

Abstract
Cholesterol synthesis and clearance by astrocytes are tightly regulated to maintain constant levels within the brain. In this context, liver X receptors (LXRs) are the master regulators of cholesterol homeostasis in the central nervous system (CNS). Increasing levels of cholesterol in astrocytes trigger LXR activation leading to the transcription of target genes involved in cholesterol trafficking and efflux, including apolipoprotein E (ApoE), cytochrome P450 enzymes, sterol regulatory binding protein (SREBP), and several ATP-binding cassette (ABC) transporter proteins. The disturbance of LXR signaling in the brain can lead to significant dysfunctions in cholesterol homeostasis, and disruptions in this pathway have been implicated in numerous neurological diseases including Alzheimer’s disease and Huntington’s disease. HIV infection of the CNS in combination with cocaine use is associated with astrocyte and neuronal energy deficit and damage. We propose that dysregulation in CNS cholesterol metabolism may be involved in the progression of HIV-associated neurocognitive disorders (HAND) and in cocaine-mediated neurocognitive impairments. We hypothesize that exposure of astrocytes to cocaine and the HIV protein Tat will disrupt LXR signaling. Alterations in these pathways will in turn, affect cholesterol bioavailability for neurons. Our data show that exposure of astrocytes to cocaine and HIV-Tat significantly decreases LXRβ levels, downstream signaling and bioavailability of cholesterol. Taken together, these data uncover novel alterations in a bioenergetic pathway in astrocytes exposed to cocaine and the HIV protein Tat. Results from these studies point to a new pathway in the CNS that may contribute to HAND in HIV+ cocaine user individuals.
Keywords: astrocytes, cholesterol, HIV, cocaine, metabolism
Introduction
The human brain has the highest content of cholesterol in the body as it contains 20-25% of the body’s total cholesterol (Björkhem and Meaney 2004; Dietschy 2009). The brain requires an intricate balance of cholesterol homeostasis to meet the high metabolic requirements of neurons and to maintain synaptodendritic connectivity (Goritz et al. 2005; Pfenninger 2009). Because the blood brain barrier prevents uptake of lipids from the periphery, neurons rely on cholesterol synthesized de novo by astrocytes for synaptogenesis, synaptic vesicle formation, and neuronal plasticity (Mauch et al. 2001; Pfrieger and Ungerer 2011; Ullian et al. 2004; van Deijk et al. 2017; Vance 2012). Additionally, cholesterol is the major component of the myelin sheath (Dietschy and Turley 2004). Neuronal fitness can be impaired under conditions of cholesterol deficiency or surplus (Ko et al. 2005; Linetti et al. 2010). Astrocytes are the primary suppliers of the cholesterol needed by both neurons and oligodendrocytes (Pfrieger and Ungerer 2011). Cholesterol levels and trafficking in the brain are tightly regulated by an elaborate feedback mechanism that coordinates biosynthesis, efflux, uptake, and release of cholesterol. Liver X Receptors (LXRs) play a central role in regulating the exchange of cholesterol between astrocytes and neurons (Courtney and Landreth 2016). LXRs are transcription factors dependent on heterodimerization with the retinoic acid receptor (RXR) and ligand binding to both receptors by oxysterols and 9-cis retinoic acid, respectively. Target genes include Apolipoprotein E (ApoE), sterol regulatory binding proteins (SREBP), acetyl CoA decarboxylase, fatty acid synthetase, GFAP, and several ATP-binding cassette transporter (ABC) proteins. In the absence of ligand agonists, LXR and RXR are bound by co-repressors to inhibit the expression of target genes. Activation of transcription occurs upon agonist binding, conformational change of the receptor complex, release of repressors and association with co-activators (Spann and Glass 2013). Apolipoprotein E (ApoE) is a major lipid carrier in the brain (Kim et al. 2009) and a variant of ApoE, ApoE4, has been widely implicated as being a risk factor in the development of Alzheimer’s disease (AD), thus supporting the importance of ApoE in maintaining proper neural functioning (Kim et al. 2009; Vance 2012). ABCa1 works in accord with ApoE as the major cholesterol transporter to regulate ApoE lipidation (Tachikawa et al. 2005). The disturbance of LXR signaling in the brain can lead to significant dysfunction in cholesterol homeostasis and has been implicated in numerous neurological diseases including AD and Huntington’s disease (HD) (Leoni and Caccia 2015; Martín et al. 2014). In this context, disruptions in peripheral cholesterol flux are also observed in HIV-infected individuals, thereby contributing to increased risk of atherosclerosis and cardiovascular disease (Bandaru et al. 2013; Feeney et al. 2013; Mujawar et al. 2006). Several studies suggest that dysregulation in CNS lipid metabolism may be involved in HIV-associated neurocognitive disorders (HAND) pathogenesis; however, the underlying mechanisms remain unclear. A major contributor to the development of HAND is the use of illicit drugs such as cocaine (Larrat and Zierler 1993). In fact, cocaine use is a major risk factor for becoming infected with HIV and together, HIV and cocaine synergize to increase cellular dysfunction in astrocytes in the brain (Buch et al. 2012; Cai et al. 2016; Dahal et al. 2015; Hauser and Knapp 2014; Reynolds et al. 2006; Yang et al. 2016). Likewise, HIV infection and cocaine use are associated with myelin loss and synaptodendritic damage (Dash et al. 2011; Fitting et al. 2013; Kim et al. 2003; Periyasamy et al. 2016), suggesting that dysregulation in CNS cholesterol metabolism may be involved in the progression of neurocognitive impairment. Several studies report toxic synergy between cocaine and HIV proteins such as Tat on cells of the CNS, including astrocytes (Gandhi et al. 2010; Wayman et al. 2015). Therefore, given the importance of astrocytes in LXR-mediated cholesterol regulation and their role in providing metabolic support to other CNS cells, we hypothesized that exposure of astrocytes to cocaine and to the HIV protein Tat would lead to disruptions in LXR signaling and its target gene expression. Alterations in these pathways would in turn affect the bioavailability of cholesterol from astrocytes to neurons and oligodendrocytes, and promote neuronal dysfunction leading to increased neurotoxicity and likely exacerbate HAND. Here, we examine the impact of cocaine and HIV-1 Tat on LXR signaling and cholesterol homeostasis in astrocytes. In combination, cocaine and HIV-1 Tat disrupted LXR signaling and its target genes leading to reduced release of ApoE-bound cholesterol by astrocytes which negatively influenced neuronal cholesterol uptake and neuronal synaptic properties.
Materials and Methods
Animals
This study used doxycycline (Dox)-inducible GFAP promoter driven HIV-1 Tat transgenic mice generously provided to the Comprehensive NeuroAIDS Center by Dr. Johnny He and C57BL/6J wild-type mice (Kim et al. 2003). Animal care and experimental procedures were conducted according to the Guide for the Care and Use of Laboratory Animals. The Institutional Animal Care and Use Committee of Temple University approved all experimental protocols. Mice were housed in single-sex groups of 4-5 animals in an animal facility with constant airflow controlled temperature (21–23°C), a 12-hour light/dark cycle, and the mice were supplied with food and water ad libitum. The mice were divided into four groups with 5 mice per group both male and female. Cocaine hydrochloride (Sigma-Aldrich, St. Louis, MO) was dissolved in sterile water, sterile-filtered, and administered at 15 mg/kg intraperitoneally (i.p.) once per day for 14 days. For Tat induction, Tat mice were injected i.p. with doxycycline hyclate (Dox) (Sigma-Aldrich) for 7 days at a dosage of 80 mg/kg/day. The control (Con) group received saline injections over the course of the experiment. The Tat only (Tat) group was administered Dox for 7 days. The cocaine only (Coc) group received 14 days of cocaine (15 mg/kg i.p.). The cocaine plus Tat group was pretreated for 7 days with Dox, followed by 14 days of cocaine. All mice received a last dose of cocaine (15 mg/kg i.p.) 24 hours before euthanasia.
Tissue Harvest
Brains from mice were quickly removed after euthanasia and placed into ice-cold 1X PBS. Brains were dissected into hippocampus, striatum and frontal cortex and stored at −80°C.
Protein Extraction from Tissue
Frozen brain tissue was homogenized by mechanical dounce disruption on ice in TNN buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.5% NP40, 1: 100 protease inhibitor cocktail; Calbiochem, San Diego, CA, USA). Tissue homogenates were centrifuged at 14,000 rpm at 4°C for 5 min. The supernatant containing proteins was collected and protein concentrations were determined using the Pierce™ 660nm Protein Assay (Thermo).
Primary Cell Culture
Human primary neurons and astrocytes were provided by the Temple University Comprehensive NeuroAIDS Center. In brief, fetal brain tissue (gestational age 16-18 weeks) was obtained from elective abortion procedures performed in full compliance with National Institutes of Health and Temple University ethical guidelines. Briefly, the tissue was washed with cold Hanks balanced salt solution (HBSS) and meninges and blood vessels were removed. For astrocytes, after tissue dissociation in trypsin, cells were plated in mixed glial growth media; DME:F12 (1:1) media supplemented with insulin, 10% FBS, Amphotericin B, L-glutamine, and gentamicin. The mixed culture was maintained under 10% CO2 for 5 days, and the media was changed to remove any cell debris. To enrich for astrocytes, flasks were placed on orbital shaker for 14-18h at 200 rpm in glial growth media. Detached cells constituted the microglial component of the culture and were removed leaving astrocytes attached. Astrocytes were cultured in glial growth media at 37°C, 5% CO2. For primary neuron isolation, brain tissue in HBSS was digested with papain (Sigma-Aldrich, St. Louis, MO) for 30 min at 37°C. The tissue was further dissociated to obtain single-cell suspensions by repeated pipetting. Neurons were plated at a density of approximately 1.8 × 106 cells/60mm dish coated with poly-D lysine in neurobasal media with B27 supplement, 5% horse serum, and gentamicin (Invitrogen). After approximately 2h, the media was changed and unattached cells were discarded. Twenty-four hours later, cultures were re-fed with a complete change of neurobasal media without horse serum. Four days later, one fourth of the media was removed and replaced with neurobasal media supplemented with fluoro-deoxyuridine (FDU) and uridine. Following FDU treatment, neurons were maintained in neurobasal media containing Glutamax and B27 supplement, with half-media changes every other day. Purity of cell types was assessed by immunolabeling for cell-type specific markers including MAP2 and neurofilament for neurons and GFAP for astrocytes. Neurons and astrocytes were cultured separately or in co-culture separated by 24mm transwell insert with 1.0μm pore polycarbonate membrane.
Cocaine and Tat Treatments
Cocaine hydrochloride (Sigma-Aldrich) was dissolved in sodium citrate buffer (pH 5.0) to increase its stability in culture during incubation. Ten millimolar stock solution and subsequent dilutions were prepared in fresh daily in medium and added directly to cell cultures. Fresh medium was used as the control vehicle during cocaine treatments. A physiologically relevant dose of 5 μM was utilized for all experiments. Recombinant Tat HIV-1 MN (ImmunoDX, 101 amino acids) was utilized for all experiments at a concentration of 50ng/ml. Cocaine and/or Tat were administered at the same time every 24h for 48h unless otherwise stated.
Cell Harvest and Protein Extraction
Following treatments, cells were washed in 1X PBS, scraped from the culture plate and collected in ice-cold PBS. The cell suspension was centrifuged at 14,000rpm for 10 min at 4°C and the supernatant was discarded. The cell pellet was then re-suspended in ice-cold RIPA lysis buffer (Thermo) containing protease and phosphatase inhibitor cocktails (Thermo), vortexed, and incubated on ice for 25 min to complete cell lysis. Samples were then centrifuged at 14,000rpm for 10 min at 4°C to separate insoluble material. The supernatant containing proteins was collected and placed into pre-chilled Eppendorf tubes and samples were stored at −80°C until protein analysis was performed.
Western Blotting
Equal amounts of protein were loaded onto pre-cast midi-gels (4–12% Bis-Tris; Invitrogen), separated by electrophoresis and transferred onto nitrocellulose membranes using the iBlot™ 2 gel transfer device (Thermo). Membranes were blocked in 5% non-fat milk or 5% bovine serum albumin (BSA) in Tris-buffered saline, 0.1% Tween-20 (TBST) for 1h before incubation with primary antibodies. Primary antibodies included: ABCa1 (1:500, Abcam), apoE (1:1000), LXRβ (1:1000), CYP46 (1:1,000), LDLR (1:500), SREBP2 (1:1000), HMCGR (1:1000), PSD95 (1:1000) (all from Abcam) and loading control GAPDH (1:1000; Santa Cruz). Membranes were incubated with primary antibodies overnight at 4°C, washed in 1X TBST, incubated with appropriate secondary anti-mouse or -rabbit antibodies (1:10,000; Thermo Scientific) for 1h, and developed with ECL Prime (Amersham Pharmacia Biotech, Piscataway, NJ). Band intensities were calculated using ImageJ software and normalized to the loading control (Rasband 1997).
Cell Viability
Cell viability was measured by the MTT assay according to manufacturer’s protocol (Sigma). Cells were seeded at 3 × 105 cells/mL in a 12-well plate and treated with cocaine and/or Tat every 24h for 48h. MTT solution (150μL) was added to each well and incubated for 2h. The culture medium was aspirated and 1mL of MTT solvent was added to each well. Cell viability was determined by the formation of formazan crystal and measured absorbance at 590nm and 620nm.
Gene Expression Analysis
To determine changes over time in relative levels of ApoE, ABCa1, and LXRβ following treated with cocaine and Tat, RNA was isolated from astrocytes (5 × 105 cells/mL) by RNeasy Mini Kit (Qiagen) according to manufacturer’s protocol and cDNA was synthesized from 500ng of RNA using the iScript™ cDNA Synthesis Kit (BioRad). qPT-PCR was performed using the following Taqman probes and primers (IDT): ABCa1 (Forward 5′-GGTGGTGTTCTTCCTCATTACT-3′ Reverse 5′-CCGCCTCACATCTTCATCTT-3′, Probe 5′-TCAGGCCCAGACCTGTAAATGCAA-3′); ApoE (Forward 5′- AGGTCACCCAGGAACTGA-3′ Reverse 5′- TTGTTCCTCCAGTTCCGATTT-3′ Probe 5′- ACGAGACCATGAAGGAGTTGAAGGC- 3′); LXRβ (Forward 5′- TTCCTCTTCCTAGGGTGGAA-3′ Reverse 5′- TTATCCCAAGGGATGAGAGC-3′Probe 5′- CCCTGGGCCGAGCCTGTAGA-3′); HMCGR (Forward 5′- TGAAGGGTTCGCAGTGATAAA-3′ Reverse 5′- CCTGGACTGGAAACGGATATAAA-3′ Probe 5′-TCTGCTAGTGTCAAATGCCTCC-3′) LDLR (Forward 5′- CTCCCGCCAAGATCAAGAAA-3′ Reverse 5′- GTTTGGAGTCAACCCAGTAGAG-3′ Probe 5′- TGGACATCTACTCGCTGGTGACTGA-3′) Beta Actin (Forward 5′- GCCCTGGACTTCGAGCAAGA-3′ Reverse 5′- GGAAGGCTGGAAGAGTGCCT-3′ Probe 5′- TGGCCACGGCTGCTTCCAGCTCC-3′). Quantitative real-time PCR was performed on the Roche LightCycler 96 using Taqman Fast Universal Master Mix (Thermo). After the addition of primers, probe, and template DNA to the master mix, PCR thermal cycle parameters were as follow: 95°C for 10 minutes, 45 cycles at 95°C for 15 seconds and 60°C for 60 seconds. Beta actin was used as a control to normalize for differences in the amounts of RNA in each sample.
ApoE Release
Conditioned media from cocaine-, Tat-, and cocaine + Tat- treated astrocytes was concentrated 20-fold using Pierce Protein Concentrator PES (Thermo) and analyzed by ELISA for levels of secreted ApoE. Any cells were removed by centrifugation and the remaining supernatants were analyzed using a human ApoE ELISA kit according to manufacturer’s instructions (Thermo). One-hundred microliters of each sample was measured in duplicate and ApoE levels were assessed by absorbance at 450 nm.
Measurement of Cellular Cholesterol
Astrocytes were seeded in six-well plates (5 × 105 cells/well) and 48h later culture media was replaced with serum-free DMEM/F12 for cocaine and Tat treatments. Serum-free conditioned medium from astrocytes was evaluated using Amplex Red Cholesterol Assay Kit (Thermo) following manufacturer’s instructions.
Fillipin III staining
To visualize intracellular cholesterol accumulation, astrocytes were plated on to glass-bottomed dishes and treated with cocaine and Tat once per day for 48h. The Cholesterol Assay Kit (Abcam) was utilized according to the manufacture’s protocol to visualize cholesterol through its interaction with Filipin III. Cells were visualized with the Leica Advanced Wide field imaging system (Leica Microsystems; Buffalo Grove, IL).
Immunocytochemical labeling
Following the treatments, cells plated on coverslips in 12-well plates were washed once with 1X PBS and fixed with 4 % formaldehyde for 15 min at room temperature. Following fixation, coverslips were washed with 1X PBS three times and cells were permeabilized with 0.2 % Triton X-100 for 15 min, washed again, and placed in blocking solution (5 % normal goat or horse serum; Vector Laboratories, Burlingame, CA) for 1h. Coverslips were then incubated in primary antibody MAP2 (1:200, Abcam) overnight at room temperature. Following incubation with primary antibodies, coverslips were rinsed 3X with PBS, and incubated with fluorescein isothiocyanate (FITC) (1:500; Vector Laboratories) or TRITC (1:500;Vector Laboratories)-conjugated secondary antibodies for 1h at room temperature in the dark. Coverslips were washed 3X with PBS, and mounted onto slides with an aqueous based mounting media containing DAPI for nuclear labeling (Vectashield; Vector Laboratories), and visualized with the Leica Advanced Widefield imaging system (Leica Microsystems; Buffalo Grove, IL). Fluorescence intensity was quantified using ImageJ Software (NIH, Rockville, MD).
Statistical analysis
All data were analyzed using either the student’s t-test or one-way analysis of variance (ANOVA) with post hoc testing where appropriate using GraphPad Prizm® (GraphPad Software Inc., San Diego, CA). Results were expressed as mean ± SEM, n ≥ 3. Values of p ≤ 0.05 were considered statistically significant.
Results
Effects of cocaine and HIV-1 Tat on LXRβ and target gene expression in astrocytes
To examine whether LXR signaling is disrupted by cocaine and/or HIV-1 Tat, we exposed astrocytes to cocaine, Tat, or both. We found that protein levels of LXRβ were significantly reduced in response to cocaine and Tat co-treatment compared to control (Figure 1A, B). LXRβ levels were unaffected in cocaine only and Tat only treated astrocytes whereas. LXRβ acts as a cholesterol sensor that is activated by binding to metabolites of cholesterol. This triggers the transcription of target genes involved in cholesterol efflux, ApoE and ABCa1. Therefore, we assessed changes in LXR downstream gene expression in response to cocaine+Tat-induced LXR deficits. Our data show that cocaine+Tat significantly decreased ApoE protein when compared to the control (Figure 1A, C). However, alone neither cocaine nor Tat had any effect. Additionally, ABCa1 was significantly reduced when exposed to Tat or to cocaine+Tat (Figure 1A, D). Taken together, these data show that cocaine and Tat have converging effects on LXR signaling that result in diminished LXR-mediated gene expression. To confirm that the reduction in LXR pathway signaling is not a result of cell death, we conducted the MTT cell viability assay. Our data show that astrocyte cell viability is unaffected by cocaine and Tat treatment (Figure 2) thereby, supporting disruption of LXR signaling by cocaine and Tat.
Figure 1. Cocaine and Tat affect the LXR signaling pathway and LXR-β downstream target gene expression in astrocytes.
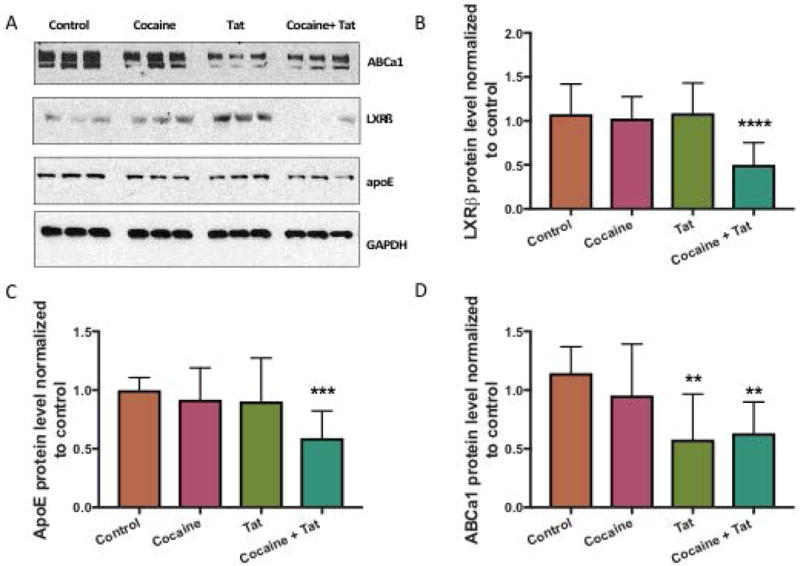
Astrocytes were exposed to cocaine (5μM), recombinant Tat (50ng/ml), or cocaine and Tat concurrently every 24 h for 48 h. A) Representative western blot of LXRβ and target genes ABCa1 and ApoE. GAPDH was used as a loading control. B) Analyses of LXRβ protein expression relative to GAPDH (****p< 0.0001) and its gene targets, C) ApoE (***p< 0.0005), D) ABCa1 (**p< 0.005). Abbreviations: LXRβ (Liver X Receptor Beta), ApoE (apolipoprotein E), ABCa1 (ATP-binding cassette transporter 1) and GAPDH (Glyceraldehyde 3-phosphate dehydrogenase). Results are expressed as mean ± SEM, n ≥ 3.
Figure 2. Cocaine and Tat do not affect astrocyte viability.
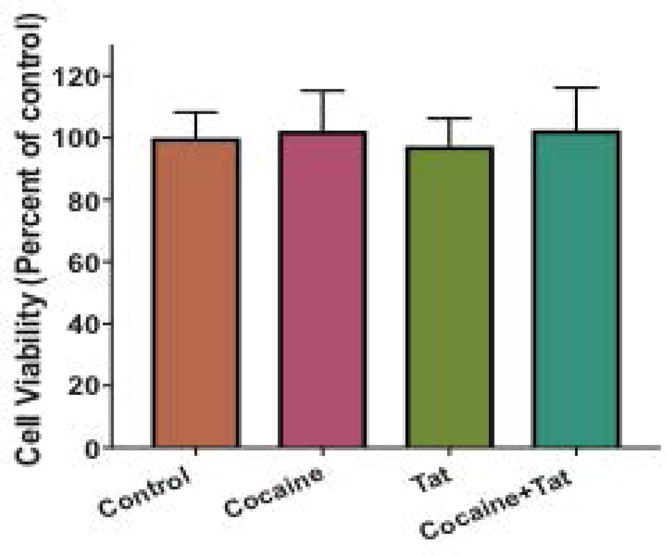
Astrocytes were treated with cocaine (5μM), recombinant Tat (50ng/ml), or cocaine and Tat concurrently every 24 h for 48 h. Astrocyte viability was assessed by the MTT assay and expressed relative to control levels. Abbreviations: MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide). Results are expressed as mean ± SEM, n ≥ 3.
Time course of LXR downstream mRNA expression reduction by cocaine and Tat in astrocytes
To better understand the combined effects of HIV-1 Tat and cocaine on astrocytes, the effects of cocaine and/or Tat were assessed after 1h, 2h, 6h, 24h, or 48h of exposure using quantitative RT-PCR (Figure 3). Cocaine+Tat treatment had minimal effects on LXRβ mRNA levels after 1h and 2h. However, by 6h LXRβ was significantly reduced by approximately 40% and this trend extended to 24h and 48h (Figure 3A). Similarly, ABCa1 mRNA expression was unaffected at 1h and 2h (Figure 3B). By 6h, ABCa1 mRNA levels decreased significantly and remained decreased through 48h. Interestingly, ApoE mRNA levels followed a cyclic pattern after cocaine+Tat treatment (Figure 3C). ApoE mRNA expression is significantly increased after 1h, but by 2h, expression returned to baseline. ApoE mRNA levels decreased significantly by 6h, but returned to normal levels at 24h. At 48h, a significant decrease in ApoE was detected. In summary, these data reveal that at the message level, the combined effects of cocaine and Tat occur early after exposure and have lasting effects.
Figure 3. Changes in LXR-β and downstream target gene mRNA expression in astrocytes over 48 hours following cocaine and Tat exposure.
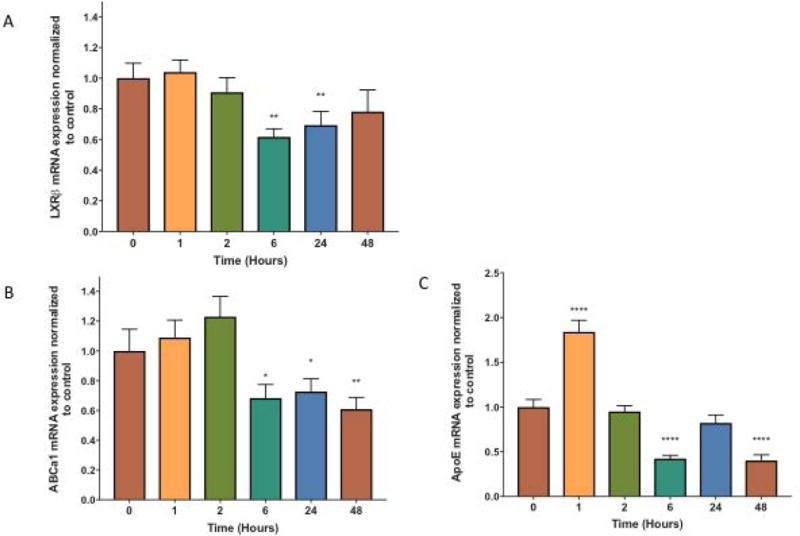
Astrocytes were treated with cocaine (5μM) and recombinant Tat (50ng/ml) concurrently for 1, 2, 6, 24, and 48 h. mRNA was extracted and reverse transcribed followed by quantitative real time PCR for A) LXRβ (p< 0.01**), B) ABCa1 (*p< 0.05, **p < 0.005), and C) ApoE (**** p< 0.0001). Results are expressed as mean ± SEM, n ≥ 3.
ApoE and Cholesterol Efflux following cocaine and Tat exposure in astrocytes
To test whether cocaine and Tat-induced dysfunction in LXR signaling results in alterations in cholesterol efflux, we assessed the media from cells incubated with media alone (control), cocaine, and/or Tat for release of ApoE and cholesterol using quantitative assays (Figure 4). The levels of secreted ApoE following cocaine or Tat alone did not change significantly when compared to control. However, astrocytes treated with both cocaine and Tat showed a decrease in ApoE levels in media following 48h exposure when compared to control (Figure 4). When assessing levels of cholesterol, our data showed that Tat, cocaine and Tat/cocaine reduced total levels of cholesterol, with Tat and Tat/cocaine reaching statistical significance (Figure 5A). Furthermore, intracellular cholesterol was reduced in all conditions but this reduction was greatest (50%) in astrocytes treated with both cocaine and Tat (Figure 5B). Similarly, cholesterol released into the astrocyte-conditioned media was reduced in all three treatment conditions. Yet, this reduction was only significant in the Tat, and cocaine/Tat groups where cholesterol levels were reduced by 83% and 73%, respectively (Figure 5C). Decreases in net cholesterol levels reflect a significant defect in cholesterol homeostasis in astrocytes when exposed to cocaine or Tat and this effect was exacerbated when astrocytes were exposed to cocaine and Tat simultaneously (Figure 5). We next utilized Filipin III to visualize intracellular accumulation of cholesterol following treatment with cocaine and Tat. Our data show that cocaine and Tat-treated astrocytes have a dramatic reduction in fluorescence intensity compared to control astrocytes (Figure 5D), thus confirming that cocaine and Tat decrease intracellular cholesterol. Defects in cholesterol synthesis and efflux will likely reduce the amount of astrocyte-derived cholesterol available to neurons.
Figure 4. ApoE release is reduced following cocaine and Tat exposure.
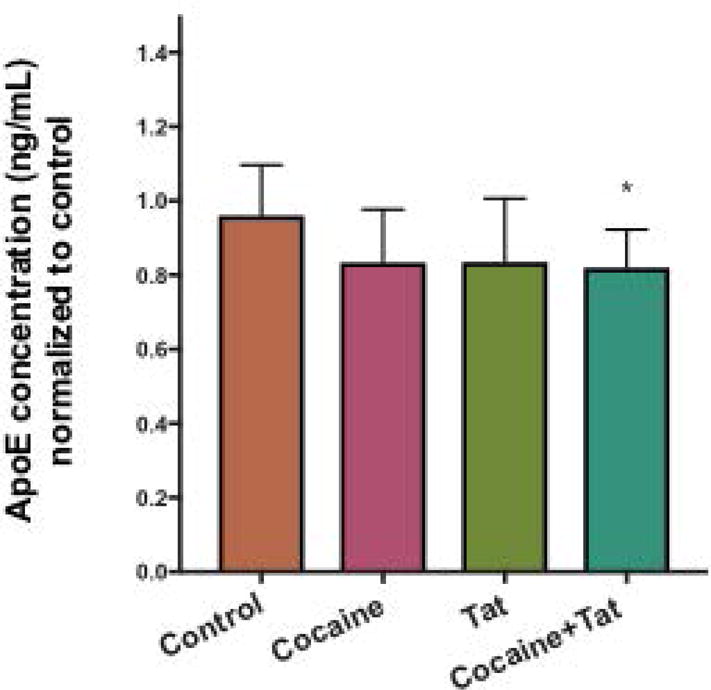
Conditioned media from astrocytes exposed to cocaine (5μM), recombinant Tat (50ng/ml), or cocaine and Tat was collected and ApoE was quantified by ELISA assay (*p<0.05). Results are expressed as mean ± SEM, n ≥ 3.
Figure 5. Cholesterol deficits in astrocytes exposed to cocaine and Tat.
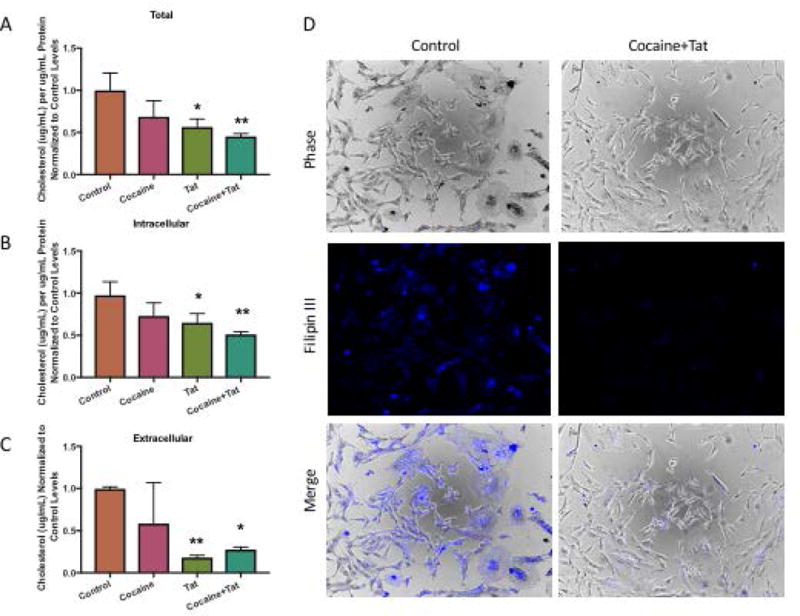
Astrocytes were cultured in serum-free media and exposed to cocaine (5μM), recombinant Tat (50ng/ml), or cocaine and Tat. A-C) Cells were harvested and conditioned media was collected. Levels of cholesterol were quantified by Amplex Red Cholesterol Assay. A) Total cholesterol (*p<, 0.02, **p<0.005), B) Intracellular cholesterol (*p< 0.05, **p <0.005), C) Extracellular cholesterol (*p<0.02, **p< 0.01). D) Astrocytes were exposed to cocaine (5μM) and Tat (50ng/ml), fixed and stained with Filipin III to visualize intracellular cholesterol. Left panels show representative images from control astrocytes. Right panels are representative images from cocaine and Tat treated astrocytes. Phase, fluorescence, and the merged images are shown.
Cocaine and Tat affect Cholesterol Biosynthesis Gene Expression
Cells monitor their cholesterol levels using membrane bound sterol regulatory element-binding proteins (SREBPs)(Eberlé et al. 2004). SREBPs are transcription factors that regulate the expression of genes important for cholesterol biosynthesis and uptake. In particular, SREBP-2 favors the cholesterol biosynthesis pathway. In response to reduced cellular cholesterol in cells, SREBP2 is cleaved into its active form that then translocates to the nucleus where it binds to sterol response elements (SREs) upstream of genes that encode proteins needed for key steps in cholesterol biosynthesis such as HMG-CoA reductase (HMGCR). This is necessary for the de novo synthesis of cholesterol. To assess the cocaine- and Tat- induced changes in cholesterol biosynthesis over time, we exposed astrocytes to cocaine and HIV-1 Tat and harvested cells at various times over the course of 72h. Our data showed that at 2h protein levels of total SREBP2 decreased significantly in astrocytes exposed to cocaine and Tat compared to control astrocytes and returned to control levels over the time course (Figure 6A, B). Interestingly, the decrease in total SREBP2 was coupled to an increase of cleaved SREBP2 at 2h and 6h (Figure 6A, C), suggesting that SREBP2 is activated under this condition. In line with increased activation of SREBP2, we observed an increase in the protein levels of its target gene, HMGCR in the cocaine and Tat treated-astrocytes at 48h (Figure 6A, D). To confirm that cleaved SREBP2 upregulates its downstream target genes, we measured the transcript levels of HMCGR and low-density lipoprotein receptor (LDLR), other target genes of SREBP2 that are involved in cholesterol uptake, and found a significant burst in transcript levels of HMCGR at 2h (Figure 6E). LDLR mRNA levels increased at 2h however, this increase was not statistically significant (Figure 6F).
Figure 6. Cocaine and Tat activate the SREBP2 signaling pathway.
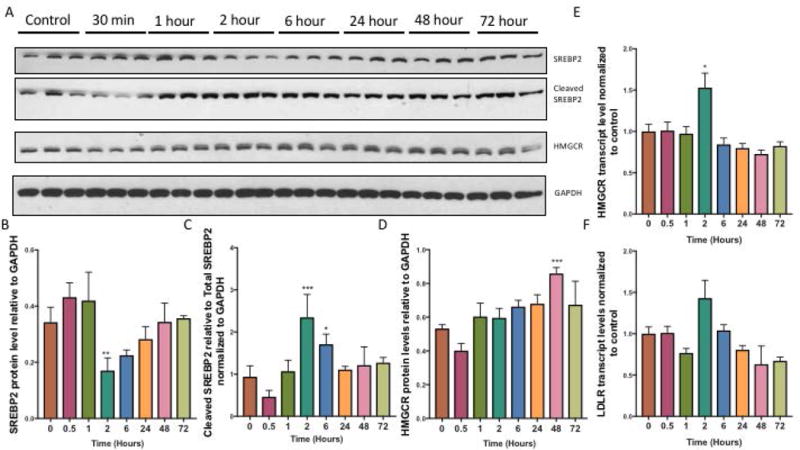
Astrocytes were treated with cocaine (5μM) and recombinant Tat (50ng/ml) over a 72h time course. A) Representative western blot of SREBP2 precursor, SREBP2 cleaved form and its target, HMGCR. GAPDH was used as a loading control. B) Quantification of precursor SREBP2 protein expression relative to GAPDH (**p<0.005), C) cleaved SREBP2 (***p<0.0005, p<0.05), and target gene D) HMGCR (***p<0.0005). E-F) mRNA was extracted and reverse transcribed followed by quantitative real time PCR for E) HMGCR (p< 0.05*), F) LDLR. Abbreviations: SREBP2 (Sterol response element binding protein 2), HMGCR (3-Hydroxy-3- Methylglutaryl-CoA Reductase) and GAPDH (Glyceraldehyde 3-phosphate dehydrogenase). Results are expressed as mean ± SEM, n ≥ 3.
Disruption in astrocyte-neuronal cholesterol crosstalk
To determine whether cocaine- and Tat-induced disruption of cholesterol supply in astrocytes influences neuronal integrity, we utilized a transwell system to co-culture human primary neurons in the lower chamber and astrocytes in the upper chamber whereby direct contact between the two cell types was prevented yet, secreted factors, such as ApoE and cholesterol could be exchanged. Astrocytes were plated onto transwell inserts, which were then placed into plates containing neurons. Cocaine and Tat treatments were then added directly to the upper chamber with the astrocytes. Levels of neuronal LDLR and CYP46a1 proteins were assessed. LDLR mediates the uptake of lipids into the cell and is upregulated in response to decreased intracellular cholesterol concentrations. Our data show that LDLR is significantly increased in neurons cultured with cocaine- and Tat- exposed astrocytes (Figure 7A, B). This suggests a need for neurons to take up more cholesterol when exposed to conditioned media from astrocytes treated with cocaine and Tat. Along these same lines, we found that CYP46a1, an enzyme that metabolizes cholesterol, is down regulated in neurons exposed to conditioned media from astrocytes treated with cocaine and Tat when compared to controls (Figure 7A, C). These data suggest that neurons respond to conditions of low cholesterol from astrocytes exposed to Tat and cocaine.
Figure 7. Cocaine- and Tat induced cholesterol dysregulation in astrocytes effects neuronal cholesterol homeostasis.
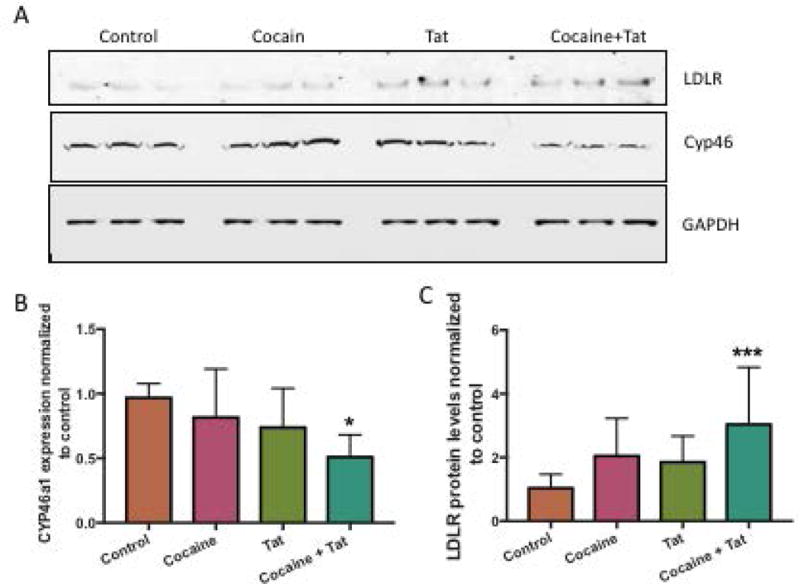
Neurons and astrocytes were co-cultured in 6-well plates separated by a 24mm transwell insert with a 1.0μm pore polycarbonate membrane. Astrocytes were seeded on top of the insert and neurons were seeded into the wells below. Astrocytes were exposed to cocaine (5μM), recombinant Tat (50ng/ml), or cocaine and Tat. Neurons were harvested following the treatments. A) Representative western blot of neuronal LDLR and CYP46a1. GAPDH was used as a loading control. B) Analyses of LDLR protein expression relative to GAPDH (*p< 0.0005) and, C) CYP46 (**p< 0.002). Abbreviations: LDLR (Low density lipoprotein receptor) and CYP46a1 (Cytochrome P450 Family 46). Results are expressed as mean ± SEM, n ≥ 3.
To assess the neurotoxic effects of culturing neurons in the presence of astrocytes that have been exposed to cocaine and Tat, we measured the protein levels of two synaptic markers, post synaptic density 95 (PSD95) and synaptophysin in the neurons (Figure 8A, B, C). Both markers were significantly reduced in neurons cultured with astrocytes that had been exposed to cocaine and Tat (Figure 8A, B, C). In addition, neurons were immunolabeled with anti-MAP2 antibody to visualize changes in neuronal connectivity (Figure 8D, E). MAP2 immunofluorescence intensity was reduced in neurons cultured with astrocytes exposed to cocaine and Tat (Figure 8D). In contrast, there was no difference in cholesterol regulatory proteins, synaptic or structural proteins in neurons exposed directly to cocaine and Tat, without the presence of astrocytes (Supplementary Figure 1). No difference in neuronal viability was observed when cultured with astrocytes in the transwells (Figure 8F). This suggests that astrocytes play a key role in the neurotoxicity-associated with cocaine and Tat and this may be mediated by the reduction in cholesterol supply.
Figure 8. Cocaine- and Tat- treated astrocytes reduce synaptic and neuronal markers.
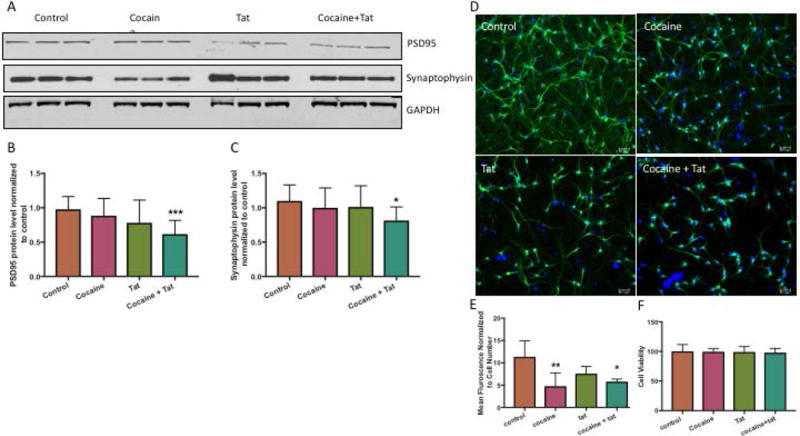
Neurons and astrocytes were co-culture in 6-well or 12-well plates separated by a transwell insert with a 1.0μm pore polycarbonate membrane. Astrocytes were seeded on top of the insert and neurons were seeded into the wells below. Astrocytes were exposed to cocaine (5μM), recombinant Tat (50ng/ml), or cocaine and Tat. Neurons were harvested following the treatments. A) Representative western blot of neuronal PSD95 and Synaptophysin. GAPDH was used as a loading control. B) Analyses of PSD95 protein expression relative to GAPDH (***p< 0.0005) and, C) Synaptophysin (*p< 0.05). D) Representative immunocytochemical labeling of neurons cultured below control (untreated), cocaine-, Tat-, and cocaine and Tat-treated astrocytes with anti-MAP2 (green) and DAPI (blue). Magnification 20×. E) Analysis of fluorescence intensity normalized for total cell number (**p<0.001, *p<0.01). Abbreviations: PSD95 (Post Synaptic Density 95) and MAP2 (Microtubule Associated Protein 2). Results are expressed as mean ± SEM, n ≥ 3.
Effect of cocaine administration on LXR signaling pathway in Tat transgenic mice
To examine whether the effects of cocaine and Tat on cholesterol homeostasis also occurs in vivo, we utilized Tat-transgenic mice in combination with a model of chronic cocaine use. Using brain-region specific homogenates from the hippocampus, our data showed a significant decrease of LXRβ in Tat-expressing mice that received cocaine (Figure 9A, B). Furthermore, downstream genes ABCa1 and ApoE were also significantly reduced in the hippocampus of Tat mice given cocaine compared to the wild type (Figure 9C, D). These results confirm in the animal model that HIV-1 Tat and cocaine disrupt LXR-β signaling. When we assessed changes in cholesterol uptake and break down, we found that LDLR was significantly increased in Tat mice that received cocaine, whereas, the cholesterol metabolizing enzyme, CYP46a1 was significantly decreased (Figure 9E, F).
Figure 9. Disruption of cholesterol homeostasis in hippocampus of Tat-transgenic mouse model of chronic cocaine abuse.
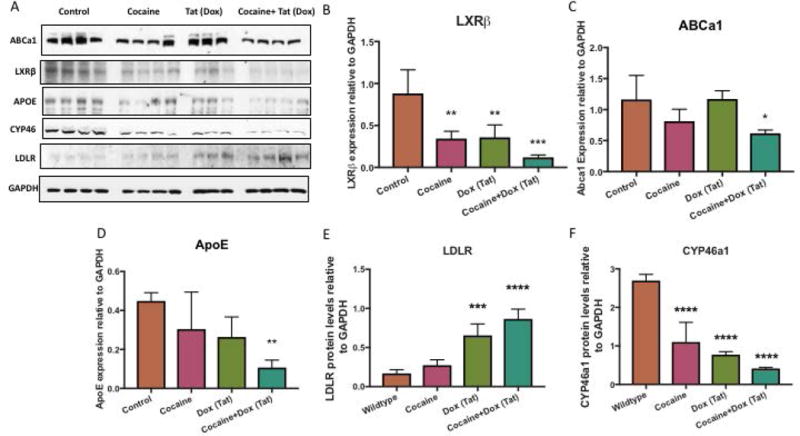
A) Representative western blot of LXRβ and target genes ABCa1 and ApoE and cholesterol uptake and metabolism LDLR and CYP46a1. GAPDH was used as a loading control B) Analyses of LXRβ protein expression relative to GAPDH (**p< 0.005, ***p< 0.0001) and its gene targets, C) ABCa1 (*p< 0.02*), D) ApoE (**p< 0.005) E) LDLR (***p<0.0005, ****p<0.0001), F) CYP46a1 (****p<0.0001). Abbreviations: WT (Wildtype), Cocaine (Cocaine only group), Tat (Tat induced by DOX group), Cocaine + Tat (Cocaine and Dox to induce Tat), LXRβ (Liver X Receptor Beta), ApoE (apolipoprotein E), ABCa1 (ATP-binding cassette transporter 1), LDLR (Low density lipoprotein receptor) and CYP46a1 (Cytochrome P450 Family 46) and GAPDH (Glyceraldehyde 3-phosphate dehydrogenase). Results are expressed as mean ± SEM, n ≥ 3.
Discussion
Since the beginning of the HIV epidemic, illicit drug use has been closely intertwined with transmission and disease progression of HIV (Larrat and Zierler 1993; Purohit et al. 2011). In particular, cocaine use is a well known exacerbating factor that contributes to neurocognitive impairment associated with HIV infection (Cai et al. 2016; Dahal et al. 2015). Numerous reports have shown that the HIV-1 Tat protein plays an additive or synergetic role in cocaine co-exposure models both in vitro and in vivo (De Simone et al. 2016; Wayman et al. 2015). However, the combined effects of cocaine and Tat on astrocytes have been largely overlooked. Astrocytes make up a significant portion of the cell population in the CNS and are crucial to the brain’s normal functioning (Bélanger et al. 2011; Eroglu and Barres 2010; Stobart and Anderson 2013). Astrocytes support neurons through a range of diverse functions including supplying key energy metabolites and growth factors. Cholesterol is one key substrate provided to neurons by astrocytes (Pfrieger and Ungerer 2011). Brain cholesterol is a major component of myelin and is crucial for cell membrane integrity and synaptic transmission. Astrocyte dysfunction and disruption in cholesterol homeostasis are increasingly recognized as contributors to a number of neurodegenerative diseases including AD, HD, Parkinson’s disease and Niemann-Pick Type C disease (Björkhem and Meaney 2004; Petrov et al. 2016). In the case of AD, several groups have shown that total brain cholesterol and brain cholesterol synthesis is reduced in patients (Martín et al. 2014; Mason et al. 1992; Mulder et al. 1998; Roher et al. 2002). In support of AD reports, low cholesterol levels have also been noted in the hippocampus of aging human brains and in hippocampal synapses of aging mice (Desai et al. 2010; Martín et al. 2014; Svennerholm et al. 1991; Thelen et al. 2006). In fact, reports of short-term memory loss have been associated with cholesterol-lowering statin therapy causing the FDA to issue a warning on this drug class. In vitro, statins have been found to induce apoptosis in rat astrocytes and neurons (März et al. 2007). Given the many roles of cholesterol in the CNS, it is clear that any disruption in the cholesterol supply to neurons can likely contribute to neurodysfunction. In agreement with human and animal studies, in vitro studies demonstrate that depletion of cholesterol has deleterious effects on neuronal function (Frank et al. 2008; Fukui et al. 2016; Thiele et al. 2000; Valenza et al. 2015). Taken together, this reveals a potential convergence point for cocaine use and HIV-1 Tat to accelerate neurocognitive impairment. However, there are no reports on the potential effects of HIV-1 Tat or cocaine on cholesterol homeostasis within the CNS.
In the present study, we assessed the individual and combined effects of cocaine and Tat on cholesterol homeostasis in astrocytes. We discovered multiple aspects of cholesterol regulation that were disrupted by cocaine and Tat in vitro and in vivo. Cholesterol homeostasis is maintained by a dynamic balance between cholesterol efflux controlled by LXR signaling and cholesterol biosynthesis triggered through SREBP2 activation (Courtney and Landreth 2016). We found that LXR signaling was reduced in astrocytes following cocaine and Tat exposure resulting in decreased expression of the lipid carrier, ApoE and the lipid transporter, ABCa1 (Figures 1, 3, and 4). In addition to altered capacity to export cholesterol, diminished levels of astrocyte-derived cholesterol were detected following cocaine and Tat exposure (Figure 5), suggesting that cholesterol biosynthesis may also be hampered under these conditions. However, cocaine- and Tat- treated astrocytes appear to respond to this deficiency by increased cleavage of SREBP2 into its active form to stimulate cholesterol biosynthesis mediated by upregulation of HMGCR expression, a rate-limiting enzyme in cholesterol generation (Figure 6). However, total cholesterol levels remained low despite the transient activation of SREBP2 indicating that the cell may be unable to compensate for Tat/cocaine reduced cholesterol supply. It has been previously reported that in hepatic cells reduced cholesterol efflux and synthesis is observed after induction of endoplasmic reticulum (ER) stress despite increased SREBP2 activity (Röhrl et al. 2014). This study demonstrated that although SREBP2 activation and HMGCR levels were increased, HMGCR activity was reduced by 70% under ER stress, which led to decreased cholesterol synthesis. There are a number of reports of ER stress induced by cocaine or Tat contributing to neurotoxicity (Desai et al. 2010; Fan and He 2016; Ma et al. 2016; Periyasamy et al. 2016). Therefore, it is possible that ER stress may be an upstream event mediating cholesterol dysregulation following cocaine and Tat exposure. Furthermore, one group demonstrated that a reduction in SREBP activity in astrocytes can contribute to impaired synaptic plasticity in the hippocampus (van Deijk et al. 2017). This supports the importance of astrocyte-derived cholesterol in synaptic health and reveals that deficiencies in SREBP alone can have a negative impact.
Because the blood brain barrier prevents uptake of cholesterol from the periphery into the CNS, neurons rely on cholesterol synthesized by astrocytes (Björkhem and Meaney 2004). Cholesterol bound to ApoE is transported out of astrocytes via ABCa1 (Pfrieger and Ungerer 2011). Once in the extracellular space, the ApoE-cholesterol complex is taken up via LDL receptors into neurons where it can be utilized for synaptogenesis, synaptic vesicle formation, and axonal plasticity (Allen et al. 2007; Mauch et al. 2001; Pfrieger 2003; Thiele et al. 2000). Reduced bioavailability of astrocyte-derived cholesterol to neurons has been shown to reduce neurite outgrowth, decrease synaptic properties, and impede synaptic activity (Fan et al. 2002; Valenza et al. 2015). Using our co-culture system, we showed that cocaine and Tat together increase LDLR expression in human primary neurons, suggesting a decrease in intracellular cholesterol and a need for neurons to increase cholesterol uptake. This could be a direct compensatory response to the diminished supply of cholesterol provided by astrocytes (Figure 7). Similarly, data that cholesterol metabolism was decreased further suggest that intracellular cholesterol was reduced in neurons exposed to cocaine- and Tat- treated astrocytes. Cholesterol is a major component of synaptic vesicles and cholesterol depletion has been shown to impair synaptic vesicle exocytosis and to block biogenesis of synaptic vesicles in cultured neurons (Linetti et al. 2010; Thiele et al. 2000). Cholesterol depletion has also been demonstrated to affect the ability of synapses to sustain synaptic transmission (Goritz et al. 2005). In the post synaptic compartment, loss of cholesterol has been reported to alter AMPA and NMDA receptor function, decrease endocytosis, and dysregulate TrkB receptor activation (Thiele et al. 2000). All together, this can result in decreased hippocampal functioning. Our in vitro data reveal that cocaine and Tat can have deleterious effects on astrocyte cholesterol supply, thereby providing compelling evidence for a novel indirect effect of cocaine- and Tat on neuronal dysfunction in HIV infected patients who use cocaine.
In ApoE- knockout mice, ApoE deficiency was associated with signs of neurodegeneration including decreased immunolabeling for the synaptodendritic markers, MAP2 and synaptophysin in the hippocampus and cortex (Masliah et al. 1995). This demonstrates the importance of ApoE in the maintenance of synaptic integrity and how alterations can lead to neurodegeneration. Additionally, this highlights the importance of cholesterol delivery to neurons by ApoE to sustain neuronal fitness. In our Tat-transgenic mice model of chronic cocaine use, we detected decreased expression of LXR, ApoE, and ABCa1 within the hippocampus. It is likely that disruption of LXR signaling within the hippocampus may hinder the astrocyte’s ability to support the cholesterol needs of neighboring neurons resulting in the loss of synaptodendritic connectivity (Figure 9).
In conclusion, our results indicate that cocaine and Tat have converging effects on cholesterol homeostasis in astrocytes by disrupting LXR signaling and diminishing cholesterol bioavailability to neurons (Figure 10). Astrocyte-neuron crosstalk and metabolite exchange is required for normal brain function. Any alterations to this tightly regulated exchange can have detrimental effects on the health of the CNS. Cocaine and Tat hinder astrocytes’ ability to provide needed metabolic support to neurons. Cholesterol production and efflux are greatly reduced following cocaine and Tat exposure. If astrocytes cannot meet neuronal cholesterol needs, it is possible that synaptic integrity may be lost, neurotransmission may begin to fail and this could manifest as neurocognitive impairments that are often reported in HIV + cocaine users. An LXR agonist may be a potential candidate to alleviate astrocyte cholesterol dysfunction. Enhancement of this pathway may attenuate neurite degeneration and synaptic defects seen in HIV infection and/or cocaine use.
Figure 10. Representation model of cholesterol dysregulation in astrocytes following cocaine and Tat exposure and its proposed impact on neurons.
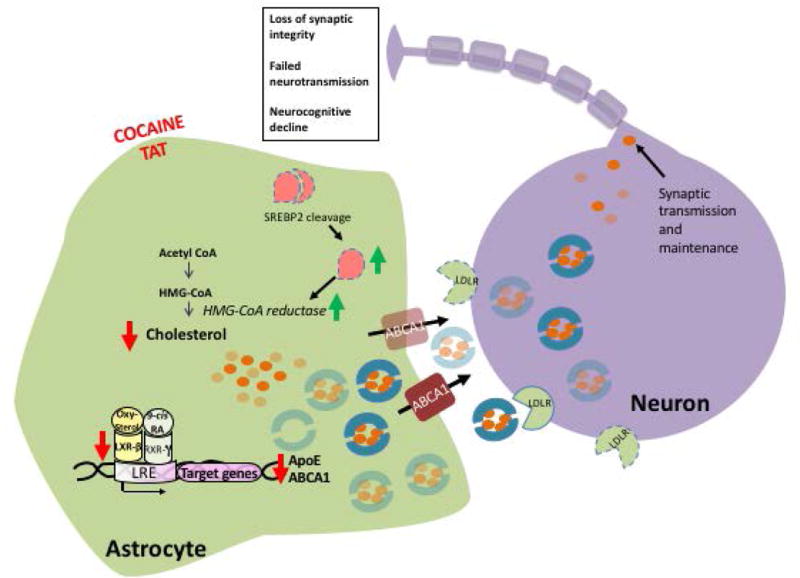
Astrocytes exposed to cocaine and Tat undergo several changes in cholesterol homeostasis. In astrocytes (left), intracellular levels of cholesterol are reduced following cocaine and Tat exposure. This diminishes activation of the LXR signaling pathway and therefore decreases expression of LXRβ target genes, ApoE and ABCa1. ApoE is a major carrier of cholesterol and forms a lipoprotein complex to export cholesterol out of the cell. ABCa1 transporters aid in lipidating and exporting these complexes into the extracellular compartment. In the presence of cocaine and Tat, there is a significant decrease in intracellular and extracellular ApoE and cellular ABCa1. In agreement with diminished intracellular cholesterol and reduced secreted ApoE, there is significantly less cholesterol released into the extracellular environment. In response to reduced cholesterol levels, SREBP2 is cleaved into is active form to stimulate cholesterol biosynthesis through the upregulation of target genes important for cholesterol synthesis including, HMCGR. Neurons (right) utilize cholesterol to maintain many functions including synaptic vesicle formation and maintenance of cell structure. To compensate for the diminished availability of astrocyte-derived cholesterol, neurons upregulated the expression of LDLR likely in an effort to bring in more lipidated ApoE particles. Deficits in cholesterol supply to neurons could have detrimental effects including loss of synaptic integrity, failed neurotransmission, and gradual cognitive decline. Key: Red arrows indicate decline, green arrows indicate increases, transparent symbols signify a reduction, dotted lines suggest upregulation. Abbreviations: LXRβ (Liver X Receptor Beta), ApoE (apolipoprotein E), ABCa1 (ATP-binding cassette transporter 1), SREBP2 (Sterol response element binding protein 2), HMGCR (3-Hydroxy-3- Methylglutaryl-CoA Reductase), LDLR (Low density lipoprotein receptor) and CYP46a1 (Cytochrome P450 Family 46), LXRE (LXR-response element), RXRγ (Retinoic acid receptor gamma).
Supplementary Material
Neurons were exposed to cocaine (5μM), recombinant Tat (50ng/ml), or cocaine and Tat concurrently every 24h for 48h. Neurons were harvested following the treatments. A) Representative western blot of neuronal LDLR and CYP46a1. Tubulin was used as a loading control. B) Analyses of LDLR protein expression relative to GAPDH and, C) CYP46. Abbreviations: LDLR (Low density lipoprotein receptor) and CYP46a1 (Cytochrome P450 Family 46). Results are expressed as mean ± SEM, n ≥ 3.
Main Points.
Liver X receptors (LXRs) are the master regulators of cholesterol homeostasis in the central nervous system.
HIV-1 infection and cocaine abuse disrupt cholesterol homeostasis.
Cocaine and HIV-1 Tat disrupt astrocyte LXR signaling leading to decreased bioavailability of cholesterol.
Acknowledgments
This work was supported by NIH funding: P01DA037830, T32MH079785 and P30MH09217.
Footnotes
The authors declare that they have no conflicts of interest.
References
- Allen JA, Halverson-Tamboli RA, Rasenick MM. Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci. 2007;8:128–40. doi: 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- Bandaru VV, Mielke MM, Sacktor N, McArthur JC, Grant I, Letendre S, Chang L, Wojna V, Pardo C, Calabresi P, et al. A lipid storage-like disorder contributes to cognitive decline in HIV-infected subjects. Neurology. 2013;81:1492–9. doi: 10.1212/WNL.0b013e3182a9565e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkhem I, Meaney S. Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol. 2004;24:806–15. doi: 10.1161/01.ATV.0000120374.59826.1b. [DOI] [PubMed] [Google Scholar]
- Buch S, Yao H, Guo M, Mori T, Mathias-Costa B, Singh V, Seth P, Wang J, Su TP. Cocaine and HIV-1 interplay in CNS: cellular and molecular mechanisms. Curr HIV Res. 2012;10:425–8. doi: 10.2174/157016212802138823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–38. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Cai Y, Yang L, Callen S, Buch S. Multiple Faceted Roles of Cocaine in Potentiation of HAND. Curr HIV Res. 2016 doi: 10.2174/1570162x14666160324125158. [DOI] [PubMed] [Google Scholar]
- Courtney R, Landreth GE. LXR Regulation of Brain Cholesterol: From Development to Disease. Trends Endocrinol Metab. 2016 doi: 10.1016/j.tem.2016.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahal S, Chitti SV, Nair MP, Saxena SK. Interactive effects of cocaine on HIV infection: implication in HIV-associated neurocognitive disorder and neuroAIDS. Front Microbiol. 2015;6:931. doi: 10.3389/fmicb.2015.00931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Gorantla S, Gendelman HE, Knibbe J, Casale GP, Makarov E, Epstein AA, Gelbard HA, Boska MD, Poluektova LY. Loss of neuronal integrity during progressive HIV-1 infection of humanized mice. J Neurosci. 2011;31:3148–57. doi: 10.1523/JNEUROSCI.5473-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simone FI, Darbinian N, Amini S, Muniswamy M, White MK, Elrod JW, Datta PK, Langford D, Khalili K. HIV-1 Tat and Cocaine Impair Survival of Cultured Primary Neuronal Cells via a Mitochondrial Pathway. J Neuroimmune Pharmacol. 2016;11:358–68. doi: 10.1007/s11481-016-9669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai RI, Paronis CA, Martin J, Desai R, Bergman J. Monoaminergic psychomotor stimulants: discriminative stimulus effects and dopamine efflux. J Pharmacol Exp Ther. 2010;333:834–43. doi: 10.1124/jpet.110.165746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietschy JM. Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biol Chem. 2009;390:287–93. doi: 10.1515/BC.2009.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietschy JM, Turley SD. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45:1375–97. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- Eberlé D, Hegarty B, Bossard P, Ferré P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie. 2004;86:839–48. doi: 10.1016/j.biochi.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–31. doi: 10.1038/nature09612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan QW, Yu W, Gong JS, Zou K, Sawamura N, Senda T, Yanagisawa K, Michikawa M. Cholesterol-dependent modulation of dendrite outgrowth and microtubule stability in cultured neurons. J Neurochem. 2002;80:178–90. doi: 10.1046/j.0022-3042.2001.00686.x. [DOI] [PubMed] [Google Scholar]
- Fan Y, He JJ. HIV-1 Tat Induces Unfolded Protein Response and Endoplasmic Reticulum Stress in Astrocytes and Causes Neurotoxicity through Glial Fibrillary Acidic Protein (GFAP) Activation and Aggregation. J Biol Chem. 2016;291:22819–22829. doi: 10.1074/jbc.M116.731828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeney ER, McAuley N, O’Halloran JA, Rock C, Low J, Satchell CS, Lambert JS, Sheehan GJ, Mallon PW. The expression of cholesterol metabolism genes in monocytes from HIV-infected subjects suggests intracellular cholesterol accumulation. J Infect Dis. 2013;207:628–37. doi: 10.1093/infdis/jis723. [DOI] [PubMed] [Google Scholar]
- Fitting S, Ignatowska-Jankowska BM, Bull C, Skoff RP, Lichtman AH, Wise LE, Fox MA, Su J, Medina AE, Krahe TE, et al. Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 Tat transgenic mice. Biol Psychiatry. 2013;73:443–53. doi: 10.1016/j.biopsych.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank C, Rufini S, Tancredi V, Forcina R, Grossi D, D’Arcangelo G. Cholesterol depletion inhibits synaptic transmission and synaptic plasticity in rat hippocampus. Exp Neurol. 2008;212:407–14. doi: 10.1016/j.expneurol.2008.04.019. [DOI] [PubMed] [Google Scholar]
- Fukui K, Ferris HA, Kahn CR. Effect of cholesterol reduction on receptor signaling in neurons. J Biol Chem. 2016;291:15910. doi: 10.1074/jbc.A115.664367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi N, Saiyed ZM, Napuri J, Samikkannu T, Reddy PV, Agudelo M, Khatavkar P, Saxena SK, Nair MP. Interactive role of human immunodeficiency virus type 1 (HIV-1) clade-specific Tat protein and cocaine in blood-brain barrier dysfunction: implications for HIV-1-associated neurocognitive disorder. J Neurovirol. 2010;16:294–305. doi: 10.3109/13550284.2010.499891. [DOI] [PubMed] [Google Scholar]
- Goritz C, Mauch DH, Pfrieger FW. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol Cell Neurosci. 2005;29:190–201. doi: 10.1016/j.mcn.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Hauser KF, Knapp PE. Interactions of HIV and drugs of abuse: the importance of glia, neural progenitors, and host genetic factors. Int Rev Neurobiol. 2014;118:231–313. doi: 10.1016/B978-0-12-801284-0.00009-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BO, Liu Y, Ruan Y, Xu ZC, Schantz L, He JJ. Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am J Pathol. 2003;162:1693–707. doi: 10.1016/S0002-9440(10)64304-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M, Zou K, Minagawa H, Yu W, Gong JS, Yanagisawa K, Michikawa M. Cholesterol-mediated neurite outgrowth is differently regulated between cortical and hippocampal neurons. J Biol Chem. 2005;280:42759–65. doi: 10.1074/jbc.M509164200. [DOI] [PubMed] [Google Scholar]
- Larrat EP, Zierler S. Entangled epidemics: cocaine use and HIV disease. J Psychoactive Drugs. 1993;25:207–21. doi: 10.1080/02791072.1993.10472272. [DOI] [PubMed] [Google Scholar]
- Leoni V, Caccia C. The impairment of cholesterol metabolism in Huntington disease. Biochim Biophys Acta. 2015;1851:1095–105. doi: 10.1016/j.bbalip.2014.12.018. [DOI] [PubMed] [Google Scholar]
- Linetti A, Fratangeli A, Taverna E, Valnegri P, Francolini M, Cappello V, Matteoli M, Passafaro M, Rosa P. Cholesterol reduction impairs exocytosis of synaptic vesicles. J Cell Sci. 2010;123:595–605. doi: 10.1242/jcs.060681. [DOI] [PubMed] [Google Scholar]
- Ma R, Yang L, Niu F, Buch S. HIV Tat-Mediated Induction of Human Brain Microvascular Endothelial Cell Apoptosis Involves Endoplasmic Reticulum Stress and Mitochondrial Dysfunction. Mol Neurobiol. 2016;53:132–142. doi: 10.1007/s12035-014-8991-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín MG, Pfrieger F, Dotti CG. Cholesterol in brain disease: sometimes determinant and frequently implicated. EMBO Rep. 2014;15:1036–52. doi: 10.15252/embr.201439225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Ge N, Alford M, Veinbergs I, Roses AD. Neurodegeneration in the central nervous system of apoE-deficient mice. Exp Neurol. 1995;136:107–22. doi: 10.1006/exnr.1995.1088. [DOI] [PubMed] [Google Scholar]
- Mason RP, Shoemaker WJ, Shajenko L, Chambers TE, Herbette LG. Evidence for changes in the Alzheimer’s disease brain cortical membrane structure mediated by cholesterol. Neurobiol Aging. 1992;13:413–9. doi: 10.1016/0197-4580(92)90116-f. [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–7. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- Mujawar Z, Rose H, Morrow MP, Pushkarsky T, Dubrovsky L, Mukhamedova N, Fu Y, Dart A, Orenstein JM, Bobryshev YV, et al. Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biol. 2006;4:e365. doi: 10.1371/journal.pbio.0040365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder M, Ravid R, Swaab DF, de Kloet ER, Haasdijk ED, Julk J, van der Boom JJ, Havekes LM. Reduced levels of cholesterol, phospholipids, and fatty acids in cerebrospinal fluid of Alzheimer disease patients are not related to apolipoprotein E4. Alzheimer Dis Assoc Disord. 1998;12:198–203. doi: 10.1097/00002093-199809000-00012. [DOI] [PubMed] [Google Scholar]
- März P, Otten U, Miserez AR. Statins induce differentiation and cell death in neurons and astroglia. Glia. 2007;55:1–12. doi: 10.1002/glia.20422. [DOI] [PubMed] [Google Scholar]
- Periyasamy P, Guo ML, Buch S. Cocaine induces astrocytosis through ER stress-mediated activation of autophagy. Autophagy. 2016;12:1310–29. doi: 10.1080/15548627.2016.1183844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov AM, Kasimov MR, Zefirov AL. Brain Cholesterol Metabolism and Its Defects: Linkage to Neurodegenerative Diseases and Synaptic Dysfunction. Acta Naturae. 2016;8:58–73. [PMC free article] [PubMed] [Google Scholar]
- Pfenninger KH. Plasma membrane expansion: a neuron’s Herculean task. Nat Rev Neurosci. 2009;10:251–61. doi: 10.1038/nrn2593. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol Life Sci. 2003;60:1158–71. doi: 10.1007/s00018-003-3018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfrieger FW, Ungerer N. Cholesterol metabolism in neurons and astrocytes. Prog Lipid Res. 2011;50:357–71. doi: 10.1016/j.plipres.2011.06.002. [DOI] [PubMed] [Google Scholar]
- Purohit V, Rapaka R, Shurtleff D. Drugs of abuse, dopamine, and HIV-associated neurocognitive disorders/HIV-associated dementia. Mol Neurobiol. 2011;44:102–10. doi: 10.1007/s12035-011-8195-z. [DOI] [PubMed] [Google Scholar]
- Rasband WS. Image J. Bethesda: NIH; 1997. Version 1.37. [Google Scholar]
- Reynolds JL, Mahajan SD, Bindukumar B, Sykes D, Schwartz SA, Nair MP. Proteomic analysis of the effects of cocaine on the enhancement of HIV-1 replication in normal human astrocytes (NHA) Brain Res. 2006;1123:226–36. doi: 10.1016/j.brainres.2006.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roher AE, Weiss N, Kokjohn TA, Kuo YM, Kalback W, Anthony J, Watson D, Luehrs DC, Sue L, Walker D, et al. Increased A beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer’s disease. Biochemistry. 2002;41:11080–90. doi: 10.1021/bi026173d. [DOI] [PubMed] [Google Scholar]
- Röhrl C, Eigner K, Winter K, Korbelius M, Obrowsky S, Kratky D, Kovacs WJ, Stangl H. Endoplasmic reticulum stress impairs cholesterol efflux and synthesis in hepatic cells. J Lipid Res. 2014;55:94–103. doi: 10.1194/jlr.M043299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spann NJ, Glass CK. Sterols and oxysterols in immune cell function. Nat Immunol. 2013;14:893–900. doi: 10.1038/ni.2681. [DOI] [PubMed] [Google Scholar]
- Stobart JL, Anderson CM. Multifunctional role of astrocytes as gatekeepers of neuronal energy supply. Front Cell Neurosci. 2013;7:38. doi: 10.3389/fncel.2013.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svennerholm L, Boström K, Helander CG, Jungbjer B. Membrane lipids in the aging human brain. J Neurochem. 1991;56:2051–9. doi: 10.1111/j.1471-4159.1991.tb03466.x. [DOI] [PubMed] [Google Scholar]
- Tachikawa M, Watanabe M, Hori S, Fukaya M, Ohtsuki S, Asashima T, Terasaki T. Distinct spatio-temporal expression of ABCA and ABCG transporters in the developing and adult mouse brain. J Neurochem. 2005;95:294–304. doi: 10.1111/j.1471-4159.2005.03369.x. [DOI] [PubMed] [Google Scholar]
- Thelen KM, Falkai P, Bayer TA, Lütjohann D. Cholesterol synthesis rate in human hippocampus declines with aging. Neurosci Lett. 2006;403:15–9. doi: 10.1016/j.neulet.2006.04.034. [DOI] [PubMed] [Google Scholar]
- Thiele C, Hannah MJ, Fahrenholz F, Huttner WB. Cholesterol binds to synaptophysin and is required for biogenesis of synaptic vesicles. Nat Cell Biol. 2000;2:42–9. doi: 10.1038/71366. [DOI] [PubMed] [Google Scholar]
- Ullian EM, Christopherson KS, Barres BA. Role for glia in synaptogenesis. Glia. 2004;47:209–16. doi: 10.1002/glia.20082. [DOI] [PubMed] [Google Scholar]
- Valenza M, Marullo M, Di Paolo E, Cesana E, Zuccato C, Biella G, Cattaneo E. Disruption of astrocyte-neuron cholesterol cross talk affects neuronal function in Huntington’s disease. Cell Death Differ. 2015;22:690–702. doi: 10.1038/cdd.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deijk AF, Camargo N, Timmerman J, Heistek T, Brouwers JF, Mogavero F, Mansvelder HD, Smit AB, Verheijen MH. Astrocyte lipid metabolism is critical for synapse development and function in vivo. Glia. 2017;65:670–682. doi: 10.1002/glia.23120. [DOI] [PubMed] [Google Scholar]
- Vance JE. Dysregulation of cholesterol balance in the brain: contribution to neurodegenerative diseases. Dis Model Mech. 2012;5:746–55. doi: 10.1242/dmm.010124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman WN, Chen L, Persons AL, Napier TC. Cortical consequences of HIV-1 Tat exposure in rats are enhanced by chronic cocaine. Curr HIV Res. 2015;13:80–7. doi: 10.2174/0929867322666150311164504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Yao H, Chen X, Cai Y, Callen S, Buch S. Role of Sigma Receptor in Cocaine-Mediated Induction of Glial Fibrillary Acidic Protein: Implications for HAND. Mol Neurobiol. 2016;53:1329–42. doi: 10.1007/s12035-015-9094-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Neurons were exposed to cocaine (5μM), recombinant Tat (50ng/ml), or cocaine and Tat concurrently every 24h for 48h. Neurons were harvested following the treatments. A) Representative western blot of neuronal LDLR and CYP46a1. Tubulin was used as a loading control. B) Analyses of LDLR protein expression relative to GAPDH and, C) CYP46. Abbreviations: LDLR (Low density lipoprotein receptor) and CYP46a1 (Cytochrome P450 Family 46). Results are expressed as mean ± SEM, n ≥ 3.