data were recorded in profile mode. Up to twenty precursors per cycle were selected for fragmentation and dynamic exclusion was set to 45 seconds. Normalized collision energy was set to 27. Data were extracted and searched against Uniprot complete Human or Mouse proteome databases (January 2013) concatenated with common contaminants using Proteome Discoverer 1.4 (Thermo Scientific) and Mascot 2.4 (Matrix Science). All cysteines were considered alkylated with acetamide. N-terminal glutamate to pyroglutamate conversion, oxidation of methionine, and protein N-terminal acetylation were allowed as variable modifications. Data were first searched using fully tryptic constraints. Matched peptides were filtered using a Percolator-based 1% false discovery rate. Spectra not being matched at a false discovery rate of 1% or better were re- searched allowing for semi-tryptic peptides. The average area of the three most abundant peptides for a matched protein was used to gauge protein amounts within and in between samples.
Exosome treatment and labeling
Five micrograms of total exosomal protein were injected into the retro-orbital venous sinus in a total volume of 100μl PBS. For education experiments, mice received 5μg of exosomes every other day, 3 times a week. For exosome-tracking experiments, purified exosomes were fluorescently labeled using PKH67 membrane dye (Sigma). Labeled exosomes were washed in 20ml of PBS, collected by ultracentrifugation, and resuspended in PBS. In experiments involving evaluation of exosome incorporation, labeled exosomes were injected 24 hours prior to tissue collection and analysis for exosome+ cells was conducted by flow cytometry or immunofluorescence. For exosome-tracking experiments, 5μg of non-tumor exosomes from normal pancreas were used as controls. Unlabeled exosomes were used as controls of signal specificity. For education experiments, retro-orbital injection of PBS or normal pancreas exosomes was used in control groups.
Illumina strand-specific RNA sequencing
To analyze the genes whose expression was altered in human KC cells as a result of in vitro education with normal pancreas, PAN02, or BxPC-3 exosomes, total RNA was isolated using the RNeasy Mini Kit (QIAGEN). Experiments were performed in triplicate. Illumina sequencing libraries were constructed by following a modified strand-specific RNA-Seq protocol70. The library was then PCR-amplified with TruSeq-indexed PCR primers and sequenced using the Illumina HiSeq2000 platform (Weill Cornell Medical College).
Human studies
Human studies were approved by the Weill Cornell Medical College, Institution Review Board #0604008488. Human peripheral blood samples were obtained from control healthy subjects and PDAC patients with liver metastasis or from patients without longstanding liver metastasis (approximately 5 years) at Weill Cornell Medical College, Oslo University Hospital, Memorial Sloan-Kettering Cancer Center, and University of Nebraska Medical Center and all pathologically confirmed. All individuals provided informed consent for blood donation on approved institutional protocols. Blood was collected in purple top tubes containing EDTA, and centrifuged at 500g for 10 min. Plasma exosomes were isolated as described above in the “Exosome Isolation, Characterization and Analyses” section.
Mouse strains and treatments
All mouse work was performed in accordance with institutional, IACUC, and AAALAS guidelines, animal protocol #0709-666A. Six week-old C57Bl/6 female mice were used for all animal experiments. For CD11b+ cell depletion, B6.FVB-Tg-(ITGAM- DTR) mice (Jackson Laboratories, stock 006000) were injected intra-peritoneally with 150ng of diphtheria toxin diluted in 100μl of PBS every other day during the second week of exosome education. As a control, wild type C57Bl/6 mice received a similar regimen of treatment. CD11b+ cell depletion from the liver was verified by immunofluorescence or flow cytometry for F4/80+ cells and CD11b+F4/80+ cells, respectively.
To generate inducible FN knockout mice, we crossed B6;129- Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J mice expressing tamoxifen-sensitive Cre under the Gt(Rosa)26or promoter (Jackson Laboratories, stock 008463) with mice homozygous for a floxed allele of fibronectin (Fnfl/fl, generously provided by Dr. Reinhard Fässler)71. Cre+/−Fnfl/fl mice were then maintained by breeding to Fnfl/fl mice. Both Cre+/−Fnfl/fl (CRE+) and Cre−/−Fnfl/fl (CRE-) control mice were treated with intra-peritoneal injections of 40μg of Tamoxifen (Sigma) per gram of mouse weight (diluted in 100μl of olive oil) every other day, for a total of 3 doses, during the second week of PAN02 exosome education. For verification of Fn deletion, liver FN expression was evaluated by immunofluorescence.
For evaluation of liver pre-metastatic niche formation formation in a spontaneous mouse model of PDAC, we used the p53 and Kras-driven mouse model of pancreatic cancer21, termed as PKCY. PKCY mice were evaluated at different ages, during the early and late stages of PanIN lesions (4–6 and 8–11 weeks, respectively), and at the PDAC stage (16–20 weeks). As controls, livers isolated from mice that do not develop pancreatic lesions (Cre transgenic lacking activating TP53 and Kras mutations, referred here as CY) were used.
For systemic TGFβ receptor inhibition, mice were treated with intra-peritoneal injections of A83-01 (Santa Cruz). Mice were administered 10μg of A83-01 (diluted in 100μl of DMSO) per gram of mouse weight for a total of 9 doses, every other day, for the three weeks of PAN02 exosome education.
Liver metastasis studies
To analyze the role of exosome education in tumor metastasis, 6–8 week-old C57Bl/6 female mice pre-educated with PDAC-derived exosomes were injected intra-splenically with 1 × 106 PAN02 mCherry cells resuspended in 30μl of Matrigel (Corning) as previously described19,72,73. Either 24 hours or 21 days later, mice were sacrificed and livers were analyzed for metastatic lesions by counting mCherry+ cells by fluorescence microscopy or measuring liver weights. All animals were monitored for abnormal tissue growth or ill effects according to AAALAS guidelines and sacrificed if excessive deterioration of animal health was observed.
GFP Bone Marrow transplantation
Bone Marrow (BM) transplantation was performed by reconstituting the BM of lethally irradiated (950 rads) C57Bl/6 female 6 week-old mice via retro-orbital injection of 5 × 106 total BM cells isolated from eGFP-transgenic mice (Jackson Laboratory). After 4 weeks, the eGFP BM-reconstituted C57Bl/6 mice were educated with PBS or PAN02 exosomes for 21 days.
Tissue processing and immunofluorescence
For histological analysis, tissues were dissected and fixed in a mix of 2% PFA and 20% sucrose solution overnight, then embedded in Tissue-tek O.C.T. (Electron Microscopy Sciences). Blocks were frozen in a dry ice and ethanol bath. For immunofluorescence, 6μm O.C.T. tissue cryosections were stained with antibodies against F4/80 (14-4801-85, 1:100, eBioscience), fibronectin (sc59826, 1:50, Santa Cruz), αSMA (CBL171, 1:500, EMD Millipore), Gr-1 (ab25377, 1:50, Abcam), CD31 (sc59906, 1:100, Santa Cruz), S100A4 (ab27957, 1:100, Abcam), epCAM (sc59906, 1:50, Santa Cruz), and TGFβ (ab66043, 1:100, Abcam). In some experiments, ECM evaluation was done by using antibodies directed to collagen I (ab6308, 1:100, Abcam), vitronectin (sc15332, 1:100, Santra Cruz), and tenascin C (ab6346, 1:100, Abcam). Secondary antibodies conjugated to Alexa Fluor 488, 555, or 594 were used (a11007, a11001, or a21424, respectively, 1:1000, Life Technologies). GFP and mCherry+ cells were detected by their intrinsic signal. Fluorescent images were obtained using a Nikon confocal microscope (Eclipse TE2000U) and analyzed using Nikon software (EZ-C1 3.6). FN and αSMA expression were quantified using ImageJ Software (NIH) by determining the ratio between the areas of FN and DAPI staining, expressed in arbitrary units (A.U).
SDS PAGE and Western blot
Equal amounts of cell lysate or exosome lysate were resuspended in 1.5X Laemmli buffer, subsequently incubated at 95°C for 5 min and centrifuged in a microcentrifuge at 10,000rpm for 5 min. Samples were separated on a Novex 4–12% Bis-Tris Plus Gel (Life technologies), and transferred onto a PVDF membrane (Milipore). The membrane was activated in 100% methanol and rinsed with ddH2O prior to transfer.
After transfer, membranes were processed for Ponceau red staining. For that, PVDF membranes were rinsed in ddH2O and immersed in 100% methanol for 5 seconds. Membranes were dried for 15 min on Whatmann paper and reactivated in 100% methanol for 5 seconds. Reactivated membranes were rinsed ddH2O and incubated with Ponceau red solution (BIORAD) for 3 mins. The de-stained membranes were then used further for antibody incubations in ready-to-use Odyssey blocking buffer (OBB, Li-COR, Part No. 927-40000) and blocked for 1 hour at room temperature (RT). Primary antibody anti-MIF (ab175189, 1:1500, Abcam) was diluted in OBB containing 0.1% Tween-20, and incubated overnight at 4°C. Membranes were then washed 4X (5 min each) with TBS containing 0.1% Tween-20 (TBS-T) at RT. Secondary antibodies conjugated to anti-rabbit IRDye 800CW (Li-COR, 1:20000) were diluted in OBB containing 0.1% Tween-20 and 0.01% SDS. Membranes were incubated with secondary antibody solutions for 1 hour at RT with boxes wrapped in aluminum foil to prevent light exposure. Afterwards membranes were washed 4 times (5 min each) with TBS-T at RT in the dark, and then briefly rinsed 2 times (2 min each) in PBS before scanning. Membranes were scanned and analyzed using an OdysseyH IR scanner using OdysseyH imaging software 3.0. Scan settings were lowest image quality, 42μm resolution, auto intensity for both 700 and 800 channels with no offset.
Flow cytometry analysis
For labeled exosome tracking and phenotypic analysis of murine organs, femurs were flushed and livers were mechanically dissociated, and single cell suspensions were filtered through a 40μm strainer. Cells were washed in PBS with 1% BSA and incubated with anti-CD11b-PerCP-Cyanine5.5 (clone M1/70, 1:100, BD Biosciences) and anti-F4/80-APC (clone BM8, 1:100, eBioscience) antibodies at pre-determined saturating concentrations. PKH67-labeled exosome positive cells were detected using blue laser excitation and 488nm emission. Data for 1,000,000 cells was acquired on a BD FACS Canto™ cytometer with Diva software (BD) and was analyzed using FlowJo™ software (TreeStar).
Statistical and Pathway Analysis
Error bars in graphical data represent means±s.e.m. Statistical significance was determined using a two-tailed Student’s t-test or by ANOVA. P < 0.05 was considered statistically significant. Statistical analyses were performed using GraphPad Prism software. Pathway analyses were performed using Ingenuity IPA software (Ingenuity Systems). No statistical method was used to predetermine sample size. The experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment.
Accession codes
The raw sequencing data for human Kupffer cells treated in vitro with PAN02 or normal pancreas exosomes (Supplementary Table 1) or BxPC-3 exosomes (Supplementary Table 2) have been deposited in the GEO database under accession number GSE66876. The raw data for proteomic analysis of PAN02 tumor exosomes presented in Table 3 and Table 4 have been deposited in the figshare database under the respective accession numbers: http://dx.doi.org/10.6084/m9.figshare.1333561 and http://dx.doi.org/10.6084/m9.figshare.1333563.
Supplementary Material
Acknowledgments
We thank D. L. Bajor (Vonderheide laboratory, University of Pennsylvania) for the gift of the R6560B cells. We thank L. Bojmar for carefully reviewing the paper. We thank S. Rudchenko and M. Barbu-Stevanovic at the Hospital for Special Surgery Fannie E. Rippel Foundation Flow Cytometry Core Facility for expert flow cytometry. We are supported by grants from the Children’s Cancer and Blood Foundation (H.P., D.L.), Manning Foundation (D.L.), Hartwell Foundation (D.L.), Champalimaud Foundation (D.L.), Fundacao para a Ciencia e a Tecnologia (D.L.), Nancy C and Daniel P Paduano Foundation (H.P., D.L.), Mary Kay Foundation (D.L.), Pediatric Oncology Experimental Therapeutic Investigator Consortium (D.L.), James Paduano Foundation (D.L., H.P.), Melanoma Research Alliance (H.P.), Sohn Conference Foundation (H.P.), Beth Tortolani Foundation (D.L., J.B.), Malcolm Hewitt Weiner Foundation (D.L.), Jose Carreras Leukemia Foundation (B.K.T.), Theodore Rapp Foundation (D.L.), American Hellenic Educational Progressive Association 5th District Cancer Research Foundation (D.L.), Charles and Marjorie Holloway Foundation (J.B.), Sussman Family Fund (J.B.), Lerner Foundation (J.B.), Breast Cancer Alliance (J.B.), and Manhasset Women’s Coalition Against Breast Cancer (J.B.).
Footnotes
AUTHOR CONTRIBUTIONS
B.C-S. developed the hypothesis, designed the experimental approach, performed experiments, analysed the data, coordinated the project and wrote the manuscript. N.M.A. conducted experiments. S.S., G.R. and T-L.S. performed immunostaining. H.Z. and Y.H. extracted RNA. B.K.T. and A.B. performed western blots. A.H., T-M.T., C.W. and Y.A. maintained mouse colonies. M.T.M. and H.M. performed proteomic analysis. J.X. and T.Z. processed samples and analysed RNA sequencing data. G.G-S. and C.W. processed human samples. P.M.G., M.A.H., K.J.L., I.M.B.L., E.H.K., A.J.O., J.H., A.D., M.J., K.M., S.K.B. andW.R.J. collected patient samples and managed clinical records. S.H.E. contributed to writing the manuscript. R.E.S., I.M., H.P. and B.Z.S. contributed to hypothesis generation, experimental design and data interpretation. J.B. coordinated the project and interpreted data. D.L. conceived the hypothesis, led the project, interpreted data and wrote the manuscript.
References
- 1.Saif MW. Pancreatic neoplasm in 2011: an update. JOP : Journal of the pancreas. 2011;12:316–321. [PubMed] [Google Scholar]
- 2.Chan A, Diamandis EP, Blasutig IM. Strategies for discovering novel pancreatic cancer biomarkers. Journal of proteomics. 2013;81:126–134. doi: 10.1016/j.jprot.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fesinmeyer MD, Austin MA, Li CI, De Roos AJ, Bowen DJ. Differences in survival by histologic type of pancreatic cancer. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2005;14:1766–1773. doi: 10.1158/1055-9965.EPI-05-0120. [DOI] [PubMed] [Google Scholar]
- 4.Arscott WT, Camphausen KA. EGFR isoforms in exosomes as a novel method for biomarker discovery in pancreatic cancer. Biomarkers in medicine. 2011;5:821. doi: 10.2217/bmm.11.80. [DOI] [PubMed] [Google Scholar]
- 5.Record M, Carayon K, Poirot M, Silvente-Poirot S. Exosomes as new vesicular lipid transporters involved in cell-cell communication and various pathophysiologies. Biochimica et biophysica acta. 2014;1841:108–120. doi: 10.1016/j.bbalip.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 6.SELA, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nature reviews. Drug discovery. 2013;12:347–357. doi: 10.1038/nrd3978. [DOI] [PubMed] [Google Scholar]
- 7.Choi DS, Kim DK, Kim YK, Gho YS. Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. Proteomics. 2013;13:1554–1571. doi: 10.1002/pmic.201200329. [DOI] [PubMed] [Google Scholar]
- 8.Martins VR, Dias MS, Hainaut P. Tumor-cell-derived microvesicles as carriers of molecular information in cancer. Current opinion in oncology. 2013;25:66–75. doi: 10.1097/CCO.0b013e32835b7c81. [DOI] [PubMed] [Google Scholar]
- 9.Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Seminars in cancer biology. 2011;21:139–146. doi: 10.1016/j.semcancer.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 10.Thakur BK, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell research. 2014 doi: 10.1038/cr.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tetta C, Ghigo E, Silengo L, Deregibus MC, Camussi G. Extracellular vesicles as an emerging mechanism of cell-to-cell communication. Endocrine. 2013;44:11–19. doi: 10.1007/s12020-012-9839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valadi H, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nature cell biology. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 13.Zoller M. Pancreatic cancer diagnosis by free and exosomal miRNA. World journal of gastrointestinal pathophysiology. 2013;4:74–90. doi: 10.4291/wjgp.v4.i4.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaplan RN, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sceneay J, Smyth MJ, Moller A. The pre-metastatic niche: finding common ground. Cancer metastasis reviews. 2013;32:449–464. doi: 10.1007/s10555-013-9420-1. [DOI] [PubMed] [Google Scholar]
- 16.Peinado H, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nature medicine. 2012;18:883–891. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hood JL, San RS, Wickline SA. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer research. 2011;71:3792–3801. doi: 10.1158/0008-5472.CAN-10-4455. [DOI] [PubMed] [Google Scholar]
- 18.Corbett TH, et al. Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer research. 1984;44:717–726. [PubMed] [Google Scholar]
- 19.Little EC, et al. Novel immunocompetent murine models representing advanced local and metastatic pancreatic cancer. The Journal of surgical research. 2012;176:359–366. doi: 10.1016/j.jss.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hingorani SR, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 21.Rhim AD, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Achyut BR, Yang L. Transforming growth factor-beta in the gastrointestinal and hepatic tumor microenvironment. Gastroenterology. 2011;141:1167–1178. doi: 10.1053/j.gastro.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayashi H, Sakai T. Biological Significance of Local TGF-beta Activation in Liver Diseases. Frontiers in physiology. 2012;3:12. doi: 10.3389/fphys.2012.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wight TN, Potter-Perigo S. The extracellular matrix: an active or passive player in fibrosis? American journal of physiology. Gastrointestinal and liver physiology. 2011;301:G950–955. doi: 10.1152/ajpgi.00132.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gressner AM, Weiskirchen R, Breitkopf K, Dooley S. Roles of TGF-beta in hepatic fibrosis. Frontiers in bioscience : a journal and virtual library. 2002;7:d793–807. doi: 10.2741/A812. [DOI] [PubMed] [Google Scholar]
- 26.Cong M, Iwaisako K, Jiang C, Kisseleva T. Cell signals influencing hepatic fibrosis. International journal of hepatology. 2012;2012:158547. doi: 10.1155/2012/158547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawelke N, et al. Fibronectin protects from excessive liver fibrosis by modulating the availability of and responsiveness of stellate cells to active TGF-beta. PloS one. 2011;6:e28181. doi: 10.1371/journal.pone.0028181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu G, et al. Gene expression and synthesis of fibronectin isoforms in rat hepatic stellate cells. Comparison with liver parenchymal cells and skin fibroblasts. The Journal of pathology. 1997;183:90–98. doi: 10.1002/(SICI)1096-9896(199709)183:1<90::AID-PATH1105>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 29.Tojo M, et al. The ALK-5 inhibitor A-83-01 inhibits Smad signaling and epithelial-to-mesenchymal transition by transforming growth factor-beta. Cancer science. 2005;96:791–800. doi: 10.1111/j.1349-7006.2005.00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duffield JS, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. The Journal of clinical investigation. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lau C, et al. Role of pancreatic cancer-derived exosomes in salivary biomarker development. The Journal of biological chemistry. 2013;288:26888–26897. doi: 10.1074/jbc.M113.452458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heinrichs D, et al. Macrophage migration inhibitory factor (MIF) exerts antifibrotic effects in experimental liver fibrosis via CD74. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:17444–17449. doi: 10.1073/pnas.1107023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barnes MA, et al. Macrophage migration inhibitory factor contributes to ethanol-induced liver injury by mediating cell injury, steatohepatitis, and steatosis. Hepatology. 2013;57:1980–1991. doi: 10.1002/hep.26169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Funamizu N, et al. Macrophage migration inhibitory factor induces epithelial to mesenchymal transition, enhances tumor aggressiveness and predicts clinical outcome in resected pancreatic ductal adenocarcinoma. International journal of cancer. Journal international du cancer. 2012 doi: 10.1002/ijc.27736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nanji AA, et al. Macrophage migration inhibitory factor expression in male and female ethanol-fed rats. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research. 2001;21:1055–1062. doi: 10.1089/107999001317205187. [DOI] [PubMed] [Google Scholar]
- 36.Shin HN, Moon HH, Ku JL. Stromal cell-derived factor-1alpha and macrophage migration-inhibitory factor induce metastatic behavior in CXCR4- expressing colon cancer cells. International journal of molecular medicine. 2012;30:1537–1543. doi: 10.3892/ijmm.2012.1141. [DOI] [PubMed] [Google Scholar]
- 37.Zhang HY, et al. Macrophage migration inhibitory factor expression correlates with inflammatory changes in human chronic hepatitis B infection. Liver international : official journal of the International Association for the Study of the Liver. 2005;25:571–579. doi: 10.1111/j.1478-3231.2005.01047.x. [DOI] [PubMed] [Google Scholar]
- 38.Adamali H, et al. Macrophage migration inhibitory factor enzymatic activity, lung inflammation, and cystic fibrosis. American journal of respiratory and critical care medicine. 2012;186:162–169. doi: 10.1164/rccm.201110-1864OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kobayashi S, Nishihira J, Watanabe S, Todo S. Prevention of lethal acute hepatic failure by antimacrophage migration inhibitory factor antibody in mice treated with bacille Calmette-Guerin and lipopolysaccharide. Hepatology. 1999;29:1752–1759. doi: 10.1002/hep.510290610. [DOI] [PubMed] [Google Scholar]
- 40.Yaddanapudi K, et al. Control of tumor-associated macrophage alternative activation by macrophage migration inhibitory factor. Journal of immunology. 2013;190:2984–2993. doi: 10.4049/jimmunol.1201650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen PF, et al. ISO-1, a macrophage migration inhibitory factor antagonist, inhibits airway remodeling in a murine model of chronic asthma. Molecular medicine. 2010;16:400–408. doi: 10.2119/molmed.2009.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Javle M, et al. Biomarkers of TGF-beta signaling pathway and prognosis of pancreatic cancer. PloS one. 2014;9:e85942. doi: 10.1371/journal.pone.0085942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ellermeier J, et al. Therapeutic efficacy of bifunctional siRNA combining TGF- beta1 silencing with RIG-I activation in pancreatic cancer. Cancer research. 2013;73:1709–1720. doi: 10.1158/0008-5472.CAN-11-3850. [DOI] [PubMed] [Google Scholar]
- 44.Gaspar NJ, et al. Inhibition of transforming growth factor beta signaling reduces pancreatic adenocarcinoma growth and invasiveness. Molecular pharmacology. 2007;72:152–161. doi: 10.1124/mol.106.029025. [DOI] [PubMed] [Google Scholar]
- 45.Melisi D, et al. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Molecular cancer therapeutics. 2008;7:829–840. doi: 10.1158/1535-7163.MCT-07-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumour microenvironment. Nature reviews. Cancer. 2013;13:788–799. doi: 10.1038/nrc3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ijichi H, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes & development. 2006;20:3147–3160. doi: 10.1101/gad.1475506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hezel AF, et al. TGF-beta and alphavbeta6 integrin act in a common pathway to suppress pancreatic cancer progression. Cancer research. 2012;72:4840–4845. doi: 10.1158/0008-5472.CAN-12-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bayon LG, et al. Role of Kupffer cells in arresting circulating tumor cells and controlling metastatic growth in the liver. Hepatology. 1996;23:1224–1231. doi: 10.1002/hep.510230542. [DOI] [PubMed] [Google Scholar]
- 50.Kruse J, et al. Macrophages promote tumour growth and liver metastasis in an orthotopic syngeneic mouse model of colon cancer. International journal of colorectal disease. 2013;28:1337–1349. doi: 10.1007/s00384-013-1703-z. [DOI] [PubMed] [Google Scholar]
- 51.Wen SW, Ager EI, Christophi C. Bimodal role of Kupffer cells during colorectal cancer liver metastasis. Cancer biology & therapy. 2013;14:606–613. doi: 10.4161/cbt.24593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grzesiak JJ, et al. Knockdown of the beta(1) integrin subunit reduces primary tumor growth and inhibits pancreatic cancer metastasis. International journal of cancer. Journal international du cancer. 2011;129:2905–2915. doi: 10.1002/ijc.25942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saito N, et al. Inhibition of hepatic metastasis in mice treated with cell-binding domain of human fibronectin and angiogenesis inhibitor TNP-470. International journal of clinical oncology. 2001;6:215–220. doi: 10.1007/pl00012108. [DOI] [PubMed] [Google Scholar]
- 54.Zvibel I, Halpern Z, Papa M. Extracellular matrix modulates expression of growth factors and growth-factor receptors in liver-colonizing colon-cancer cell lines. International journal of cancer. Journal international du cancer. 1998;77:295–301. doi: 10.1002/(sici)1097-0215(19980717)77:2<295::aid-ijc20>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 55.Porembka MR, et al. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer immunology, immunotherapy : CII. 2012;61:1373–1385. doi: 10.1007/s00262-011-1178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamamoto M, et al. TSU68 prevents liver metastasis of colon cancer xenografts by modulating the premetastatic niche. Cancer research. 2008;68:9754–9762. doi: 10.1158/0008-5472.CAN-08-1748. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Y, Davis C, Ryan J, Janney C, Pena MM. Development and characterization of a reliable mouse model of colorectal cancer metastasis to the liver. Clinical & experimental metastasis. 2013;30:903–918. doi: 10.1007/s10585-013-9591-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seubert B, et al. Tissue inhibitor of metalloproteinases (TIMP)-1 creates a premetastatic niche in the liver through SDF-1/CXCR4-dependent neutrophil recruitment in mice. Hepatology. 2015;61:238–248. doi: 10.1002/hep.27378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kato R, et al. A new type of antimetastatic peptide derived from fibronectin. Clinical cancer research : an official journal of the American Association for Cancer Research. 2002;8:2455–2462. [PubMed] [Google Scholar]
- 60.Bissell DM. Therapy for hepatic fibrosis: revisiting the preclinical models. Clinics and research in hepatology and gastroenterology. 2011;35:521–525. doi: 10.1016/j.clinre.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 61.Korpal M, Kang Y. Targeting the transforming growth factor-beta signalling pathway in metastatic cancer. European journal of cancer. 2010;46:1232–1240. doi: 10.1016/j.ejca.2010.02.040. [DOI] [PubMed] [Google Scholar]
- 62.Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gu G, Brown JR, Melton DA. Direct lineage tracing reveals the ontogeny of pancreatic cell fates during mouse embryogenesis. Mechanisms of development. 2003;120:35–43. doi: 10.1016/s0925-4773(02)00330-1. [DOI] [PubMed] [Google Scholar]
- 70.Zhong S, et al. High throughput illuma strand-specific RNA sequencing library preparation. Cold Spring Harbor Protocols. 2011;2011:940–949. doi: 10.1101/pdb.prot5652. [DOI] [PubMed] [Google Scholar]
- 71.Sakai T, et al. Plasma fibronectin supports neuronal survival and reduces brain injury following transient focal cerebral ischemia but is not essential for skin-wound healing and hemostasis. Nat Med. 2001;7:324–330. doi: 10.1038/85471. [DOI] [PubMed] [Google Scholar]
- 72.Suemizu H, et al. A versatile technique for the in vivo imaging of human tumor xenografts using near-infrared fluorochrome-conjugated macromolecule probes. PLoS ONE. 2013;8:e82708. doi: 10.1371/journal.pone.0082708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morikawa K, Walker SM, Jessup JM, Fidler IJ. In vivo selection of highly metastatic cells from surgical specimens of different primary human colon carcinomas implanted into nude mice. Cancer Research. 1988;48:1943–1948. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.