

Abstract
Aciniform silk protein (AcSp1) is the primary component of wrapping silk, the toughest of the spider silks due to combined high tensile strength and extensibility. Argiope trifasciata AcSp1 contains a core repetitive domain with at least 14 homogeneous 200-amino acid units (“W” units). Upon fibrillogenesis, AcSp1 converts from an α-helix-rich soluble state to a mixed α-helical/β-sheet conformation. Solution-state nuclear magnetic resonance (NMR) spectroscopy allowed demonstration of variable local stability within the W-unit, but comprehensive characterization was confounded by spectral overlap, exacerbated by decreased chemical shift dispersion upon denaturation. Here, 19F-NMR spectroscopy, in the context of a single W-unit (W1), is applied to track changes in structure and dynamics. Four strategic positions in the W-unit were mutated to tryptophan and biosynthetically labeled with 5-fluorotryptophan (5F-Trp). Simulated annealing-based structure calculations implied that these substitutions should be tolerated while circular dichroism (CD) spectroscopy and 1H-15N chemical shift displacements indicated minimal structural perturbation in W1 mutants. Fiber formation by W2 concatemers containing 5F-Trp substitutions in both W-units demonstrated retention of functionality, a somewhat surprising finding in light of sequence conservation between species. Each 5F-Trp-labeled W1 exhibited a unique 19F chemical shift, linewidth, longitudinal relaxation time constant (T1), and solvent isotope shift. Perturbation to 19F chemical shift and nuclear spin relaxation parameters reflected changes in conformation and dynamics at each 5F-Trp site upon addition of urea and dodecylphosphocholine (DPC). 19F-NMR spectroscopy allowed unambiguous localized tracking throughout titration with each perturbant, demonstrating distinct behavior for each perturbant not previously revealed by heteronuclear NMR experiments.
Graphical abstract
Spider silks have long generated significant interest as biomaterials because of their exceptionally high tensile strength, extensibility, toughness, and biocompatibility.1–3 Correspondingly, spider silks have many potential applications, including functioning as drug delivery vehicles,4 in nerve regeneration,5 and in tendon repair.6 Spiders produce up to seven different protein-based silks, each with distinct mechanical properties.7 Aciniform, or wrapping, silk is the toughest spider silk (i.e., it absorbs maximal mechanical energy prior to failing), and is used by spiders to wrap and immobilize prey and to build sperm webs, egg cases and stabilimenta.8 The toughness of aciniform silk per unit weight surpasses even the toughest synthetic fibers available to date, such as nylon, Kevlar and high-tensile steel9 but it is as light as cotton or nylon.10
Aciniform silk is composed primarily of the protein aciniform spidroin 1 (AcSp1). For AcSp1 proteins characterized to date, sizes depend upon the spider species and range from ~300–430 kDa, comprising a core repetitive domain flanked by short N- and C-terminal domains.8 In Argiope trifasciata, the AcSp1 core repetitive domain is composed of a 200-amino acid unit (referred to here as the “W” unit, for wrapping) repeated at least 14 times.8,11 Using solution-state nuclear magnetic resonance (NMR) spectroscopy, we recently demonstrated that a single W unit (W1) consists of an ellipsoidally shaped, compactly folded five α-helix bundle flanked by intrinsically disordered N- and C-terminal tails.12 Concatemers of two to four W units (W2, W3 and W4) have been shown to be highly modular in terms of both structure and dynamics, with the linked C- and N-terminal tails of neighboring subunits retaining intrinsic disorder and giving rise to a “beads-on-a-string” architecture.12
Recombinant AcSp1 concatemers comprising at least two W units (i.e., W2 and larger) can be induced to form silk-like fibers upon application of shear force.13 The fact that fibers can be pulled directly from W2 solution-state NMR samples implies that the observed beads-on-a-string concatemeric conformation acts as the soluble precursor for fibrillogenesis.12 Fiber formation is accompanied by a concomitant secondary structural transition, with partial - but not complete - α-helix to β-sheet conversion.12,13 This process can be inhibited by the addition of the zwitterionic detergent dodecylphosphocholine (DPC) above its critical micelle concentration (CMC ≈ 1.1 mM14) or by the chaotropic reagents urea or guanidinium chloride.12
In the course of titrations with these detergent and reagents, the W-unit exhibited both localized and progressive denaturation. Specifically, the fifth helix denatured prior to the remainder of the globular core alongside clear perturbation to the region of the core underneath this helix in the folded state. We therefore hypothesized that this localized denaturation may provide an intermediate state en route to fiber formation, with the remainder of the globular helices retaining their secondary structure, given that the fibrous state exhibits loss of only a portion of the total helical content.12 However, mapping of this structural transition employing chemical shift perturbation in heteronuclear 1H, 13C and/or 15N correlation NMR experiments proved to be of limited utility due to spectral overlap exacerbated by loss of chemical shift dispersion during W1 denaturation.
The 19F nucleus is an outstanding NMR probe of local environment in proteins.15–18 19F chemical shifts are extremely sensitive to van der Waals and electrostatic interactions, exhibiting ~100 times more sensitivity than the corresponding 1H nuclei upon replacement.19 The 19F chemical shift,18 longitudinal relaxation time constant (T1),18 and solvent isotope shift (SIS) difference between H2O and D2O environments20 are good indicators of the extent of burial of the nucleus within a protein. In general, more buried residues are associated with more downfield chemical shifts, more rapid T1 relaxation, and smaller values of SIS.18,20 To date, a single study has employed 19F-NMR to investigate silk, using 4-fluorophenylalanine-labeled Bombyx mori silkworm fibroin model peptides as a probe for side chain mobility.21 This technique has not yet, to our knowledge, been applied to spider silk.
The exquisite modularity of AcSp1 makes W1 an ideal model system to better understand the structural transition, orientation and self-assembly of W units that occur during fiber formation. Here, we have investigated the sensitivity and utility of probes of local conformational environment within W1 using 19F-NMR spectroscopy. Conveniently, the sequence of W1 is devoid of tryptophan,8,13 allowing for straightforward background-free site-directed mutagenesis. 19F-labeling of tryptophan, in particular the incorporation of 5-fluorotryptophan (5F-Trp), is inexpensive and efficient.22 This has allowed for straightforward position-specific incorporation of 19F probes.
Four residues have been chosen for site-directed tryptophan mutagenesis of W1 herein. Three were selected as probes of the globular domain: one in the most readily denatured helix 5 region, F146, potentially involved in conformational change to β-strand during fibrillogenesis; a mildly exposed phenylalanine in helix 3 on the surface of the globular domain, F90, possibly enhancing protein-protein interactions during fibrillogenesis; and, a buried, albeit charged residue, R36, within the center of the folded protein in the region most perturbed upon helix 5 unfolding. The fourth residue, Y169, is fully solvent exposed and located in the intrinsically disordered, C-terminal tail providing a probe that acts herein effectively as an internal control of an unperturbed 5F-Trp site. Using 5-fluoroindole,22 we biosynthetically 19F-labeled the tryptophan residues at each of these four positions (R36W, F90W, F146W and Y169W) in W1 mutants. Through NMR and circular dichroism (CD) spectroscopy in combination with simulated-annealing structural calculations, we show that tryptophan mutation and 19F-labeling do not affect the global fold of the protein. Furthermore, W2 proteins containing 5F-Trp in both W-units at each of the four substitutions are capable of silk-like fiber formation in the same manner as wild type W2. Each fluorinated mutant provides distinct 19F chemical shift, T1 relaxation time, and SIS values reflective of burial or solvent exposure and is sensitive to changes in local conformational environment and dynamics upon addition of urea or DPC. To our knowledge, this work represents the first employment of 19F labels in a spider silk alongside the first report of mutagenesis of an aciniform silk protein.
EXPERIMENTAL PROCEDURES
Simulated W1 Trp mutant structural ensembles and analysis
A single round of structure calculation was performed using Xplor-NIH 2.3823,24 for each W1 Trp mutant protein in identical manner to the final round of structural calculation for W1 (PDB entry 2MU312) prior to water refinement with the following two modifications: (1) the Xplor-NIH sequence input file was modified appropriately to convert R36, F90, F146, or Y169 to a Trp; and, (2) nuclear Overhauser effect (NOE) distance constraints to the native side chain were removed, leading to the removal of the following number of restraints: 49 (R36W), 36 (F90W), 14 (F146W), or 8 (Y169W). The per-residue and per-atom solvent accessible surface area (SASA) were calculated by the GETAREA web script (http://curie.utmb.edu/getarea.html) based upon the program FANTOM.25 SASA values were averaged for the 20 lowest energy members of each 100 member mutant W1 ensemble and weighted as a function of total Trp surface area26 as for W1.12
W1 and W2 mutagenesis, expression, 5F-Trp-labeling, and purification
W1 Trp mutant genes were produced using previously described protocols for W113 with slight modifications. In brief, individual tryptophan mutations were introduced on a previously descried pDW1 plasmid (pDrive vector containing the W1 gene) through inverse PCR27 with mutagenic primers (Table S1). W2 Trp mutant plasmids (pDW2) were produced using the same cloning strategy as previously described13, by ligation (using T4 DNA ligase) of two DNA fragments prepared from each of the above pDW1 Trp mutant plasmids through restriction endonuclease digestions (BseRI and BamHI; BsgI and BamHI). pDW1 (or pDW2) Trp mutant plasmids were then digested by BsaI and BfuAI (New England Biolabs, Ipswich, MA), and the resulting W1 (or W2) Trp mutant genes were inserted downstream of the His6-SUMO coding sequence in a modified pET-32 expression plasmid (EMD Millipore, Billerica, MA, USA) that was digested by the same enzymes.
Each expression plasmid encoding a His6-SUMO-W1 (or W2) Trp mutant was introduced into E. coli BL21 (DE3) (Novagen, Darmstadt, Germany) using standard transformation protocols. The resulting E. coli cells containing the correct plasmid were grown in liquid LB medium with ampicillin (50 μg/mL) to OD600 ~ 0.6. For 19F labeling, cells were then collected by centrifugation and re-suspended in M9 minimal growth medium28 containing 1 g/L (NH4)2SO4 (for 15N labeling, 15(NH4)2SO4 (Cambridge Isotope Laboratories, Tewksbury, MA)), 2 g/L D-glucose, 4 mL/L of 1M MgSO4, and 1.8 ml/L of 1 mM FeSO4. Following incubation in minimal medium at 37 °C for ~30 min, 60 mg/L of 5-fluoroindole (Sigma-Aldrich, Oakville, ON) in 1 mL dimethyl sulfoxide was added for biosynthetic 19F-labeling of tryptophan.22 Expression of a given 19F-labeled W1 (or W2) Trp mutant protein was then induced through addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a concentration of 0.8 mM. After overnight induction at ~22 °C, protein purification and SUMO cleavage were performed as described previously for wild type W1.29
Circular dichroism (CD) spectroscopy
Far-ultraviolet (UV) CD spectra were acquired on a J-810 spectropolarimeter (Jasco, Easton, MD) with a scan speed of 20 nm/min, response time of 1 s, acquisition interval of 1 nm, and a bandwidth of 2 nm. Spectra of 10–30 μM 5F-Trp-labeled W1 Trp mutants in 20 mM sodium acetate buffer, pH 5.0 were collected at 22 ± 2 °C from 260 to 195 nm in a quartz cuvette (0.05 cm path length; Hellma Analytics, Plainview, NY). A blank solution containing the same buffer was measured under the same experimental conditions. For each sample, the blank solution was subtracted from the average of two scans, and the blank subtracted spectrum was converted to mean residue ellipticity on the basis of concentration determined by ultraviolet spectroscopy at 210 nm (estimated as ℰ210 = 270,858 M−1 cm−1 for W1 and 541,716 M−1 cm−1 for W2) in the same 0.05 cm path length quartz cuvette.
NMR characterization of W1 mutants
Samples of individual W1 Trp and 5F-Trp-labeled mutant proteins (0.06–0.2 mM), 2,2,2-trifluoroethanol-d2 (TFE, 0.1 mM), and 5-fluoroindole (0.1 mM) were prepared in the buffer (20 mM CD3COONa, 1 mM 2,2-dimethyl-2-silapentane-5-sulfonic acid (DSS), 1 mM NaN3; pH 5.0; H2O:D2O at 9:1 (v:v), “NMR buffer”) previously employed for W112,29 and W212 chemical shift assignment, structural and hydrodynamic characterization. For 19F SIS effect measurement, the buffer for a given protein or 5-fluoroindole sample was exchanged to NMR buffer containing H2O:D2O at 1:9 (v:v). TFE was included as a reference for 19F experimental setup standard in all samples, but not as a chemical shift standard (referencing detailed below).
NMR experiments were performed on an 11.7 T Avance spectrometer (Bruker Canada, Milton, ON) using a double-resonance 5-mm BBFO SmartProbe at 30 °C. 1D 1H experiments (16–32 scans, sweep width 16 ppm, acquisition time 2 s, recycle delay 1–2 s) were performed with water suppression using presaturation or excitation sculpting.30 2D 1H-15N heteronuclear single quantum coherence (HSQC) experiments (16 scans, sweep width 26/14 ppm in F1/F2, size of fid 128/1024 in F1/F2, recycle delay 1 s) were performed using sensitivity improvement31 and decoupling during acquisition. 1D 19F experiments (128–5120 scans, sweep width 90 ppm centered at −105.0 ppm, acquisition time 387 ms, recycle delay 1–2 s) were performed with or without 1H decoupling during acquisition. Inversion-recovery 19F T1 experiments (320–1136 scans, sweep width 50 ppm centered at −124.5 ppm, acquisition time 695 ms, recycle delay 5 s) were acquired with a series of 10 recovery delays (0.01, 0.03, 0.06, 0.10, 0.15, 0.25, 0.50, 1.00, 1.50 and 2.00 s) with or without 1H decoupling during acquisition.
NMR data were processed and analyzed using Bruker TopSpin 3.1 and, for T1 analysis, Bruker Dynamics Center. 1H chemical shifts were referenced directly to DSS at 0.0000 ppm. 19F and 15N chemical shifts were indirectly referenced to the DSS frequency at 0.0000 ppm in the 1H 1D spectrum, following IUPAC32 and IUBMB33 standards, respectively. 19F chemical shifts and linewidths were determined using the Lorentz/Gauss Deconvolution in the Line Shape Fitting module of Topspin 3.1. 19F SIS values were obtained as the difference between the chemical shift in 1:9 H2O:D2O and that in 9:1 H2O:D2O (i.e., δ(D2O) – δ(H2O)). T1 relaxation time constants were determined from exponential fits of the observed signal intensities as a function of delay for the inversion-recovery experiment, using the relationship:
| (1) |
where I(t) is the observed signal intensity at delay time t, I(0) is the intensity of the full signal, and A is a normalization factor. Perturbation to structure by insertion of 5F-Trp were assessed through analysis of perturbation of 1H-15N HSQC peak positions through a gyromagnetic ratio-weighted combined chemical shift (CCS)34 comparison to wild type W1, as detailed previously.12
Fiber formation assays
Hand-drawn fibers were pulled from solutions of individual W2 5F-Trp-labeled mutants (R36W, F90W, F146W or Y169W on both W units) either in 50 mM potassium phosphate buffer (pH 7.5) or in 20 mM sodium acetate buffer (pH 5.0), using a pipette tip as described previously13 for wild type W2–4. In brief, 10-μL aliquots of protein solution (at ~19 μM) were deposited on clean glass slides and ~1 cm long fibers were drawn and put directly back on the slide. Fibers were imaged using an inverted optical microscope (Axiovert 200M, Carl Zeiss Canada, Toronto, ON; 40× objective and 10× ocular).
W1 mutant titrations
Titrations were carried out using mixtures of either all four or of two of the four W1 5F-Trp-labeled mutants (individual protein concentrations of 0.06 mM–0.23 mM) prepared in the acetate buffer detailed above. Independent DPC (0, 0.5, 0.7, 1.0 1.3, 2.5, 3.7, 5.0, 10 and 20 mM; dilution negligible) and urea (0, 0.5, 1.0, 1.2, 1.4, 2.0, 2.3, 2.5, 2.7, 2.9, 3.1, 3.3, 3.5, 3.7 and 4.0 M; total W1 concentration ~0.19–0.32 mM at 4.0 M urea endpoint) titrations were carried out. 1D 1H and 19F spectra were acquired at each titration point and 19F inversion-recovery experiments were carried out at 0, 1.3 and 20.0 mM DPC and at 0 and 4.0 M urea, with T1 values determined as for individual proteins using Bruker Dynamics Center.
19F T1 and T2 predictions
Two interactions are generally held to dominate 19F spin relaxation: heteronuclear dipolar coupling of the dilute 19F nucleus with surrounding protons (dip)18,35 and chemical shift anisotropy (CSA).36,37 Each contribution may be considered independently, with a given relaxation rate provided through the sum of the components:
| (2) |
| (3) |
where R1 and R2 are the longitudinal and transverse relaxation rates, respectively; T1 and T2 the longitudinal and transverse relaxation time constants, respectively; and, the bracketed subscripts indicate the contribution from a given relaxation mechanism. The formalism employed to estimate the dipolar and CSA contributions to eqs. 1–2 is detailed in the Appendix.
RESULTS
Putative structural ensembles of W1 Trp mutants
Structural ensembles were calculated for each W1 Trp mutant (positions in wild type W1 in Figure 1A) in the same manner as for wild type W112 employing all NMR restraints except distance constraints involving the mutated side chain. Despite the increased steric bulk of the Trp side chain, the total energies observed for the resulting 20-member structural ensembles were similar in magnitude to W1. Both backbone and heavy atom root-mean-square deviations (RMSDs) of the ensembles of mutant structures, relative to wild type W1, are comparable in magnitude to the RMSD for the wild type W1 ensemble itself (Table S2). Furthermore, none of the mutant structural ensembles exhibited substantially increased NOE constraint violations relative to the converged W1 ensemble (Table S2). As a whole, these structure calculations imply that W1 should be able to accommodate mutation to Trp at each of the four selected locations without significant structural perturbation. Importantly, the 19F probe in each mutant (blue spheres, Figure 1B–E) should experience a different local environment.
Figure 1.

(A) Superposed solution NMR structural ensemble of W1 (PDB entry 2MU312) showing side chains only for residues mutated to tryptophan. Putative structural ensembles for (B) R36W, (C) F90W, (D) F146W and (E) Y169W tryptophan mutants (20 lowest energy members of 100 member ensembles superposed onto the lowest energy structure; calculated using wild type W1 NMR restraints with removal of distance restraints involving the mutated side chain). 5-fluoro moieties are represented by blue spheres.
Relative ensemble-averaged SASA values were predicted for each Trp residue, with SASA ranked as R36W < F90W < F146W < Y169W (Table 1). Notably, the SASA values were only subtly perturbed relative to those of the corresponding residues in wild type W1, with three mutants increasing in relative SASA (2% for R36W, 11% for F90W, and 12% for Y169W) and one decreasing (−4.5% for F146W). Following the relative degrees of surface exposure and packing, increased relative SASA corresponds directly to increased variability of Trp side chain positioning over the putative structural ensembles (Figures 1B–E). Localized solvent accessibility at the C-4 (Cε2), C-5 (Cζ3), and C-6 (Cη2) positions was used to infer potential for solvent exposure in proximity to the 5F position (Table 1); even in the most protected R36W position, ~25% of the ensemble members exhibited a SASA of ~6–9 Å2 at both the C-5 and C-6 positions.
Table 1.
Solvent accessible surface area (SASA) relative to total Trp side chain area and for indicated carbon in each W1 mutant (average ± average deviation over 20-member structural ensembles).
| R36W | F90W | F146W | Y169W | |
|---|---|---|---|---|
| Predicted side chain SASA (%) | 4±4 | 30±3 | 71±14 | 93±10 |
| C-4 SASA (Å2) | 0.0±0.0 | 1.2±1.1 | 14.4±5.7 | 17.4±6.9 |
| C-5 SASA (Å2) | 1.7±2.5 | 8.0±5.6 | 30.4±8.2 | 33.2±6.0 |
| C-6 SASA (Å2) | 2.8±3.2 | 24.1±7.5 | 34.7±4.3 | 36.3±3.0 |
Properties of 5F-Trp-labeled W1 tryptophan mutants
The expression levels of each His6-SUMO-W1 Trp mutant fusion protein (hereafter referred to by mutation as R36W, F90W, F146W or Y169W) in the presence of 5-fluoroindole were similar to that of His6-SUMO-W1 containing wild type W1 in M9 minimal medium (~80 mg/L of cell culture). All fusion proteins were soluble after cell lysis and efficiently purified to >90% purity (Figure S1). After cleaving His6-SUMO from the fusion proteins using SUMO protease, the resulting 5F-Trp-labeled W1 Trp mutants (~0.1 mM in solution) were stable at 4 °C for at least two weeks.
Far-UV CD spectra of the four W1 5F-Trp-labeled mutants and wild type W1 were practically identical (Figure 2). All mutants display the expected12 characteristic negative α-helical bands at ~208 and ~222 nm convoluted by features from disordered and β-turn structures. Consistent with the CD spectroscopy results, 1D 1H NMR spectra were highly similar for each mutant relative to wild type W1 (Figure S2).
Figure 2.
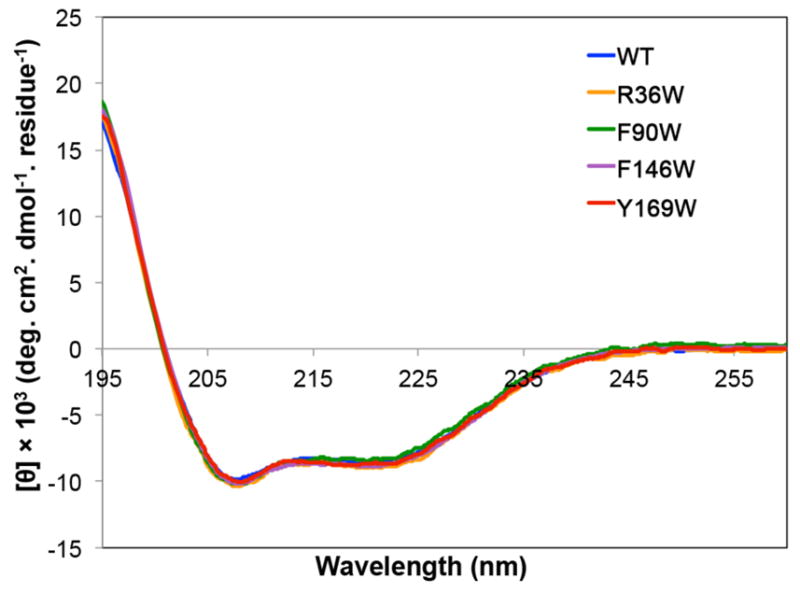
Far-UV CD spectra of fluorinated W1 tryptophan mutants (5F-Trp-labeled at R36W, F90W, F146W and Y169W) and wild type (WT) W1.
2D 1H-15N HSQC spectra provided greater insight into the extent and localization of structural perturbation. The same general peak pattern (or backbone “fingerprint”) was observed for each mutant relative to wild type W1 (Figure 3), with varying degrees of perturbation apparent. Minimal spectral perturbation in the globular core was observed for R36W, F146W, and Y169W (Figure 3), with each mutant exhibiting a distinct subset of 1H-15N HSQC cross-peaks that was perturbed relative to W1. CCS perturbations relative to the assigned W1 chemical shifts29 demonstrated that the cross-peaks that exhibit the highest degree of perturbation correspond to residues close in sequence and/or space (Figures 3, S3, S5 and S6). The F90W mutant, conversely, exhibited a higher degree of chemical shift perturbation, not necessarily localized to the site of mutation (Figures 3 and S4).
Figure 3.

2D 1H-15N HSQC spectra of indicated W1 5F-Trp-labeled mutants overlaid on wild type (WT) W1. Note that some side chain 1H-15N cross-peaks are aliased differently in W1 WT vs. W1 mutants (WT W1 data were acquired previously using a different field strength and sweep witdth12). Combined chemical shift34 perturbations as a function of position are mapped onto the lowest energy member of each calculated mutant protein structural ensemble, with the thickness of the cylinder representing the degree of 1H-15N HSQC peak displacement. Positions corresponding to cross-peaks that broadened beyond detection under experimental conditions are colored in magenta and ambiguous cross-peaks are colored in yellow.
Fiber formation by 5F-Trp-labeled W2 Trp mutants
The expression level of each double Trp mutant His6-SUMO-W2 fusion protein labeled with 5F-Trp was similar to that of the wild type His6-SUMO-W2 fusion protein in M9 minimal medium (~20–40 mg/L of cell culture), but different degrees of solubility were evident after cell lysis (wild type and F90W: ~80% soluble; R36W, F146W and Y169W: ~20% soluble). Only the soluble fraction of the cell lysate was subjected to immobilized metal affinity purification, allowing selective binding of the hexahistidine (His6) tag of the fusion protein. Cleavage of His6-SUMO from each fusion protein was carried out using SUMO protease, and the purity of each 5F-Trp-labeled W2 mutant following reverse purification to remove the His6-SUMO was >90% (Figure S7). All 5F-Trp-labeled W2 mutants (~0.02 mM) were stable in both acetate and phosphate buffers at 4 °C for at least two weeks.
The ability to manually draw fibers from each W2 double-mutant, with 5F-Trp at the same position in each W unit, was tested in both phosphate13 and acetate12 buffer. Each mutant exhibited similar fiber-forming capability to wild type W2 (Figure 4). Therefore, despite varying degrees of backbone chemical shift perturbation (Figures 3 and S3–S6), implying varying degrees of structural perturbation, each of the tryptophan mutants retains full ability to form fibers.
Figure 4.

Optical micrographs showing manually drawn fibers formed by wild type (WT) and mutant W2 proteins in indicated buffer (scale bars: 20 μm).
19F-NMR properties of W1 mutants
Each 5F-Trp-labeled W1 mutant exhibited a distinct 19F chemical shift, ranging from −123.64 ppm to −126.27 ppm (Figure 5; Table 2). The residue predicted to be most buried, R36W, exhibited the most downfield chemical shift, while the mildly exposed F90W exhibited the most upfield chemical shift. In comparison, 5-fluoroindole under identical conditions exhibited a chemical shift between those of F90W and the next most upfield W1 mutant, Y169W, the 5F-Trp residue predicted to be most solvent exposed (Figure 1 and Table 1). Upon exchange from 10% to 90% D2O, each W1 mutant exhibited an SIS (Figure 5, Table 2; 5-fluoroindole and TFE shown for comparison), with the largest magnitude of SIS being observed for R36W. An upfield SIS was observed for every 19F species except F90W, which exhibited a downfield shift.
Figure 5.
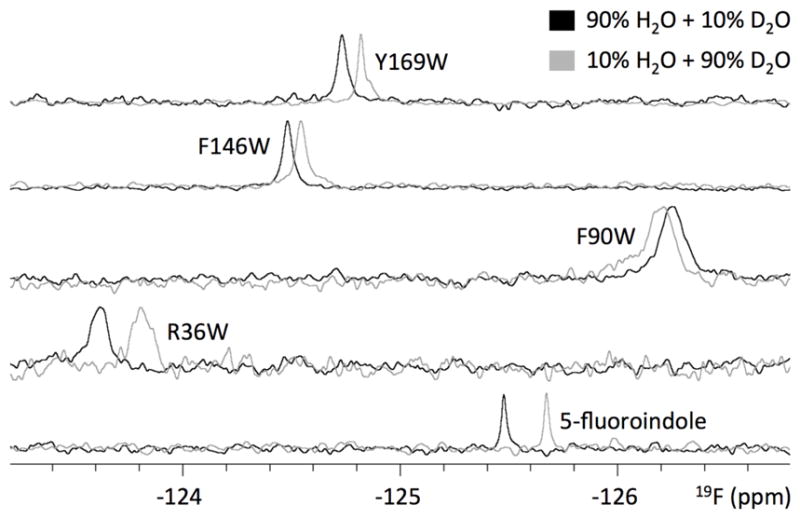
1D 19F-NMR spectra of W1 5F-Trp-labeled mutants and free 5-fluoroindole as observed in indicated H2O/D2O mixture (W1 mutants: 512 scans; 5-fluoroindole: 128 scans).
Table 2.
9F chemical shifts (±0.01 ppm; δH2O in 90%H2O+10%D2O and δD2O in 10%H2O+90%D2O), SIS (Δ(δD2O - δH2O)), T1 relaxation time constant, and linewidth for 5F-Trp-labeled W1 mutants alongside TFE and 5-fluoroindole controls.
| R36W | F90W | F146W | Y169W | TFE | 5-fluoroindole | |
|---|---|---|---|---|---|---|
| δH2O (ppm) | −123.64 | −126.27 | −124.48 | −124.73 | −76.53 | −125.48 |
| δD2O (ppm) | −123.81 | −126.23 | −124.54 | −124.82 | −76.64 | −125.67 |
| SIS (ppm) | −0.17 | +0.04 | −0.06 | −0.09 | −0.11 | −0.19 |
| T1 (s) | 0.64±0.18 | 0.44±0.06 | 0.45±0.05 | 0.61±0.07 | ||
| Linewidth (Hz) | 38.2±0.9 | 44.8±0.9 | 25.3±1.1 | 19.5±0.7 |
Each W1 mutant also exhibited a spin relaxation behavior (Table 2). The T1 arising from CSA (eq. 11, Appendix) was calculated to be 20.6 s. Consistent with the study of protein J by Post et al.,38 this value is ~25–50 times larger than our observed T1 values, implying that T1 is largely dominated here by dipole-dipole interactions (eq. 9, Appendix). Under isotropic tumbling conditions without a contribution from significant internal motion, the T1 values (eq. 2) for buried vs. solvent exposed 5-fluoro moieties were calculated as 0.529 s and 0.507 s, respectively. For comparison, CSA is predicted to dominate for T2 relaxation time constant (eq. 12, Appendix), adding ~11 Hz in linewidth in addition to ~4 Hz contribution from dipole-dipole interactions (eq. 10, Appendix). Quantitative determination of T2 values was not carried out both due to the strong CSA dependence of T2 (values at multiple field strengths would optimally be used to quantify dipolar vs. CSA contributions) and due to instrumental limitations in employing Carr-Purcell-Meiboom-Gill (CPMG) pulse trains for accurate T2 measurement.
Following titration-induced W1 perturbation
Distinct chemical shift and linewidth behavior of each 5F-Trp means that mixtures of W1 mutants are amenable for detailed characterization of titration-induced perturbation. 19F-NMR behavior of the mutant W1 species in mixtures was indistinguishable from that of each individual species, indicating that mutant W1 species did not interact with or affect each other (Figure S8).
Urea-induced W1 denaturation gave rise to distinct behavior at each 5F-Trp position. R36W and F90W exhibited the largest chemical shift changes and, as with the SIS effect (Figure 5), the changes were opposite in sign (Figure 6B). Both of these 19F probes behaved very similarly, with a urea concentration-dependent perturbation in the opposite direction prior to broadening beyond detection before a final, dramatic change in chemical shift (Figure 6C). F146W, conversely, transitioned in a unidirectional manner towards its final state. Each 19F probe converged on the essentially unchanged F169W chemical shift, with a jump to this chemical shift at a distinct urea level (Figure 6C): first R36W, then F146W, and, finally, F90W. Following the jump, each probe exhibited a consistent, narrowed linewidth (Figure S9A for 4 M urea endpoint) while T1 values became indistinguishable from Y169W at all sites at 4 M urea (Figure S9B).
Figure 6.

Titration of wild type W1 followed at indicated backbone amide site by heteronuclear NMR (A, D; combined chemical shift34 perturbation based on our previously reported data12) and of indicated 5F-Trp-labeled mutants followed by 19F NMR, with absolute (B, E) and normalized (C, F) 19F chemical shift changes illustrated (spectra shown in Figure S8). W1-Avg. represents average from all assigned residues (± average deviation). Dashed lines are used to join titration points when signals could not be tracked due to severe line broadening.
DPC-induced 19F transitions were generally unidirectional and, with the exception of R36W, gradual (Figure 6E–F). As with urea, each probe exhibited differences in DPC concentration-dependent perturbation, with R36W affected first, followed by F146W, and, finally, F90W. Although moderately perturbed, F146W became highly similar to the unchanged Y169W chemical shift, while R36W and F90W converged upon a distinct chemical shift that was upfield by ~0.6 ppm (Figure 6E). The R36W signal broadened beyond detection after the first DPC titration point, reappearing and remaining unchanged at its final, upfield shift over 2.5–20 mM DPC (Figure S8). At 1.3 mM DPC (just above the CMC), F146W becomes significantly broadened but its T1 is unchanged; Y169W, conversely, is only slightly broadened and T1 is effectively unchanged (Figure S9). At 20 mM DPC, all three of these 19F probes exhibited decreased linewidth and increased T1 (Figure S9), thus becoming similar to Y169W.
DISCUSSION
Mutation tolerance of W unit
The tolerance to mutation within the W unit, exhibited through minimal structural perturbation in W1 and ability to form fibers by the corresponding W2 concatemers, is remarkable. The gene and, correspondingly, primary structuring of W units within AcSp1 are exceptionally homogeneous, with ~99.9% similarity at the DNA level for the A. trifasciata sequence.8 Even more strikingly, Chaw et al. demonstrated that the ~150 amino acid structured globular domain, annotated on the basis of our chemical shift assignments12,29, exhibits ~84% pairwise identity over three Argiope and one Araneous species.11 Conversely, as might be anticipated from structural constraints, the intrinsically disordered linker region exhibits only ~54% pairwise identity alongside indels leading to variable length.11 Notably, R36, F90 and Y146 all exhibit 100% conservation over these species. It is somewhat surprising, therefore, that the W unit is tolerant to mutation at all of these sites, particularly at R36 given the extreme modification of chemical character in an Arg to Trp substitution. It is certainly possible that one of more of these substitutions would affect the efficiency of fiber spinning or of protein storage at a high concentration in the spider aciniform gland, parameters not detectable in our hand-pulling assay. As a whole, however, the mutation tolerance of the W unit bodes well for rational engineering of modifications in this class of protein.
Y169W behavior
Residue Y169 is highly exposed, with Y169W correspondingly predicted to have 93% SASA. 19F chemical shift and SIS values are fully consistent with this positioning (Table 2). Slow exchange on the NMR time scale is also apparent, with multiple distinct 19F chemical shifts that vary in intensity as a function of conditions (Figure S8). The Y169W spin relaxation behavior is consistent with solvent exposure, exhibiting a narrow linewidth throughout both urea and DPC titrations (perhaps anomalously narrow at 4 M urea in comparison at 5.4 Hz) and a T1 that remains unchanged at 4 M urea (Figure S9). Notably, the T1 of Y169W is longer than anticipated (eq. 2). This would be consistent with relaxation arising from a spectral density with components originating both from molecular tumbling at the global correlation time of 7.9 ns and from internal motion at some other local correlation time (e.g., as described in the Lipari-Szabo model-free formalism39). Assuming a generalized order parameter (S2) of ~0.5–0.6 consistent with a surface exposed Trp side chain and with the heteronuclear NOE (hetNOE) enhancement factor of −0.23 for the 1H-15N backbone amide at Y169,12 a sub-ns effective correlation time would be required to recapitulate this in the model-free approach. However, slow ring flipping is likely giving rise to more complicated motional relationships than can be described by internal motion using the Lipari-Szabo model-free approach, such as the requirement for an exchange contribution.40 In short, Y169W can be considered an internal 19F-NMR control for the other mutants, in that its behavior is generally unperturbed and consistent throughout with a solvent exposed 5F-Trp. This also provides a potentially highly valuable NMR probe for future studies tracking fiber formation and internal conformation by 19F-NMR, since perturbation would be expected upon fibrillogenesis.
R36W behavior
Our previous finding that R36 is positioned in the W1 protein core was unexpected, but very well supported by the NOE restraint network.12 The structural and functional tolerance to Trp mutation implies that basic side chain functionality is not essential, at least in an in vitro fibrillogenesis setting. Consistent with a buried positioning (4% SASA), R36W exhibits the most downfield chemical shift of the four probes. Its linewidth is also ~double that of Y169W, consistent with a buried positioning. However, its T1 is longer than predicted by eq. 2 and comparable to that of Y169W. Presuming an S2 of ~0.8–0.9, consistent with a Trp side chain restricted through its location within in the protein core and consistent with the 1H-15N hetNOE enhancement of 0.62 observed at R36 in W1,12 local motional fluctuations in the sub-ns range are implied under model-free analysis to explain the lengthened T1. Notably, fluctuations of this nature would also lead to a line narrowing relative to eq. 3 - this corresponds nicely to the fact that the less buried F90W probe has a broader linewidth than R36W.
The observed SIS at R36W is anomalous - it exhibits the largest SIS of all four probes, an upfield shift nearly as large as that of free 5-fluoroindole. It is notable that the putative mutant protein structure has two cavities leading from surface to the apparently buried R36W (Figure S10). Correspondingly, a subset of ensemble members exhibits solvent exposure in proximity to the 5F position (Table 1). This region of W1 exhibits atypical 1H, 15N and 13C chemical shifts29 alongside evidence of intermediate exchange dynamics on the NMR time scale.12 This is also consistent with the high frequency motion implied by the 19F spin relaxation behavior of R36W. In the putative structure, the W36 side chain is positioned in close proximity (~4.5–7 Å in many ensemble members) to the side chain of R50 (Figure S10). Although not in an ideal “T-shaped” orientation, a cation-π interaction may still occur between the 6-membered ring of W36 and the guanidino moiety of R50.41 None of the other three 5F-Trp probes are in close proximity to charged moieties. A combination of solvent accessibility through cavities, proximity to a charged group, and/or structural dynamics poorly reflected in the NOE- and chemical shift-derived NMR ensemble of W1 may therefore all contribute to a large SIS value for this buried residue.
Consistent with urea and DPC titrations followed by heteronuclear NMR at the backbone (demonstrated for R36 in Figures 6A, D and for neighboring residues in Figure S11), R36W was the first position to undergo substantial titrant-induced perturbation. In 1H-15N HSQC-based titrations, individual backbone amide cross-peaks could not be tracked beyond loss of ~15% of α-helical character (2.5 M urea).12 While this was sufficient to show greater than average perturbation at R36, it was not possible to follow it through to its final fate upon denaturation. Here, we observed a surprising sensitivity of R36W to urea relative to the backbone data in that the 19F probe underwent severe broadening and was actually restored as a narrow line, at its final chemical shift, before 2.5 M urea. This is illustrative of distinct differences in behavior at the side chain vs. main chain in this region.
F90W behavior
An unusual upfield chemical shift is observed for F90W, falling above even 5-fluoroindole. Notably, its SIS is also opposite in direction to all of the other 5F-Trp probes. Despite these issues, F90W exhibits the broadest line of the four probes, consistent with a relatively buried positioning (30% SASA) and a T1 shorter than predicted by eq. 2. In wild type W1, F90 exhibited well below average perturbation under titration by both urea and DPC (Figures 6A, D and S11). Correspondingly, F90W was the last residue to change in both 19F-NMR titrations. Under urea titration, the jump in chemical shift for F90W occurs in the same concentration regime where the final α-helical character is lost according to CD spectroscopy,12 indicating that this portion of the globular domain is relatively unperturbed until late in the denaturation process. Overall, the titration behavior of F90W is consistent with a buried side chain.
Charged groups in close proximity, such as neighboring carboxyls, have been noted in the literature to cause significant upfield chemical shifts.18 Although electric field-induced 19F chemical shifts for the 5F-Trp and 6F-Trp species may be relatively minimal on a protein surface,42 buried residues experiencing a fixed dipole within the protein interior or residues interacting with solvent may exhibit significant and apparently anomalous chemical shift perturbation.42,43 The relatively high SASA at the C-6 position on the F90W indole ring (Table 1) would support the possibility of solvent-induced changes in shielding. Short T1 values may also be indicative of a strongly bound water molecule in close proximity to the 19F, enhancing dipolar relaxation over and above the effect of the nearest (~2.6 Å) side chain ring proton (i.e., reducing the value of rmin in eq. 9, Appendix). If solvent exposure were the only consideration, however, one would expect an upfield shift when H2O was replaced with D2O rather than the observed downfield shift. Notably, F90W also exhibits the only downfield shift upon denaturation of all four 5F-Trp probes upon titration with either urea or DPC. The most probable explanation for a downfield shift upon solvent exchange or titration is that the 5F moiety is experiencing an electric field-induced chemical shift perturbation from the protein itself with only minor influence from solvent.
F146W behavior
The F146W side chain is predicted to be mostly solvent exposed (71% SASA), with a corresponding upfield chemical shift and narrow linewidth. Its SIS also follows the expected trend of an upfield shift, with a lesser magnitude than that of Y169W. Unlike Y169W, however, slow exchange is not apparent and the observed T1 is shorter than predicted by eq. 2 for a solvent exposed 5F-Trp. This enhanced longitudinal relaxation is consistent with a relatively solvent exposed positioning in a setting which minimizes internal motion, as would be expected in its context in helix 5 of the W1 globular domain.
Under urea titration, F146 in wild type W1 is perturbed at a level slightly above average (Figure 6A), while other residues in helix 5 are significantly more perturbed (Figure S11). Following individual residues in helix 5 through the DPC is challenging due to its Ser-rich sequence (Figure S11), with much of the helix being unassignable at 20 mM DPC even through triple-resonance experiments. Despite this behavior, F146 itself is relatively unperturbed, in keeping with a general trend of the perturbations along helix 5 being maximal for residues in close proximity to the W1 helical bundle and minimal for those, like F146, that are solvent exposed. Under 19F-NMR titration, F146W transitions to its final state at urea and DPC concentrations intermediate to R36W and F90W. Its dynamics are much more notably affected, with a significant line broadening at 1.3 mM DPC and restoration of linewidth at 20 mM DPC. Hence, although the chemical environment at F146W remains relatively consistent throughout titration, its dynamics are quite sensitive to the denaturation of helix 5 observed in heteronuclear experiments.
Urea denaturation vs. DPC perturbation
Comparing and contrasting the two titrations, urea denaturation appears to dramatically affect the folded region of the 5F-Trp-labeled W1 mutant proteins by effecting global denaturation, with 19F chemical shifts demonstrating differences in local solvent exposure/burial over the course of the structural transition. In contrast, DPC-induced perturbation of W1 is gradual, with the exception of R36W. Given that urea is a chaotropic agent while DPC is an amphiphilic lysophospholipid analog, a difference in their denaturing behavior is to be expected.
We presume that the urea-denatured W1 is completely unfolded, as reflected by highly similar 19F chemical shifts and T1 values alongside narrowed lines for all four probes beside a lack of ellipticity arising from α-helical (or β-strand) secondary structuring in CD spectroscopy. Unlike our previous study, use of 19F-NMR allowed us to track denaturation of the protein to completion. Early denaturation of helix 5 (residues 135–149) is consistent with the observed perturbation to R36W. New insight into this process comes from the fact that the 5F-position in the R36W side chain is affected prior to the backbone and transitions to its final chemical shift and linewidth at a point when the backbone is only starting to be perturbed at an above average level. This implies that loosening of helix 5 away from the remainder of the globular bundle occurs even more readily than previously thought.
F90W is located in helix 3 with its side chain packed against helix 1. This residue remains practically unperturbed until near the titration endpoint, implying the late unfolding of this region of W1, corresponding to the final ~20% of helicity according to CD. Although helix 5 denaturation and the resulting perturbation to the residues underneath it (residues 26–39) were apparent previously, the long-lived stability of the helix 1/helix 3 region as well as the perturbation and rearrangement implied in the vicinity of the R36 side chain could not be resolved.
The DPC-induced perturbation to W1 appears much more subtle, in that CD spectra are hardly changed despite the loss of fiber formation ability.12 This appears likely to be due to adsorption of DPC molecules to the W unit based upon the hydrophobicity of the residues and favorable phosphocholine head group-protein moiety interactions. This is consistent with the fact that the two 19F probes from the globular core, R36W and F90W, converge to a chemical shift ~0.6 ppm upfield from that converged upon by the two solvent exposed probes proximal to and in the intrinsically disordered C-terminal tail, F146W and Y169W. Such behavior is also consistent with dodecyl sulfate-ubiquitin complexes characterized by Shaw et al., where detergent molecules exhibited preferential binding to hydrophobic regions with a positive surface potential without the necessity for salt-bridge formation.44 Similar preferential binding of DPC to the W1 globular domain may take place, although the surface potential dependence may not be so clear cut given the zwitterionic nature of the phosphocholine moiety vs. the anionic sulfate moiety.
The role of helix 5 in fiber formation has already been postulated,12 and is fully reflected by the perturbation of R36W and F146W 19F chemical shifts and relaxation behavior over the course of both urea and DPC titrations. In all likelihood, DPC becomes associated with helix 5 residues as part of the mechanism by which fiber formation is prevented. F90W as a probe of helix 3 in the globular domain suggests that although this region is initially unperturbed, it becomes much more perturbed in the later stages of protein denaturation. Taken together, these data are consistent with a fibrillogenesis pathway requiring helix 5 unfolding, whereas loss of the core structuring of the globular domain cannot be accommodated.
Summary
Spider silk proteins are extremely difficult to study at a high resolution. To our knowledge, this work represents the first example of incorporation of 19F probes in this class of protein. Our work suggests that strategic mutagenesis of single residues to tryptophan does not alter the global fold of the W1 protein, nor does it inhibit fiber formation by W2, despite high conservation at these sites in a variety of species. 5F-Trp-labeled W1 mutants exhibited distinct 19F chemical shifts and T1 relaxation times that are reflective of the local environment of each fluorine probe, and are interesting in terms of solvent exposure, local protein-19F interactions, and the effects of side chain dynamics. Correspondingly, perturbation of W1 by addition of urea and DPC, in distinct manner, is reflected in corresponding changes of 19F chemical shifts, T1 relaxation times, and linewidths. 19F probes allow for more precise tracking of transitions in both conformation and dynamics within the W unit as perturbants are added, which in turn provides additional clues as to the nature of the structural transition required for fiber formation.
Supplementary Material
Acknowledgments
Funding Source Statement: This work was supported by Discovery and Research Tools and Instruments Grants from the Natural Sciences and Engineering Research Council of Canada (NSERC; to JKR and XQL); a Leaders Opportunity Fund award from the Canadian Foundation for Innovation (to JKR); and, a Dalhousie Medical Research Foundation Capital Equipment Grant (to JKR and XQL). JKR is supported by a Canadian Institutes for Health Research New Investigator Award; and, KEO was supported by NSERC Undergraduate Student Research Awards and MLT by an NSERC Doctoral Postgraduate Scholarship.
We thank Dr. Mike Lumsden for 11.7 T NMR spectrometer support at the Nuclear Magnetic Resonance Research Resource Facility (NMR3, Dalhousie University), Dr. David Waisman for CD spectropolarimeter access and Dr. Stephen Bearne for comments on the manuscript.
ABBREVIATIONS
- 5F-Trp
5-fluorotryptophan
- AcSp1
aciniform spidroin 1
- CCS
combined chemical shift
- CD
circular dichroism
- CMC
critical micelle concentration
- CSA
chemical shift anisotropy
- dip
dipolar
- DPC
dodecylphosphocholine
- DSS
4,4-dimethyl-4-silapentane-1-sulfonic acid
- HSQC
heteronuclear single quantum coherence
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- PCR
polymerase chain reaction
- RMSD
root-mean-square deviation
- SASA
solvent accessible surface area
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SIS
solvent isotope shift
- SUMO
small ubiquitin-like modifier protein
- T1
longitudinal relaxation time constant
- T2
transverse relaxation time constant
- TFE
2,2,2-trifluoroethanol
- W
AcSp1 (“wrapping” silk) repeat unit
- WT
wild type
APPENDIX
The dipolar and CSA contributions to T1 and T2 (eqs. 3 and 4) were estimated as follows. To calculate the contribution of heteronuclear dipolar coupling to relaxation of the 19F nucleus by the surrounding protons, we followed the formalism of Hull and Sykes,18 as shown to be appropriate for a dilute 19F spin interacting with several 1H nuclei,45 assuming isotropic motion with a rotational correlation time (τc) of 7.9 ns46 the dipolar contributions to T1 and T2 for a spin i (19F) by unlike spins j (1H) are given by:
| (4) |
| (5) |
where γx is a gyromagnetic ratio (γi = 2.5176×104 rad−1·s−1·G−1; γj = 2.6753×104 rad−1·s−1·G−1); ħ is the reduced Planck constant; and, Σrij−6 is the sum over all internuclear distances between spin i and all interacting spins j. F1 and F2 are given by:
| (6) |
| (7) |
where ωi and ωj are the angular frequencies of the given nuclei (ωi = 2π × 470.59 MHz; ωj = 2π × 500.13 MHz) and the spectral density function at angular frequency ω, J(ω), is given by:
| (8) |
To account for the surrounding proton bath,38,47 the sum in eqs. 4 and 5 can be approximated as an integral over a sphere containing protons at a defined density (ρ). T1 and T2 for spin i can then be written as:
| (9) |
| (10) |
where rmin is defined as the distance from the nearest proton neighbor (assumed, unless otherwise noted, to be 2.6 Å, corresponding to the nearest protons at positions 4 and 6 on the 5-fluoroindole ring). The value of ρ for a buried site was taken as 5.73×1022 mL−1 (as for the 29 kDa globular J protein38,48) and that for a solvent-exposed site approximated as 5.99×1022 mL−1 (based on the density of pure H2O at 30 °C49 with a population of 90% 1H).
The contributions to T1 and T2 relaxation for spin i from CSA36,37 are given by (with prefactors appropriately modified to use J(ω) as defined in eq. 8):
| (11) |
| (12) |
where the components of the traceless chemical shift tensor for nucleus i are δx′, δy′, and δz′ with the asymmetry parameter, η, defined as
| (13) |
Values of δz′ = 49.3 ppm and η = −0.397, based on single crystal 19F-NMR analysis of 5-fluoro-DL-tryptophan,50 were employed.
Footnotes
Supporting Information Available.
Table S1: Oligonucleotide primers for site-directed mutagenesis; Table S2: Details pertaining to simulated annealing structure calculations for W1 mutants; Figure S1: W1 protein expression and purification elaborated by SDS-PAGE; Figure S2: 1H-NMR spectra of 5F-Trp mutants; Figures S3–S6: Chemical shift displacements for each W1 mutant relative to WT; Figure S7: SDS-PAGE of purified W2 mutants; Figures S8–S9: 19F-NMR spectra (Figure S8) and relaxation properties (Figure S9) under titration with urea and DPC; Figure S10: R36W positioning; Figure S11: 1H-15N chemical shift displacements for WT W1 in vicinity of each mutant under titration with urea and DPC.
References
- 1.Gosline JM, Guerette PA, Ortlepp CS, Savage KN. The mechanical design of spider silks: From fibroin sequence to mechanical function. J Exp Biol. 1999;202:3295–3303. doi: 10.1242/jeb.202.23.3295. [DOI] [PubMed] [Google Scholar]
- 2.Vollrath F, Porter D. Spider silk as a model biomaterial. Appl Phys A Mater Sci. 2006;82:205–212. [Google Scholar]
- 3.Tokareva O, Jacobsen M, Buehler M, Wong J, Kaplan DL. Structure-function-property-design interplay in biopolymers: Spider silk. Acta Biomater. 2014;10:1612–1626. doi: 10.1016/j.actbio.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pritchard EM, Valentin T, Panilaitis B, Omenetto F, Kaplan DL. Antibiotic-releasing silk biomaterials for infection prevention and treatment. Adv Funct Mater. 2013;23:854–861. doi: 10.1002/adfm.201201636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wendt H, Hillmer A, Reimers K, Kuhbier JW, Schafer-Nolte F, Allmeling C, Kasper C, Vogt PM. Artificial skin--culturing of different skin cell lines for generating an artificial skin substitute on cross-weaved spider silk fibres. PLoS One. 2011;6:e21833. doi: 10.1371/journal.pone.0021833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hennecke K, Redeker J, Kuhbier JW, Strauss S, Allmeling C, Kasper C, Reimers K, Vogt PM. Bundles of spider silk, braided into sutures, resist basic cyclic tests: Potential use for flexor tendon repair. PLoS One. 2013;8:e61100. doi: 10.1371/journal.pone.0061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scheibel T. Spider silks: Recombinant synthesis, assembly, spinning, and engineering of synthetic proteins. Microb Cell Fact. 2004;3:14. doi: 10.1186/1475-2859-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayashi CY, Blackledge TA, Lewis RV. Molecular and mechanical characterization of aciniform silk: Uniformity of iterated sequence modules in a novel member of the spider silk fibroin gene family. Mol Biol Evol. 2004;21:1950–1959. doi: 10.1093/molbev/msh204. [DOI] [PubMed] [Google Scholar]
- 9.Fu C, Shao Z, Fritz V. Animal silks: Their structures, properties and artificial production. Chem Commun. 2009:6515–6529. doi: 10.1039/b911049f. [DOI] [PubMed] [Google Scholar]
- 10.Agnarsson I, Dhinojwala A, Sahni V, Blackledge TA. Spider silk as a novel high performance biomimetic muscle driven by humidity. J Exp Biol. 2009;212:1990–1994. doi: 10.1242/jeb.028282. [DOI] [PubMed] [Google Scholar]
- 11.Chaw RC, Zhao Y, Wei J, Ayoub NA, Allen R, Atrushi K, Hayashi CY. Intragenic homogenization and multiple copies of prey-wrapping silk genes in Argiope garden spiders. BMC Evol Biol. 2014;14:31. doi: 10.1186/1471-2148-14-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tremblay ML, Xu L, Lefevre T, Sarker M, Orrell KE, Leclerc J, Meng Q, Pezolet M, Auger M, Liu XQ, Rainey JK. Spider wrapping silk fibre architecture arising from its modular soluble protein precursor. Sci Rep. 2015;5:11502. doi: 10.1038/srep11502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu L, Rainey JK, Meng Q, Liu XQ. Recombinant minimalist spider wrapping silk proteins capable of native-like fiber formation. PLoS One. 2012;7:e50227. doi: 10.1371/journal.pone.0050227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palladino P, Rossi F, Ragone R. Effective critical micellar concentration of a zwitterionic detergent: A fluorimetric study on n-dodecyl phosphocholine. J Fluoresc. 2010;20:191–196. doi: 10.1007/s10895-009-0537-0. [DOI] [PubMed] [Google Scholar]
- 15.Ge X, MacRaild CA, Devine SM, Debono CO, Wang G, Scammells PJ, Scanlon MJ, Anders RF, Foley M, Norton RS. Ligand-induced conformational change of Plasmodium falciparum AMA1 detected using 19F NMR. J Med Chem. 2014;57:6419–6427. doi: 10.1021/jm500390g. [DOI] [PubMed] [Google Scholar]
- 16.Kitevski-LeBlanc JL, Evanics F, Prosser RS. Approaches for the measurement of solvent exposure in proteins by 19F NMR. J Biomol NMR. 2009;45:255–264. doi: 10.1007/s10858-009-9359-2. [DOI] [PubMed] [Google Scholar]
- 17.Evanics F, Bezsonova I, Marsh J, Kitevski JL, Forman-Kay JD, Prosser RS. Tryptophan solvent exposure in folded and unfolded states of an SH3 domain by 19F and 1H NMR. Biochemistry. 2006;45:14120–14128. doi: 10.1021/bi061389r. [DOI] [PubMed] [Google Scholar]
- 18.Hull WE, Sykes BD. Fluorotyrosine alkaline phosphatase19F nuclear magnetic resonance relaxation times and molecular motion of the individual fluorotyrosines. Biochemistry. 1974;13:3431–3437. doi: 10.1021/bi00714a002. [DOI] [PubMed] [Google Scholar]
- 19.Kitevski-LeBlanc JL, Prosser RS. Current applications of F-19 NMR to studies of protein structure and dynamics. Prog Nucl Magn Reson Spectrosc. 2012;62:1–33. doi: 10.1016/j.pnmrs.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 20.Hull WE, Sykes BD. Fluorine-19 nuclear magnetic resonance study of fluorotyrosine alkaline phosphatase: the influence of zinc on protein structure and a conformational change induced by phosphate binding. Biochemistry. 1976;15:1535–1546. doi: 10.1021/bi00652a027. [DOI] [PubMed] [Google Scholar]
- 21.Asakura T, Suita K, Kameda T, Afonin S, Ulrich AS. Structural role of tyrosine in Bombyx mori silk fibroin, studied by solid-state NMR and molecular mechanics on a model peptide prepared as silk I and II. Magn Reson Chem. 2004;42:258–266. doi: 10.1002/mrc.1337. [DOI] [PubMed] [Google Scholar]
- 22.Crowley PB, Kyne C, Monteith WB. Simple and inexpensive incorporation of 19F-tryptophan for protein NMR spectroscopy. Chem Commun. 2012;48:10681–10683. doi: 10.1039/c2cc35347d. [DOI] [PubMed] [Google Scholar]
- 23.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 24.Schwieters CD, Kuszewski JJ, Clore GM. Using Xplor-NIH for NMR molecular structure determination. Prog Nucl Magn Reson Spectrosc. 2006;48:47–62. [Google Scholar]
- 25.Fraczkiewicz R, Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J Comput Chem. 1998;19:319–333. [Google Scholar]
- 26.Samanta U, Bahadur RP, Chakrabarti P. Quantifying the accessible surface area of protein residues in their local environment. Protein Eng. 2002;15:659–667. doi: 10.1093/protein/15.8.659. [DOI] [PubMed] [Google Scholar]
- 27.Dominy CN, Andrews DW. Site-directed mutagenesis by inverse PCR. Methods Mol Biol. 2003;235:209–223. doi: 10.1385/1-59259-409-3:209. [DOI] [PubMed] [Google Scholar]
- 28.Marley J, Lu M, Bracken C. A method for efficient isotopic labeling of recombinant proteins. J Biomol NMR. 2001;20:71–75. doi: 10.1023/a:1011254402785. [DOI] [PubMed] [Google Scholar]
- 29.Xu L, Tremblay ML, Meng Q, Liu XQ, Rainey JK. 1H, 13C and 15N NMR assignments of the aciniform spidroin (AcSp1) repetitive domain of Argiope trifasciata wrapping silk. Biomol NMR Assign. 2012;6:147–151. doi: 10.1007/s12104-011-9344-z. [DOI] [PubMed] [Google Scholar]
- 30.Hwang TLS, AJ Water suppression that works. Excitation sculpting using arbitrary waveforms and pulsed field gradients. J Magn Reson A. 1995;112:275–279. [Google Scholar]
- 31.Palmer AG, Cavanagh J, Wright PE, Rance M. Sensitivity improvement in proton-detected 2-dimensional heteronuclear correlation NMR spectroscopy. J Magn Reson. 1991;93:151–170. [Google Scholar]
- 32.Harris RK, Becker ED, De Menezes SMC, Granger P, Hoffman RE, Zilm KW. Further conventions for NMR shielding and chemical shifts (IUPAC recommendations 2008) Pure Appl Chem. 2008;80:59–84. doi: 10.1002/mrc.2225. [DOI] [PubMed] [Google Scholar]
- 33.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 34.Schumann FH, Riepl H, Maurer T, Gronwald W, Neidig KP, Kalbitzer HR. Combined chemical shift changes and amino acid specific chemical shift mapping of protein-protein interactions. J Biomol NMR. 2007;39:275–289. doi: 10.1007/s10858-007-9197-z. [DOI] [PubMed] [Google Scholar]
- 35.Solomon I. Relaxation processes in a system of two spins. Phys Rev. 1955;99:559–565. [Google Scholar]
- 36.Abragam A. The Principles of Nuclear Magnetism. Clarendon Press; Oxford, UK: 1961. [Google Scholar]
- 37.Hull WE, Sykes BD. Fluorotyrosine alkaline phosphatase: Internal mobility of individual tyrosines and the role of chemical shift anisotropy as a 19F nuclear spin relaxation mechanism in proteins. J Mol Biol. 1975;98:121–153. doi: 10.1016/s0022-2836(75)80105-7. [DOI] [PubMed] [Google Scholar]
- 38.Post JF, Cottam PF, Simplaceanu V, Ho C. Fluorine-19 nuclear magnetic resonance study of 5-fluorotryptophan-labeled histidine-binding protein J of Salmonella typhimurium. J Mol Biol. 1984;179:729–743. doi: 10.1016/0022-2836(84)90164-5. [DOI] [PubMed] [Google Scholar]
- 39.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 40.Clore GM, Szabo A, Bax A, Kay LE, Driscoll PC, Gronenborn AM. Deviations from the simple 2-parameter model-free approach to the interpretation of N-15 nuclear magnetic relaxation of proteins. J Am Chem Soc. 1990;112:4989–4991. [Google Scholar]
- 41.Gallivan JP, Dougherty DA. Cation-pi interactions in structural biology. Proc Natl Acad Sci U S A. 1999;96:9459–9464. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lian C, Le H, Montez B, Patterson J, Harrell S, Laws D, Matsumura I, Pearson J, Oldfield E. Fluorine-19 nuclear magnetic resonance spectroscopic study of fluorophenylalanine- and fluorotryptophan-labeled avian egg white lysozymes. Biochemistry. 1994;33:5238–5245. doi: 10.1021/bi00183a029. [DOI] [PubMed] [Google Scholar]
- 43.Pearson JG, Oldfield E, Lee FS, Warshel A. Chemical shifts in proteins: A shielding trajectory analysis of the fluorine nuclear magnetic resonance spectrum of the Escherichia coli galactose binding protein using a multipole shielding polarizabilit-local reaction field-molecular dynamics approach. J Am Chem Soc. 1993;115:6851–6862. [Google Scholar]
- 44.Shaw BF, Schneider GF, Arthanari H, Narovlyansky M, Moustakas D, Durazo A, Wagner G, Whitesides GM. Complexes of native ubiquitin and dodecyl sulfate illustrate the nature of hydrophobic and electrostatic interactions in the binding of proteins and surfactants. J Am Chem Soc. 2011;133:17681–17695. doi: 10.1021/ja205735q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hull WE, Sykes BD. Dipolar nuclear spin relaxation of F-19 in multispin systems - Application to F-19 labeled proteins. J Chem Phys. 1975;63:867–880. [Google Scholar]
- 46.Tremblay ML. PhD Thesis. Dalhousie University; Halifax, NS: 2015. [Google Scholar]
- 47.Bloembergen N, Purcell EM, Pound RV. Relaxation effects in nuclear magnetic resonance absorption. Phys Rev. 1948;73:679–712. [Google Scholar]
- 48.Lever JE. Purification and properties of a component of histidine transport in Salmonella typhimurium The histidine-binding protein J. J Biol Chem. 1972;247:4317–4326. [PubMed] [Google Scholar]
- 49.Lide DR, editor. CRC Handbook of Chemistry and Physics. 89. CRC Press; Boca Raton, FL: 2008. [Google Scholar]
- 50.Zhao X, DeVries JS, McDonald R, Sykes BD. Determination of the 19F NMR chemical shielding tensor and crystal structure of 5-fluoro-DL-tryptophan. J Magn Reson. 2007;187:88–96. doi: 10.1016/j.jmr.2007.03.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.