

Abstract
Chemical cross-linking combined with mass spectrometric analysis has become an important technique for probing protein three-dimensional structure and protein–protein interactions. A key step in this process is the accurate identification and validation of cross-linked peptides from tandem mass spectra. The identification of cross-linked peptides, however, presents challenges related to the expanded nature of the search space (all pairs of peptides in a sequence database) and the fact that some peptide-spectrum matches (PSMs) contain one correct and one incorrect peptide but often receive scores that are comparable to those in which both peptides are correctly identified. To address these problems and improve detection of cross-linked peptides, we propose a new database search algorithm, XLSearch, for identifying cross-linked peptides. Our approach is based on a data-driven scoring scheme that independently estimates the probability of correctly identifying each individual peptide in the cross-link given knowledge of the correct or incorrect identification of the other peptide. These conditional probabilities are subsequently used to estimate the joint posterior probability that both peptides are correctly identified. Using the data from two previous cross-link studies, we show the effectiveness of this scoring scheme, particularly in distinguishing between true identifications and those containing one incorrect peptide. We also provide evidence that XLSearch achieves more identifications than two alternative methods at the same false discovery rate (availability: https://github.com/COL-IU/XLSearch).
Keywords: mass spectrometry, chemical cross-linking, machine learning, structural biology, ribosome
Graphical abstract
Introduction
In recent years, chemical cross-linking combined with mass spectrometry (XL–MS) has become a powerful tool in structural biology,1–4 in particular for investigating protein three-dimensional (3D) structures,5,6 determining subunit arrangement in large protein complexes7,8 and for characterizing physical interactions between proteins at a whole-cell level.9,10 In a typical cross-linking experiment, a chemical reagent (called the linker) with two reactive groups is introduced to a protein mixture, subsequently leading to the formation of covalent bonds between specific amino acid residues (e.g., lysine) and the linker. Because the reactive groups are separated by a spacer arm of a specific length, only those residue pairs, either from a single protein or two interacting proteins, whose distances in 3D space are within the length of the spacer arm may be linked. After proteolytic digestion, the resulting sample containing cross-linked peptides is analyzed using liquid chromatography–tandem mass spectrometry (LC–MS/MS) and the cross-linked peptides identified in the experiment and then generates a set of distance constraints between linked residues. When combined with conventional techniques such as X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy, these distance constraints can help model the 3D structure of large proteins or protein complexes,11–13 particularly for disordered regions where structural details cannot be easily resolved.
A critical step in the analysis pipeline is the identification of cross-linked peptides through database searching. Here, a computer algorithm is used to score the similarity between each experimental spectrum and theoretical spectra of peptide pairs that match its measured mass-to-charge ratio. The best peptide pair receiving a sufficiently high score against each experimental spectrum is considered to be identified. Several algorithms have been implemented and successfully applied to the identification of cross-linked peptides.14–18 Nevertheless, statistical and computational challenges remain. First, the search space for the identification of cross-linked peptides grows quadratically with the size of the peptide database. The increased search space typically leads to longer search times and a greater chance of incorrect identifications due to random matches; this problem arises similarly in the identification of post-translationally modified peptides19 or semitryptic peptides.20 Secondly, the fragment spectra of cross-linked peptides may contain a larger variety of product ions, including those that contain the linker and those resulting from single or double fragmentation sites. Finally, candidate peptide-spectrum matches (PSMs) in a cross-linking experiment contain a class of incorrect identifications in which one of the two cross-linked peptides is correctly identified and the other is not. In these semifalse PSMs, the fragmentation in the correctly identified individual peptide may explain a large number of observed product ions in the MS/MS spectra, leading to score distributions that are comparable to those of correct PSMs but distinctly different from the scores of cross-link candidates for which both of the peptides in a PSM are false.15
The occurrences of semifalse PSMs in cross-linked peptide identification are partially caused by the scoring schemes used in the identification algorithms. A common practice in a scoring scheme is to measure the matching quality between theoretical fragmentation of the entire cross-linked peptide pair and an observed MS/MS spectrum. As a result, the number of fragment ions supporting each of the two linked peptides individually may be insufficient. To address this issue, some identification algorithms require that a correct PSM contain a minimum number of fragment ions to be observed from of each of the two linked peptides.21,22 Similarly, a recent study proposed to measure peptide-spectrum matching quality separately for each of the cross-linked peptides and use the lower of the two scores as the matching score for the entire cross-linked peptide.16 Despite their success in improving the confidence of identified cross-linked peptides, these methods are largely based on empirical scoring schemes, and their effectiveness may not generalize to a broad range of experimental platforms and protocols (e.g., those using different cross-linking reagents).
In this paper, we propose a database search algorithm XLSearch for identifying cross-linked peptides from their MS/MS spectra. XLSearch is based on a novel scoring function that estimates the joint posterior probability that both of the cross-linked peptides are correctly identified given a set of descriptors of the candidate PSM. The joint probability is computed from the outputs of two binary classification models that approximate the posterior probabilities of one of the two identified peptides being true, given that the other peptide is true or false, respectively. In addition, the joint posterior probabilities can be used to estimate the marginal probabilities of each of the two cross-linked peptides to be correctly identified. We confirm that the lower marginal probability of these two peptides is a stronger indicator of the correctness of the entire PSM than the higher marginal probability but also demonstrates that both are inferior to the joint posterior score. Finally, using the data from two previous cross-linking studies, we evaluate the performance of XLSearch and show a larger number of identified cross-linked peptides, compared to xQuest14,15 and Kojak,17 that are the current state-of-the-art.
Materials and Methods
Identification of Cross-Linked Peptides through Database Searching
We consider the problem of identifying cross-linked peptides using database searching. First, protein sequences in a given database are in silico digested and paired, resulting in a collection of hypothetical cross-linked peptides C = {(αi, βi)}, where α and β represent sequences of the two cross-linked peptides, respectively. Next, each experimental MS/MS spectrum S is compared against a subset of cross-linked peptides in C, whose mass (the sum of the masses of the two peptides plus the mass of the linker) is within the mass tolerance of the precursor ion mass of S. An MS/MS spectrum Si and its best-scoring candidate peptide pair constitute a peptide-spectrum match (PSM), denoted as αi, βi, Si. Because each algorithm considers a different scoring scheme between experimental spectra and theoretical spectra of the peptides in C, the set of PSMs is specific to the search algorithm.
Unlike the conventional peptide identification, in which a PSM is either true (correctly identified) or false (incorrectly identified), in cross-linked peptide identification, we can think of three categories of PSMs: true–true, in which both peptides are correctly identified; true–false, in which one peptide is correctly identified but the other is not; and false–false, in which neither peptide is correctly identified. Although all true–false and false–false PSMs constitute incorrect identifications, separating true–false from true–true PSMs is more challenging as they could contain an individual peptide that contributes a significant number of shared peaks between theoretical and experimental spectra. Thus, a powerful scoring scheme should be able to incorporate this type of information and distinguish both true–false and false–false PSMs from the true–true PSMs.
Scoring PSMs Involving Cross-Linked Peptides
We seek to express the score for each PSM (α, β, S) resulting from a database search as the joint posterior probability P(α = T, β = T|x) that both peptides are correctly identified given the feature vector x describing α, β, S. Using Bayes' rule,
| (1) |
| (2) |
We can also express marginal posterior probabilities of correct identification as
| (3) |
| (4) |
Solving equations 3 and 4 for P(α = T|x) and P(β = T|x), it follows that
| (5) |
| (6) |
After the substitution of equations 5 and 6 into 1 and 2, P(α = T, β = T|x) can be expressed solely in terms of the conditional probabilities
| (7) |
| (8) |
It is straightforward to show from here that P(α = T, β = T|x) is bound by the lesser of P(α = T|x) and P(β = T|x).
In equations 7 and 8, the conditional probabilities P(α = T|β = T,x), P(α = T|β = F,x), P(β = T|α = T,x), and P(β = T|α = F,x) indicate the probability of α (or β) being the true peptide, given that β (or α) is the true–false peptide, whereas the marginal probabilities P(α = T|x) and P(β = T|x) represent the probabilities that the peptide α (or β) is true, whether or not the other peptide β (or α) is true. Note that both joint probability P(α = T, β = T|x) and marginal probabilities P(α = T|x) (or P(β = T|x)) are in fact posterior probabilities with respect to the set of features x.
For gaining an intuitive understanding of how the conditional probabilities are related to the marginal probabilities and the joint probability, consider a true–false PSM (α = T, β = F,x). It follows that P(α = T|β = F,x) ≈ 1 and P(β = T|α = T,x) ≈ 0. Substituting P(α = T|β = F,x) and P(β = T|α = T,x) into equations 5 and 6, we have P(α = T|x) ≈ 1 and P(β = T|x) ≈ 0 regardless of the values of P(α = T|β = T,x) and P(β = T|α = F,x), which eventually leads to P(α = T, β = T|x) ≈ 0. Similar results can be shown for other cases (i.e., (α = T, β = T,x), (α = F, β = T,x), and (α = F, β = F,x)). Finally, we note that the identification events for peptides α and β are not conditionally independent given x. In certain situations, α and β may share theoretical fragment ions, causing the experimental peaks that match fragment ions in one peptide to also match the ions from the other peptide. Therefore, the knowledge about one peptide's correct identification for this PSM may significantly influence the identification outcome for the other peptide.
Estimating Posterior Probabilities Using Binary Classifiers
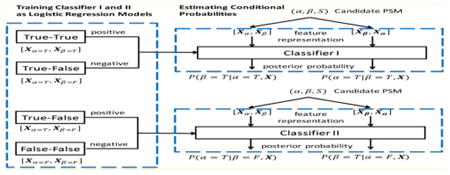
The conditional probabilities used in equations 7 and 8 can be regarded as posterior probabilities that the peptide in question is true under the condition that the other peptide is true or false, respectively. Given that the outputs from well-trained binary classification models can approximate posterior probabilities,23 we trained two logistic regression-based models to learn P(α = T|β = T,x) and P(α = T|β = F,x), respectively. As illustrated in Figure 1a, true–true and true–false PSMs are used to train Classifier I, where all training examples (PSMs) contain a correct β peptide. This model is used to classify whether α is correct as well. Likewise, true-false and false–false PSMs are used to train Classifier II, in which all training examples (PSMs) contain an incorrect β peptide. This model is used to classify whether α is correct.
Figure 1.
Schematic diagrams showing (a) how the classifiers are trained and used to estimate the posterior probabilities; (b) how the true–true PSMs are used to derive the true–false and false–false PSMs for constructing the training data; and (c) the composition of target–target, target–decoy, and decoy–decoy hits in terms of true–true, true–false, and false–false PSMs.
For the computation of the features representing a particular candidate PSM (α, β, S), the set of theoretical ions are first derived from the potentially cross-linked peptide (α and β). Based on the matching between the theoretical ions and peaks in the experimental spectrum S, we compute features of the PSMs including the numbers of cleavage sites supported by matched experimental peaks for both α and β as well as the percentage of matched experimental peaks (see Table 1 for details). This results in 8-dimensional feature vectors xα and xβ that describe the quality of matching for α and β, separately. In the case when α and β share a set of theoretical fragment ions matched to the experimental peaks, these peaks are (conservatively) counted only for the peptide containing more matched peaks overall.
Table 1. Summary of the Theoretical Ions and the Features Computed from PSMa.
| fragment site | productions | |
|---|---|---|
| 1 | backbone | b, b – water, b – ammonia |
| a, a– water, a – ammonia | ||
| y, y – water, y – ammonia | ||
| 2 | backbone closest to C-terminus | b + water, a + water |
| 3 | between lysine side | precursor, precursor – water, precursor – ammonia |
| chain and the linker | precursor + linker, precursor + linker-water precursor + linker – ammonia, precursor + linker – double water | |
| features | ||
| 1 | number of cleavage sites supported by a matched prefix ion (i.e., b and a) | |
| 2 | number of cleavage sites supported by a matched suffix ion (i.e., y) | |
| 3 | length of longest consecutive cleavage sites supported by a matched prefix ion | |
| 4 | length of longest consecutive cleavage sites supported by a matched suffix ion | |
| 5 | binary value indicating if a precursor ion (e.g., precursor + linker) matched an experimental peak | |
| 6 | percentage of experimental peaks that can be assigned | |
| 7 | percentage of total experimental peak intensities that can be assigned | |
| 8 | peptide length | |
Given PSM (α, β, S), the masses of theoretical ions are first computed by assuming a single cleavage at each peptide bond for α and β, which leads to the regular b, y, and a ions. In addition, we consider the ions resulting from the dissociation of the bond between the side chain of the cross-linked residue and the linker (e.g., the bond between lysine's amino group and DEST). The matching between the theoretical ions and the peaks in S are represented as feature vectors for α and β separately, giving rive to xα and xβ (each having a length of eight).
Given Classifiers I and II and feature vectors xα and xβ, the posterior probabilities are computed as shown in Figure 1(a). Specifically, P(β = T|α = T,x) is the output of Classifier I on the input x = [xα, xβ], P(α = T|β = T,x) is the output of Classifier I on the input [xβ, xα], P(α = T|β = F,x) is the output of Classifier II on the input [xα, xβ], and P(β = T|α = F,x) is the output of Classifier II on the input [xβ, xα]. The computed posterior probabilities are further calibrated to reconcile the differences in class priors of the PSMs used as training data and the PSMs from a specific database search.24 Briefly, we adjusted the expectation-maximization approach by Saerens et al.24 to our problem as follows: Classifiers I–II are first applied on the PSMs from the database search to obtain the uncalibrated posterior probabilities that are then used to estimate the new class priors. These new class priors are subsequently used to calibrate the posterior probabilities and the updates are performed until convergence (see the Supplementary Methods section). Finally, the posterior probabilities from Equations 7 and 8 are computed using the calibrated posterior probabilities.
Estimating False Discovery Rates
A target–decoy approach was adopted for the identification of cross-linked peptides.15,25 Specifically, the target sequence database was combined with a decoy database containing the reversed sequence of each protein from the target database. Searching spectra against this database result in (1) target–target hits, where both peptides are from target proteins; (2) target–decoy hits, where one peptide is from a target protein and the other is from a decoy protein; and (3) decoy–decoy hits, where both peptides are from decoy proteins.
We estimate the false discovery rate (FDR) using the formula from Walzthoeni et al.15 as
where nTT, nTD, and nDD denote the numbers of the three types of database hits, respectively. The FDR was estimated at the level of unique peptide pairs as well as at the level of PSMs, and both measures were used to determine the target–target crosslink identifications in our benchmarking experiments. In addition, we estimated the FDR separately for intraprotein and interprotein cross-links.15–17 This is because the number of theoretical combinations of intraprotein peptide pairs is usually far smaller than the number of interprotein peptide pairs. As a result, the validation of the two types of cross-linked peptides would require different score cutoffs, as the likelihood of encountering a random intraprotein or interprotein cross-links are typically different.
In addition to the target–decoy method, the identified cross-linked peptides were further validated using the protein structural models available in the Protein Data Bank.26 In these cases, the peptides α and β from each identified PSM were mapped to the structural model of the desirable protein complex, and the distance between the cross-linked residues was calculated between their α-carbon atoms. We then computed the percentage of the identified PSMs at a particular FDR threshold of which the distance exceeds the expected value. These PSMs were subject to manual inspection.
Data Sets
We used a large data set containing 80 811 MS/MS spectra from a previous cross-linking experiment on the E. coli ribosome (PRIDE: PXD003381).27 The E. coli ribosome is a large ribonucleoprotein complex composed of a small and large subunit containing three rRNA molecules and over 50 proteins. Its 3D structure was probed using an amidinating cross-linker diethyl suberthioimidate (DEST), a bifunctional reagent with an 11 Å spacer arm that reacts with the amino group on lysine side chains or protein N-termini. The cross-linked proteins were tryptically digested, and the resulting sample was fractionated and analyzed through a routine LC–MS/MS workflow. The fragment ion spectra were acquired using an LTQ Orbitrap XL mass spectrometer from Thermo Scientific.
To further validate our method, we considered another publicly available data set from a cross-linking study28 on the Human transcription factor TFIID (PRIDE: PXD001454). TFIID contains TATA-binding proteins (TBP) and a number of TBP-associated factors (TAFs). The sample containing the TAFs was cross-linked using the bifunctional reagent bissulfosuccinimidyl-suberate (BS3). The cross-linked peptides were analyzed on an LTQ Orbitrap Velos mass spectrometer coupled with an UltiMate 3000 Rapid Separation LC system, which yielded a 45 331 MS/MS spectra.
Results and Discussion
XLSearch was first applied to the E. coli data set to examine the score distributions of PSMs and spatial separation of the identified cross-linked peptides in the 3D structure of the E. coli ribosome. Then, the performance of XLSearch was compared with alternative search algorithms xQuest and Kojak on both E. coli and Human TAF data sets.
Constructing Training Sets for Classification Models
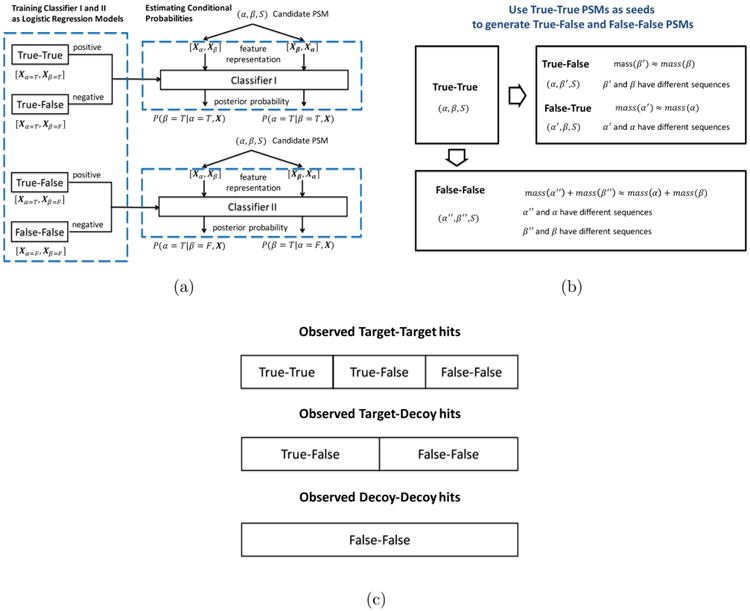
The fragment ion spectra of the E. coli data set were used to construct the true–true, true–false and false–false PSMs for training Classifiers I and II. The initial set of true–true PSMs was obtained by searching the experimental spectra against the forward sequences of the E. coli ribosomal proteins using a stringent rule-based algorithm. This algorithm simply computes the feature vectors xα and xβ of the candidate PSMs for each query tuple (α, β, S). The PSMs are then filtered to keep only those for which the number of cleavage sites supported by matched prefix (b and a) and suffix (y) ions are no less than 20% of the length of α and β. This criterion ensures that the identification of both peptides is supported by a sufficient number of matched peaks (note, however, that the XLSearch software also provides a functionality that allows a user to supply a set of confident true–true identifications as seeds for training purposes). For the spectra containing more than one candidate PSM satisfying this criterion, only the PSM with the highest percent total intensity of matched peaks is retained.
Based on the set of true–true PSMs, the sets of true–false and false–false PSMs are constructed based on the procedure illustrated in Figure 1b. Specifically, two sets of PSMs {(α, β′, S)} and {(α′, β, S)} are derived from each PSM (α, β, S) in the true–true set, in which the peptides α′ and β′ have the same precursor masses as α and β, respectively, but are sampled from the set of proteins of another organism (i.e., mouse). Because (α, β′) and (α′, β) are still paired with the same spectrum S, they can be used to mimic the set of semifalse PSMs occurring during the database search. Likewise, {(α″, β″, S)} are derived from (α, β, S), in which the sum of the precursor masses of α′′ and β′′ equals that of α and β but have different sequences than their counterparts. They are used to mimic the false–false PSMs. Using this approach, we can use one true–true PSM to construct multiple true–false and false–false PSMs.
By applying this algorithm, we obtained a training set consisting of 169 true–true, 8568 true–false, and 91 242 false–false PSMs and used them to train Classifiers I and II as logistic regression models. To estimate the prediction accuracy of these models, we performed a 10-fold cross-validation. The area under the ROC curve (AUC) values of 0.969 and 0.988 were observed for Classifiers I and II, respectively, as illustrated in the ROC curves (Supplementary Figure S6). Although the classifiers were trained using significantly more negative than positive data, the false positive rates were reasonably small (0.18 and 0.05 for Classifiers I and II, respectively) at a high sensitivity level (0.95).
Analysis of the E. coli Ribosomal Protein Data Set
We searched the fragment ion spectra against a target–decoy database containing 56 E. coli ribosomal proteins, obtained from UniProt,29 and their reversed sequences (112 proteins in total). The search algorithm first performed in silico digestion of the protein sequences, resulting in a total of 3146 tryptic peptides with up to two missed cleavage sites containing at least four amino acid residues and an internal lysine (i.e., not on the C-terminus). The database search was conducted on the whole set of hypothetical cross-linked peptides, which were preindexed according to their masses so that a subset of cross-linked peptides consistent with the precursor mass is compared with the query spectrum. Cross-linking was considered to occur on protein N-terminus residues and internal lysine. Mass tolerance of 5 ppm and 0.01 Da were used for matching the precursor and fragment ions, respectively. Cysteine residues were not modified because the protein sample was not reduced and alkylated. Only the top-scoring PSMs were reported for each MS/MS spectrum after the search.
Distinguishing Power of the Scoring Scheme
The search against the target–decoy database reported a total of 50 562 spectra identified with at least one candidate cross-linked peptide. Among those, 13 535 were target–target hits, 24 522 were target–decoy hits, and 12 505 were decoy–decoy hits. In the rare cases in which an individual peptide appeared in both target and decoy databases, it was considered to be only from the target database. The compositions of the three types of database hits in terms of true–true, true–false, and false–false identifications is schematically illustrated in Figure 1c.
We first examined the score distributions of target-target, target–decoy, and decoy–decoy hits. As shown in Figure 2a, in the high-score range, we observed a larger proportion of Target–Target hits than Target-Decoy and Decoy–Decoy hits. Interestingly, the discrepancy in score distribution between Target–Target and Target-Decoy hits appears to be more conspicuous than that between Target-Decoy and Decoy–Decoy hits (Figure 2b), which is likely accounted for by the discrepancy in the unknown score distribution between true positives and semifalse hits. This implies that the joint probability has the potential to distinguish true positives from semifalse hits, even though the latter contains one correctly identified individual peptide.
Figure 2.
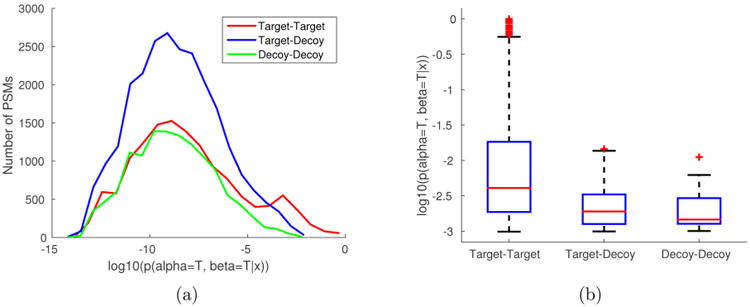
Score distribution of the PSMs that result from database searching against the target-decoy database of E. coli ribosomal proteins. (a) The histogram of the joint probability scores of PSMs where (1) both peptides are in the target database (target–target), (2) one peptide is in the target and the other is in the decoy database (target–decoy), and (3) both peptides are in the decoy database (decoy–decoy). (b) The box plot of top-thousand PSMs divided into three groups: target–target (794), target–decoy (162), and decoy–decoy (44). The averages are 5.59·10−2, 2.76·10−3, and 2.42·10−3, respectively.
In addition to the joint probability of cross-linked peptides, we also estimated the marginal probabilities for each individual peptide, as shown in equations 5 and 6. We distinguish between the individual peptide that comes with the higher marginal probability and the one with the lower marginal probability and show the scatter plot of the marginal probabilities versus the corresponding joint probability in Figure 3a. We observe that, in addition to being a tighter upper bound of the joint probability, the lower marginal probability seems to be better correlated with the joint probability than the higher marginal probability (Pearson's correlation coefficients of 0.882 and 0.237, respectively). Moreover, for many PSMs, the difference between the higher and lower marginal probability appears to be large. This implies that the individual peptides in these PSMs were identified with different confidence. If a scoring scheme simply calculates the sum of the match quality measures as the score of the entire cross-linked peptide, the confidence of individual peptides may not be guaranteed. In contrast, as the joint probability is bounded by the lower marginal probability that is independently estimated, the semifalse PSMs would not likely receive a high joint probability, even if they contain a (true) peptide identified with high confidence. Figure 3c illustrates an example of a semifalse identification in which the marginal probability of the false peptide ( QEALELAAKVR) is considerably lower than that of the true peptide ( EKPTWLEVDAGK). This resulted in a low joint probability for the entire cross-linked peptide.
Figure 3.
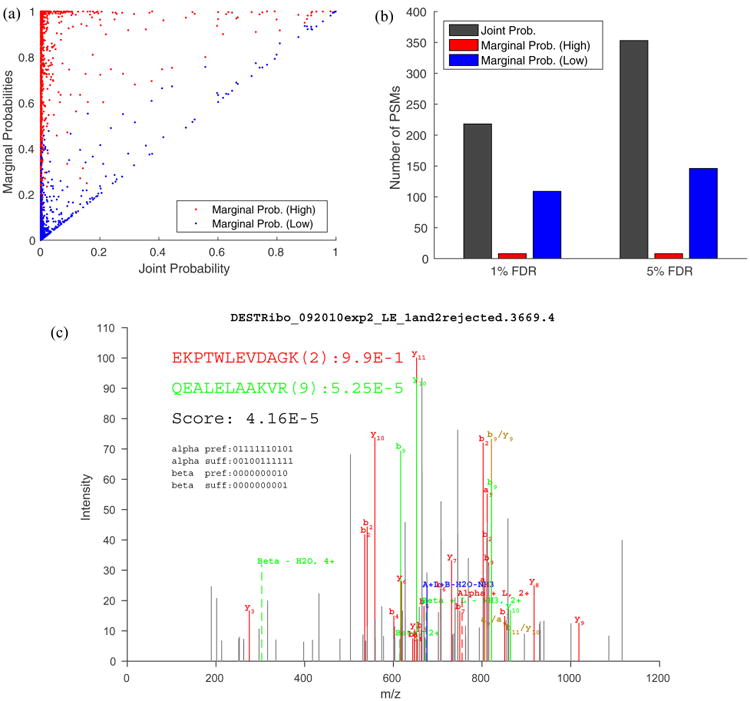
Marginal probabilities estimated for each individual peptide in a PSM. (a) Scatter plot of the higher (red) and lower (blue) marginal probability versus the corresponding joint probability for each PSM. The Pearson correlation coefficient between the marginal probabilities and the joint probabilities are 0.237 (red) and 0.882 (blue), respectively. (b) The numbers of cross-link PSMs identified at the PSM-level FDR using the target–decoy approach. The score associated with each PSMs equals (1) the joint probability of the PSM (gray), (2) higher marginal probability (red), and (3) lower marginal probability (blue). Notice that the lower marginal probability resulted in more identifications than the higher one, and the joint probability is superior to either marginal probability. (c) The spectrum identified as a correct (red) peptide cross-linked with an incorrect (green) peptide with marginal probabilities of 0.99 and 5.25·10−5, respectively. The peptide depicted in red is from the forward sequence of S4 of the small subunit of the E. coli ribosome and is extensively covered by fragment ion assignment. The peptide depicted in green is from the reversed sequence of S4 and has only two fragment ion assignments. The decoy peptide (in green) has the same amino acid composition as the true peptide (i.e., VKAALELAEQR).
The effectiveness of the marginal probabilities can be further demonstrated by examining the numbers of PSMs identified using each marginal probability in place of the joint probability as the score of the entire cross-linked peptides. As illustrated in Figure 3b, more cross-linked peptides were identified using the lower marginal probability at both 1% and 5% FDR (109 versus 8 and 146 versus 8) cutoff, consistent with the hypothesis that the lower marginal probability represents a better indicator of a true PSM than the higher marginal probability. Nevertheless, the joint probability achieved overall more identifications than either of the marginal probabilities (218 versus 109 versus 8 and 353 versus 146 versus 8), indicating that the higher or lower marginal probability may be too loose or too stringent, respectively, compared with the joint probability in assessing the overall confidence of cross-linked peptide identification.
Spatial Separation of the Identified Cross-Linked Peptides
The peptide sequences from the identified cross-linked peptides were mapped to the structural model of the E. coli ribosome (PDB ID: 4KIX, 4KIY).30 We computed the Cα – Cα distances for each identified pair of cross-linked residues and counted the number of cross-linked residues whose distance exceeded 24.0 Å (i.e., the length of the DEST spacer arm plus twice of the length of the lysine side chain). As shown in Table 2 for intraprotein cross-links, the percentages of target–target hits (at the PSM or unique peptide pair level) with distance greater than the expected value is generally comparable to the FDRs estimated from the target–decoy approach. However, this number is found be to significantly larger for interprotein cross-links. One reason for this might be the small size of the data set as well as the fact that the Cα – Cα distances were computable for only a subset (60–80%) of the identified PSMs. In addition, some of the distance-violating target–target hits may not be false positives (see below).
Table 2. Comparing Cross-Link Identifications Validated Using the Target–Decoy Approach against the Structural Modela.
| FDR | target–target hits | ||||
|---|---|---|---|---|---|
|
| |||||
| total | distance computable | distance ≥24.0 Å | percentage | ||
| 1% | intraprotein | 132 (36) | 105 (28) | 1 (1) | 0.95% (3.57%) |
| interprotein | 84 (25) | 59 (15) | 3 (3) | 5.08% (20.0%) | |
| 5% | intraprotein | 134 (38) | 107 (30) | 1 (1) | 0.93% (3.33%) |
| interprotein | 87 (27) | 62 (17) | 5 (5) | 8.06% (29.4%) | |
The total numbers of PSMs, the numbers of PSMs for which the Cα –Cα distance were computable, and the numbers of PSMs for which the Cα – Cα distance exceeded 24.0 Å are illustrated. The numbers in the parentheses correspond to the unique peptide pairs. The FDR was estimated at the unique peptide-pair level.
At 1% FDR, 132 intraprotein and 84 interprotein PSMs were identified. These PSMs correspond to 55 unique cross-linked sites (Table 3), of which 32 were intraprotein and 23 were interprotein. The larger number of intraprotein peptides was expected because the search space for intraprotein cross-links (54 289) was significantly smaller than that for interprotein cross-links (4 899 089). Therefore, the likelihood that the score of a decoy match exceeds a certain threshold would be lower for the identification of intraprotein cross-linked peptides.
Table 3. Unique Cross-Linked Sites Corresponding to the 132 Intraprotein and 84 Interprotein PSMs Identified at 1% FDR (at the Unique Peptide Pair Level) by Searching against the E. coli Ribosomal Proteins (See Table 4)a.
| proteins | linkage | distance (Å) | PSMs |
|---|---|---|---|
| intraprotein cross-links | |||
| L1–L1 | K14–K6 | NA | 2 |
| L1-L1 | K205–K54 | NA | 3 |
| L1–L1 | K54–K167 | NA | 5 |
| L14–L14 | K44–K54 | 5.5 | 8 |
| L14–L14 | K54–K111 | 25.6 | 1 |
| L17–L17 | K78–K121 | NA | 1 |
| L18–L18 | K63–K76 | 16.5 | 2 |
| L19–L19 | K63–K87 | 14.2 | 1 |
| L2–L2 | K59–K207 | 20.1 | 1 |
| L20–L20 | K85–K78 | 10.2 | 9 |
| L25–L25 | K25–K10 | 13.3 | 3 |
| L33–L33 | K10–K50 | 8.8 | 1 |
| L33–L33 | K37–K50 | 9.0 | 1 |
| L33–L33 | K50–K33 | 7.4 | 7 |
| L4–L4 | K166–K132 | 14.3 | 1 |
| L6–L6 | K44–K6 | 11.9 | 1 |
| L7–L7 | K108–K60 | NA | 3 |
| L9–L9 | K42–K57 | NA | 8 |
| S14–S14 | K19–K47 | 17.9 | 2 |
| S14–S14 | K23–K47 | 16.0 | 3 |
| S17–S17 | K39–K30 | 9.0 | 1 |
| S19–S19 | K17–K21 | 5.9 | 8 |
| S19–S19 | K17–K28 | 13.5 | 1 |
| S20–S20 | K16–K19 | 5.3 | 11 |
| S20–S20 | K49–K34 | 10.1 | 2 |
| S20–S20 | K69–K76 | 11.3 | 2 |
| S3–S3 | K108–K147 | 14.3 | 2 |
| S3–S3 | K49–K86 | 16.8 | 20 |
| S3–S3 | K86–K45 | 14.0 | 4 |
| S3–S3 | K86–K79 | 11.3 | 3 |
| S4–S4 | K167–K156 | 16.1 | 10 |
| S7–S7 | K149–K171 | NA | 5 |
| interprotein cross-links | |||
| L14–L31B | K114–K30 | NA | 1 |
| L17–L32 | K35–K53 | 16.7 | 1 |
| L25–L16 | K83–K127 | 12.5 | 3 |
| L27–L18 | K62–K17 | 15.7 | 1 |
| L28–L9 | K44–K35 | 15.8 | 2 |
| L28–L9 | K44–K42 | 22.7 | 15 |
| L28–L9 | K44–K57 | NA | 8 |
| L28–L9 | K62–K42 | 12.5 | 5 |
| L6–L7 | K29–K71 | NA | 2 |
| L6–L7 | K86–K60 | NA | 2 |
| L7–L11 | K71–K100 | NA | 1 |
| S1–L2 | K150–K68 | NA | 1 |
| S14–L3 | K12–K106 | 148.4 | 1 |
| S14–S10 | K76–K11 | 17.8 | 2 |
| S15–S17 | K71–K81 | 17.6 | 2 |
| S17–S8 | K30–K87 | 12.3 | 20 |
| S19–S14 | K17–K47 | 12.1 | 4 |
| S21–S12 | K40–K44 | 46.4 | 1 |
| S6–S18 | K106–K30 | NA | 8 |
| S7–S1 | K149–K279 | NA | 1 |
| S8–S2 | K69–K26 | 15.9 | 1 |
| S9–S1 | K100–K279 | NA | 1 |
| S9–S12 | K100–K44 | 85.0 | 1 |
The “Linkage” column indicates the indices of the lysine residues in the protein sequences.
Despite the agreement with the structural model of E. coli ribosomes for most of the identified cross-linked peptides, one intraprotein and three interprotein cross-linked peptides were observed to have Cα – Cα distance exceeding the expected value (Table 3). The distance of K54–K111 in ribosomal protein L14 appears to be on the borderline, which may be accounted for by the conformational flexibility of the proteins. However, the distances between the pairs of cross-linked residues K40–K44, K100–K44, and K12–K106 that connect the ribosomal proteins S21 to S12, S9 to S12, and S14 to L3, respectively, are much greater than expected and thus are unlikely caused by conformational flexibility. We examined the fragment ion spectra identified as the corresponding cross-linked peptides and found that the majority of peak were matched with theoretical fragmentation and both peptides were extensively supported by fragment ion assignments (Supplementary Figures S1–S5). In fact, E. coli ribosomes may form polysomes during translation such that residues on different ribosomes may be in close proximity.31 The structural basis of these cross-links is worth further investigation.
Comparative Evaluation
We evaluated the performance of XLSearch using the E. coli ribosome and Human TAF protein data sets and compared it with two alternative searching algorithms, xQuest and Kojak. The spectra from the E. coli data set were searched against a database containing 56 E. coli ribosomal proteins as described earlier. The spectra of the human data set were searched against a database containing eight Human TAF proteins appended with an additional 50 randomly selected human protein sequences to simulate a more complex sample. For XLSearch, the logistic regression models trained on the E. coli data set were directly used for scoring PSMs from the Human TAF protein search. Except for a fixed modification of carbamidomethylation on cysteine that was only used to analyze the human data, the same parameters were used in both searches: minimum peptide length = 4, maximum missed cleavage sites = 2, residues for cross-link = lysine, allow cross-link on peptide C-terminus = false, allow cross-link on protein N-terminus = true, MS1 mass tolerance = 5 ppm, and MS2 mass tolerance = 0.01 Da. We used the latest version of xQuest (v2.1.1) and Kojak (v1.4.2) for the comparative evaluation. The parameters of xQuest and Kojak were adjusted to match those used in XLSearch (for a complete set of parameters, see Supplementary Tables S6–S8). The FDR was estimated separately for intraprotein and interprotein cross-links at both the PSM and unique peptide pair levels.
Table 4 and Figure 4 illustrate the cross-linked peptides identified by the different search algorithms. As expected, we observe that the numbers of target–target hits identified with FDR at the unique peptide pair level are less than or equal to those at the PSM level. This is because the target–target hits are more likely to be dominated by a few cross-links with many PSMs as compared with decoy hits (as is the case in Table 3). Thus, counting only the number of unique cross-linked peptides would typically give rise to a more conservative estimate of FDR.
Table 4. Numbers of PSMs and Unique Cross-Linked Peptides (in the Parentheses) Identified by XLSearch, xQuest, and Kojak, Separated into Intraprotein and Interprotein Cross-Linksa.
| FDR | methods | E. coli | Expanded E. coli | Human | Expanded Humanb | Expanded Humanc | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| intra | inter | intra | inter | intra | inter | intra | inter | intra | inter | ||
| FDR estimated at PSM level | |||||||||||
| 1% | XLSearch | 134 (38) | 84 (25) | 192 (51) | 115 (33) | 66 (33) | 115 (60) | 74 (36) | 116 (61) | 74 (36) | 119 (63) |
| xQuest | 32 (10) | 38 (10) | 14 (4) | 2 (1) | 40 (21) | 52 (30) | 19 (7) | 18 (10) | 19 (7) | 19 (11) | |
| Kojak | 47 (15) | 23 (8) | 50 (15) | 22 (9) | 63 (23) | 42 (23) | 8 (4) | 7 (5) | 8 (4) | 8 (6) | |
| 5% | XLSearch | 225 (59) | 128 (34) | 260 (67) | 130 (35) | 185 (79) | 231 (113) | 158 (71) | 208 (106) | 158 (71) | 218 (110) |
| xQuest | 108 (35) | 103 (25) | 14 (4) | 2 (1) | 244 (89) | 251 (120) | 44 (21) | 59 (33) | 44 (21) | 60 (34) | |
| Kojak | 54 (15) | 25 (9) | 64 (16) | 25 (9) | 78 (29) | 46 (27) | 61 (22) | 39 (22) | 61 (22) | 45 (28) | |
| FDR estimated at unique peptide pair level | |||||||||||
| 1% | XLSearch | 132 (36) | 84 (25) | 192 (51) | 113 (32) | 66 (33) | 88 (50) | 74 (36) | 109 (58) | 74 (36) | 112 (60) |
| xQuest | 32 (10) | 38 (10) | 14 (4) | 2 (1) | 40 (21) | 52 (30) | 19 (7) | 18 (10) | 19 (7) | 19 (11) | |
| Kojak | 47 (15) | 23 (8) | 50 (15) | 22 (9) | 63 (23) | 42 (23) | 8 (4) | 7 (5) | 8 (4) | 8 (6) | |
| 5% | XLSearch | 134 (38) | 87 (27) | 197 (53) | 115 (33) | 161 (70) | 210 (109) | 103 (49) | 164 (85) | 103 (49) | 170 (87) |
| xQuest | 32 (10) | 38 (10) | 14 (4) | 2 (1) | 44 (21) | 53 (30) | 40 (21) | 52 (30) | 40 (21) | 53 (31) | |
| Kojak | 47 (15) | 23 (8) | 50 (15) | 22 (9) | 71 (25) | 46 (27) | 8 (4) | 7 (5) | 8 (4) | 8 (6) | |
FDR was estimated at both the PSM level and the unique peptide pair level and separately for intraprotein and interprotein cross-links. The corresponding Venn diagrams are shown in Figure 4. The searches were performed against 56 ribosomal proteins (E. coli), 56 ribosomal and 34 associated proteins (Expanded E. coli), eight TAF proteins (Human), eight TAF proteins, and 50 arbitrary proteins (Expanded Human).
Note that for the search against the Expanded Human database, we separately listed the target–target PSMs that do not contain peptides from the 50 arbitrary proteins, and.
Note that for the search against the Expanded Human database, we separately listed the target–target PSMs that include peptides from the 50 arbitrary proteins.
Figure 4.
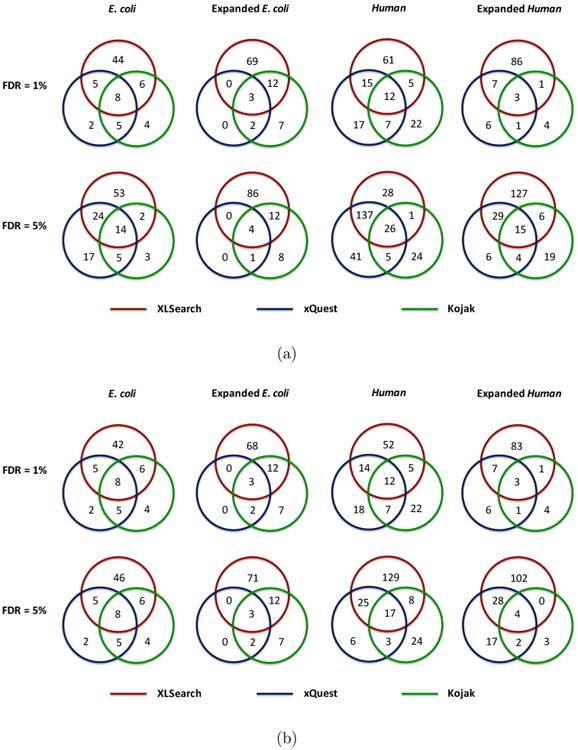
Venn diagram illustration of the comparison of the unique cross-linked peptides identified by XLSearch, xQuest, and Kojak, respectively. The FDR was estimated at both the PSM level (a) and the unique peptide pair level (b). For the target–target PSMs identified in the search against the Expanded Human database, those with at least one individual peptide from the 50 randomly selected proteins were excluded in this figure (see Table 4).
For the E. coli ribosome data set, XLSearch identified 65 unique cross-linked peptides with 5% FDR (at unique cross-linked peptide level), of which 19 overlapped with the results from xQuest or Kojak and 46 were unique to XLSearch (Figure 4b). In comparison, there were a total of 11 cross-linked peptides that were unique to xQuest or Kojak. We found that the majority of these peptides were also identified by XLSearch at a higher FDR level (Supplementary Table S1). A pair of the 46 cross-linked peptides unique to the XLSearch and three of the 11 cross-links that it missed were observed to contain no fragment ion assignment to one of the two individual peptides. These peptides were typically short (with 4–6 amino acid residues) and, thus, were not as informative as longer peptides that would result in more fragment ions. Although they were observed in the identification results of all algorithms, the peptides in the XLSearch-identified cross-linked peptides were found to be generally longer than those in xQuest or Kojak. Furthermore, the cross-links identified by XLSearch at 1% FDR contained longer individual peptides than those identified at 5% FDR (Supplementary Figure S7). This is expected because short peptides generally provide fewer fragment ion assignments, and, therefore, cross-links containing short peptides would likely receive relatively lower scores.
In addition, the spectra from the E. coli data set were also searched against an expanded database containing 34 additional proteins that likely interact with the ribosome. We refer to these proteins as the ribosome-associated proteins; see the Supplementary Methods section for the procedure that was used to determine these proteins.. This resulted in a number of identifications involving at least one ribosome-associated protein (Table 5). As compared with the search against the 56 ribosomal proteins, the size of the target search space for intraprotein and interprotein cross-links increases by 134% (from 54 289 to 127 314) and 224% (from 4 899 089 to 15 898 976). However, the number of identifications were observed to increase only moderately. This can be explained by a deficiency of stable complexes between ribosomal and associated proteins and an increased likelihood for decoy matches in the expanded database that prevented more crosslinks from being identified. As shown in Table 4, the numbers of identifications by xQuest was actually reduced in the expanded database search compared with XLSearch, suggesting that XLSearch may be less susceptible to the increased likelihood of decoy matches in larger databases.
Table 5. Unique Cross-Links Identified at 1% FDR (at the Unique Cross-Linked Peptide Level) That Involve Peptides from Ribosome Associated Proteins.
| peptides | proteins | matches |
|---|---|---|
| associated protein–associated protein | ||
| KLDEVR(1)-KWLEER(1) | C9QZG0, uncharacterized protein; C9QZG0, uncharacterized protein | 14 |
| KDAEVR(1)-KIEAALADK(1) | C9QRL6, ribosome-recycling factor; C9QRL6, ribosome-recycling factor | 2 |
| ALDDVKK(6)-YVELKAR(5) | C9QR42, regulatory protein; C9QR42, regulatory protein | 2 |
| FLADNYGKTVSR(8)-TFVIDKAGY(6) | C9R1A9, ribonuclease; C9R1A9, ribonuclease | 1 |
| AEKALDDVK(3)-YVELKAR(5) | C9QR42, regulatory protein; C9QR42, regulatory protein | 1 |
| NKELMDNMR(2)-TNELKADEER(5) | C9QQ82, trigger factor; C9QQ82, trigger factor | 2 |
| IAGLEVKR(7)-QATKDAGR(4) | C9QSQ5, chaperone protein DnaK; C9QSQ5, chaperone protein DnaK | 1 |
| associated protein–ribosomal protein | ||
| GGKR(3)-AVAERALEKGIK(9) | C9QTY9, translation initiation factor; ribosomal L18 | 1 |
| EAKLK(3)-IKLVSSAGTGHFYTTTK(2) | C9R115, signal recognition particle protein; ribosomal L33 | 1 |
| MKGGKR(1)-ADGKVK(4) | C9QTY9, translation initiation factor; ribosomal L27 | 1 |
The search of spectra from the Human data set against the database containing eight TAF and 50 arbitrary proteins resulted in 94 and 134 unique cross-linked peptides by XLSearch at 1% and 5% FDR levels, respectively (these numbers do not include target–target PSMs containing peptides from the arbitrary human proteins) (Figure 4b). In this case, we were unable to validate the identified cross-linked peptides using protein structure data as the 3D structure of the whole Human TAF protein complex is not available. Instead, we compared our results with the manually validated identifications in the original publication.28 Out of the 59 unique cross-linked peptides reported in the paper, XLSearch identified 45 and 49 at 1% and 5% FDRs, respectively (Supplementary Figure S8).
Note that the 50 randomly added human protein sequences were introduced to simulate the characterization of a more complex sample. For the comparison purposes, we carried out another search against a restricted database that contains only the eight TAF proteins (Table 4 and Figure 4). We observe that xQuest reported significantly more cross-linked peptides at the same FDR levels compared with the expanded database, and the numbers of identifications by XLSearch are similar. This again illustrates that XLSearch appears to be less sensitive to the increased number of decoy peptides that may prevent more target peptides from being identified.
Software
XLSearch is on open-source software implemented in Python. It takes experimental MS/MS spectra, protein sequence database, and search parameters as input. The best matching cross-linked peptide is reported for each query spectrum, and the fragment ion assignments for experimental peaks will be shown. The user can opt to use the pretrained logistic regression models or to in-sample train the models using the available data. As of now, XLSearch has only been evaluated on high-resolution LC–MS/MS platforms such as Orbitrap-MS. It is freely available at https://github.com/COL-IU/XLSearch.
Conclusions
In this study, we developed a database search algorithm for identifying cross-linked peptides in a tandem mass spectrometry experiment. We were motivated by the thought that the identification events of two individual peptides in a cross-linked peptide are typically not independent. Hence, we proposed a rigorous probabilistic scoring method that takes into account the dependence of the score of one peptide on the score of the other peptide and employed a machine learning approach to estimate the posterior joint probability that both peptides in the cross-link are correctly identified.
XLSearch is able to perform in-sample training and can therefore incorporate different linker reagents, although it depends on the initial set of confidently identified true–true peptides. In this work, two data sets were available, each containing a relatively small number of true–true peptides. For this reason, we engineered a small set of features and used a linear classifier to approximate posterior probabilities. As a larger number of cross-link data sets becomes available, it will be possible to pretrain classification models for each experimental platform and use them either as off-the-shelf models or to provide an initial set of peptides to be used in the training phase.
XLSearch was evaluated on the E. coli ribosome and the human TAF data sets. Our initial experiments provide evidence that its performance is reliable and favorable to xQuest and Kojak using high-resolution MS/MS data. However, further experiments will be necessary to more thoroughly characterize each of these tools.
Supplementary Material
Acknowledgments
This work was supported by the NIH grant R01GM103725.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.6b00004.
Supplementary Table S1: The cross-linked peptides missed by XLSearch at 5% FDR in the search against the E. coli ribosomal proteins. Supplementary Figures S1–S5: the annotated fragment ion spectra of the five interprotein cross-linked peptides whose Cα – Cα distance exceeds 24.0 Å as shown in Table 2. Supplementary Figure S6: The ROC curves resulting from the 10-fold cross-validation of Classifiers I and II trained using spectra from the E. coli data set. Supplementary Figure S7: The distributions of the lengths of the shorter individual peptide in a cross-link. The distribution are separated into cross-link identifications unique to XLSearch and those unique to xQuest or Kojak. Supplementary Figure S8: Venn diagrams illustrating the overlap of the unique cross-link identification between XLSearch, xQuest, Kojak, and the validated results for the search against the Expanded Human database. Supplementary Figure S9: Scatter plot of the joint probabilities of the same set of PSMs using the classifiers trained on all data or the classifiers trained in a 10-fold cross-validation process. Additional details on calibrating posterior probabilities through estimation of class priors in unlabeled data; comparative evaluation of XLSearch, xQuest, and Kojak; and experimental procedure for identifying proteins associated with E. coli ribosomal proteins. (PDF)
Supplementary Table S2: List of 56 E. coli ribosomal proteins, 34 ribosome-associated proteins, eight human TAF proteins, and 50 arbitrary proteins used in this study. (XLSX)
Supplementary Table S3: List of PSMs identified by XLSearch at 1% and 5% FDR as in Table 4. (XLSX)
Supplementary Table S4: List of PSMs identified by xQuest at 1% and 5% FDR as in Table 4 (XLSX)
Supplementary Table S5: List of PSMs identified by Kojak at 1% and 5% FDR as in Table 4. (XLSX)
Supplementary Table S6: List of all top-hits for all the searches reported by XLSearch and the corresponding list of parameters. (XLSX)
Supplementary Table S7: List of all top-hits for all the searches reported by xQuest and the corresponding list of parameters. (XLSX)
Supplementary Table S8: List of all top-hits for all the searches reported by Kojak and the corresponding list of parameters. (XLSX)
Notes: The authors declare no competing financial interest.
References
- 1.Sinz A. Chemical cross-linking and mass spectrometry for mapping three-dimensional structures of proteins and protein complexes. J Mass Spectrom. 2003;38:1225–1237. doi: 10.1002/jms.559. [DOI] [PubMed] [Google Scholar]
- 2.Sinz A. Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrom Rev. 2006;25:663–682. doi: 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- 3.Rappsilber J. The beginning of a beautiful friendship: cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes. J Struct Biol. 2011;173:530–540. doi: 10.1016/j.jsb.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Merkley ED, Cort JR, Adkins JN. Cross-linking and mass spectrometry methodologies to facilitate structural biology: finding a path through the maze. J Struct Funct Genomics. 2013;14:77–90. doi: 10.1007/s10969-013-9160-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young MM, Tang N, Hempel JC, Oshiro CM, Taylor EW, Kuntz ID, Gibson BW, Dollinger G. High throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry. Proc Natl Acad Sci U S A. 2000;97:5802–5806. doi: 10.1073/pnas.090099097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh P, Panchaud A, Goodlett DR. Chemical cross-linking and mass spectrometry as a low-resolution protein structure determination technique. Anal Chem. 2010;82:2636–2642. doi: 10.1021/ac1000724. [DOI] [PubMed] [Google Scholar]
- 7.Maiolica A, Cittaro D, Borsotti D, Sennels L, Ciferri C, Tarricone C, Musacchio A, Rappsilber J. Structural analysis of multiprotein complexes by cross-linking, mass spectrometry, and database searching. Mol Cell Proteomics. 2007;6:2200–2211. doi: 10.1074/mcp.M700274-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Zheng C, Yang L, Hoopmann MR, Eng JK, Tang X, Weisbrod CR, Bruce JE. Cross-linking measurements of in vivo protein complex topologies. Mol Cell Proteomics. 2011;10:M110–006841. doi: 10.1074/mcp.M110.006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaake RM, Wang X, Burke A, Yu C, Kandur W, Yang Y, Novtisky EJ, Second T, Duan J, Kao A, Guan S, Vellucci D, Rychnovsky SD, Huang L. A new in vivo cross-linking mass spectrometry platform to define protein-protein interactions in living cells. Mol Cell Proteomics. 2014;13:3533–3543. doi: 10.1074/mcp.M114.042630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang L, Tang X, Weisbrod CR, Munske GR, Eng JK, von Haller PD, Kaiser NK, Bruce JE. A photocleavable and mass spectrometry identifiable cross-linker for protein interaction studies. Anal Chem. 2010;82:3556–3566. doi: 10.1021/ac902615g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lauber MA, Rappsilber J, Reilly JP. Dynamics of ribosomal protein S1 on a bacterial ribosome with cross-linking and mass spectrometry. Mol Cell Proteomics. 2012;11:1965–1976. doi: 10.1074/mcp.M112.019562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murakami K, Elmlund H, Kalisman N, Bushnell DA, Adams CM, Azubel M, Elmlund D, Levi-Kalisman Y, Liu X, Gibbons BJ, Levitt M, Kornberg RD. Architecture of an RNA polymerase II transcription pre-initiation complex. Science. 2013;342:1238724. doi: 10.1126/science.1238724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen VQ, Ranjan A, Stengel F, Wei D, Aebersold R, Wu C, Leschziner AE. Molecular architecture of the ATP-dependent chromatin-remodeling complex SWR1. Cell. 2013;154:1220–1231. doi: 10.1016/j.cell.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rinner O, Seebacher J, Walzthoeni T, Mueller L, Beck M, Schmidt A, Mueller M, Aebersold R. Identification of cross-linked peptides from large sequence databases. Nat Methods. 2008;5:315–318. doi: 10.1038/nmeth.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walzthoeni T, Claassen M, Leitner A, Herzog F, Bohn S, Forster F, Beck M, Aebersold R. False discovery rate estimation for cross-linked peptides identified by mass spectrometry. Nat Methods. 2012;9:901–903. doi: 10.1038/nmeth.2103. [DOI] [PubMed] [Google Scholar]
- 16.Trnka MJ, Baker PR, Robinson PJ, Burlingame A, Chalkley RJ. Matching cross-linked peptide spectra: only as good as the worse identification. Mol Cell Proteomics. 2014;13:420–434. doi: 10.1074/mcp.M113.034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoopmann MR, Zelter A, Johnson RS, Riffle M, MacCoss MJ, Davis TN, Moritz RL. Kojak: Efficient analysis of chemically cross-linked protein complexes. J Proteome Res. 2015;14:2190–2198. doi: 10.1021/pr501321h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang B, et al. Identification of cross-linked peptides from complex samples. Nat Methods. 2012;9:904–906. doi: 10.1038/nmeth.2099. [DOI] [PubMed] [Google Scholar]
- 19.Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat Biotechnol. 2003;21:255–261. doi: 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- 20.Alves P, Arnold RJ, Clemmer DE, Li Y, Reilly JP, Sheng Q, Tang H, Xun Z, Zeng R, Radivojac P. Fast and accurate identification of semi-tryptic peptides in shotgun proteomics. Bioinformatics. 2008;24:102–109. doi: 10.1093/bioinformatics/btm545. [DOI] [PubMed] [Google Scholar]
- 21.Chen Z, Jawhari A, Fischer L, Buchen C, Tahir S, Kamenski T, Rasmussen M, Lariviere L, Bukowski-Wills JC, Nilges M, Cramer P, Rappsilber J. Architecture of the RNA polymerase II-TFIIF complex revealed by cross-linking and mass spectrometry. EMBO J. 2010;29:717–726. doi: 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalisman N, Adams CM, Levitt M. Subunit order of eukaryotic TRiC/CCT chaperonin by cross-linking, mass spectrometry, and combinatorial homology modeling. Proc Natl Acad Sci U S A. 2012;109:2884–2889. doi: 10.1073/pnas.1119472109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richard MD, Lippmann RP. Neural network classifiers estimate Bayesian a posteriori probabilities. Neural Computation. 1991;3:461–483. doi: 10.1162/neco.1991.3.4.461. [DOI] [PubMed] [Google Scholar]
- 24.Saerens M, Latinne P, Decaestecker C. Adjusting the outputs of a classifier to new a priori probabilities: a simple procedure. Neural Computation. 2002;14:21–41. doi: 10.1162/089976602753284446. [DOI] [PubMed] [Google Scholar]
- 25.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 26.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lauber MA, Reilly JP. Structural analysis of a prokaryotic ribosome using a novel amidinating cross-linker and mass spectrometry. J Proteome Res. 2011;10:3604–3616. doi: 10.1021/pr200260n. [DOI] [PubMed] [Google Scholar]
- 28.Trowitzsch S, et al. Cytoplasmic TAF2-TAF8-TAF10 complex provides evidence for nuclear holo-TFIID assembly from preformed submodules. Nat Commun. 2015;6:6011. doi: 10.1038/ncomms7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O'Donovan C, Redaschi N, Yeh LS. The universal protein resource (UniProt) Nucleic Acids Res. 2005;33:D154–9. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pulk A, Cate JH. Control of ribosomal subunit rotation by elongation factor G. Science. 2013;340:1235970. doi: 10.1126/science.1235970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsai YC, Du D, Domínguez-Malfavon L, Dimastrogiovanni D, Cross J, Callaghan AJ, García-Mena J, Luisi BF. Recognition of the 70S ribosome and polysome by the RNA degradosome in Escherichia coli. Nucleic Acids Res. 2012;40:10417. doi: 10.1093/nar/gks739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.