

Summary
Background
Haemopoietic stem-cell transplantation (HSCT) eradicates host haemopoiesis before venous infusion of haemopoietic stem cells (HSCs). The pathway to cellular recovery has been difficult to study in human beings because of risks associated with interventions during aplasia. We investigated whether 18F-fluorothymidine (18F-FLT) imaging was safe during allogenic HSCT and allowed visualisation of early cellular proliferation and detection of patterns of cellular engraftment after HSCT.
Methods
Eligible patients were aged 18–55 years, had high-risk haematological malignancies. All patients underwent myeloablation followed by HSCT. The imaging primary endpoint was detection of early subclinical engraftment after HSCT with 18F-FLT PET or CT. Imaging was done 1 day before and 5 or 9, and 28 days, and 1 year after HSCT. This study is registered with ClinicalTrials.gov, number NCT01338987.
Findings
Between April 1, 2014, and Dec 31, 2015, 23 patients were enrolled and assessable for toxic effects after completing accrual. 18F-FLT was not associated with any adverse events or delayed engraftment. 18F-FLT imaging objectively identified subclinical bone-marrow recovery within 5 days of HSC infusion, which was up to 20 days before engraftment became clinically evident. Quantitatively, 18F-FLT intensity differed significantly between myeloablative infusion before HSCT and subclinical HSC recovery (p=0.00031). 18F-FLT biodistribution over time revealed a previously unknown path of cellular recovery of haemopoiesis in vivo that mirrored fetal ontogeny.
Interpretation
18F-FLT allowed quantification and tracking of subclinical bone-marrow repopulation in human beings and revealed new insights into the biology of HSC recovery after HSCT.
Funding
National Institutes of Health, Ben’s Run/Ben’s Gift, Albert and Elizabeth Tucker Foundation, Mex Frates Leukemia Fund, Jones Family fund, and Oklahoma Center for Adult Stem Cell Research.
Introduction
Although the homing of haemopoietic stem cells (HSCs) to bone marrow has been studied in detail in mice,1 the pathway to engraftment in human beings is poorly understood. HSC transplantation (HSCT) involves eradication of resident host haemopoiesis followed by venous infusion of HSCs. Typically, after 14–28 days, peripheral blood shows evidence of new haemopoiesis, with release of donor monocytes and neutrophils, and by day 28 the posterior superior iliac crest shows evidence of donor haemopoiesis. Whether HSCs travel directly to the iliac crest bone-marrow space or first home to other organs in human beings is unknown and has been difficult to study because invasive techniques, such as biopsy, carry a substantial risk of infection during the period of aplasia before neutrophil recovery.
Various imaging methods can reveal cellular events in bone marrow, but available techniques have had poor sensitivity and specificity.2–4 For example, 18F-fluorodeoxyglucose (18F-FDG) PET shows bone-marrow proliferation, but non-specific uptake due to inflammation leads to poor correlation between signal changes and bone-marrow events.4 The investigational imaging agent 18F-fluorothymidine (18F-FLT) detects haemopoietic cell division with pre cision.5–10 18F-FLT is a thymidine analogue that enters cells via transporters during the S phase of cell division and is temporarily trapped via phosphorylation of thymidine kinase 1, which is found only in haemopoietic stem cells, lymphocytes, and cancer cells.11 In rodents, 18F-FLT imaging has quanti tatively shown migration of newly infused HSCs to bone marrow within 4 days of HSCT, which is significantly earlier than can be detected with bone-marrow biopsy.12 These data suggest that 18F-FLT could be used to study the preclinical recovery of haemopoiesis in human beings.
We did a pilot study to investigate whether 18F-FLT would have a favourable safety profile in the peri-HSCT period, could identify subclinical HSC engraftment, and could characterise and quantify the pattern of preclinical migration of HSCs and the pace of engraftment.
Methods
Study design and participants
We did a planned prospective, open-label, pilot study in a subpopulation of patients enrolled in a study at the University of Oklahoma and National Institutes of Health to investigate whether 18F-FLT imaging could safely identify cellular recovery and assess the effects of leuprorelin on immune reconstitution (both primary endpoints). Eligible patients were aged 18–55 years and had high-risk acute leukaemia, chronic myeloid leukaemia resistant to tyrosine-kinase inhibitors, chronic myelo monocytic leukaemia, or myelo dysplastic syndrome for which allogeneic HSCT is the standard of care. Other inclusion criteria were Lansky or Karnofsky scores greater than 60%, life expectancy longer than 3 months, organ function sufficient to tolerate myeloablative HSCT (total bilirubin <2.5 mg/dL [<42.8 μmol/L], transaminase concentrations less than five times the upper limit of normal, creatinine clearance >60 mL/min per 1.73 m2 [>1.00 mL/s per m2], ejection fraction ≥50%, diffusing capacity of the lung for carbon monoxide adjusted for alveolar volume and haemoglobin at least 50%, and forced expiratory volume in 1 s at least 60% of predicted), absence of HIV infection, active hepatitis B, uncontrolled infections, psychiatric disorders that might compromise study adherence, pregnancy, and previous exposure to 18F-FLT, and intolerance of leuprorelin. Previous autologous transplantation was permitted. Any experimental treatments had to have been completed more than 2 weeks before study entry. Patients were considered consecutively. If 18F-FLT imaging could not be done for technical or clinical reasons on or close to the designated days, the patients were excluded from this cohort.
The study was approved by the institutional review board at each institution and the NIH Radiation Safety Committee, and was done in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines. All patients provided written informed consent before enrolment. Additionally, the University of Oklahoma Institutional Review Board approved a protocol (number 6432) to confirm the sequence of engraftment shown by 18F-FLT imaging with autopsy evidence of donor haemopoiesis in lymphoid organs from patients not enrolled in this study who underwent sex-mismatched HSCTs and died within 10 days of infusion.
Procedures
Patients underwent myeloablative conditioning with 1200 cGy total-body irradiation and 120 mg/kg cyclophosphamide followed by infusion of HLA 7/8-matched or 8/8-matched HSCs from related or unrelated donor bone marrow or peripheral blood stem cells (PBSCs). We used a clacineurin inhibitor and mini-methotrexate regimen (10 mg/m2 on day 1 and 5 mg/m2 on days 3, 6, and 11 after HSCT) for graft-versus-host disease prophylaxis. Engraftment was defined as the first day of 3 consecutive days with an absolute neutrophil count greater than 0.5 × 109/L after HSCT. Relapse of leukaemia before engraftment was censored. No patients received growth factors before engraftment.
All imaging was done at the NIH Clinical Center in the Molecular Imaging Program, National Cancer Institute’s Center for Cancer Research (Bethesda, MD, USA). Patients were scheduled to undergo imaging with simultaneous 18F-FLT PET and CT (PET/CT) 1 day before and 5 or 9 days, 28 days, and 1 year after HSCT (appendix p 1) and with 18F-FDG at 28 days and 1 year after HSCT.
18F-FLT PET/CT was done with a Gemini TF Time of Flight scanner (Philips, Cleveland, OH, USA) after administration of 0.07 mCi/kg (maximum 3.00 mCi/kg) 18F-FLT (Investigational New Drug approval 111119; Cardinal Health, Greenbelt, MD, USA). Low-dose CT was done for attenuation correction. Immediately after infusion of 18F-FLT, dynamic emission studies over the thoracic (n=17) or lumbar (n=6) regions were done for 45 min in the following sequence: four at 30 s, eight at 1 min, eight at 2 min, and two at 5 min. After dynamic imaging, static imaging of the whole body from the base of the skull to the tibia was done 1 h and 2 h after radiotracer injection. Images were reconstructed on the Extended Brilliance Workspace (Philips), and 18F-FLT PET/CT images were registered, displayed, and analysed with MIM software (version 6.1).
The following sites were analysed on static fused 18F-FLT PET/CT images: thorax (thoracic spine 1–12), lumbar region (lumbar spine 1–5), cervical region, bilateral pelvic wings, bilateral iliac crests, bilateral humeri and femurs, sternum, spleen, liver, and fat. Circular three-dimensional regions of interest were applied to static or dynamic images by reviewers unaware of clinical outcomes (appendix p 1). Regions of interest were 1 cm3 for all sites in static images. We compared 18F-FLT standardised uptake values (SUVs) by time to engraftment. Dynamic images focused at the level of the thorax were analysed by applying regions of interest of 5 cm3 to the liver, 3 cm3 to the spleen, and 1 cm3 to three vertebral marrow spaces within the field of view. Representative images were chosen from those obtained on designated imaging days and from bone-marrow and PBSC graft recipients.
Adverse events were assessed continuously for longer than 1 year and classified as being attributable or not attributable to 18F-FLT. Additionally, we assessed immediate toxic effects related to 18F-FLT, which is a concern owing to the concurrent administration of toxic medications and preceding myeloablation. In all patients we measured creatinine concentrations, liver function, and bilirubin concentrations before and within 24 h after imaging.
To corrobrate the findings on engraftment shown by 18F-FLT imaging, splenic and liver tissues were tested from two patients not enrolled in the study who received HSCs from sex-mismatched donors and who died within 10 days of infusion. Paraffin-embedded samples obtained at autopsy were stained with haematoxylin and eosin and assessed by immunohistochemistry with rabbit polyclonal antibody (DAKO, Carpentaria, CA, USA) against human c-Kit (approved symbol SCFR) and by XY fluorescence in-situ hybridisation.
Outcomes
The imaging primary endpoint of the study was detection of subclinical engraftment by 18F-FLT imaging. The secondary endpoints were the anatomical sites of early cell proliferation, the pace of engraftment, and safety early after HSCT. The second primary endpoint of the study, immune reconstitution associated with leuprorelin after allogeneic HSCT, is not discussed in this Article.
Statistical analysis
Initially, the trial was powered to include 15 patients to achieve 13 assessable patients who had undergone imaging on days 5 and had a known engraftment date. A correlation coefficient was estimated in a pilot fashion: a 0.1 one-sided, one-tailed test to determine whether the correlation coefficient had a value of 0.85 rather than a null value of 0.50 with 80% power. Patients could be removed from study for refusal to continue therapy, loss to follow-up, and death. An interim safety analysis of adverse events attributed to 18F-FLT was planned for the first seven patients enrolled, with planned stopping of the 18F-FLT arm of the protocol if two or more did not have engraftment and needed a back-up product. Recovery of leukaemia before engraftment was censored because leukaemia cells upregulate thymidine kinase 1 and might alter images around the time of relapse.10 An additional eight assessable patients permitted comparison of scans done on day 9 with those done on day 5 and exploration of differences between bone-marrow and PBSC sources of HSCs. All patients, irrespective of relapse, would be included in assessment of 18F-FLT toxic effects and non-relapsed patients would be included in the primary and non-relapse secondary analyses.
We used the Wilcoxon’s signed rank test to do pairwise analyses. To determine the dichotomised effect of the median thoracic SUV on the difference in days between engraftment and measurement of 18F-FLT, we used a cutoff point 2 SD above bone-marrow myeloablation threshold (SUV=1.4), and days between SUV groups (<1.4 vs ≥1.4) and time to engraftment were compared with the exact Wilcoxon rank sum test. The Page’s L test for trend was used to investigate whether the mean slopes of the median SUVs (from 1 day before to day 5–12 after HSCT) in osseous structures and organ sites were ranked in a consistent pattern. We used mixed models to compare mean of the median SUVs across timepoints, test for linearity of bone-marrow Ki across timepoints, and test for non-linearity of liver Ki and spleen Ki across timepoints. We did all analyses with SAS version 9.4. All p values are two tailed, except those for Page’s L test, which are one tailed, with an α of 0.05. This study is registered with ClinicalTrials.gov, number NCT01338987.
Role of the funding source
The funders of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. The corresponding author had access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Between April 1, 2011, and Dec 31, 2015, we enrolled 23 patients who underwent 18F-FLT imaging, completing accrual goals. Patients underwent 18F-FLT imaging and were assessable for safety endpoints (table). The median follow-up after HSCT was 4.3 years. 18F-FLT imaging was well tolerated, with no attributed adverse events, including delay of engraftment. The median time to engraftment was 15 days (IQR 13–19) for recipients of PBSC grafts and 20 days (17–22) for recipients of bone-marrow grafts. All patients underwent 18F-FLT and 18F-FDG imaging during the planned visits except for one patient who had a scan on day 6 and one who had a scan on day 12 for technical or clinical reasons. Additionally, one patient who had a missing scan from days 5–12 and three patients had missing scans from day 28 for technical or clinical reasons. Two patients were excluded from the analysis because relapse before day 28 might have altered 18F-FLT uptake.10
Table.
Characteristics of recipients, indications for transplantation, and donor characteristics
| Data (n=23) | |
|---|---|
| Recipients’ characteristics | |
|
| |
| Age (years) | 33.7 (26.9–39.4) |
| Sex | |
| Women | 13 (57%) |
| Men | 10 (43%) |
| Ethnicity | |
| Hispanic | 17 (74%) |
| Non-Hispanic white | 2 (9%) |
| African American black | 2 (9%) |
| Asian | 1 (4%) |
| Native American | 1 (4%) |
|
| |
| Indications for transplantation | |
|
| |
| Acute lymphocytic leukaemia, B cell | |
| Ph positive CR1 | 3 (13%) |
| CR1, MLL positive | 1 (4%) |
| CR2, Ph positive (p190+) | 2 (9%) |
| CR2, Ph negative | 1 (4%) |
| CR3, Ph negative | 1 (4%) |
| Ambiguous leukaemia CR2 | 1 (4%) |
| Acute myeloid leukaemia | |
| CR1 | 1 (4%) |
| CR1, FLT3 mutation, ITD | 1 (4%) |
| CR1, secondary | 1 (4%) |
| CR2 | 5 (22%) |
| CR3 (chronic myeloid leukaemia) | 1 (4%) |
| Refractory | 3 (13%) |
| Myelodysplastic syndrome or acute myeloid leukaemia refractory | 1 (4%) |
| Chronic myeloid leukaemia | 1 (4%) |
|
| |
| Donor characteristics | |
|
| |
|
| |
| Age (years) | 36.1 (25.0–39.4) |
| Related donors | |
| 8/8-matched HSCs | 17 (74%) |
| 7/8-matched HSCs | 1 (4%) |
| Unrelated donors | |
| 8/8-matched HSCs | 4 (17%) |
| 7/8-matched HSCs | 1 (4%) |
| Stem-cell source | |
| Bone marrow | 11 (48%) |
| Peripheral blood | 12 (52%) |
| CD34 stem-cell dose (× 106 cells per kg) | 4.3 (3.1–5.1) |
Data are median (IQR) or number of patients (%). Ph=Philadelphia chromosome (BCR-ABL fusion gene). CR=complete remission. MLL=mixed-lineage leukaemia. ITD=internal tandem duplication. HSCs=haemopoietic stem cells.
18F-FLT imaging revealed subclinical engraftment after HSCT. Compared with 18F-FDG imaging, 18F-FLT imaging allowed better visualisation of the bone marrow of all patients (figure 1A). Of the 21 patients without relapse, 18F-FLT imaging consistently revealed bone-marrow uptake by day 5, with increasing intra-patient SUVs as a function of time from transplantation (figure 1B, appendix p 2). To determine the pattern of uptake objectively across bone-marrow sites, we compared SUVs for patients imaged 1 day before and 5–6 and 28 days after HSCT (n=15) with those in pateints imaged 1 day before and 9–12 and 28 days after HSCT (n=5). We found a consistent pattern of activation of bone marrow, with the most pronounced initial uptake in the thoracic spine, followed by the remainder of the axial skeleton and sternum, and finally the extremities (figure 1C). This pattern was consistent irrespective of whether patients underwent imaging on days 5–6 or 9–12, although the increase in SUVs was slightly greater at the later imaging timepoint. The magnitude of 18F-FLT uptake differed significantly between 1 day before HSC infusion and 28 days after infusion (after clinical engraftment in most patients; p<0.0001, figure 1D).
Figure 1. Imaging of osseous sites after HSCT.
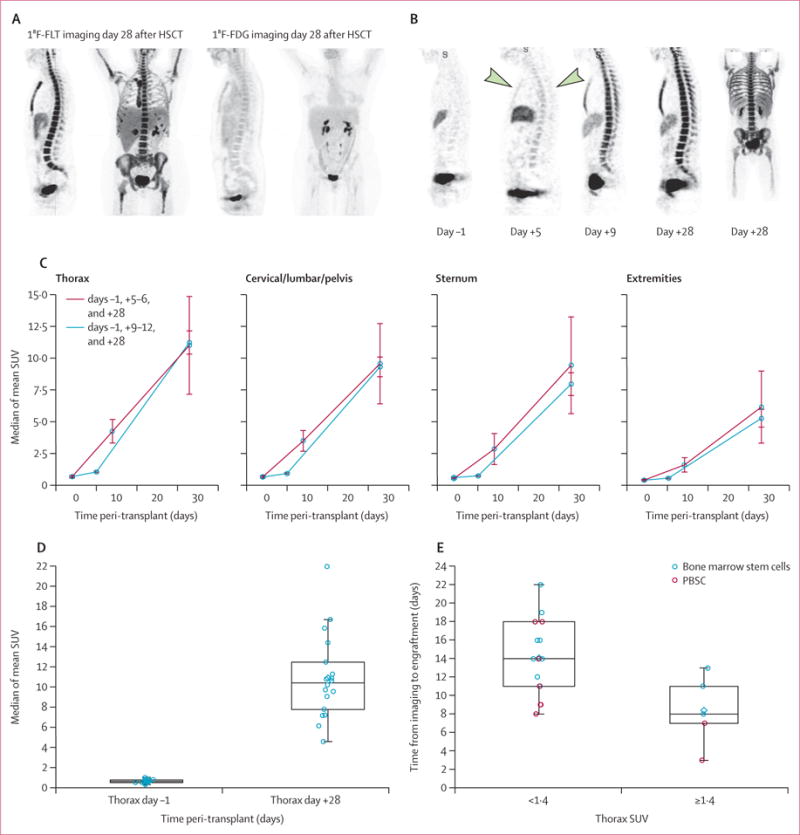
(A) Representative 18F-FLT and 18F-FDG images done 28 days after HSCT. (B) Representative 18F-FLT images in the sagittal plane 1 day before and on days 5, 9, and 28 after HSCT. The images for 1 day before and 9 and 28 days after HSCT are from one patient (who received PBSCs) and the image for day 5 is from a different patient (who received bone-marrow stem cells) because patients did not undergo imaging on days 5 and 9. The arrows highlight the increased uptake in thoracic spine on day 5 after HSCT. The day 28 image is a maximum intensity projection to permit views of the extremities and full spine simultaneously; all images are scaled equally. (C) Mapping of osseous recovery by comparison of mean (SE) slopes of median SUVs in 15 patients imaged on days 5–6 and five imaged on days 9–12. In all patients slopes were lower for the cervical, lumbar, and pelvic bones, the sternum, and the extremities than for the thoracic spine (ptrend<0.0001). The mean of the slopes on days 5–12 to day 28 after HSCT was consistently greater than that for the slopes from 1 day before to 5–12 days after HSCT: thorax p=0.0324; cervical, lumbar, and pelvic bones p=0.0094; sternum p=0.0023; and extremities p=0.0012. (D) The median thoracic SUV increased significantly from 1 day before to 28 days after HSCT (p<0.0001, n=18). (E) Association between SUV and time to engraftment. The time between 18F-FLT imaging and clinical engraftment was longer if the median was <1.4 (p=0.0075) and shorter if the median thoracic SUV was ≥1.4. Data are medians with 25th and 75th percentiles. HSCT=haemopoietic stem-cell transplantation. 18F-FLT=18F-fluorothymidine. 18F-FDG=18F-fluorodeoxyglucose. SUV=standardised uptake value. PBSC=peripheral blood stem cells.
18F-FLT uptake was associated with the pace of cellular recovery. On early (day 5–12) 18F-FLT images, patients with median thoracic spine SUVs of 1.4 or greater had shorter time to engraftment than those with SUVs lower than 1.4 (<8 days vs 14 days, p=0.0075; figure 1E). Between 1 day before and 5–6 days after HSCT, the median of the mean thoracic SUV rose from 0.61 to 1.09 (p=0.00031), as is consistent with early subclinical engraftment. By comparison, uptake in fat, a non-proliferative tissue, was unchanged over time (p=0.96). By day 28 after HSCT, dynamic analyses (Ki) showed that bone-marrow uptake of 18F-FLT was indicative of site-specific cellular expansion and exceeded the background circulation of 18F-FLT in peripheral blood (p<0.0001, figure 2A, B). These data suggest that 18F-FLT imaging is a precise indicator of subclinical cellular homing to marrow spaces, bone-marrow uptake exceeds unbound circulating radioisotope, quantification of 18F-FLT uptake is associated with haemopoietic cellular recovery, and the technique reveals a consistent pattern of bone-marrow repopulation.
Figure 2. Changes in 18F-FLT uptake over time.
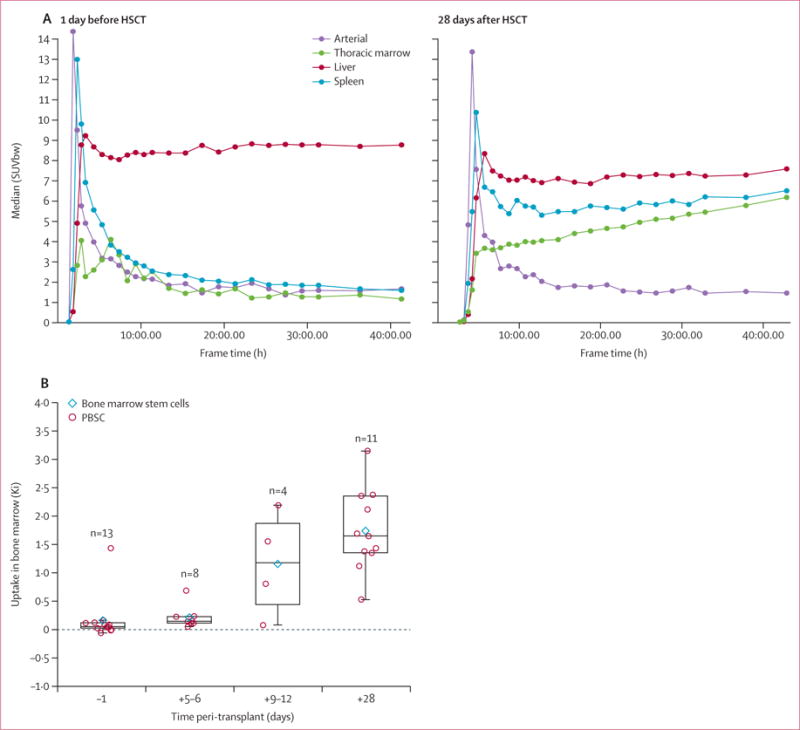
(A) Representative time activity curves for medullary space, blood, liver, and spleen obtained by dynamic 18F-FLT imaging in one patient. By day 28 after HSCT, bone-marrow uptake of 18F-FLT had exceeded background circulation in peripheral blood. (B) Uptake of 18F-FLT in thoracic bone-marrow increased significantly over time across all timepoints (ptrend<0.0001) and between 1 day before and 28 days after HSCT (p=0.002) and between 5 and 28 days after HSCT (p=0.0049). Data are medians with 25th and 75th percentiles. 18F-FLT=18F-fluorothymidine. 1HSCT=haemopoietic stem-cell transplantation. SUVbw=standardised uptake value by bodyweight. PBSC=peripheral blood stem cells.
18F-FLT imaging revealed the sequence of haemopoietic cell migration after HSCT. 18F-FLT uptake peaked in the liver and spleen on days 5–6 and decreased thereafter (figure 3A, B). Dynamic image analysis with compartmental modeling to assess tracer sequestration and cellular metabolism showed that 18F-FLT uptake in the spleen exceeded the background circulation of unbound 18F-FLT early after HSCT (figure 3C). In the liver, the Ki showed increased proliferation by days 5–6, followed by decreased uptake and a return to baseline by day 28. However, because 18F-FLT is metabolised by the liver, the Ki reflects liver cellular proliferation and metabolism of circulating 18F-FLT. By contrast, the spleen showed increased early uptake that fell between days 9–12 and day 28 (figure 3C).
Figure 3. Pattern of haemopoietic engraftment over time after HSCT.
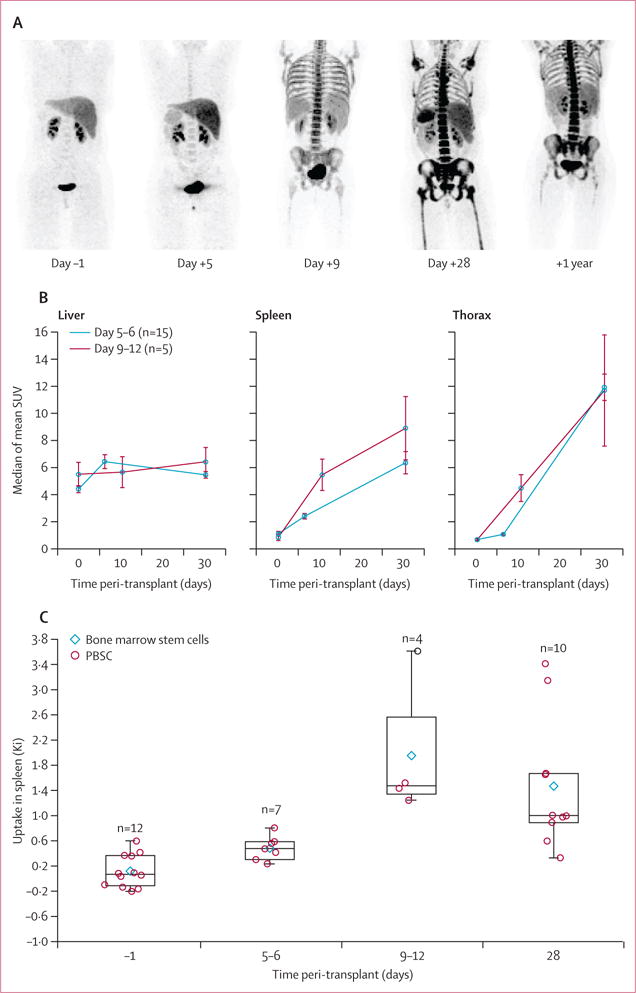
(A) Representative maximum intensity projection 18F-FLT images from the posterior coronal view 1 day before and 5, 9, and 28 days and 1 year after HSCT. (B) Comparison of mean (SE) slopes of median SUVs for liver, spleen, and thorax in 20 patients. The slopes between 1 day before and 5–12 days after HSCT were lower for the spleen and liver than for the thorax (ptrend=0.0005). In the liver, the average slope from 1 day before to 5–12 days after HSCT was greater than that from days 5–12 to day 28 after HSCT (p=0.0095), and with the early timepoint showed a plateau after days 5–6. (C) The rate of uptake of 18F-FLT in the spleen was significantly altered over time across all timepoints (ptrend=0.0088). Data are medians with 25th and 75th percentiles. 18F-FLT=18F-fluorothymidine. HSCT=haemopoietic stem-cell transplantation. SUV=standardised uptake value. PBSC=peripheral blood stem cells.
The temporal pattern of engraftment identified by 18F-FLT showed early liver and spleen engraftment, followed by thoracic spine, the remainder of the axial spine, sternum, and the medullary spaces of the distal extremity (figures 3A, 4). Between 28 days and 1 year, haemopoiesis waned in the extremities, suggesting a return to the normal adult pattern involving only the bone marrow of the axial skeleton. The mean extremity SUV derived from eight patients was 6.72 (SD 3.81) at 28 days and 3.64 (SD 2.13) at 1 year (p=0.0078).
Figure 4. Comparison of patterns of engraftment after HSCT and in fetal ontogeny.
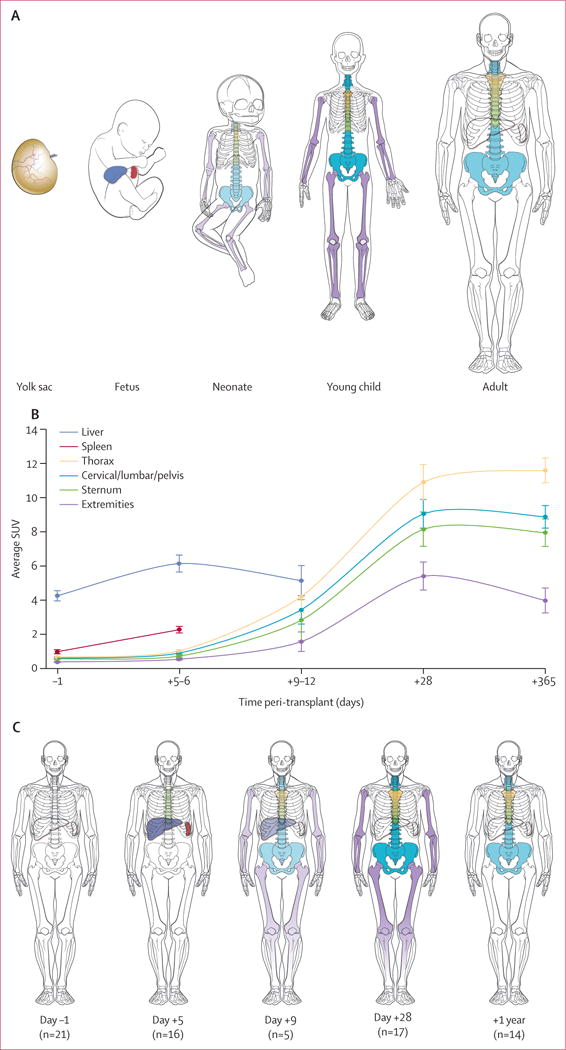
(A) Pattern of fetal ontogeny. (B) Means (SEs) of median SUVs for each organ, based on data from 21 HSCT recipients imaged with 18F-fluorothymidine, and (C) pattern of HSC engraftment in these patients. Because the sternum overlies the thoracic spaces, only the upper one-third is coloured and the thoracic vertebral bodies are shown in the position of the lower two-thirds. Splenic uptake reflected secondary lymphoid expansion after day 28 and, therefore, these data were included only up to day 28. HSCT=haemopoietic stem-cell transplantation. SUV=standardised uptake value.
To assess whether the increased 18F-FLT uptake in lymphoid organs early after HSCT represented donor haemopoiesis, we tested splenic and liver tissue samples obtained at autopsy from two patients who died within 10 days of transplantation on a separate protocol. XY fluorescence in-situ hybridisation confirmed that 99% of haemopoietic cells in the spleen were of donor origin (appendix p 2). In the liver tissue, we found CD34-positive cells and cells of donor origin identified by sex mismatch. In contrast, haemopoiesis was minimal in the bone marrow at that time (appendix p 2).
Discussion
18F-FLT PET/CT imaging with the investigational radioisotope 18F-FLT characterised and quantified subclinical HSC homing and repopulation after HSCT. These processes have not been previously accessible for study in human beings. Our approach was safe and had no associated adverse events. In our study, time to engraftment was consistent with that in previous reports,13 which was crucial for the use of 18F-FLT in HSCT because it suggests that the radiation exposure early after infusion had no harmful effects on HSC engraftment. 18F-FLT uptake clearly showed early haemopoietic cell proliferation in bone marrow. 18F-FLT SUVs showed subclinical haemopoietic settling and proliferation in organs 5 days after HSC infusion, in stark contrast to clinical neutrophil recovery, which can take 28 days or longer. Increases in 18F-FLT uptake over time did not reflect alterations in liver metabolism; uptake in non-proliferating tissues (eg, fat) was unchanged. Dynamic imaging done in single fields of view captured SUV increases over time in specific organ spaces and showed a rapid fall-off of concentrations in blood vessels as the unbound circulating agent was cleared by the renal system. This finding confirms cell proliferation in haemopoietic organs (ie, medullary spaces, liver, and spleen) after HSCT. There was minimal variation in bone-marrow uptake at early timepoints (ie, days 5–12), which suggests that early bone-marrow proliferation occurs at a similar rate in patients after HSCT. The iliac crest (the standard-of-care site for bone-marrow biopsy) is one of the later sites of bone-marrow settling, which suggests that other less accessible sites might better indicate early engraftment.
The intensity of 18F-FLT uptake was associated with pace of engraftment, with a clinically meaningful difference seen in the time remaining for early compared with late recovery (8 vs 14 days) by the median SUV threshold of 1.4. More patients with early engraftment had received PBSC grafts, which are known to clinically engraft more rapidly than bone-marrow grafts. However, 18F-FLT classified patients who engrafted faster than expected irrespective of donor source.13 18F-FLT, therefore, can show the presence and pace of subclinical engraftment and might serve as a biomarker of haemopoietic recovery.
18F-FLT imaging revealed a consistent and previously unknown pattern of sequential organ uptake after HSCT in human beings, trafficking first to the liver and spleen, then the thoracic spine, and follwed by the axial skeleton and, ultimately, the distal extremities, reclaiming areas that are normally dormant in healthy adults. Clinical convention has suggested that donor HSCs travel directly to the bone-marrow spaces after HSCT, but the pattern could not previously be assessed objectively in human beings.14,15 In rodents the mechanism of engraftment has been long established, with studies showing a milieu conducive to HSC survival in the liver and spleen early after HSCT,16 but this engraftment pattern was presumed to be distinct from that in human beings despite a similar upregulation of cytokine and chemokines after HSCT preparative regimens.17,18 Our data suggest that human HSCs take the same path of engraftment as is seen in mice, which mirrors human fetal ontogeny (figure 4).16,19–21 Consistent with this mechanism, 18F-FLT imaging 1 year after HSCT revealed that haemopoiesis in the extremities had diminished, which was presumably due to a return to the normal dormant state in human adults. This conclusion is supported by the evidence of subclinical migration of HSCs to liver and splenic tissues before clinical engraftment, as shown in the autopsy tissues of sex-mismatched HSCT recipients.
Persistent increased uptake of 18F-FLT was seen 1 year after allogeneic HSCT, with SUVs being around three times higher than those in normal controls.22 Although similar findings have been shown with 18F-FDG in recipients of autologous HSCT, long-term follow-up imaging with either 18F-FLT or 18F-FDG after allogeneic HSCT has not been studied.23
Our findings suggest that 18F-FLT could be useful to assess subclinical engraftment in populations at the highest risk of non-engraftment, including recipients of cord-blood transplants in which cell doses might be inadequate to recover haemopoiesis.24,25 We were, however, unable to assess this theory in our study because no patients had graft failure, and the need to prove safety for an investigational agent in allogeneic HSCT recipients precluded assessment for this purpose. 18F-FLT imaging might also be useful to visualise subclinical engraftment in studies in which major mismatches or minimum cell doses adversely affect engraftement.26,27
Limitations of this work include that it was a pilot study with a small population and, although patients were accrued at two sites, imaging was done at one site. The ability to confirm imaging findings by invasive methods was constrained by ethical considerations. Furthermore, although our data suggest that the liver is a site of HSC migration, 18F-FLT is trapped in the liver by glucuronidation5 and, therefore, future studies are needed to confirm this finding. Additionally, the 18F-FLT SUV threshold of 1.4 we applied was obtained in our limited dataset and should also be confirmed in a larger cohort and other transplant settings. Given how well 18F-FLT was tolerated, we contend that assessments could be extended to patients at high risk of graft failure, such as those receiving alternative donor sources. An important strength of this study is that our imaging method can be used at most major centres that have PET/CT capabilities.
We have shown a novel way to visualise and assess subclinical haemopoietic engraftment with 18F-FLT imaging, suggesting that 18F-FLT could be used as a biomarker of haemopoiesis. 18F-FLT imaging enabled safe, comprehensive, and non-invasive assessment of the entire bone-marrow compartment and showed the pattern of cellular recovery in adults after transplantation of bone-marrow or PBSC HSCT. Our data will be useful for preclinical and clinical investigations of radiation exposure effects, diagnosis and treatment responses of primary bone-marrow disorders, and loss of bone-marrow function (ie, aplastic anaemia or graft failure).
Supplementary Material
Research in context.
Evidence before this study
No method is approved to assess subclinical haemopoiesis after haemopoietic stem-cell transplantation (HSCT) because of the risk of infection associated with invasive biopsy during pancytopenia and poor specificity of 18F-fluorodeoxyglucose imaging to interrogate the bone-marrow space. We searched MEDLINE on Oct 1, 2017, with the search terms “18F-FLT” and “marrow”, unrestricted by date or language. We found no clinical trials of 18F-fluorothymidine (18F-FLT) imaging in the context of allogeneic HSCT. Among the 50 studies retrieved, 23 used 18F-FLT to assess tumours (only one in leukaemia), six assessed the toxic effects of 18F-FLT, two studied biodistribution, and two investigated chemical properties. The remaining 19 studies involved the bone-marrow compartment: two were reviews, four were animal studies (one of which was authored by our group, showed that 18F-FLT could identify subclinical engraftment after HSCT, and formed the basis for this clinical study), and 13 assessed bone-marrow function in human beings, but only six of these were published before we started this trial. Of these six publications, three (one published by our group) assessed the effect of radiation exposure on bone-marrow proliferation, five studied imaging of aplastic anaemia, and one used 18F-FLT to assess the proliferation of bone marrow more than 6 months after autologous HSCT. Among the seven clinical studies published after this study started, none studied 18F-FLT in the context of allogeneic HSCT or used this radioisotope to map early events after HSCT.
Added value of this study
To our knowledge, this is the first study to use 18F-FLT in the context of allogeneic HSCT. We found that simultaneous 18F-FLT PET and CT was safe and could reveal engraftment early after HSCT, in time to permit expeditious second HSCT. Furthermore, 18F-FLT imaging could show HSC trafficking in vivo and revealed a previously unknown pattern of engraftment that mirrors fetal ontogeny.
Implications of all the available evidence
18F-FLT enables objective assessment of important biological processes related to haemopoiesis. Use of this radioisotope has the potential to answer questions about altered haemopoiesis in the context of high proliferative or infiltrative states (eg, leukaemia and after immunotherapy), absent haemopoiesis (eg, aplastic anaemia or medication toxicity), or autoimmune attack of bone-marrow production.
Acknowledgments
This work was supported in part by the intramural research programme of the National Cancer Institute, National Institutes of Health (NIH). KMW was supported by Ben’s Run/Ben’s Gift and Albert L and Elizabeth T Tucker Foundation. JH-C was supported by National Institute of General Medical Sciences (U54GM104938), Mex Frates Leukemia Fund, Jones Family fund, and Stephenson Cancer Center Biostatistics and Research Design Core. The Oklahoma Center for Adult Stem Cell Research (OCASCR) did the imaging. For care of the study and other support, we thank Hahn Khuu, Bazetta Blacklock-Schuver, Frances Hakim, WWAMI Regional Medical Education Program, Jeremy Rose, Dennis Hickstein, James N Kochenderfer, Jennifer Wilder, Tracey Chinn, Daniel H Fowler, Tiffani N Taylor, Amy Chai, Jennifer Mann, Jennifer Hsu, and Steven Z Pavletic. We thank Ann Morris for providing editorial review, which was supported by study funding. This project was funded wholely or partly by federal funds from the National Cancer Institute, NIH, under contract number HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organisations imply endorsement by the US Government.
Footnotes
Contributors
This study was developed and designed by KMW and JH-C. The trial principal investigator was KMW, and the site principal investigator was JH-C. The Investigational New Drug application was held by KK. Programmatic oversight and quality assurance were provided by PC and REG. KMW, JH-C, LL, JG-B, DNA, GS, CGK, PC, and REG cared for the transplantation patients and were responsible for data collection and data integrity. Data analysis was done by KMW, JH-C, QD, CTN, JPH, FIL, and SA. Immunohistochemistry was done by SL and TS. SKV did the statistical analyses. KMW and JH-C wrote the first draft of the Article, and CMB and PC contributed with substantial editing. All authors reviewed the manuscript, made the decision to submit for publication, and vouched for the accuracy and completeness of the data and fidelity to the protocol.
Declaration of interests
We declare no competing interests.
See Online for appendix
Contributor Information
Kirsten M Williams, Children’s Research Institute, Children’s National Health System, Washington, DC, USA.
Jennifer Holter-Chakrabarty, Stephenson Cancer Center, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA.
Liza Lindenberg, Molecular Imaging Branch, National Cancer Institute, and National Institutes of Health, Bethesda, MD, USA.
Quyen Duong, Stephenson Cancer Center, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA.
Prof Sara K Vesely, Stephenson Cancer Center, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA.
Chuong T Nguyen, School of Electrical and Computer Engineering, University of Oklahoma, Norman, OK, USA.
Prof Joseph P Havlicek, School of Electrical and Computer Engineering, University of Oklahoma, Norman, OK, USA.
Karen Kurdziel, Molecular Imaging Branch, National Cancer Institute, and National Institutes of Health, Bethesda, MD, USA.
Juan Gea-Banacloche, Division of Infectious Diseases, Mayo Clinic Arizona, AZ, USA.
Frank I Lin, Cancer Imaging Program, National Cancer Institute, and National Institutes of Health, Bethesda, MD, USA.
Daniele N Avila, Experimental Transplantation and Immunology Branch, National Cancer Institute, and National Institutes of Health, Bethesda, MD, USA.
George Selby, Stephenson Cancer Center, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA.
Christopher G Kanakry, Experimental Transplantation and Immunology Branch, National Cancer Institute, and National Institutes of Health, Bethesda, MD, USA.
Prof Shibo Li, Stephenson Cancer Center, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA.
Teresa Scordino, Stephenson Cancer Center, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA.
Stephen Adler, Clinical Research Directorate/Clinical Monitoring Research Program, Leidos Biomedical Research, National Cancer Institute Campus at Frederick, Frederick, MD, USA.
Prof Catherine M Bollard, Children’s Research Institute, Children’s National Health System, Washington, DC, USA.
Peter Choyke, Molecular Imaging Branch, National Cancer Institute, and National Institutes of Health, Bethesda, MD, USA.
Ronald E Gress, Experimental Transplantation and Immunology Branch, National Cancer Institute, and National Institutes of Health, Bethesda, MD, USA.
References
- 1.Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci USA. 1999;96:3120–25. doi: 10.1073/pnas.96.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwee TC, Fijnheer R, Ludwig I, et al. Whole-body magnetic resonance imaging, including diffusion-weighted imaging, for diagnosing bone marrow involvement in malignant lymphoma. Br J Haematol. 2010;149:628–30. doi: 10.1111/j.1365-2141.2010.08093.x. [DOI] [PubMed] [Google Scholar]
- 3.Sambuceti G, Brignone M, Marini C, et al. Estimating the whole bone-marrow asset in humans by a computational approach to integrated PET/CT imaging. Eur J Nucl Med Mol Imaging. 2012;39:1326–38. doi: 10.1007/s00259-012-2141-9. [DOI] [PubMed] [Google Scholar]
- 4.Marini C, Podesta M, Massollo M, et al. Intrabone transplant of cord blood stem cells establishes a local engraftment store: a functional PET/FDG study. J Biomed Biotechnol. 2012;2012:767369. doi: 10.1155/2012/767369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shields AF, Grierson JR, Dohmen BM, et al. Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nat Med. 1998;4:1334–36. doi: 10.1038/3337. [DOI] [PubMed] [Google Scholar]
- 6.Vanderhoek M, Juckett MB, Perlman SB, Nickles RJ, Jeraj R. Early assessment of treatment response in patients with AML using [18F]FLT PET imaging. Leuk Res. 2011;35:310–16. doi: 10.1016/j.leukres.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sambuceti G, Massollo M, Marini C, et al. Trafficking and homing of systemically administered stem cells: the need for appropriate analysis tools of radionuclide images. Q J Nucl Med Mol Imaging. 2013;57:207–15. [PubMed] [Google Scholar]
- 8.Agool A, Slart RH, Thorp KK, et al. Effect of radiotherapy and chemotherapy on bone marrow activity: a 18F-FLT-PET study. Nucl Med Commun. 2011;32:17–22. doi: 10.1097/mnm.0b013e328340798c. [DOI] [PubMed] [Google Scholar]
- 9.Agool A, Slart RH, Kluin PM, de Wolf JT, Dierckx RA, Vellenga E. F-18 FLT PET: a noninvasive diagnostic tool for visualization of the bone marrow compartment in patients with aplastic anemia: a pilot study. Clin Nucl Med. 2011;36:286–89. doi: 10.1097/RLU.0b013e31820aa1a1. [DOI] [PubMed] [Google Scholar]
- 10.Buck AK, Bommer M, Juweid ME, et al. First demonstration of leukemia imaging with the proliferation marker 18F-fluorodeoxythymidine. J Nucl Med. 2008;49:1756–62. doi: 10.2967/jnumed.108.055335. [DOI] [PubMed] [Google Scholar]
- 11.Schwartz JL, Tamura Y, Jordan R, Grierson JR, Krohn KA. Monitoring tumor cell proliferation by targeting DNA synthetic processes with thymidine and thymidine analogs. J Nucl Med. 2003;44:2027–32. [PubMed] [Google Scholar]
- 12.Awasthi V, Holter J, Thorp K, Anderson S, Epstein R. F-18-fluorothymidine-PET evaluation of bone marrow transplant in a rat model. Nucl Med Commun. 2010;31:152–58. doi: 10.1097/MNM.0b013e3283339f92. [DOI] [PubMed] [Google Scholar]
- 13.Anasetti C, Logan BR, Confer DL. Peripheral-blood versus bone marrow stem cells. N Engl J Med. 2013;368:288. doi: 10.1056/NEJMc1214025. [DOI] [PubMed] [Google Scholar]
- 14.Srour EF, Jetmore A, Wolber FM, et al. Homing, cell cycle kinetics and fate of transplanted hematopoietic stem cells. Leukemia. 2001;15:1681–84. doi: 10.1038/sj.leu.2402256. [DOI] [PubMed] [Google Scholar]
- 15.Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci USA. 1999;96:3120–25. doi: 10.1073/pnas.96.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kieusseian A, Brunet de la Grange P, Burlen-Defranoux O, Godin I, Cumano A. Immature hematopoietic stem cells undergo maturation in the fetal liver. Development. 2012;139:3521–30. doi: 10.1242/dev.079210. [DOI] [PubMed] [Google Scholar]
- 17.Kollet O, Shivtiel S, Chen YQ, et al. HGF, SDF-1, and MMP-9 are involved in stress-induced human CD34+ stem cell recruitment to the liver. J Clin Invest. 2003;112:160–69. doi: 10.1172/JCI17902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sefc L, Psenak O, Sykora V, Sulc K, Necas E. Response of hematopoiesis to cyclophosphamide follows highly specific patterns in bone marrow and spleen. J Hematother Stem Cell Res. 2003;12:47–61. doi: 10.1089/152581603321210136. [DOI] [PubMed] [Google Scholar]
- 19.Christensen JL, Wright DE, Wagers AJ, Weissman IL. Circulation and chemotaxis of fetal hematopoietic stem cells. PLoS Biol. 2004;2:E75. doi: 10.1371/journal.pbio.0020075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samokhvalov IM. Deconvoluting the ontogeny of hematopoietic stem cells. Cell Mol Life Sci. 2014;71:957–78. doi: 10.1007/s00018-013-1364-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kordes C, Haussinger D. Hepatic stem cell niches. J Clin Invest. 2013;123:1874–80. doi: 10.1172/JCI66027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agool A, Glaudemans AW, Boersma HH, Dierckx RA, Vellenga E, Slart RH. Radionuclide imaging of bone marrow disorders. Eur J Nucl Med Mol Imaging. 2011;38:166–78. doi: 10.1007/s00259-010-1531-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woolthuis C, Agool A, Olthof S, et al. Auto-SCT induces a phenotypic shift from CMP to GMP progenitors, reduces clonogenic potential and enhances in vitro and in vivo cycling activity defined by 18F-FLT PET scanning. Bone Marrow Transplant. 2011;46:110–15. doi: 10.1038/bmt.2010.75. [DOI] [PubMed] [Google Scholar]
- 24.Danby R, Rocha V. Improving engraftment and immune reconstitution in umbilical cord blood transplantation. Front Immunol. 2014;5:68. doi: 10.3389/fimmu.2014.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laughlin MJ, Eapen M, Rubinstein P, et al. Outcomes after transplantation of cord blood or bone marrow from unrelated donors in adults with leukemia. N Engl J Med. 2004;351:2265–75. doi: 10.1056/NEJMoa041276. [DOI] [PubMed] [Google Scholar]
- 26.Horwitz ME, Chao NJ, Rizzieri DA, et al. Umbilical cord blood expansion with nicotinamide provides long-term multilineage engraftment. J Clin Invest. 2014;124:3121–28. doi: 10.1172/JCI74556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Popat U, Mehta RS, Rezvani K, et al. Enforced fucosylation of cord blood hematopoietic cells accelerates neutrophil and platelet engraftment after transplantation. Blood. 2015;125:2885–92. doi: 10.1182/blood-2015-01-607366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.