

Abstract
Live regulatory T cells (Treg cells) suppress antitumor immunity, but how Treg cells behave in the metabolically abnormal tumor microenvironment remains unknown. Here we show that tumor Treg cells undergo apoptosis, and such apoptotic Treg cells abolish spontaneous and PD-L1-blockade-mediated antitumor T cell immunity. Biochemical and functional analyses show that adenosine, but not typical suppressive factors such as PD-L1, CTLA-4, TGF-β, IL-35, and IL-10, contributes to apoptotic Treg-cell-mediated immunosuppression. Mechanistically, apoptotic Treg cells release and convert a large amount of ATP to adenosine via CD39 and CD73, and mediate immunosuppression via the adenosine and A2A pathways. Apoptosis in Treg cells is attributed to their weak NRF2-associated antioxidant system and high vulnerability to free oxygen species in the tumor microenvironment. Thus, the data support a model wherein tumor Treg cells sustain and amplify their suppressor capacity through inadvertent death via oxidative stress. This work highlights the oxidative pathway as a metabolic checkpoint that controls Treg cell behavior and affects the efficacy of therapeutics targeting cancer checkpoints.
Extensive studies have been conducted to define the development, conversion, stability, and regulatory mechanisms of CD4+Foxp3+ Treg cells in homeostasis and a variety of disease models1–10. It is well known that Treg cells are recruited into the tumor microenvironment and act as one of the major immunosuppressors dampening spontaneous tumor-associated antigen (TAA)-specific T cell immunity4–6, as well as immunotherapy-induced and active-vaccination-induced antitumor immunity5,6. However, how Treg cells behave in the metabolically abnormal tumor microenvironment remains unknown.
The Warburg effect is an important metabolic feature in many types of cancer11. Recent studies indicate that glycolysis regulates T cell activation and effector function12,13. Given that glucose, among other nutrients, is poorly replenished in tumors, it is assumed that T cell glycolytic metabolism is altered as a result of the Warburg effect in the tumor microenvironment13–16. In support of this, poor glycolysis can alter effector memory T cell function in the tumor microenvironment14,16. In addition, oxygen-sensing prolyl-hydroxylase proteins17, potassium ions released from necrotic cells18, and abnormal zinc metabolism19 can impair effector T cell function in the tumor microenvironment. These findings underscore the significance of the metabolic regulation of memory T cells in tumors.
The homeostatic balance between Treg cells and T helper cells may be metabolically regulated in mice20–23. However, Treg cells adopt memory and effector phenotypes in the human tumor microenvironment4,24. It is unknown whether Treg cells are subject to glycolytic regulation in tumors. Furthermore, oxidative stress is an additional metabolic feature in the tumor microenvironment. Recent studies have shown that myeloid dendritic cells (DCs) are phenotypically and functionally altered by oxidative stress in the tumor microenvironment25. However, it is unknown whether oxidative stress alters Treg cell phenotype and function in tumors.
To address these questions, we examined the phenotypic and functional nature of Treg cells in the tumor microenvironment in human ovarian cancer and in several types of mouse cancer, and investigated the mechanisms and roles of metabolism in shaping the biological behaviors of Treg cells. We observed that Treg cells were highly apoptotic in the tumor microenvironment, and that apoptotic Treg cells achieved superior suppressor function via an oxidative-stress-associated mechanism. Furthermore, we found that oxidative stress, rather than glycolysis, was the metabolic mechanism that controlled tumor Treg cell functional behavior and tempered the therapeutic efficacy of immune checkpoint therapy.
RESULTS
High Treg cell apoptosis in the tumor microenvironment
A previous study showed that Treg cells are recruited into the human tumor microenvironment and inhibit TAA-specific T cell immunity4. However, it is unknown how Treg cells behave in the metabolically abnormal tumor microenvironment. To investigate this, we used polychromatic flow cytometry analysis (Supplementary Fig. 1a) to analyze cell proliferation and apoptosis in primary Foxp3+ Treg cells and conventional Foxp3−CD4+ T cells in human ovarian cancer tissues. We found that Treg cells expressed higher levels of the cell cycle protein Ki67 than conventional T cells did in the human ovarian cancer microenvironment (Supplementary Fig. 1b). In addition, flow cytometry analyses showed that Treg cells underwent substantial apoptosis compared with Foxp3− conventional T cells in primary and metastatic ovarian cancer tissues, as determined on the basis of cleaved caspase-3 expression (Fig. 1). Immunofluorescence staining demonstrated the colocalization of Foxp3 and cleaved caspase-3 in ovarian cancer tissues (Fig. 1c and Supplementary Fig. 1c). We quantified proapoptotic and antiapoptotic gene transcripts in human ovarian-cancer-infiltrating Treg cells and conventional T cells. Treg cells expressed high levels of proapoptotic gene transcripts (Fig. 1d) and low levels of antiapoptotic gene transcripts (Fig. 1e) compared with conventional T cells in the same human ovarian cancers. We obtained similar results in mice with ID8 ovarian cancer, MC38 colon cancer, and B16 melanoma (Fig. 1f,g). Furthermore, using gene set enrichment analysis, we found enriched expression of apoptotic genes in infiltrating Treg cells in mouse B16 melanoma (Fig. 1h and Supplementary Tables 1 and 2) in the GEO database (GSE55705)26. In mouse Treg cells cultured with MC38 tumor media, we detected increased expression of proapoptotic genes (Supplementary Fig. 1d) and decreased expression of antiapoptotic genes (Supplementary Fig. 1e). Thus, the data indicate that Treg cells are highly apoptotic in the tumor microenvironment.
Figure 1.
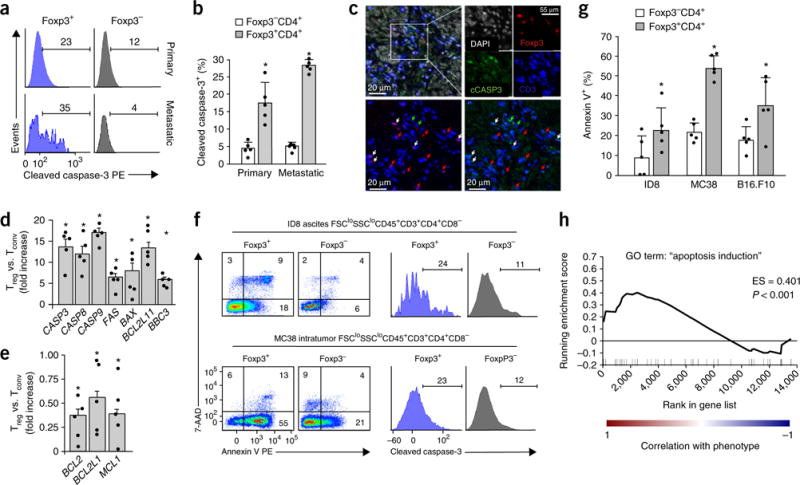
Treg cells are highly apoptotic in the tumor microenvironment. (a,b) Apoptotic Foxp3+ Treg cells and Foxp3− conventional T cells in human primary and metastatic ovarian cancers. Fresh human ovarian-cancer-infiltrating T cells were stained for viability, surface markers, Foxp3, and cleaved caspase-3. Polychromatic flow cytometry analysis was used to analyze cleaved caspase-3 expression in live CD45+CD3+CD4+ cells. The representative histogram (a) and summarized data (b) show the relative amount of cleaved caspase-3+ cells. Data in b are the mean and s.e.m. for n = 5 subjects. PE, phycoerythrin. (c) Cleaved caspase-3+Foxp3+ Treg cells in human ovarian cancer tissues. Ovarian cancer tissue sections were stained for CD3 (blue), Foxp3 (red), cleaved caspase-3 (cCASP3; green), and nuclei (DAPI; white). The upper left image shows merged staining, and the white rectangle outlines the region of interest with high T cell infiltration. Single fluorescence channels for each antigen and DAPI are shown in the upper right. For detailed analysis (bottom), we overlaid the same region with the CD3 channel (blue) and the Foxp3 channel (red) (lower left) or with the CD3 channel (blue) and the cleaved caspase-3 channel (green) (lower right). The arrows indicate the same cells in both of the bottom images. Red arrows, Foxp3+ cells; green arrows, cleaved caspase-3+ cells; white arrows, triple-positive (Foxp3+cCASP3+CD3+) cells. (d,e) The expression of proapoptotic (d) and antiapoptotic (e) genes in human ovarian-cancer-infiltrating Treg cells. Results are shown as the fold change in expression of each gene in Treg cells after normalization to expression in conventional T cells (Tconv) isolated from the same cancer tissue. Data shown are the mean and s.e.m. of n = 5 individual subjects. (f,g) Apoptotic Treg cells and conventional T cells in mouse tumor tissues. We prepared single-cell suspensions from mouse ID8 ovarian cancer, MC38 colon cancer, and B16 melanoma and stained the cells for annexin V, 7-AAD, and cleaved caspase-3. The representative plots in f show the percentages of apoptotic cells in Foxp3+CD4+ and Foxp3−CD4+ T cells as assessed on the basis of annexin V or cleaved caspase-3 expression. Numbers in corners indicate the percentage of cells in the gate. The results shown in g are the mean percentages (+ s.e.m.) of annexin V+ cells among all cells in the group; n = 5 mice. (h) Gene set enrichment analysis of apoptosis genes in tumor-associated Treg cells versus conventional T cells from the GSE55705 data set. The enrichment score (ES) and P value are reported for the GO term “apoptosis induction” and were calculated by GSEA with weighted enrichment statistics and ratio of classes for the metric as input parameters. Images and data are pooled from 5 experiments (b,d,e) or from 1 experiment with 5 animals (g) or are representative of single experiments with 5 subjects (a,f), 10 subjects (c), or 2 subjects per group (h). *P < 0.05, Wilcoxon test (b) or Student’s t-test (g); *P < 0.01, Wilcoxon test (d,e).
Apoptotic Treg cells mediate superior immunosuppression
Foxp3 controls Treg cell development and functional integrity1,2. Live Treg cells inhibit TAA-specific T cell activation4,6,27. It is unknown whether apoptotic Treg cells are functionally suppressive. To test this, we isolated infiltrating Treg cells from human ovarian cancer samples, induced their apoptosis with anti-Fas monoclonal antibody (mAb) (Supplementary Fig. 2a), and examined their suppressor activity. Apoptotic Treg cells inhibited the expression of interferon-γ (IFN-γ) and tumor necrosis factor (TNF) in CD4+ and CD8+ T cells (Fig. 2a). When we compared the suppressor activities of live and apoptotic infiltrating Treg cells and apoptotic conventional T cells in human ovarian cancer, we found that apoptotic Treg cells were more efficient than live Treg cells at inhibiting T cell IL-2 production (Fig. 2b). Thus, human-cancer-associated apoptotic Treg cells are potent immune suppressors.
Figure 2.
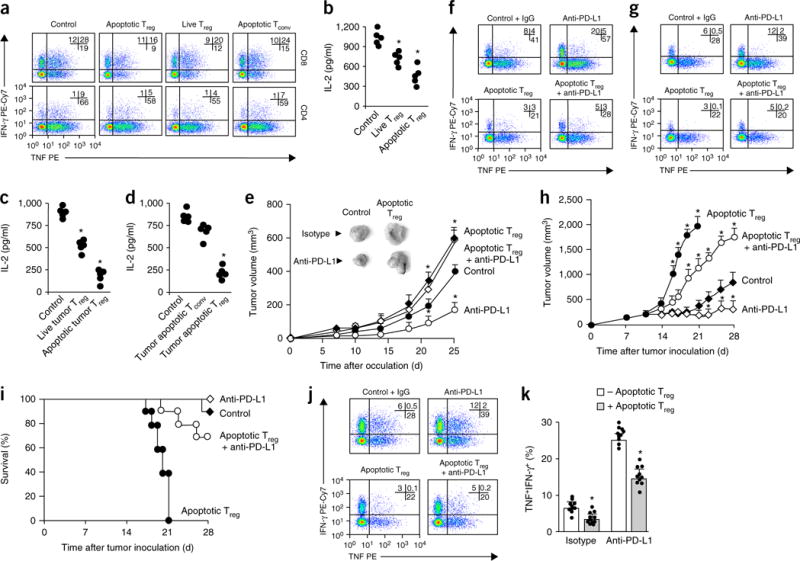
Apoptotic Treg cells are immunosuppressive. (a) Immunosuppression mediated by human ovarian-cancer-associated apoptotic Treg cells. CD3+CD4+CD25hi Treg cells were enriched and sorted from fresh ovarian cancer tissues. We used anti-Fas mAb to induce apoptosis in Treg cells and conventional T (Tconv) cells. Apoptotic Treg cells, live Treg cells, and apoptotic Tconv cells were cultured with T cells at a ratio of 1:2 in the presence of anti-CD3 and anti-CD28. Shown are representative results of flow cytometry analysis of the expression of IFN-γ and TNF in CD4+ and CD8+ T cell gates. (b) Immunosuppression by human ovarian-cancer-associated live Treg cells and apoptotic Treg cells. We measured T cell IL-2 by ELISA on day 3. (c) Immunosuppression mediated by mouse MC38-associated live and apoptotic Treg cells. Mouse GFP+Foxp3+ Treg cells were sorted from MC38 tumor tissues and treated with anti-Fas mAb to induce apoptosis. T cell immunosuppressive assays included equal numbers of live Treg cells and apoptotic Treg cells. We measured T cell IL-2 by ELISA on day 3. (d) Immunosuppression mediated by mouse MC38-associated apoptotic Treg cells and apoptotic conventional T cells. Mouse GFP+Foxp3+ Treg cells and GFP−Foxp3− T cells were enriched and sorted from MC38 tumor tissues and treated with anti-Fas mAb to create apoptotic Treg cells and conventional T cells. T cell immunosuppressive assays included equal numbers of apoptotic Treg cells and apoptotic conventional T cells. We measured T cell IL-2 by ELISA on day 3. In b,c,d, each individual data point represents a single donor (n = 5), and significant differences are relative to the control. (e) The effect of apoptotic Treg cells on MC38 tumor growth in vivo. C57BL/6 mice were inoculated subcutaneously with MC38 tumor cells or MC38 tumor cells plus radiation-induced apoptotic Treg cells. On days 7 and 14 the mice were treated with anti-PD-L1 or isotype control antibody. Tumor volume was measured at the indicated time points. The images in the upper left show representative tumors at the endpoint of the experiment. Data shown are the mean and s.d. from n = 10 mice per group. (f,g) Cytokine expression as measured by flow cytometry in tumor-draining lymph nodes (f) and tumor tissue (g) at the endpoint of the experiments. The cells were gated on the CD45+CD3+ population. n = 5 mice per group. (h,i) The effect of apoptotic Treg cells on B16-F10 tumor growth in a TAA-specific T cell transfusion model. Experiments are described in Supplementary Figure 6g. Rag2−/− mice were inoculated subcutaneously with B16-F10 cells, and after 7 d PMEL-specific T cells were transferred intravenously. Mice received intratumoral Treg cells and mAb treatment. Tumor growth (h) and mouse survival rate (i) were monitored. The data in h are the mean ± s.d. of n = 10 mice per group. Survival curves with initial animal numbers were considered as 100% (i). (j,k) The effect of apoptotic Treg cells on effector cytokine expression in PMEL-specific T cells. We used flow cytometry to analyze cytokine expression in tumor-infiltrating CD45+CD3+CD8+ T cells. Data are presented as representative dot plots (h) and quantification of polyfunctional TNF+IFN-γ+ populations (i). The data in i are the mean and s.d. of n = 10 mice per group. Data are from 4 experiments (a), 1 experiment with 5 donors (b–d), or 1 experiment with 10 animals per group (e–k). In a,f,g,j, numbers in upper right corners show the percentage of cells in the indicated quadrants. PE, phycoerythrin; Cy7, indotricarbocyanine. *P < 0.05 versus control, Wilcoxon test (b,d) or Student’s t-test (c,e,h,k).
Next, we carried out similar experiments with Treg cells from healthy mice and tumor-bearing mice. As expected, in the in vitro T cell suppressive assay, live Treg cells from healthy mice inhibited T cell cytokine production (Supplementary Fig. 2b,c). We induced Treg cell apoptosis with irradiation, serum starvation, fluorouracil, and anti-Fas mAb. Similar to what we observed in human apoptotic Treg cells (Fig. 2b), regardless of how Treg cell apoptosis was induced, mouse apoptotic Treg cells were superior at suppressing the expression of T cell TNF and IL-2 compared with identical amounts of normal live Treg cells (Supplementary Fig. 2b,c). We further compared the suppressor activities of live and apoptotic mouse MC38-tumor-infiltrating Treg cells. Again, apoptotic mouse tumor-infiltrating Treg cells were more potent suppressors than live Treg cells (Fig. 2c). Furthermore, mouse apoptotic tumor-infiltrating Treg cells, rather than apoptotic tumor-infiltrating conventional T cells, strongly inhibited T cell IL-2 production (Fig. 2d). Thus, mouse tumor-associated apoptotic Treg cells are powerful immune suppressors.
In addition to in vitro and ex vivo suppressor activity, we studied whether apoptotic Treg cells suppress tumor immunity in vivo, using three systems: (i) spontaneous tumor immunity, (ii) tumor immunity induced by PD-L1-checkpoint blockade, (iii) and adoptive TAA-specific T-cell-transfusion-mediated tumor immunity. In the first model, we treated mice carrying MC38 cancer cells with apoptotic Treg cells. We found that tumor volumes were larger in mice that received apoptotic Treg cells compared with those in control mice (Fig. 2e). Then, we examined the potential effect of apoptotic Treg cells on anti-PD-L1 therapy in this model. PD-L1 blockade reduced tumor growth (Fig. 2e) and enhanced the expression of effector T cell cytokines in tumor-draining lymph nodes (Fig. 2f) and tumor tissue (Fig. 2g). However, apoptotic Treg cells abolished these antitumor effects mediated by anti-PD-L1 (Fig. 2e–g). Thus, apoptotic Treg cells inhibit spontaneous and checkpoint-blockade-induced antitumor immunity.
Finally, we explored the role of apoptotic Treg cells in two adoptive TAA-specific T cell transfer models. We transfused MHC class I–restricted (ovalbumin (OVA)-specific; OT-I) T cell receptor–transgenic T cells into ID8-OVA-bearing (otherwise wild-type) mice and then treated the mice with apoptotic Treg cells and live Treg cells. Both apoptotic Treg cells and live Treg cells promoted tumor growth (Supplementary Fig. 2d) and inhibited effector T cell cytokine expression (Supplementary Fig. 2e,f). However, apoptotic Treg cells demonstrated superior suppressor activity compared with that of live Treg cells (Supplementary Fig. 2d–f). In addition, we transferred T cell receptor–transgenic CD8+ T cells that specifically recognized the melanoma antigen PMEL into B16-F10-bearing Rag2−/− mice. We treated the mice with apoptotic Treg cells and anti-PD-L1 mAb (Supplementary Fig. 2g). Again, we found that apoptotic Treg cells suppressed PMEL-specific T cell immunity and disabled the antitumor effect of PD-L1 blockade, as shown by increased tumor growth (Fig. 2h), shortened mouse survival (Fig. 2i), and reduced numbers of IFN-γ+TNF+ polyfunctional effector T cells (Fig. 2j,k). Thus, apoptotic Treg cells were superior suppressors in multiple in vitro and in vivo models.
Apoptotic Treg cells make suppressive nonprotein factor(s)
Next, we studied the cellular and molecular mechanisms by which apoptotic Treg cells suppress T cell activation. Myeloid cells, including macrophages, can process apoptotic cells and may mediate immunosuppression in the tumor microenvironment28,29. To explore whether apoptotic Treg cell–mediated immunosuppression depends on macrophages, we tested the effect of apoptotic Treg cells on tumor immune response with macrophage depletion in an MC38 model. As expected, the addition of apoptotic Treg cells and the depletion of macrophages promoted and inhibited tumor growth, respectively (Fig. 3a). Accordingly, polyfunctional effector T cell cytokine expression was suppressed after the addition of apoptotic Treg cells and enhanced after macrophage depletion (Fig. 3b,c). Apoptotic Treg cells remained immunosuppressive after macrophage depletion (Fig. 3a–c). These observations suggest that macrophages are dispensable for apoptotic Treg cell–mediated immunosuppression in this system.
Figure 3.
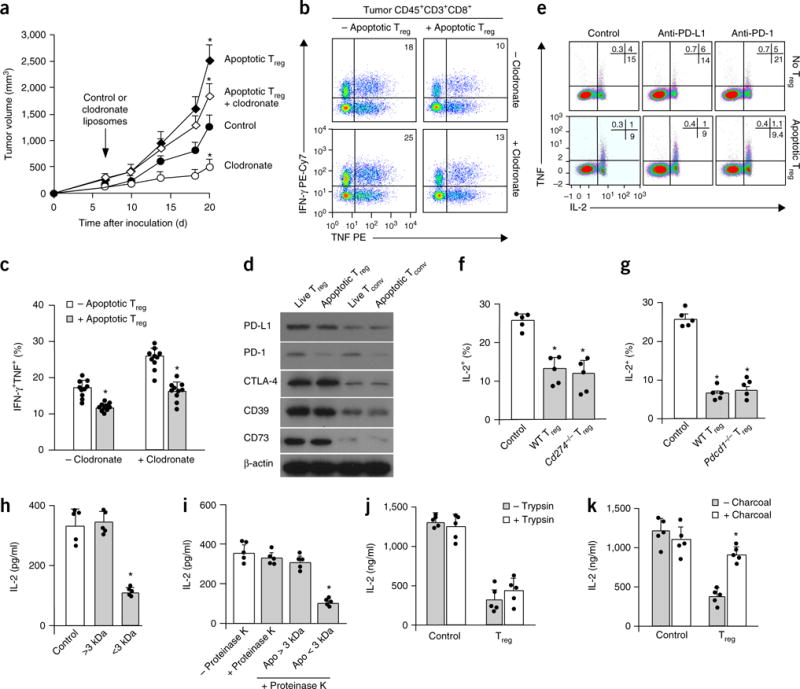
Apoptotic Treg cells mediate immunosuppression via soluble factor(s). (a–c) The effect of macrophage depletion on apoptotic Treg cell–mediated suppression. Mice bearing MC38 and apoptotic Treg cells were treated with clodronate-loaded liposomes. The plots show tumor volume (a) and numbers of tumor-infiltrating effector T cells (b,c). Data in a and c are the mean and s.d. of n = 10 mice per group. *P < 0.05 versus the control, Student’s t-test. (d) The expression of immunosuppression-associated molecules in live and apoptotic T cell subsets. The indicated mouse T cell subsets were immunoblotted with antibodies to the indicated proteins. (e) The effect of PD-L1 or PD-1 blockade on apoptotic Treg cell–mediated T cell suppression. Apoptotic Treg cells and apoptotic conventional T cells were induced by irradiation. Apoptotic Treg cells and apoptotic conventional T cells were cultured with normal T cells at a ratio of 1:2 in the presence of anti-CD3. We measured the expression of T cell TNF and IL-2 by flow cytometry. (f,g) Immunosuppression mediated by PD-L1−, PD-1−, and wild-type (WT) apoptotic Treg cells. Apoptotic Treg cells from Cd274−/− (f), Pdcd1−/− (g), and wild-type mice were cultured with normal T cells at a ratio of 1:2 in the presence of anti-CD3. We measured the expression of T cell TNF and IL-2 by flow cytometry. Data shown are the mean and s.e.m. (n = 5 mice). *P < 0.05 compared with controls, Wilcoxon test. (h) Immunosuppression mediated by low-molecular-weight components in apoptotic Treg cell–derived supernatants. Treg cells were treated with anti-Fas mAb for 6 h. Treg cell supernatants were passed through a 100-kDa cutoff filter and divided into two fractions with a 3-kDa cutoff filter. The two fractions were collected for T cell immunosuppression assays. T cell IL-2 was detected on day 3 by ELISA. The data shown are the mean and s.d. (n = 5 mice). *P < 0.05 versus the control, ANOVA with Dunett’s post-test. (i,j) Immunosuppression mediated by nonprotein elements in apoptotic Treg cell supernatants. Apoptotic Treg cell supernatants (apo) were treated with proteinase K (i) or trypsin (j) and subsequently used for immunosuppression assays. T cell IL-2 was detected on day 3 by ELISA. The data shown are the mean and s.d. (n = 5 mice). ANOVA with Dunett’s post-test, *P < 0.05. (k) The effect of the removal of small lipid-like factors on immunosuppression mediated by apoptotic Treg cell supernatants. Apoptotic Treg cell supernatants were mixed with charcoal-treated dextran to remove small lipid-like molecules and were subsequently used for immunosuppression assays. T cell IL-2 was detected on day 3 by ELISA. The data shown are the mean and s.d. (n = 5 mice). Wilcoxon test, *P < 0.05 versus the control. Data are representative of 10 (d) or 5 (e) experiments. In b and e, numbers in the upper right show the percentage of cells in the indicated quadrant.
On the molecular level, live Treg cells express Foxp3 and can mediate immunosuppression via multiple modes of action5,30,31. Immunoblotting showed high levels of immunosuppression-associated membrane molecules, including PD-L1, PD-1, CTLA-4, CD39, and CD73, in apoptotic Treg cells compared with the amounts in conventional T cells, regardless of their live status (Fig. 3d). PD-1 and PD-L1 blockade with specific antibodies had no effect on the suppressive properties of apoptotic Treg cells (Fig. 3e). Apoptotic Treg cells from PD-L1-deficient (Cd274−/−) (Fig. 3f), PD-1-deficient (Pdcd1−/−) (Fig. 3g), and wild-type mice suppressed T cell IL-2 expression to similar degrees (Fig. 3f,g). CTLA-4 blockade did not change the suppressive activity of apoptotic Treg cells (Supplementary Fig. 3a–c). In addition to membrane proteins, it is possible that, similarly to live Treg cells, apoptotic Treg cells may release soluble factors including cytokines to mediate T cell suppression. To test this possibility, we collected apoptotic Treg cell–derived supernatants via ultracentrifugation. The supernatants from apoptotic Treg cells inhibited T cell IL-2 expression in a dose-dependent manner (Supplementary Fig. 3d). However, neutralization of the immunosuppressive cytokine TGF-β (Supplementary Fig. 3e–g), IL-35 subunit EBI3 (Supplementary Fig. 3h–j), and IL-10 (Supplementary Fig. 3k–m) with blocking antibodies did not rescue this suppression. Thus, apoptotic Treg cells release unknown soluble immunosuppressive factor(s), and TGF-β, IL-35, and IL-10 are not involved in apoptotic Treg cell–mediated T cell suppression.
To identify the soluble suppressive factor(s), we divided the apoptotic Treg cell supernatants into two fractions with a 3-kDa molecular-weight-cutoff filter. We observed that the fraction above 3 kDa did not suppress T cell IL-2 production, whereas the fraction below 3 kDa did (Fig. 3h). This molecular weight is consistent with the observations that PD-L1, PD-1, CTLA-4, IL-10, TGF-β, and IL-35 were not involved in the suppressive function of apoptotic Treg cells (Fig. 3e–g and Supplementary Fig. 3). Supernatants from apoptotic Treg cells remained immunosuppressive after treatment with proteinase K (Fig. 3i) or trypsin (Fig. 3j). However, incubation with dextran-coated charcoal resulted in a loss of immunosuppression (Fig. 3k). The data suggest that the soluble immunosuppressive factors may be small molecules, rather than proteins or large peptides.
Apoptotic Treg cells mediate suppression via the A2A pathway
We attempted to define the molecular nature of apoptotic Treg cell–derived suppressive supernatants. Live mouse Treg cells express CD39 and CD73 and can convert ATP to adenosine32. Live human cancer-infiltrating Treg cells express CD39 and CD73 and inhibit effector T cells through an adenosinergic pathway33. We noticed that mouse apoptotic Treg cells expressed CD39 and CD73 (Fig. 3d) and released a nonprotein small immunosuppressive molecule (or molecules) (Fig. 3h–k and Supplementary Fig. 3). Thus, we hypothesized that CD39 and CD73 in apoptotic Treg cells remain enzymatically active and could mediate adenosine production, and that the nonprotein small immunosuppressive molecule might be adenosine. To test this hypothesis, we induced apoptosis in mouse Treg cells with anti-Fas mAb and kinetically measured adenosine in the supernatants. A colorimetric assay detected high amounts of adenosine at 6 h in the apoptotic Treg cell supernatants, but not in live Treg cell medium (Fig. 4a). Mass spectrometry (Supplementary Fig. 4a,b) detected a kinetic increase in adenosine levels in apoptotic Treg cell supernatants (Supplementary Fig. 4b,c). The amounts of adenosine also peaked at hour 6 of Treg cell apoptosis (Supplementary Fig. 4c). Then, we examined the potential suppressive role of adenosine in apoptotic Treg cell supernatants. The addition of exogenous adenosine to mouse T cell cultures led to reduced production of TNF and IL-2 (Fig. 4b,c). A2A is a receptor for adenosine in T cells34 and mediates IL-2 suppression in particular35. We observed that the A2A inhibitor ZM241385 abolished T cell effector cytokine suppression mediated by apoptotic Treg cell supernatants (Fig. 4d,e). We extended these mouse studies to human ovarian cancer. Again, the supernatants of human ovarian-cancer-infiltrating apoptotic Treg cells inhibited T cell activation. The effect was abrogated by ZM241385 (Fig. 4f,g). Thus, apoptotic Treg cells mediate immunosuppression via adenosine and the A2A pathway.
Figure 4.
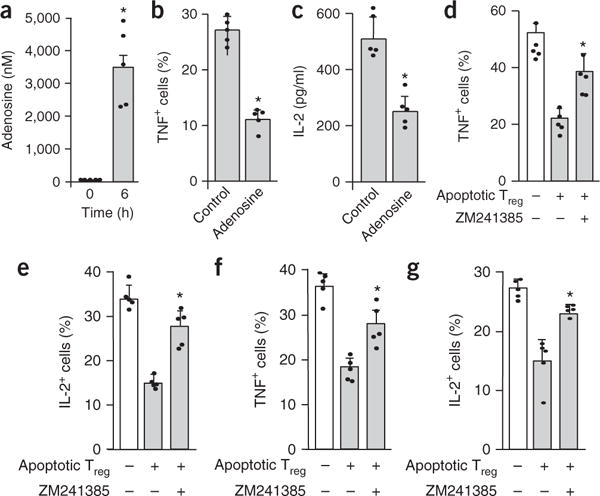
Apoptotic Treg cells mediate immunosuppression via adenosine. (a) Adenosine in apoptotic Treg cell supernatants. Mouse Treg cells were treated with anti-Fas mAb for 6 h. (b,c) The effect of exogenous adenosine on T cell TNF (b) and IL-2 (c) expression. Mouse T cells were activated with anti-CD3 and anti-CD28 in the absence (control) or presence of adenosine (5 μM). T cell cytokines were analyzed by flow cytometry on day 3. (d,e) The effect of the A2A inhibitor ZM241385 on mouse apoptotic Treg cell–mediated immunosuppression. Mouse T cells were activated with anti-CD3 and anti-CD28 in the presence of apoptotic Treg cell supernatants for 3 d. ZM241385 was added into the culture. Expression of the T cell cytokines TNF (d) and IL-2 (e) was analyzed by flow cytometry on day 3. (f,g) The effect of ZM241385 on human cancer-associated apoptotic Treg cell–mediated immunosuppression. Human T cells were activated with anti-CD3 and anti-CD28 in the presence of human ovarian-cancer-associated apoptotic Treg cell supernatants for 3 d. ZM241385 was added into the culture. Expression of the T cell cytokines TNF (f) and IL-2 (g) was analyzed by flow cytometry on day 3. All data are representative of 1 experiment with cells acquired from 5 different animals and are shown as the mean and s.d. of n = 5 mice.*P < 0.05, paired Student’s t-test (a–c) or ANOVA with Dunett’s post-test (d–g). In d–g, significant differences are versus the untreated control group.
Apoptotic Treg cells release ATP and metabolize it to adenosine
Next, we explored how apoptotic Treg cells produce a large amount of adenosine in the absence of exogenous ATP. Gene set enrichment analysis demonstrated that mouse B16-melanoma-infiltrating Treg cells showed active expression of genes associated with purine and pyrimidine metabolism (GSE55705)26 (Supplementary Fig. 5a,b), suggesting high energy metabolism. We hypothesized that apoptotic Treg cells generate, release, and consequently metabolize to adenosine high levels of ATP via apoptotic-cell-associated CD39 and CD73, and ultimately suppress T cell activation via adenosine in the tumor. To test this hypothesis, we induced apoptosis in mouse Treg cells and conventional T cells by anti-Fas treatment (Fig. 5a), serum starvation (Fig. 5b), or irradiation (Fig. 5c). After the induction of apoptosis, Treg cells, but not conventional T cells, released a large amount of ATP (Fig. 5a,b), possibly owing to the higher ATP content in Treg cells (Supplementary Fig. 5c). Apoptotic cells can release small molecules via pannexin-1-dependent channels36. We found that treatment with pannexin-1 inhibitor (probenecid or carbenoxolone) prevented apoptotic Treg cells from releasing ATP into supernatants (Supplementary Fig. 5d). Thus, apoptotic Treg cells release ATP via pannexin-1-dependent channels.
Figure 5.
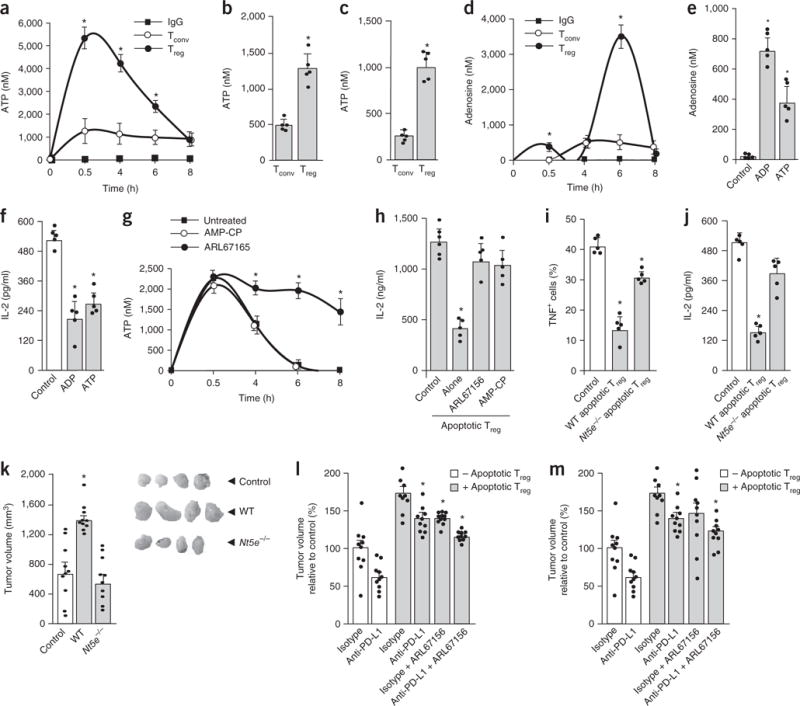
Apoptotic Treg cells release ATP and generate high levels of adenosine. (a) Kinetics of ATP release by apoptotic Treg cells. Mouse Treg cells and conventional T cells (Tconv) were treated with anti-Fas mAb or isotype IgG. ATP in the supernatant was measured by colorimetric assay. Mean ± s.d., n = 6. *P < 0.05, Student’s t-test between Treg cells and conventional T cells at each time point. (b,c) ATP released by mouse apoptotic Treg cells under serum starvation (b) or after irradiation (c) in the presence of ectonuclease inhibitor ARL67156. The ATP concentration in apoptotic Treg supernatants was measured at 16 h. Mean and s.d., n = 5. *P < 0.05, Student’s t-test. (d) Kinetics of adenosine generation by apoptotic Treg cells. Mouse Treg cells and conventional T cells were treated with anti-Fas mAb or isotype IgG in the presence of the adenosine deaminase inhibitor EHNA. Adenosine was measured in the supernatant. Mean ± s.d., n = 6. *P < 0.05, Student’s t-test between Treg cells and conventional T cells at each time point. (e,f) Adenosine generated from exogenous ADP and ATP by apoptotic Treg cells. Exogenous ADP or ATP (5 μM) was added to fresh medium with apoptotic Treg cells for 6 h. Adenosine was measured in the culture supernatants (e). Supernatants were used for immunosuppression assays (f). Mean and s.d., n = 5. *P < 0.05 versus control, Wilcoxon test. (g) The effect of ARL67165 or AMP-CP on ATP release by apoptotic Treg cells. Mouse Treg cells were treated for 8 h with anti-Fas mAb with or without ARL67165 or AMP-CP. ATP was measured in the supernatants. Mean ± s.d., n = 5. *P < 0.05 versus the untreated control, Wilcoxon test. (h) The effect of ARL67165 or AMP-CP on apoptotic Treg cell–mediated immunosuppression. Mouse Treg cells were cultured for 8 h with anti-Fas mAb with or without ARL67165 or AMP-CP. The culture supernatants were used for T cell immunosuppression. IL-2 was measured on day 3 by ELISA. Mean and s.d., n = 8. *P < 0.05 versus control, ANOVA with Dunett’s post-test. (i,j) Immunosuppression mediated by wild-type (WT) and Nt5e−/− apoptotic Treg cells. Apoptotic Treg cells from wild-type and Nt5e−/− mice were used for T cell immunosuppressive assays. TNF expression was measured by flow cytometry (i), and IL-2 was detected by ELISA (j). Mean and s.d., n = 5. *P < 0.05 versus control, Wilcoxon test. (k) The effect of Nt5e−/− apoptotic Treg cells on tumor growth in vivo. MC38 tumors were inoculated subcutaneously with wild-type or Nt5e−/− apoptotic Treg cells. The images show representative tumors at the endpoint of the experiment. Mean and s.e.m., n = 10. Student’s t-test, *P < 0.05 versus control. (l,m) The effects of CD39 and A2A inhibitors on tumor growth. MC38-bearing mice were treated with the CD39 inhibitor ARL67165 (5 mg/kg, 5 d per week) or the A2A inhibitor ZM241385 (1 mg/kg, 5 d per week) with anti-PD-L1 in the presence of apoptotic Treg cells. Mean and s.e.m., n = 10. *P < 0.05 compared with isotype control in the presence of the apoptotic Treg cell group, ANOVA with Dunnett’s post hoc test. All data in a–j were generated in single experiments; n values for those panels indicate the number of animals used for cell isolation. Data in k–m are from 1 experiment with 10 animals per group.
The amount of ATP released from apoptotic Treg cells peaked between 0.5 and 2 h and decreased to baseline at 6–8 h after apoptosis (Fig. 5a). This suggests that ATP may be metabolized and converted into adenosine. In line with this, the measured amounts of adenosine inversely correlated with those of ATP and peaked at 6 h after apoptosis in supernatants from apoptotic Treg cells (Fig. 5d). To determine the ability of apoptotic Treg cells to generate adenosine, we incubated apoptotic Treg cells in medium containing exogenous ADP or ATP. We detected higher amounts of adenosine in culture medium containing apoptotic Treg cells than in the control samples (Fig. 5e). We collected the supernatants from these experiments and filtered them with a 3-kDa-cutoff filter. Again, these supernatants mediated potent suppression in T cell cytokines (Fig. 5f). The data suggest that apoptotic Treg cells can convert ADP and ATP to adenosine.
Apoptotic Treg cells express CD39 and CD73 (Fig. 3d). To directly determine whether CD39 and CD73 are functional in apoptotic Treg cells, we treated apoptotic Treg cells with a CD39 inhibitor, ARL67156, and a CD73 inhibitor, AMP-CP. As expected, ARL67156, but not AMP-CP, prevented ATP degradation (Fig. 5g). Furthermore, CD39 and CD73 inhibitors rescued IL-2 expression that was inhibited by apoptotic Treg cells (Fig. 5h). To genetically validate our observation, we tested the suppressive activity of apoptotic Treg cells from CD73-deficient (Nt5e−/−) mice. Live Nt5e−/− and wild-type Treg cells contained similar amounts of intracellular ATP (Supplementary Fig. 5e). Nt5e−/− and wild-type apoptotic Treg cells released similar amounts of ATP into the medium (Supplementary Fig. 5f). However, Nt5e−/− apoptotic Treg cells generated less adenosine (Supplementary Fig. 5g) and were less immunosuppressive than wild-type apoptotic Treg cells (Fig. 5i,j). Next, we treated MC38-tumor-bearing mice with apoptotic Nt5e−/− T cells and apoptotic wild-type Treg cells. We found that tumor volumes were larger in mice that received apoptotic wild-type Treg cells than in those administered apoptotic Nt5e−/− Treg cells (Fig. 5k), which indicated that apoptotic Nt5e−/− Treg cells were less suppressive than apoptotic Nt5e+/+ Treg cells. In addition, we tested whether ARL67156 (Fig. 5l) and ZM241385 (Fig. 5m) affect apoptotic Treg cell–mediated immunosuppression in MC38-tumor-bearing mice with or without PD-L1 blockade. As shown by changes in tumor volume, the two inhibitors partially subverted the immunosuppressive effect of apoptotic Treg cells and additionally improved the therapeutic outcome of PD-L1 blockade (Fig. 5l,m). Together, our data suggest that apoptotic Treg cells release high levels of ATP and metabolize it to adenosine via the CD39 and CD73 axis, and suppress T cells through interaction with adenosine and A2A receptors.
Oxidative stress induces Treg cell apoptosis in tumors
Finally, we explored why Treg cells undergo apoptosis in the tumor microenvironment. Effector T cells are affected by glycolytic metabolism in the tumor microenvironment13,14,16. To explore whether tumor Treg cell apoptosis can be attributed to poor glycolysis, we treated normal Treg cells and conventional T cells with or without glucose and the hexokinase inhibitor 2-deoxy-D-glucose. Glucose restriction and 2-deoxy-D-glucose treatment induced apoptosis in conventional T cells (Supplementary Fig. 6a), but not in Treg cells (Supplementary Fig. 6b). Oxidative stress disrupts DC function in ovarian cancer25. Thus, we tested whether oxidative stress induces Treg cell apoptosis via reactive oxygen species (ROS) in the tumor microenvironment. To this end, we treated human Treg cells with ovarian cancer ascites. We found that treatment with ovarian cancer ascites and hydrogen peroxide (H2O2) induced potent apoptosis in Treg cells, but not in conventional T cells (Fig. 6a and Supplementary Fig. 6c). Treg cell apoptosis was reversed by treatment with the ROS scavenger N-acetyl-cysteine (Fig. 6a). Ovarian cancer ascites (Fig. 6b,c) and H2O2 treatment (Fig. 6d,e) led to higher expression of proapoptotic genes (Fig. 6b,d) and lower expression of antiapoptotic genes (Fig. 6c,e) in Treg cells compared with controls. Furthermore, we detected high concentrations of the major ROS component superoxide in ovarian cancer ascites (Supplementary Fig. 6d). Ovarian-cancer-infiltrating Treg cells showed high mitochondrial activity compared with that in conventional T cells (Supplementary Fig. 6e) and produced higher amounts of intracellular ROS (Supplementary Fig. 6f). Thus, Treg cells are relatively more sensitive to oxidative stress in the tumor microenvironment than conventional T cells are, and undergo potent apoptosis.
Figure 6.
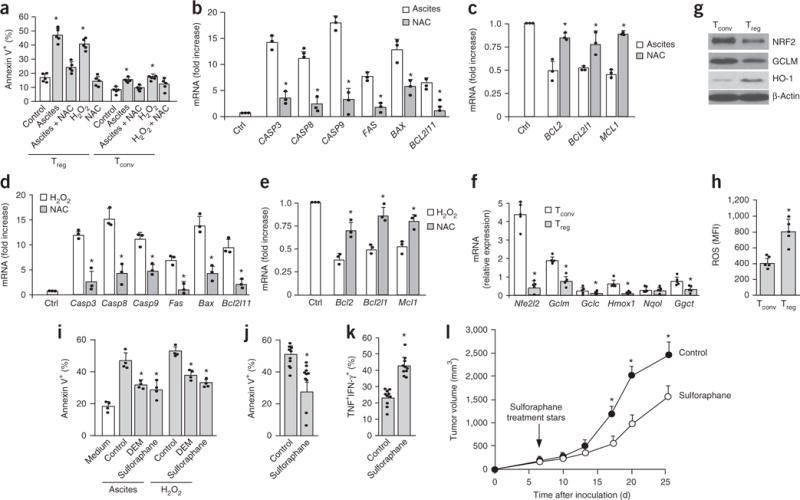
Oxidative stress induces Treg cell apoptosis in the tumor environment. (a) The effect of ovarian cancer ascites on human Treg cell apoptosis. Mouse Treg cells and conventional T cells (Tconv) were cocultured with 50% human ovarian cancer ascites or hydrogen peroxide for 24 h. Additional cultures were treated with N-acetyl-cysteine (NAC) as a free radical scavenger. Annexin V+ Treg cells and Tconv cells were analyzed by flow cytometry. n = 5. (b,c) The effect of ovarian cancer ascites on proapoptotic and antiapoptotic gene transcripts in Treg cells. Treg cells were exposed to ascites or ascites plus NAC after 24 h. Proapoptotic (b) and antiapoptotic (c) transcripts were quantified by real-time PCR. n = 3. (d,e) The effect of hydrogen peroxide on proapoptotic and antiapoptotic transcripts in Treg cells. Treg cells were exposed to hydrogen peroxide or hydrogen peroxide plus NAC after 24 h. Proapoptotic (d) and antiapoptotic (e) mouse gene transcripts were quantified by real-time PCR. n = 3. (f,g) Expression of mouse NRF2 and NRF2-associated gene transcripts (f) and proteins (g) in T cell subsets as determined by real-time PCR and immunoblotting, respectively. n = 5. (h) The expression of intracellular ROS in Treg cells. Treg cells and conventional T cells were cultured overnight. Amounts of intracellular ROS were measured by flow cytometry. n = 5. MFI, mean fluorescence intensity. (i) The effect of NRF2 inducers on Treg cell apoptosis in vitro. Mouse Treg cells were cultured for 24 h with 50% ascites or medium containing H2O2. NRF2 inducers diethylmaleate (DEM; 100 μM) and sulforaphane (10 μM) were added to the culture for 24 h. Controls were treated with vehicle (DMSO). Apoptosis was measured by flow cytometry with annexin V. n = 4. (j–l) The effect of sulforaphane on tumor immunity. MC38-bearing mice were treated with sulforaphane (25 mg/kg, 5 d per week) or vehicle (DMSO). Tumor Treg cell apoptosis (j) and CD8+ T cell polyfunctional cytokine expression (k) were analyzed by flow cytometry. Tumor volume was monitored (l). n = 10. All data are shown as the mean and s.d. and are from 1 experiment with 10 animals per group (i–l) or from single experiments with cells isolated from individual animal for each data point (a–h). *P < 0.05 versus control, Wilcoxon test (b–e), paired Student’s t-test (f,g), Student’s t-test (i–l), or Mann–Whitney U-test (a,h).
Next, we wondered why Treg cells are more vulnerable to oxidative stress. The cellular antioxidant system is largely regulated by the transcription factor NRF2 (Nfe2l2) and its associated genes37,38. We quantified the expression of Nfe2l2 and NRF2-associated gene transcripts including Gclm, Gclc, Hmox1, Nqo1, and Ggct in normal Treg cells and conventional T cells. The levels of the majority of these antioxidant gene transcripts were lower in mouse (Fig. 6f) and human (Supplementary Fig. 6g) Treg cells than in conventional T cells. Immunoblotting showed lower amounts of NRF2, GLCM, and heme oxygenase (HO-1) proteins in mouse (Fig. 6g) and human (Supplementary Fig. 6h) Treg cells than in conventional T cells. Concentrations of intracellular ROS were higher in Treg cells than in conventional T cells (Fig. 6h). Next, we tested the influence of the NRF2 inducers diethylmaleate and sulforaphane on Treg cell apoptosis in the presence of ovarian cancer ascites. Both NRF2 inducers reduced Treg cell apoptosis compared with that in the control (Fig. 6i). Furthermore, we treated MC38-tumor-bearing mice with sulforaphane. We found that treatment with sulforaphane reduced Treg cell apoptosis (Fig. 6j), increased effector T cell cytokine expression (Fig. 6k), and inhibited tumor growth (Fig. 6l) compared with that observed in the control. Taken together, these data indicate that oxidative stress results in apoptosis of Treg cells via ROS in the tumor microenvironment, which in turn increases their suppressive action.
DISCUSSION
Treg cells are recruited into the tumor microenvironment. It is thought that live, but not apoptotic, Treg cells mediate immune suppression1,2,4–6. In this work, we studied Treg cells in human ovarian cancer and in several mouse tumors. We found that Treg cells underwent potent apoptosis in human and mouse tumor microenvironments. To our surprise, apoptotic Treg cells were more efficient than live Treg cells at suppressing T cell activation in vitro and in vivo. More important, we demonstrated that the therapeutic efficacy of PD-L1 blockade was abolished by apoptotic Treg cells in tumor-bearing mouse models. The majority of cancer patients remain unresponsive to therapies based on PD-L1 and PD-1 signaling blockade39, and we wondered whether this clinical unresponsiveness (or therapeutic resistance) is associated with high degrees of Treg cell apoptosis in the tumor microenvironment. Furthermore, the targeting of Treg cells is considered a potential therapeutic strategy for cancer treatment. However, clinical trials with Treg cell depletion have not been translated into therapeutic efficacy in people with cancer40–43. This raises the possibility that therapeutic induction of Treg cell apoptosis may sustain and/or amplify, rather than disable, Treg-cell-mediated immunosuppression.
PD-L1, CTLA-4, TGF-β, IL-10, and IL-35 are well-defined immunosuppressive components for live Treg cells1–6. Our biochemical analysis and in vivo functional studies demonstrated that these molecules are not involved in the suppressor activity of apoptotic Treg cells. Live Treg cells express the ectoenzymes CD39 and CD73 and can convert extracellular ATP to adenosine, and in turn mediate immunosuppression via adenosine and the A2A receptor signaling pathway32. We observed that apoptotic Treg cells release a large amount of ATP and efficiently convert ATP into immunosuppressive adenosine via CD39 and CD73 in vitro and in vivo. Therefore, it can be assumed that CD39 and CD73 remain enzymatically active in apoptotic Treg cells. Furthermore, apoptotic Treg cells released a large amount of ATP into the extracellular space via pannexin-1 channels. In line with our observation, mouse apoptotic thymocytes release low-molecular-mass compounds via pannexin-1 channels36. Thus, apoptotic Treg cells can self-supply and release ATP, and convert it to adenosine in the local environment. We propose a novel immune-evasion mechanism: tumor-infiltrating Treg cells inadvertently die to mediate, sustain, and amplify powerful suppression in the tumor microenvironment.
It has been reported that Foxp3 is a proapoptotic factor for Treg cells in mouse thymus, unless some surviving factors such as γ-chain cytokines are present44. We have explored the potential molecular mechanism of Treg cell apoptosis in the tumor microenvironment. Several metabolic checkpoint molecules, including AMPK, mTORC1 (refs. 20,21), and HIF-1α (refs. 22,23), may physiologically regulate Treg cell function and the balance between Treg cells and effector T cells in mouse models. However, how Treg cells (particularly human tumor Treg cells) behave in the metabolically abnormal tumor microenvironment remains unknown. Glycolysis profoundly alters effector T cell phenotype and function in the tumor microenvironment14–16. We observed that Treg cells were not as sensitive as effector memory T cells to glycolytic restriction. Instead, we found that Treg cells were vulnerable to oxidative stress in the tumor microenvironment. Our data show that free oxygen species target mitochondria and induce apoptosis in Treg cells, consequently triggering the suppressive cascade of Treg cells in the tumor microenvironment. Thus, our current study complements recent reports on the metabolic control of memory T cells14–16 and DCs25 and extends to Treg cells in human and mouse tumor microenvironments.
In summary, apoptotic Treg cell–mediated immunosuppression may be a major mode of action for Treg cells in cancer and serve as a potential mechanism for checkpoint-blockade resistance. Furthermore, Treg cell apoptosis triggered by oxidative stress is a novel tumor immune-evasion mechanism in the tumor microenvironment, and targeting tumor metabolism is a potential therapeutic strategy for cancer treatment.
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
ONLINE METHODS
Ovarian cancer patients, cancer tissues, ascites, and cells
People diagnosed with high-grade serous ovarian carcinomas were recruited for this study, and informed consent was obtained from all participants. The use of human subjects for this study was approved by the University of Michigan Institutional Review Board. We used clinical samples from people who had received no prior anticancer therapies. Fresh tumor tissues and tumor ascites were processed into single-cell suspensions for phenotype and functional studies. Tumor ascites were centrifuged, and cell-free fractions were frozen at −80 °C.
Cell transfection
ID8 cells were transfected with plasmid pCI-neo-mOVA (Addgene plasmid #25099)45. Lipofectamine 2000 (Invitrogen) was used for delivery according to the vendor’s instructions. The cells were selected with geneticin (Invitrogen), and ovalbumin expression was confirmed by flow cytometry and used for in vivo mouse experiments.
Animals
Animal experiments were approved by the University of Michigan Institutional Review Board. Wild-type female C57BL/6J, C57BL/6 Foxp3−GFP+ (C57BL/6-Tg(Foxp3-GFP)90Pkraj/J), B6.SJL-PtprcaPepcb/BoyJ, C57BL/6-Tg(TcraTcrb)1100Mjb/J, B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J, and Nt5e−/− C57BL/6 mice (8–10 weeks old) were obtained from the Jackson Laboratories. PD-L1-deficient (Cd274−/−) and PD-1-deficient (Pdcd1−/−) C57BL/6 mice were originally provided by Dr. Lieping Chen at Yale University and Dr. Tasuku Honjo at Kyoto University, respectively.
In vivo tumor experiments
For tumor growth experiments, the animals were inoculated intraperitoneally (i.p.) with 1.5 × 106 ID8 ovarian cancer cells, or subcutaneously with 106 MC38 cells or 3 × 105 B16-F10 cells. When tumor cells were coinjected with apoptotic or live T cells, the ratio was 1:1.
For the PMEL-specific mouse model, T cell transfer was accomplished by a single intravenous injection of 106 PMEL-specific CD8+ T cells. Donor CD8+ T cells were collected from spleens and lymph nodes of B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J mice and expanded with plate-bound anti-CD3 and soluble anti-CD28 in the presence of recombinant mouse IL-2. Expanded T cells were transferred to mice bearing B16-F10 tumors. Animals were treated as described in Supplementary Figure 2g.
For the OVA-specific model, ID8-OVA-Luc tumor cells were inoculated into B6.SJL-PtprcaPepcb/BoyJ mice, which are congenic B6 animals that express CD45.1 antigen. After 7 d, the mice received 106 OVA-specific CD45.2+CD8+ T cells i.p. Animals were treated as described in Supplementary Figure 2d–f. 10 min after the injection of 150 mg/kg body mass D-luciferin (Promega), the bioluminescence of luciferase-expressing tumors was assessed with the IVIS 200 Spectrum imaging system (PerkinElmer).
Treatments with anti-PD-L1 or isotype control antibody (both from Bio X Cell) were done between days 7 and 20 (20 mg/kg i.p.).
Macrophages were depleted with clodronate liposome (Liposoma). Liposomes containing clodronate or control liposomes were injected into mice on days 4, 6, 8, and 10 (200 μl i.p.).
All tumor cell lines used in the experiments were routinely tested for mycoplasma contamination by PCR assay. Randomization of animals was not used in experiments. No blinding was done.
Cell culture and biochemical reagents
RPMI 1640 medium supplemented with 10% FBS (Invitrogen) was used for the majority of the experiments. For cultures without glucose, the cells were treated with RPMI 1640 medium without glucose (Sigma-Aldrich) and charcoal-treated FBS (Invitrogen). Probenecid, carbenoxolone, diethylmaleate, L-sulforaphane, N-acetyl-cysteine, H2O2, ZM241385, AMP-CP, ARL67165, EHNA, ADP, ATP, and adenosine were obtained from Sigma-Aldrich. Cell lines were obtained from the US National Institutes of Health or ATCC. All cell lines were routinely tested for mycoplasma contamination.
Isolation and culture of Treg cells and conventional T cells
Single-cell suspensions were prepared from mouse and human tissues including tumor tissues, tumor ascites, and tumor-draining lymph nodes as previously reported46–49. Mouse Foxp3+GFP+ Treg cells were enriched and sorted from spleen and lymph nodes and/or were generated in the presence of IL-2 (10 ng/ml) and TGF-β (10 ng/ml). Human T cells were isolated from healthy donor buffy coats (Carter BloodCare) and purified directly with RosetteSep human T cell enrichment kits (StemCell Technologies). Treg cells from human tumors were isolated with a commercially available Treg cell isolation kit (StemCell) and confirmed by Foxp3 staining.
Induction of Treg cell apoptosis
Treg cells and conventional T cells (5 × 106 cells/ml) were incubated with anti-Fas mAb (10 μg/ml; clone Jo-1; eBioscience) for 15–30 min, washed, and suspended for further experiments. Apoptotic Treg cells were also induced by 70-Gy irradiation and cultured serum-free for 24 h and with 5-fluorouracil for 24 h as indicated. For adenosine measurement, erythro-9-(2-hydroxy-3-nonyl) adenine (EHNA), an inhibitor of adenosine deaminase, was used to prevent adenosine degradation over time.
Apoptotic Treg cell–derived supernatants
Apoptotic Treg cell suspensions were centrifuged at 500g for 5 min, 18,000g for 1 h, and 12,000g overnight.
Immunosuppression assay
CD4+Foxp3− responder T cells were cocultured with live Treg cells or apoptotic Treg cells or with apoptotic Treg cell–derived supernatants in 96-well round-bottom plates at a ratio of 1:2 in the presence of anti-CD3 (2.5 μg/mL) and irradiated antigen-presenting cells. In some experiments, anti-PD-L1 (eBioscience), anti-PD-1 (eBioscience), anti-TGF-β (R&D), anti-CTLA-4 (BD), anti-IL-10 (eBioscience), and anti-EBI3 (Millipore) blocking antibodies were added into the culture at a final concentration of 10 μg/ml. Cells and culture supernatants were collected for cytokine quantification by flow cytometry analysis and ELISA.
Depletion of proteins and small molecules from Treg cell supernatants
To remove proteins from apoptotic or control Treg cell supernatants, we applied proteinase K or trypsin treatment for 1 h at 37 °C. Next, we inactivated enzymes by boiling samples for 5 min and removed precipitates by centrifugation at 10,000g for 10 min at 4 °C. For the removal of small molecules including lipids, hormones, and nucleotides, media were treated with dextran-coated charcoal (DCC). Briefly, 0.5% (m/v) DCC suspension was mixed with the supernatants at a volume ratio of 5:1 and incubated at room temperature for 20 min. Next, DCC was removed by centrifugation at 7,000g, 30 min, 4 °C. The supernatants were collected for further experiments.
Small-molecule assays
For the ATP assay, pellets of Treg cells and conventional T cells were directly suspended in 1 M perchloric acid and used for ATP assays or stored at −80 °C for further testing. Media were deproteinized with 4 M perchloric acid for further use. A colorimetric ATP measurement kit (Pierce) was used according to the manufacturer’s instructions. To measure adenosine, we used adenosine deaminase (Sigma Aldrich) to transform adenosine to xanthine in HClO4-deproteinized media, and then we measured xanthine by colorimetric assay (BioVision).
Adenosine was also detected by liquid chromatography coupled with tandem mass spectrometry. Cell culture medium was collected and analyzed freshly in an LC–triple quadrupole mass spectrometer (Agilent 6490 series) with a jet stream electrospray ionization source (Agilent). Chromatography was carried out on a Phenomenex Luna NH2 column (1 × 150 mm) and Agilent 1290 series liquid chromatograph. Mobile phase A was 5 mM ammonium acetate in LC-MS-grade water, pH adjusted to 9.9 with ammonium hydroxide. Mobile phase B was acetonitrile. 200 μl of extraction solvent was added to 200 μl of medium and vertexed briefly. A series of calibration standards (adenosine: 0, 0.01, 0.03, 0.1, 0.3, 1, and 5 μg/ml) were prepared along with the samples. The following transitions were used to identify and quantify adenosine: m/z 266.1 → m/z 134.1. Data were processed by MassHunter workstation software, version B.06. Experiments were performed with biological replicates. Each data point was acquired from cells harvested from different animals.
ELISA
T cell culture supernatants were collected at the time points indicated in the relevant figures. T cell effector cytokines were detected with ELISA kits (R&D).
Real-time PCR
Total RNA was isolated from Trizol-lysed (Invitrogen) samples according to the manufacturer’s instructions. A reverse-transcriptase reaction was performed with oligo-dT primers and AMV polymerase (Invitrogen). Real-time PCR reactions were carried out with the Fast-SYBR kit (Applied Biosystems) and the primers listed in Supplementary Table 3.
Flow cytometry
Antibodies were from BD Biosciences, eBioscience, or Invitrogen. For human cells, we used anti-CD45 (clone HI30), anti-CD3 (SK7), anti-CD4 (RPA-T4), anti-cCASP3 (9H19L2), anti-Foxp3 (PCH101), anti-TNF (MAb11), anti-IL-2 (MQ1-17H12), and anti-IFN-γ (4S.B3). Mouse cells were stained with anti-CD45 (30-F11), anti-CD45.1 (A20), anti-CD45.2 (104), anti-CD3 (500A2), anti-CD4 (RM4-5), anti-CD8 (53-6.7), anti-cCASP3 (9H19L2), anti-Foxp3 (FJK-16s), anti-TNF (MP6-XT22), anti-IL-2 (JES6-5H4), and anti-IFN-γ (XMG1.2). For surface staining, the cells were incubated with antibodies for 20 min, washed, and fixed in Fix/Perm solution (BD Biosciences). After being washed with Perm/Wash buffer (BD Biosciences), the cells were stained intracellularly for 30 min, washed, and fixed in 4% formaldehyde (Sigma Aldrich). In the case of apoptosis detection in cell culture, the cells were stained with human recombinant annexin V (BD Biosciences) followed by 7-ADD. In the case of tissue samples, the cells were stained for viability with blue fluorescent fixable dye (Life Technologies) followed by annexin V and Foxp3. All samples were read on an LSR II cytometer (BD) and analyzed with FACS DIVA software v. 8.0 (BD Biosciences).
Immunofluorescence staining
Tumor tissues were fixed in 10% formalin and embedded in paraffin for immunofluorescence staining. Multiplexed fluorescence staining was done with the OpalTM 4-plex staining system (PerkinElmer). Tissue sections were stained with anti-CD3 (clone PS1, mouse IgG2a; Novocastra/Leica), anti–cleaved caspase-3 (rabbit polyclonal IgG; Cell Signaling), and anti-Foxp3 (clone 259D, mouse IgG1; BioLegend). The secondary antibodies used were goat anti-mouse IgG2-α conjugated with Alexa Fluor 488 or goat anti-rabbit IgG (H+L) conjugated with Alexa Fluor 568. Staining slides were analyzed with the Mantra Quantitative Pathology Workstation (PerkinElmer).
Immunoblotting
After centrifugation cells were dissolved in cell lysis buffer (50 mm Tris-HCl, pH 7.4, 1% Triton X-100, 0.2% sodium deoxycholate, 0.2% SDS, 1 mM sodium EDTA) supplemented with protease inhibitors (5 μg/ml leupeptin, 5 μg/ml aprotinin, and 1 mM phenylmethylsulfonyl fluoride) and protein concentrations were determined with Bio-Rad protein assay reagent. The lysates were boiled for 5 min in Laemmli sample buffer (50 mm Tris-HCl, pH 6.8, 12.5% glycerol, 1% SDS, 0.01% bromophenol blue, 5% 2-mercaptoethanol). Then they were analyzed by SDS–PAGE followed by western blotting with primary antibodies as follows: anti-human/mouse NRF2 (clone 1808Y; Abcam), anti-human/mouse GCLM (rabbit polyclonal; Abcam), anti-mouse/human PD1 (clone 7A11B1; Thermo Fisher Scientific), anti-mouse PD-L1 (goat polyclonal; R&D Systems), anti-mouse CTLA-4 (clone 63828; R&D Systems), anti-mouse CD39 (clone 495826; R&D Systems), and anti-mouse CD73 (clone sc-32299; Santa Cruz Biotechnology). Proper secondary HRP-conjugated anti-rat, anti-rabbit (both from Vector Laboratories), or anti-goat (Santa Cruz Biotechnology) antibodies were used as secondary reagents.
Bioinformatics
Publicly available microarray data were analyzed with GSEA software v. 2.0, which is freely available from the Broad Institute (http://www.broad.mit.edu/gsea/)50.
Statistics
Statistical analysis was done with R v. 3.0.2. For comparison of two groups, we used two-sided Student’s t-test or Mann–Whitney U-test, depending on the data distribution (Shapiro–Wilk test) and homogeneity of variance (Fisher’s test or Levene’s test). Parametrical or nonparametrical ANOVA tests accompanied by two-sided post hoc analysis were used for multiple-comparison experiments.
Data availability
A Life Sciences Reporting Summary for this paper is available.
Supplementary Material
Acknowledgments
This work was supported (in part) by the US National Institutes of Health (grants CA217540, CA123088, CA099985, CA156685, CA171306, CA190176, CA193136, CA211016, and 5P30CA46592 to W.Z.), the Ovarian Cancer Research Fund, and the Marsha Rivkin Center for Ovarian Cancer Research (W.Z.; I.K.). We are grateful to L. Carter and X. Hu for critical discussions about the A2A pathway. We thank D. Postiff, M. Vinco, R. Craig, and J. Barikdar at the Tissue and Molecular Pathology Core for their assistance. We thank C. Ruan and S. Bridges at the Metabolomics Core for their support. Cd274−/− mice and Pdcd1−/− mice were provided by L. Chen (Yale University, New Haven, Connecticut, USA) and T. Honjo (Kyoto University, Kyoto, Japan), respectively.
Footnotes
AUTHOR CONTRIBUTIONS
T.M., Wei Wang, I.K., and W.Z. designed the experiments. T.M., I.K., and W.Z. wrote the paper. Wei Wang, T.M., J.C., S.W., L.V., and I.K. performed the in vivo tumor experiments. L.V., W.S., I.K., and J.R.L. provided and processed clinical specimens and performed immunohistochemical and pathological analysis. T.M., Wei Wang, H.Z., I.S., and Weimin Wang performed the immunological and biochemical assays. I.K., L.Z., T.M., C.L., Wei Wang and W.Z. analyzed data.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
References
- 1.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 2.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 3.Chen W, et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Curiel TJ, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 5.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 6.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 7.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-β controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, et al. A critical function for TGF-β signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol. 2008;9:632–640. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- 9.Maruyama T, et al. Control of the differentiation of regulatory T cells and TH17 cells by the DNA-binding inhibitor Id3. Nat Immunol. 2011;12:86–95. doi: 10.1038/ni.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ouyang W, et al. Novel Foxo1-dependent transcriptional programs control Treg cell function. Nature. 2012;491:554–559. doi: 10.1038/nature11581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gubser PM, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol. 2013;14:1064–1072. doi: 10.1038/ni.2687. [DOI] [PubMed] [Google Scholar]
- 13.Chang CH, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang CH, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho PC, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. 2015;162:1217–1228. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao E, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol. 2016;17:95–103. doi: 10.1038/ni.3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clever D, et al. Oxygen sensing by T cells establishes an immunologically tolerant metastatic niche. Cell. 2016;166:1117–1131. doi: 10.1016/j.cell.2016.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eil R, et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature. 2016;537:539–543. doi: 10.1038/nature19364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singer M, et al. A distinct gene module for dysfunction uncoupled from activation in tumor-infiltrating T cells. Cell. 2016;166:1500–1511. doi: 10.1016/j.cell.2016.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delgoffe GM, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeng H, et al. mTORC1 couples immune signals and metabolic programming to establish Treg-cell function. Nature. 2013;499:485–490. doi: 10.1038/nature12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi LZ, et al. HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dang EV, et al. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saito T, et al. Two FOXP3+CD4+ T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. 2016;22:679–684. doi: 10.1038/nm.4086. [DOI] [PubMed] [Google Scholar]
- 25.Cubillos-Ruiz JR, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161:1527–1538. doi: 10.1016/j.cell.2015.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arvey A, et al. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat Immunol. 2014;15:580–587. doi: 10.1038/ni.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ko K, et al. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med. 2005;202:885–891. doi: 10.1084/jem.20050940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cui TX, et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity. 2013;39:611–621. doi: 10.1016/j.immuni.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wan S, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147:1393–1404. doi: 10.1053/j.gastro.2014.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shevach EM. CD4+CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol. 2002;2:389–400. doi: 10.1038/nri821. [DOI] [PubMed] [Google Scholar]
- 31.Vignali DAA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deaglio S, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kryczek I, et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood. 2009;114:1141–1149. doi: 10.1182/blood-2009-03-208249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang S, Apasov S, Koshiba M, Sitkovsky M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood. 1997;90:1600–1610. [PubMed] [Google Scholar]
- 35.Erdmann AA, et al. Activation of Th1 and Tc1 cell adenosine A2A receptors directly inhibits IL-2 secretion in vitro and IL-2-driven expansion in vivo. Blood. 2005;105:4707–4714. doi: 10.1182/blood-2004-04-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chekeni FB, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564–571. doi: 10.1038/nrc3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2:e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barnett B, Kryczek I, Cheng P, Zou W, Curiel TJ. Regulatory T cells in ovarian cancer: biology and therapeutic potential. Am J Reprod Immunol. 2005;54:369–377. doi: 10.1111/j.1600-0897.2005.00330.x. [DOI] [PubMed] [Google Scholar]
- 41.Dannull J, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115:3623–3633. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Attia P, Maker AV, Haworth LR, Rogers-Freezer L, Rosenberg SA. Inability of a fusion protein of IL-2 and diphtheria toxin (denileukin diftitox, DAB389IL-2, Ontak) to eliminate regulatory T lymphocytes in patients with melanoma. J Immunother. 2005;28:582–592. doi: 10.1097/01.cji.0000175468.19742.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kurose K, et al. Phase Ia study of FoxP3+ CD4 Treg depletion by infusion of a humanized anti-CCR4 antibody, KW-0761, in cancer patients. Clin Cancer Res. 2015;21:4327–4336. doi: 10.1158/1078-0432.CCR-15-0357. [DOI] [PubMed] [Google Scholar]
- 44.Tai X, et al. Foxp3 transcription factor is proapoptotic and lethal to developing regulatory T cells unless counterbalanced by cytokine survival signals. Immunity. 2013;38:1116–1128. doi: 10.1016/j.immuni.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang J, et al. Kupfer-type immunological synapse characteristics do not predict anti-brain tumor cytolytic T-cell function in vivo. Proc Natl Acad Sci USA. 2010;107:4716–4721. doi: 10.1073/pnas.0911587107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang W, et al. Effector T cells abrogate stroma-mediated chemoresistance in ovarian cancer. Cell. 2016;165:1092–1105. doi: 10.1016/j.cell.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng D, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249–253. doi: 10.1038/nature15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Curiel TJ, et al. Blockade of B7-H1 improves myeloid dendritic cell–mediated antitumor immunity. Nat Med. 2003;9:562–567. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 49.Zou W, et al. Stromal-derived factor-1 in human tumors recruits and alters the function of plasmacytoid precursor dendritic cells. Nat Med. 2001;7:1339–1346. doi: 10.1038/nm1201-1339. [DOI] [PubMed] [Google Scholar]
- 50.Subramanian A, et al. Gene Set Enrichment Analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A Life Sciences Reporting Summary for this paper is available.