

Abstract
Diabetic macroangiopathy, atherosclerosis secondary to diabetes mellitus (DM), causes cerebro-cardiovascular diseases, which are major causes of death in patients with DM and significantly reduce their quality of life. The alterations in vascular homeostasis due to endothelial and vascular smooth muscle cell dysfunction are the main features of diabetic macroangiopathy. Although multiple metabolic abnormalities that characterize diabetes are involved in the progression of atherosclerosis in patients with DM, it may be said that prolonged exposure to hyperglycemia and insulin resistance clustering with other risk factors such as obesity, arterial hypertension, and dyslipidemia play crucial roles. Laboratory and clinical researches in the past decades have revealed that major biochemical pathways involved in the development of diabetic macroangiopathy are as follows: overproduction of reactive oxygen species, increased formation of advanced glycation end-products (AGEs) and activation of the AGEs-receptor for AGE axis, polyol and hexosamine flux, protein kinase C activation, and chronic vascular inflammation. Among them, oxidative stress is considered to be a key factor.
Keywords: Diabetes, Atherosclerosis, Cardiovascular disease, Oxidative stress, Advanced glycation end-products (AGEs)
Introduction
Diabetes mellitus (DM) causes a lot of complications. Diabetic macroangiopathy, atherosclerosis secondary to DM, especially may cause cerebral vascular disorder, ischemic heart disease, peripheral arterial disease, or other vascular diseases, which are major causes of death in patients with DM and significantly reduce their quality of life1–3).
Although many factors are involved in the progression of atherosclerosis in patients with DM, it may be said, indeed, that the two most important factors are insulin resistance and hyperglycemia4). A large number of basic studies have demonstrated that insulin resistance in vascular cells plays an important role in the progression of atherosclerosis5–7). Insulin resistance in the liver and muscle is not only a major cause of the onset and progression of DM, but also plays a role as a risk factor for the onset and progression of other atherosclerotic conditions such as hypertension and dyslipidemia. Accordingly, patients with insulin resistance often have multiple risk factors that induce the progression of atherosclerosis through various mechanisms8). Patients with insulin resistance may also have obesity or excessive visceral fat, resulting in an abnormal adipocytokine profile9–11). The connection of insulin resistance to DM and atherosclerosis is very complex and involves many factors. As there have been excellent review articles dealing with this subject, the present article does not detail it. The present article describes major mechanisms of how hyperglycemia accelerates the progression of atherosclerosis.
Process of the Development of Atherosclerotic Lesions
Endothelial cells produce and release a variety of bioactive substances to control and maintain the function and structure of intact vessels through balancing between oxidation and anti-oxidation, and inflammation and anti-inflammation in the vascular wall; proliferation and anti-proliferation of vascular smooth muscle cells; dilation and contraction of vessels; and coagulation and fibrinolysis of blood12). Accordingly, increased low-density lipoprotein (LDL) cholesterol levels, hyperglycemia, oxidative stress, and smoking may cause vascular endothelial dysfunction that may result in atherosclerosis12).
It is believed that the formation of atherosclerotic lesions is triggered by a local inflammation in the vascular wall that is induced by dyslipidemia, specifically high LDL-cholesterol levels, and high remnant lipoprotein levels, as well as other various disease factors. The process is considered as follows13–15): (1) Vascular endothelial cells injured by oxidative stress or other factors express adhesion molecules and release cytokines and chemokines. (2) The chemokines attract monocytes from blood circulation to the injured area, and monocytes attach to the endothelium through interaction with adhesion molecules. (3) Monocytes penetrate the subendothelial space, differentiate, and mature into macrophages that release cytokines. When LDL cholesterol levels are high, LDL cholesterol infiltrates the subendothelial space and is retained in the intima where it is oxidized or otherwise modified. (4) Macrophages take up and accumulate oxidized LDL cholesterol, leading to foam cell formation and atherogenesis. (5) Oxidized lipids trigger the secretion of various growth factors by the endothelium. Vascular smooth muscle cells of the media transform and migrate into the intima where they proliferate and actively produce extracellular matrix. These transformed vascular smooth muscle cells also take up oxidized LDL cholesterol and transform to form cells that contribute to atherogenesis. (6) On the other hand, the proliferation of vascular smooth muscle cells and an increase in extracellular matrix may cause intimal thickening and sclerosis (Fig. 1).
Fig. 1.
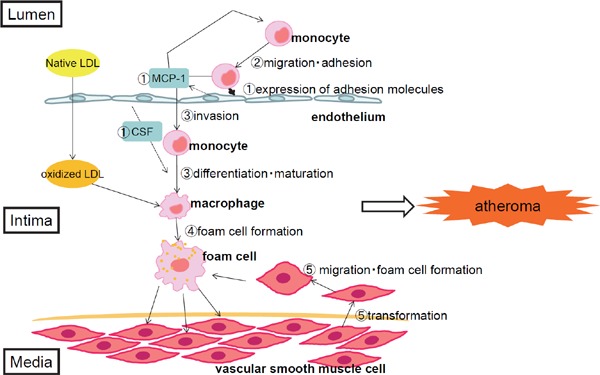
Process of the formation of atherosclerotic lesions.
(1) Vascular endothelial cells injured by oxidative stress or other factors express adhesion molecules and release cytokines and chemokines. (2) The chemokines attract monocytes from blood circulation to the injured area, and monocytes attach to the endothelium through interaction with adhesion molecules. (3) Monocytes penetrate the subendothelial space, differentiate, and mature into macrophages that release cytokines. When LDL cholesterol levels are high, LDL cholesterol infiltrates the subendothelial space and is retained in the intima where it is oxidized or otherwise modified. (4) Macrophages take up and accumulate oxidized LDL cholesterol, leading to foam cell formation and atherogenesis. (5) Oxidized lipids trigger the secretion of various growth factors by the endothelium. Vascular smooth muscle cells of the media transform and migrate into the intima where they proliferate and actively produce extracellular matrix. These transformed vascular smooth muscle cells also take up oxidized LDL cholesterol and transform to form cells that contribute to atherogenesis. (6) On the other hand, the proliferation of vascular smooth muscle cells and an increase in extracellular matrix may cause intimal thickening and sclerosis.
Abbreviations: LDL, low-density lipoprotein; MCP-1, Monocyte chemoattractant protein-1; CSF, colony stimulating factor.
As described above, atherosclerotic lesions are formed through complex interactions of various factors, and DM accelerates all these interactions. It is known that levels of small dense LDL cholesterol are high in patients with DM. Small dense LDL cholesterol particles are very atherogenic as they circulate in the blood at higher levels since they have lower affinity to LDL receptors, have greater propensity for transport into the subendothelial space due to their small size, and are more likely to be oxidized or otherwise degraded due to their low anti-oxidant content16).
Increased Formation of Advanced Glycation End-Products (AGEs) and Activation of the AGEs-RAGE Axis
Reducing sugars such as glucose bind nonenzymatically to proteins in the body. This reaction, called protein glycation, is accelerated when hyperglycemia, oxidative stress, and/or inflammatory reactions are present. In the early stage of protein glycation, the aldehyde group of sugar reacts with amino acids to produce Schiff's base, which undergoes a series of modifications to form Amadori rearrangement products. Hemoglobin A1c (HbA1c) and glycoalbumin are known as major products in the early stage of protein glycation. The early-stage protein glycation products undergo complex reactions, such as oxidation, dehydration, and condensation to form advanced glycation end products (AGEs) that have a dark color, fluorescence, and a molecular cross-linking potential and other features characteristic of aged tissues. AGEs are not a single chemical entity, but a collective term of different substances that share the above-mentioned characteristics.
The findings of the Diabetes Control and Complications Trial in patients with type 1 DM and its follow-up study called the Epidemiology of Diabetes Interventions and Complications Study, as well as the United Kingdom Prospective Diabetes Study in patients with type 2 DM, have indicated that the risk of diabetic vascular complications remained higher in patients receiving conventional treatment in whom blood glucose control was inadequate than those receiving intensive treatment to ensure strict blood glucose control even when the level of blood glucose control no longer differed between the two patient populations after the study17, 18). This persistent increase in the risk of diabetic cardiovascular complications after an exposure to high glucose levels for a certain period of time is called “metabolic memory” or “legacy effect.” As AGEs are formed more frequently at high blood glucose levels and are not easily metabolized, they are accumulated more in those with a longer history of inadequate blood glucose control and accelerate the progression of vascular disorders. The ”metabolic memory” phenomenon is best explained by the formation of AGEs.
The authors determined skin autofluorescence (AF), a measure of skin AGEs levels, in Japanese patients with type 1 DM and their gender- and age-matched healthy individuals to investigate the relationship between skin AF and the risk of diabetic complications. The findings indicated that skin AF values were significantly higher in patients with type 1 DM than healthy controls, and mean HbA1c over the past 10 years was an independent determinant of skin AF values. It was also found that skin AF was an independent risk factor for carotid atherosclerosis19).
AGEs are involved in each step of atherosclerosis. AGEs accelerate the migration of monocytes to the subendothelial space and its transformation to macrophages by promoting the expression of adhesion molecules on endothelial cells. Glycated LDL cholesterols are modified to oxidized LDL cholesterol or AGE-modified LDL cholesterol which are recognized by scavenger receptors on macrophages and lead macrophages to foam cells20). AGEs also stimulate macrophages to release cytokines. AGEs reduce the expression of ATP-binding membrane cassette transporter-A1 (ABCA1) and ABCG1 on monocytes and thereby inhibit reverse cholesterol transport. In addition, AGEs enhance vasoconstriction by increasing endothelin-1 levels, reduce vasodilation by decreasing nitric oxide levels, and stimulate AGE-modification of extracellular matrix to accelerate the progression of atherosclerosis.
As AGEs promote autocrine production of vascular endothelial growth factor (VEGF) and thereby induce pathological neovascularization, AGEs may promote neovascularization in plaques and be involved in the bleeding and instability of plaques. Moreover, AGEs promote clot formation by activating the coagulation system through accelerating the production of tissue factors and by suppressing fibrinolysis through promoting the synthesis of plasminogen activator inhibitor 1 (PAI-1). These findings indicate that AGEs are closely involved in the pathophysiology of atherothrombotic diseases.
AGEs are believed to be involved in the onset and progression of atherosclerosis through multiple mechanisms. One is a direct cytotoxic mechanism through modifications and structural changes of proteins and extracellular matrix by glycation and cross-linking. Another is a mechanism controlling cell response to cell-surface receptors that recognize AGEs as specific ligands. Oxidative stress that occurs during the formation of AGEs is also involved in cell and tissue damage21) (Fig. 2).
Fig. 2.
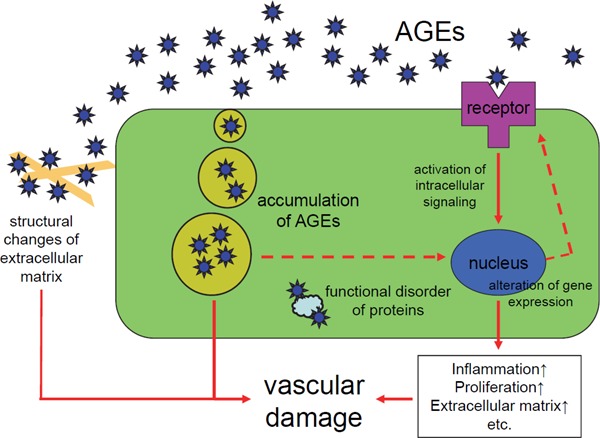
Role of AGEs in the vascular damage.
AGEs are believed to be involved in the onset and progression of atherosclerosis through multiple mechanisms. One is a direct cytotoxic mechanism through modifications and structural changes of proteins and extracellular matrix by glycation and cross-linking. Another is a mechanism controlling cell response to cell-surface receptors that recognize AGEs as specific ligands. Oxidative stress that occurs during the formation of AGEs is also involved in cell and tissue damage.
Abbreviation: AGEs, advanced glycation end-products.
Cell surface receptors that recognize AGEs include the receptor for AGE (RAGE), galectin-3, scavenger receptor class A, CD36, scavenger receptor class B type I, and lectin-like oxidized LDL receptor-1 recognize AGEs22). Among them, the activation of RAGE is an important step of atherogenesis. RAGE is found on the cell surface of endothelial cells, monocytes, macrophages, vascular smooth muscle cells, pericytes, mesangial cells, and other cells that play important roles in the onset and progression of diabetic complications. RAGE expression is especially high on atherosclerotic plaques in patients with DM23–26).
When AGEs bind to RAGE, several intracellular signaling pathways such as extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), p38, and phosphoinositide 3-kinase (PI3K) are activated27) to promote the activity of nuclear factor-κB (NF-κB) and cAMP response element binding protein28). This process is mediated by reactive oxygen species (ROS)29) that are produced in endothelial cells and macrophages through the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase30). AGEs binding to RAGE on endothelial cells induce the expression of adhesion molecules and the secretion of cytokines and growth factors through activating the above-mentioned pathways31, 32). AGEs also stimulate monocytes to induce the production and secretion of various cytokines such as tumor necrosis factor-α (TNF-α) and interleukin (IL)-633). The AGE-RAGE axis also induces the migration and proliferation of vascular smooth muscle cells and the production of extracellular matrix through activating transforming growth factor-β (TGF-β)34, 35).
In a RAGE knockout mouse model, atherosclerosis progresses slowly, and the size of myocardial infarction after experimental coronary artery ligation is small. In studies where soluble RAGE was administered to animals as a ligand decoy, it inhibited the AGE-induced ERK phosphorylation and the expression of VEGF36), reduced the size of atherosclerotic lesions in the aorta of ApoE knockout diabetes mice with DM37), and inhibited the formation of neoendothelium after femoral arterial injury in mice38). These findings indicate an important role of AGEs-RAGE axis in the progression of atherosclerosis and the onset of cardiovascular diseases.
Activation of the Polyol Pathway
The polyol pathway consists of just two steps. The rate-limiting enzyme of the first step is aldose reductase (AR) that requires nicotinamide adenine dinucleotide phosphate reduced form as a coenzyme to reduce glucose to sorbitol. Then sorbitol is oxidized to fructose via sorbitol dehydrogenase and a coenzyme, nicotinamide adenine dinucleotide (NAD).
In tissues where glucose uptake is mediated by insulin-independent glucose transporters such as glucose transporter 1, hyperglycemia is more likely to cause metabolic disorders. Glucose taken into the cell has a high affinity to glucokinase and undergoes extensive phosphorylation, but about 5% of glucose is not phosphorylated, but metabolized directly through the polyol pathway into sorbitol and fructose. However, when blood glucose levels are high, the percentage of glucose metabolized through the polyol pathway increase four- to five-fold.
It is believed that the activation of the polyol pathway induces, directly and indirectly, vascular damage for the following reasons: (1) Fructose and its metabolites, e.g., triose phosphate, methylglyoxal, fructose 3-phosphate, and 3-deoxyglucosone, are potent glycating agents. When their production is accelerated, more AGEs are produced39). (2) In the polyol pathway where NADPH is converted to NADP and NAD is converted to NADH, the depletion of NADPH decreases the production of reduced glutathione, which accelerates oxidative stress. (3) An increase in NADH levels induces an increase in glycerol-3-phosphate levels, which activates protein kinase C (PKC) (Fig. 3).
Fig. 3.
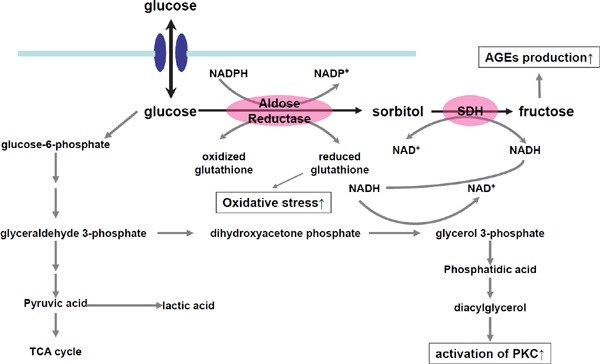
Role of the polyol pathway in the vascular damage.
The activation of the polyol pathway induces vascular damage for the following reasons: (1) Fructose and its metabolites, e.g., triose phosphate, methylglyoxal, fructose 3-phosphate, and 3-deoxyglucosone, are potent glycating agents. When their production is accelerated, more AGEs are produced. (2) In the polyol pathway where NADPH is converted to NADP and NAD is converted to NADH, the depletion of NADPH decreases the production of reduced glutathione, which accelerates oxidative stress. (3) An increase in NADH levels induces an increase in glycerol-3-phosphate levels, which activates PKC.
Abbreviations: SDH, sorbitol dehydrogenase; AGEs, advanced glycation end-products; PKC, protein kinase C.
The importance of activating the polyol pathway in the progression of atherosclerosis has been investigated in animal studies of AR inhibitors. In a study of diabetic ApoE knockout mice, atherosclerosis was accelerated when the polyol pathway was activated via overexpression of human AR (hAR) and reduced when the polyol pathway was blocked by AR inhibitors40). In a study of rats with balloon-injured carotid arteries, AR inhibitors suppressed neointimal production after balloon injury41).
Activation of Protein Kinase C (PKC)
PKC is a serine/threonine kinase activated by calcium and diacylglycerol (DAG), a phospholipid metabolite, among other substances, and plays an important role in intracellular signaling pathways stimulated by different substances including cytokines, growth factors, and vasoactive substances42). PKC has many isoforms that are classified by structure and activation mechanism into classical (or conventional) PKC (cPKC), novel PKC (nPKC), and atypical PKC (aPKC)43).
When blood glucose levels are high, an excessive amount of glucose is taken up into cells, where de novo synthesis of DAG from glucose via the glycolytic system is accelerated. Since in the presence of high glucose levels, the polyol pathway and poly (ADP-ribose) polymerase are activated and the NADH/NAD+ ratio increases, the NAD-dependent glycolysis from glyceraldehyde 3-phosphate (GAP), an aldose, to pyruvic acid is inhibited, while levels of dihydroxyacetone phosphate (DHAP), a ketose, are elevated, which results in an increased production of DAG44). DAG activates cPKC and nPKC. Experiments in diabetic animal models have demonstrated that an increase in DAG levels in the heart, aorta, and renal glomeruli correlate with the activity of cPKC (α, β1, β2) and nPKC (δ, ε)45). Elevated oxidative stress associated with DM is also an important activator for PKCs46).
Findings of studies using diabetic animal models have indicated that some PKC isoforms are activated in different organs such as the aorta, heart, retina, and renal glomeruli, and the type of PKC isoforms activated by oxidative stress differs by cell type. The activation of PKCβ is important in vascular disorders.
PKC activation causes many abnormal changes related to atherosclerosis, such as an increase in vascular permeability47), activation of NADPH oxidase48), endothelial dysfunction, and impaired vasodilation due to decreased NO production49); activation of intracellular signaling pathways, such as Akt, ERK, and p38 MAPK; modified expression of transcription factors, such as early growth response protein 1 (Egr-1), NFκB, and specificity protein 1 (SP1); increased expression of cytokines and growth factors such as VEGF, ICAM-1, ET-1, and PAI-1, among others; apoptosis and increased production of extracellular matrix.
It has been reported that in ApoE knockout mice, the inhibition of PKCβ by LY333531, a PKCβ inhibitor, or knockout of PKCβ gene inhibits the formation of atherosclerotic lesions50).
Elevation of O-GlcNAc Protein Modifications
Glycosylation is a reaction where a carbohydrate molecule is added to a protein or fat. O-linked N-acetylglucosamine (O-GlcNAc) is a form of protein glycosylation where a single sugar, N-acetylglucosamine (GlcNAc), is added to serine and threonine residues on nuclear and cytoplasmic proteins. More than 1,000 proteins are known to be O-GlcNAcylated. As O-GlcNAcylation is known to compete with phosphorylation for the same serine/threonine residues, it is believed to inhibit phosphorylation temporarily. It is thus considered that O-GlcNAcylation contributes to protein–protein associations and the stability of protein complexes through affecting multiple signaling pathways, and that abnormal O-GlcNAcylation may be involved in some diseases, including DM and cancer.
High blood glucose levels lead to high intracellular glucose levels, leading to increased levels of fructose-6-phosphate and its metabolite glucosamine-6-phosphate that is converted by glutamine:fructose-6-phosphate amidotransferase, the rate-limiting enzyme. Glucosamine-6-phosphate is converted to N-acetylglucosamine-6-phosphate (GlcNAc-6-P) and finally to UDP-N-acetylglucosamine (UDP-GlcNAc). O-GlcNAc transferase (OGT) uses UDP-GlcNAc as the substrate for the attachment of N-acetylglucosamine to proteins. Reversely, N-acetylglucosaminidase (O-GlcNAcase or OGA) removes N-acetylglucosamine from O-GlcNAcylated proteins.
These indicate that the progression of diabetic macrovasculopathy relates to O-GlcNAcylation levels. In a study of diabetic ApoE knockout mice, the progression of O-GlcNAcylation in coronary endothelial cells and vascular smooth muscle cells causes decreased levels of NF-κB inhibitory protein A20 to accelerate atherosclerosis51). In another study, coronary endothelial cells isolated from type 1 diabetic mice have low levels of OGA expression and high levels of OGT expression with impaired endothelium-dependent vasodilatation, but OGA overexpression reversed coronary endothelial cell dysfunction52).
O-GlcNAcylation levels are higher in endothelial cells in carotid plaques obtained from patients with DM than those from patients without DM. In human coronary endothelial cells, endothelial nitric oxide synthase (eNOS) is activated by phosphorylation of serine 1177 on eNOS. When blood glucose levels are high, O-GlcNAcylation at eNOS at serine 1177 occurs, which inhibits eNOS activation53, 54). Prolonged increase in O-GlcNAcylation leads to poor myocardial performance55).
Enhancement of Oxidative Stress
An increase in oxidative stress accelerates the progression of atherosclerosis and increases the risk of cardiovascular events by inducing inflammatory reactions, endothelial dysfunction, thrombogenic tendency, plaque instability, and the migration, proliferation, and transformation of smooth muscle cells56). In patients with DM, oxidative stress is elevated due to glucose autoxidation, enhanced glycation, activated AGEs-RAGE axis, enhanced polyol pathway, impaired glutathione redox cycle, and activated PKCs, among others (Fig. 4).
Fig. 4.
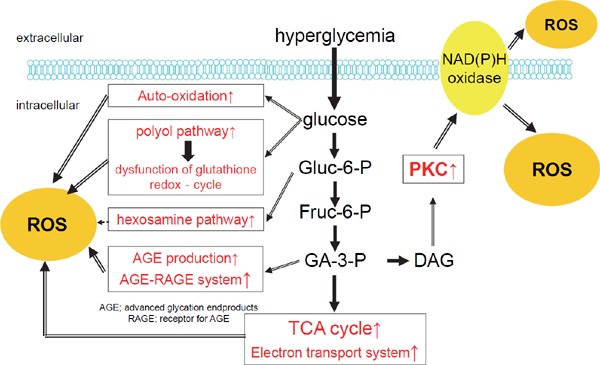
Elevation of oxidative stress in diabetes
In patients with diabetes mellitus, oxidative stress is elevated due to glucose autoxidation, enhanced glycation, activated AGEs-RAGE axis, enhanced polyol pathway, impaired glutathione redox cycle, and activated PKCs, among others.
Abbreviations: AGEs, advanced glycation end-products; PKC, protein kinase C; ROS, reactive oxygen species.
The production of ROS in mitochondria is also increased in patients with DM. Even in normal physiological situations, superoxides are generated as byproducts of oxidative phosphorylation in the mitochondrial electron transport chain, but when blood glucose levels are high, glycolysis is enhanced, which consequently increases the flow of electrons to the mitochondrial electron transport chain and the production of superoxides in cells. As normalizing mitochondrial superoxide may reverse the increased production of AGEs, activated hexosamine pathway, and activated PKC, it is considered that the abnormal intracellular metabolism is closely associated with oxidative stress57).
As mitochondrial DNA is not protected by histones and is located near the inner mitochondrial membrane that contains the enzymes of the electron transfer system, it is susceptible to oxidative damage caused by ROS. Mitochondrial DNA injury decreases the production of ATP and thereby suppresses cellular functions. The processes of mitochondrial fusion, fission, biogenesis, and mitophagy that determine mitochondrial dynamics are important in maintaining appropriate cellular bioenergetics and oxidative stress homeostasis58). An increased mitochondrial fission/fusion ratio causes a decrease in energy production efficiency and an increase in ROS production. In coronary endothelial cells isolated from diabetic mice the expression of mitochondrial fusion promoting genes is decreased while that of mitochondrial fission promoting genes is increased with elevated susceptibility to mitochondrial fragmentation and augmented oxidative stress in cytoplasm and mitochondria. Administration of a superoxide scavenger to diabetic mice led to a decrease in mitochondrial fragmentation through decreasing ROS59).
When nuclear factor E2 related factor 2 (Nrf2), a transcription factor, is activated by ROS or reactive nitrogen species, it enters the nucleus of a cell to modulate the expression of oxidative stress resistance genes such as those encoding glutathione synthesis enzymes, glutathione S-transferase, heme oxygenase-1, and thioredoxin reductase60, 61). Since Nrf2-deficient mice showed enhanced neointimal hyperplasia in a wire injury model, Nrf2 appears to be related to atherosclerosis62).
Nrf2 is regulated by Kelch-like ECH-associated protein 1 (Keap1), a sensor of oxidative stress, and is held inactive by Keap1 under normal cellular conditions. Under oxidative stress conditions, Nrf2 dissociates from Keap1 and promotes the transcription of target genes through anti-oxidant responsive elements (AREs)63). A heterodimer of Nrf2, a basic-region-leucine zipper (bzip) transcription factor, with small Maf binds to AREs to strongly activate gene expression64). Sulforaphane, a substance found in broccoli, blocks the breakdown of Nrf2 through its interaction with Keap165). In diabetic mice, sulforaphane recovered Nrf2 levels in the aorta and the expression of Nrf2-dependent antioxidative genes, and prevented wall hypertrophy, fibrosis, inflammatory reactions, apoptosis, and cell proliferation in the aorta66).
ROS react with components of the body, such as fats, proteins, and nucleic acids to degenerate them. ROS induce erroneous expression of many genes through their direct effects or by promoting AGE production or activating PKCs, which leads to the onset and progression of complications. Genes that are known to be affected by ROS include genes coding (1) catalase and other anti-oxidant enzymes, (2) heme oxygenase-1, metallothionein-1 and other stress-response proteins, and (3) VCAM-1 and other cell adhesion molecules, VEGF, monocyte chemoattractant protein-1 (MCP-1), and other cellular growth factors and cytokines. Under oxidative stress, these genes express themselves with the assistance of activated transcription factors, such as NF-κB, AP-1, and serum response factor (SRF) that are regulated by the intracellular redox status (Fig. 5).
Fig. 5.
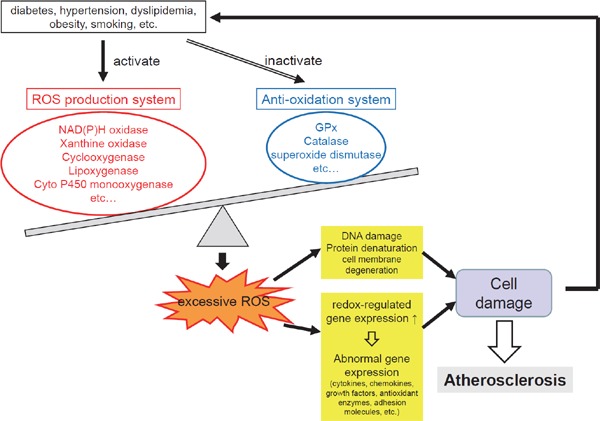
Role of oxidative stress in the vascular damage
The production of ROS is increased in patients with diabetes, hypertension, dyslipidemia, obesity, smoking habit, etc. ROS react with components of the body such as fats, proteins, and nucleic acids to degenerate them. ROS induce erroneous expression of many genes through expression of redox-regulated genes, which leads to the onset and progression of atherosclerosis.
Abbreviation: ROS, reactive oxygen species.
Activation of pro-oncogene under oxidative stress plays an important role in the proliferation of vascular smooth muscle cells. The authors have indicated a possible involvement of the activation of proto-oncogene Pim-1 by oxidative stress in the pathogenesis of atherosclerosis67). PDGF and RAGE/STA3 signaling pathways are upstream of the activation of Pim-168, 69).
Chronic Inflammation
Atherosclerosis is a metabolic disorder and also a chronic inflammatory disorder. Studies have gradually revealed how inflammation develops and persists in arteries.
The relationship between infections and atherosclerosis has been pointed out by epidemiological studies70–74). Substances that induce immune responses and inflammation during infection include pathogen-associated molecular patterns (PAMPs), a group of molecules released by microorganisms in the host, and damage-associated molecular patterns (DAMPs), a group of intrinsic molecules released by damaged tissues or necrosing cells. DAMPs include AGEs, cholesterol, and uric acid. Toll-like receptors found on the surface of macrophages, endothelial cells, and smooth muscle cells recognize PAMPs and DAMPs, and activate NFκB and thereby promote the production of inactive nucleotide-binding oligomerization domain-like receptors 3 (NLRP3) and pro-IL-β75). PAMPs and DAMPs promote the formation of the active NLRP3 inflammasome that consists of NLRP3, an apoptosis-associated speck-like protein containing caspase recruitment domain (ASC), and pro-caspase-1. In the inflammasome, molecules are brought into close proximity, resulting in the activation of pro-caspase-1, which converts pro-IL-β into mature active IL-β to maintain inflammation76). Cells that activated the inflammasome die and release DAMPs, which cause further inflammation.
It has been reported that patients with type 2 DM have high NLRP3 levels in monocytes, an elevated activity of the inflammasome, and high levels of IL-1β and IL-18 in peripheral blood77). Studies have pointed out the progression of atherosclerosis in DM involves inappropriate persistent inflammation induced through excessive inflammasome activation by PAMPs and DAMPs78).
Unlike other cell types, neutrophils release nuclear chromatin into the extracellular space79). This extracellular chromatin, called neutrophil extracellular traps (NETs), stays local to capture bacteria. These sticky “nets” consisting of nucleic acids, proteins, and proteases released from neutrophils catch pathogenic bacteria and never release them. Bacteria trapped by NETs undergo phagocytosis by neutrophils and macrophages and are also killed by NETs that have bactericidal properties80). Neutrophils die immediately after releasing NETs. This process is called NETosis and is attracting attention as a new type of cell death that differs from necrosis and apoptosis81, 82).
NETs is an excellent mechanism to eliminate bacteria but may induce excessive inflammation through the release of intracellular components, e.g., nucleic acids, proteins, and proteases, which act as intrinsic ligands affecting natural immunity and tissue damaging enzymes. In fact, it has been revealed that NETs are also involved in noninfectious conditions, such as chronic inflammation, autoimmune disorders, atherosclerosis, and thrombosis.
NETs promote inflammation through stimulating macrophages to release pro-IL-1β, among other mechanisms. Histones, a component of NETs, induce platelets aggregation, and NETs provide a scaffold for thrombus formation, which induces the formation of platelet thrombi83). Also, neutrophil elastase and cathepsin G, components of NETs, decompose tissue factor pathway inhibitor to promote blood coagulation and thrombus growth84).
Triple-knockout mice that lack ApoE as well as the two major enzymes found in NETs, neutrophil elastase, and proteinase-3, had decreased NETosis, lower IL-1β levels in the blood, and substantially smaller atherosclerotic formation than ApoE-knockout mice had85).
Recent studies have reported that hyperglycemia may promote NETosis86), and levels of NETosis markers are high in patients with type 2 DM87), which suggests the involvement of excessive NETosis in the progression of atherosclerosis in DM.
Summary
Since cardiovascular risk burden is not still totally relieved by intensive glycemic control associated with optimal multifactorial treatment, mechanism-based therapeutic strategies are in highly required. In this context, as described above, a number of basic studies using animal models and cell cultures have conducted, and then, revealed that major biochemical pathways involved in the development of diabetic macroangiopathy are as follows: overproduction of ROS, increased formation of advanced glycation end-products (AGEs) and activation of the AGEs-RAGE axis, polyol and hexosamine flux, PKC activation, and chronic vascular inflammation.
However, these findings from laboratory researches have several limitations that should be recognized. Firstly, atherosclerotic lesions created in rodent models differ substantially from complex lesions found in patients with coronary atherosclerosis, and are similar to foam cell accumulation and fatty streaks that are found in the early stage of atherosclerosis in humans. Secondly, findings from cell cultures have also limitations since crosstalk between many cell types plays an important role in the process of atherogenesis. Thirdly, as immortalized cell lines commonly used in laboratory researches may differ from primary cells in regard to substrates, including glucose, being metabolized, findings found in cell lines may reflect those in primary cells. Thus, although experimental work has helped to unravel some of the principles of atherosclerosis pathophysiology, another investigative dimension, where basic scientists and clinicians communicate closely to provide feedback in both directions, may be required before full understanding of mechanism of the development of atherosclerosis in patients with DM.
Funding Source
None.
Conflict of Interest Disclosures
The author holds an endowed chair (Department of Metabolism and Atherosclerosis) established by funds from Kowa Pharmaceutical Co. has received research funds from MSD and lecture fees from Arkray Co. Ltd., Astellas Pharma Inc., Boehringer Ingelheim, Daiichi Sankyo Inc., Dainippon Sumitomo Pharma Co., Eli Lilly, Kowa Pharmaceutical Co., Kyowa Hakko Kirin Co. Ltd., Mitsubishi Tanabe Pharma Co., MSD, Novo Nordisk Pharma, Ono Pharmaceutical Co., Takeda Pharmaceutical Co., Sanofi-Aventis, and Shionogi & Co.
References
- 1). Stamler J, Vaccaro O, Neaton JD, Wentworth D. Diabetes, other risk factors, and 12-year cardiovascular mortality for men screened in the Multiple Risk Factor Interventional trial. Diabetes Care 1993; 16: 434-444 [DOI] [PubMed] [Google Scholar]
- 2). Haffner SM, Lehto S, Rönnemaa T, PyöräläK Laakso, M Mortality. from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339: 229-234 [DOI] [PubMed] [Google Scholar]
- 3). The Emerging Risk Factors Collaboration Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet 2010; 375: 2215-2222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4). Bornfeldt KE, Tabas I. Insulin Resistance, Hyperglycemia, and Atherosclerosis. Cell Metab 2011; 14: 575-585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5). Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal Relationships Between Insulin Resistance and Endothelial Dysfunction. Molecular and Pathophysiological Mechanisms. Circulation. 2006; 113: 1888-1904 [DOI] [PubMed] [Google Scholar]
- 6). Cubbon RM, Rajwani A, Wheatcroft SB. The impact of insulin resistance on endothelial function, progenitor cells and repair. Diab Vasc Dis Res 2007; 4: 103-111 [DOI] [PubMed] [Google Scholar]
- 7). Liang CP, Han S, Senokuchi T, Tall AR. The Macrophage at the Crossroads of Insulin Resistance and Atherosclerosis. Circ Res 2007; 100: 1546-1555 [DOI] [PubMed] [Google Scholar]
- 8). DeFronzo RA. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: the missing links. The Claude Bernard Lecture 2009. Diabetologia 2010; 53: 1270-1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9). Matsuzawa Y, Shimomura I, Nakamura T, Keno Y, Kotani K, Tokunaga K. Pathophysiology and Pathogenesis of Visceral Fat Obesity. Obes Res 1995; Suppl 2: 187S-194S [DOI] [PubMed] [Google Scholar]
- 10). Hsueh WA, Law R. The central role of fat and effect of peroxisome proliferator-activated receptor-gamma on progression of insulin resistance and cardiovascular disease. Am J Cardiol 2003; 92: 3J-9J [DOI] [PubMed] [Google Scholar]
- 11). Hajer GR, van Haeften TW, Visseren FLJ. Adipose tissue dysfunction in obesity, diabetes, and vascular diseases. European Heart Journal 2008; 29: 2959-2971 [DOI] [PubMed] [Google Scholar]
- 12). Anderson TJ. Assessment and Treatment of Endothelial Dysfunction in Humans. J Am Coll Cardiol 1999; 34: 631-638 [DOI] [PubMed] [Google Scholar]
- 13). Ross R. Atherosclerosis – An inflammatory Disease. N Engl J Med 1999; 340: 115-126 [DOI] [PubMed] [Google Scholar]
- 14). Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation 2002; 105: 1135. [DOI] [PubMed] [Google Scholar]
- 15). Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature 2011; 473: 317-325 [DOI] [PubMed] [Google Scholar]
- 16). Bernesis KK, Krauss RM. Metabolic origins and clinical significance of LDL heterogeneity. J Lipid Res 2002; 43: 1363-1379 [DOI] [PubMed] [Google Scholar]
- 17). Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, Raskin P, Zinman B, Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005; 353: 2643-2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18). Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359: 1577-1589 [DOI] [PubMed] [Google Scholar]
- 19). Osawa S, Katakami N, Kuroda A, Takahara M, Sakamoto F, Kawamori D, Matsuoka TA, Matsuhisa M, Shimomura I. Skin autofluorescence is associated with early-stage atherosclerosis in patients with type 1 diabetes. J Atheroscler Thromb 2017; 24: 312-326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20). Jinnouchi Y, Sano H, Nagai R, Hakamata H, Kodama T, Suzuki H, Yoshida M, Ueda S, Horiuchi S. Glycoaldehide-modified low density lipoprotein leads macrophages to foam cells via the macrophage scavenger receptor. J Biochem 1998; 123: 1208-1217 [DOI] [PubMed] [Google Scholar]
- 21). Mullarkey CJ, Edelstein D, Brownlee M. Free radical generation by early glycation products: a mechanism for accelerated atherogenesis in diabetes. Biochem Biophys Res Commun 1990; 173: 932-939 [DOI] [PubMed] [Google Scholar]
- 22). Schmidt AM, Yan SD, Yan SF, Stern DM. The biology of the receptor for advanced glycation end products and its ligands. Biochem Biophys Acta. 2000; 1498: 99-111 [DOI] [PubMed] [Google Scholar]
- 23). Ritthaler U, Deng Y, Zhang Y, Greten J, Abel M, Sido B, Allenberg J, Otto G, Roth H, Bierhaus A. Expression of receptors for advanced glycation end products in peripheral occlusive vascular disease. Am J Pathol 1995; 146: 688-694 [PMC free article] [PubMed] [Google Scholar]
- 24). Cipollone F, Fazia M, Iezzi A, Zucchelli M, Pini B, De Cesare D, Ucchino S, Spigonardo F, Bajocchi G, Bei R, Muraro R, Artese L, Piattelli A, Chiarelli F, Cuccurullo F, Mezzetti A. Suppression of the functionally coupled cyclooxygenase-2/prostaglandin E synthase as a basis of simvastatin-dependent plaque stabilization in humans. Circulation 2003; 107: 1479-1485 [DOI] [PubMed] [Google Scholar]
- 25). Tanji N, Markowitz GS, Fu C, Kislinger T, Taguchi A, Pischetsrieder M, Stern D, Schmidt AM, D'Agati VD. Expression of advanced glycation end products and their cellular receptorRAGEin diabetic nephropathy and non-diabetic renal disease. J Am Soc Nephrol 2000; 11: 1656-1666 [DOI] [PubMed] [Google Scholar]
- 26). Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, Nowygrod R, Neeper M, Przysiecki C, Shaw A. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol 1993; 143: 1699-1712 [PMC free article] [PubMed] [Google Scholar]
- 27). Yan SF, Ramasamy R, Naka Y, Schmidt AM. Glycation, inflammation, and RAGE. Circ Res 2003; 93: 1159-1169 [DOI] [PubMed] [Google Scholar]
- 28). Kislinger T, Fu C, Huber B, Qu W, Taguchi A, Du Yan S, Hofmann M, Yan SF, Pischetsrieder M, Stern D, Schmidt AM. N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J Biol Chem 1999; 274: 31740-31749 [DOI] [PubMed] [Google Scholar]
- 29). Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS, Pinsky D, Stern D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem 1994; 269: 9889-9897 [PubMed] [Google Scholar]
- 30). Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab 2001; 280: E685-E694 [DOI] [PubMed] [Google Scholar]
- 31). Schmidt AM1, Hori O, Chen JX, Li JF, Crandall J, Zhang J, Cao R, Yan SD, Brett J, Stern D. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest 1995; 96: 1395-1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32). Basta G, Lazzerini G, Massaro M, Simoncini T, Tanganelli P, Fu C, Kislinger T, Stern DM, Schmidt AM, De Caterina R. Advanced glycation end products activate endothelium through signal-transduction receptor RAGE: a mechanism for amplification of inflammatory responses. Circulation 2002; 105: 816-822 [DOI] [PubMed] [Google Scholar]
- 33). Vlassara H., Brownlee M., Manogue K. R., Dinarello C. A., Pasagian A. Cachectin/TNF and IL-, induced by glucose-modified proteins: Role in normal tissue remodeling. Science 1988; 240: 1546-1548 [DOI] [PubMed] [Google Scholar]
- 34). Higashi T, Sano H, Saishoji T, Ikeda K, Jinnouchi Y, Kanzaki T, Morisaki N, Rauvala H, Shichiri M, Horiuchi S. The receptor for advanced glycation end products mediates the chemotaxis of rabbit smooth muscle cells. Diabetes 1997; 46: 463-472 [DOI] [PubMed] [Google Scholar]
- 35). Wolf YG, Rasmussen LM, Ruoslahti E. Antibodies against transforming growth factor-beta 1 suppress intimal hyperplasia in a rat model. J Clin Invest 1994; 93: 1172-1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36). Yonekura H, Yamamoto Y, Sakurai S, Petrova RG, Abedin MJ, Li H, Yasui K, Takeuchi M, Makita Z, Takasawa S, Okamoto H, Watanabe T, Yamamoto H. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem J 2003; 370: 1097-1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37). Bucciarelli LG, Wendt T, Qu W, Lu Y, Lalla E, Rong LL, Goova MT, Moser B, Kislinger T, Lee DC, Kashyap Y, Stern DM, Schmidt AM. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation 2002; 106: 2827-2835 [DOI] [PubMed] [Google Scholar]
- 38). Sakaguchi T1, Yan SF, Yan SD, Belov D, Rong LL, Sousa M, Andrassy M, Marso SP, Duda S, Arnold B, Liliensiek B, Nawroth PP, Stern DM, Schmidt AM, Naka Y. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J Clin Invest 2003; 111: 959-972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39). Tsukushi S, Katsuzaki T, Aoyama I, Takayama F, Miyazaki T, Shimokata K, Niwa T. Increased erythrocyte 3-DG and AGEs in diabetic hemodialysis patients: role of the polyol pathway. Kidney Int 1999; 55: 1970-1976 [DOI] [PubMed] [Google Scholar]
- 40). Vedantham S, Noh H, Ananthakrishnan R, Son N, Hallam K, Hu Y, Yu S, Shen X, Rosario R, Lu Y, Ravindranath T, Drosatos K, Huggins LA, Schmidt AM, Goldberg IJ, Ramasamy R. Human aldose reductase expression accelerates atherosclerosis in diabetic apolipoprotein E-/- mice. Arterioscler Thromb Vasc Biol. 2011; 31: 1805-1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41). Ruef J, Liu SQ, Bode C, Tocchi M, Srivastava S, Runge MS, Bhatnagar A. Involvement of aldose reductase in vascular smooth muscle cell growth and lesion formation after arterial injury. Arterioscler Thromb Vasc Biol 2000; 20: 1745-1752 [DOI] [PubMed] [Google Scholar]
- 42). Newton AC. Regulation of the abc kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003; 370: 361-371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43). Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008; 88: 1341-1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44). Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes 1998; 47 859-866 [DOI] [PubMed] [Google Scholar]
- 45). Schmitz-Peiffer C, Biden TJ. Protein kinase C function in muscle, liver, and beta-cells and its therapeutic implications for type 2 diabetes. Diabetes 2008; 57: 1774-1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46). Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000; 404: 787-790 [DOI] [PubMed] [Google Scholar]
- 47). Lynch JJ, Ferro TJ, Blumenstock FA, Brockenauer AM, Malik AB. Increased endothelial albumin permeability mediated by protein kinase C activation. J Clin Invest. 1990; 85: 1991-1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48). Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000; 49: 1939-1945 [DOI] [PubMed] [Google Scholar]
- 49). Cosentino F, Eto M, De Paolis P, van der Loo B, Bachschmid M, Ullrich V, Kouroedov A, Delli Gatti C, Joch H, Volpe M, Luscher TF. High glucose causes upregulation of cyclooxygenase-2 and alters prostanoid profile in human endothelial cells: role of protein kinase C and reactive oxygen species. Circulation. 2003; 107: 1017-1023 [DOI] [PubMed] [Google Scholar]
- 50). Harja E, Chang JS, Lu Y, Leitges M, Zou YS, Schmidt AM, Yan SF. Mice deficient in PKCbeta and apolipoprotein E display decreased atherosclerosis. FASEB J. 2009; 23: 1081-1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51). Shrikhande GV, Scali ST, da Silva CG, Damrauer SM, Csizmadia E, Putheti P, Matthey M, Arjoon R, Patel R, Siracuse JJ, Maccariello ER, Andersen ND, Monahan T, Peterson C, Essayagh S, Studer P, Guedes RP, Kocher O, Usheva A, Veves A, Kaczmarek E, Ferran C. O-glycosylation regulates ubiquitination and degradation of the antiinflammatory protein A20 to accelerate atherosclerosis in diabetic ApoE-null mice. PLoS One. 2010; 5: e14240. 10.1371/journal.pone.0014240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52). Makino A, Dai A, Han Y, Youssef KD, Wang W, Donthamsetty R, Scott BT, Wang H, Dillmann WH. O-GlcNAcase overexpression reverses coronary endothelial cell dysfunction in type 1 diabetic mice. Am J Physiol Cell Physiol. 2015; 309: C593-C599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53). Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest. 2001; 108: 1341-1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54). Federici M, Menghini R, Mauriello A, Hribal ML, Ferrelli F, Lauro D, Sbraccia P, Spagnoli LG, Sesti G, Lauro R. Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation. 2002; 106: 466-472 [DOI] [PubMed] [Google Scholar]
- 55). Clark RJ, McDonough PM, Swanson E, Trost SU, Suzuki M, Fukuda M, Dillmann WH. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J Biol Chem 2003; 278: 44230-44237 [DOI] [PubMed] [Google Scholar]
- 56). Giugliano D, Ceriello A, Paolisso G. Oxidative stress and diabetic vascular complications. Diabetes Care 1996; 19: 257-267 [DOI] [PubMed] [Google Scholar]
- 57). Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000; 404: 787-790 [DOI] [PubMed] [Google Scholar]
- 58). Vasquez-Trincado C, Garcia-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA, Lavandero S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol. 2016; 594: 509-525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59). Makino A, Scott BT, Dillmann WH. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia. 2010; 53: 1783-1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60). Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med 2004; 10: 549-557 [DOI] [PubMed] [Google Scholar]
- 61). Chen XL, Kunsch C: Induction of cytoprotective genes through Nrf2/antioxidant response element pathway: A new therapeutic approach for the treatment of inflammatory diseases. Curr Pharm Des 2004; 10: 879-891 [DOI] [PubMed] [Google Scholar]
- 62). Ashino T, Yamamoto M, Yoshida T, Numazawa S. Redox-sensitive transcription factor Nrf2 regulates vascular smooth muscle cell migration and neointimal hyperplasia. Arterioscler Thromb Vasc Biol 2013; 33: 760-768 [DOI] [PubMed] [Google Scholar]
- 63). Itoh K, Wakabayashi N, Katoh Y, Ishii K, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the aminoterminal Neh2 domain. Genes Dev 1999; 13: 76-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64). Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 1997; 236: 313-322 [DOI] [PubMed] [Google Scholar]
- 65). Xue M, Qian Q, Adaikalakoteswari A, Rabbani N, Babaei-Jadidi R, Thornalley PJ. Activation of NF-E2-related factor-2 reverses biochemical dysfunction of endothelial cells induced by hyperglycemia linked to vascular disease. Diabetes. 2008; 57: 2809-2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66). Wang Y, Zhang Z, Sun W, Tan Y, Liu Y, Zheng Y, Liu Q, Cai L, Sun J. Sulforaphane attenuation of type 2 diabetes-induced aortic damage was associated with the upregulation of Nrf2 expression and function. Oxid Med Cell Longev. 2014; 2014: 123963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67). Katakami N, Kaneto H, Hao H, Umayahara Y, Fujitani Y, Sakamoto K, Gorogawa S, Yasuda T, Kawamori D, Kajimoto Y, Matsuhisa M, Yutani C, Hori M, Yamasaki Y. Role of pim-1 in smooth muscle cell proliferation. J Biol Chem 2004; 279: 54742-54749 [DOI] [PubMed] [Google Scholar]
- 68). Willert M, Augstein A, Poitz DM, Schmeisser A, Strasser RH, Braun-Dullaeus RC. Transcriptional regulation of Pim-1 kinase in vascular smooth muscle cells and its role for proliferation. Basic Res Cardiol 2010; 105: 267-277 [DOI] [PubMed] [Google Scholar]
- 69). Meloche J, Paulin R, Courboulin A, Lambert C, Barrier M, Bonnet P, Bisserier M, Roy M, Sussman MA, Agharazii M, Bonnet S. RAGE-dependent activation of the oncoprotein Pim1 plays a critical role in systemic vascular remodeling processes. Arterioscler Thromb Vasc Biol 2011; 31: 2114-2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70). Melnick JL, Adam E, Debakey ME. Possible role of cytomegalovirus in atherogenesis. JAMA 1990; 263: 2204-2207 [PubMed] [Google Scholar]
- 71). Chiu B, Viira E, Tucker E, Fong I. W. Chlamydia pneumoniae, cytomegalovirus, and herpes simplex virus in atherosclerosis of the carotid artery. Circulation 1997; 96: 2144-2148 [DOI] [PubMed] [Google Scholar]
- 72). Kuo CC, Campbell LA, Grayston JT. Is infection with Chlamydia pneumoniae a causative agent in atherosclerosis? Mol Med Today. 1998; 4: 426-430 [DOI] [PubMed] [Google Scholar]
- 73). Cook PJ, Honeybourne D, Lip GYH, Beevers G, Wise R, Davies P. Chlamydia pneumoniae antibody titers are significantly associated with acute stroke and transient cerebral ischemia: the West Birmingham Stroke Project. Stroke 1998; 29: 404-410 [DOI] [PubMed] [Google Scholar]
- 74). Kiechl S, Egger G, Mayr M, Wiedermann CJ, Bonora E, Oberhollenzer F, Muggeo M, Xu Q, Wick G, Poewe W, Willeit J. Chronic infections and the risk of carotid atherosclerosis: prospective results from a large population study. Circulation 2001; 103: 1064-1070 [DOI] [PubMed] [Google Scholar]
- 75). Rubartelli A, Lotze MT. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends in Immunology 2007; 28: 429-436 [DOI] [PubMed] [Google Scholar]
- 76). Martinon F, Burns K. Tschopp. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-1β. Mollecular Cell 2002; 10; 417-426 [DOI] [PubMed] [Google Scholar]
- 77). Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013; 62: 194-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78). Zimmer S, Grebe A, Latz E. Danger signaling in atherosclerosis. Circ Res 2015; 116: 323-340 [DOI] [PubMed] [Google Scholar]
- 79). Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science 2004; 303: 1532-1535 [DOI] [PubMed] [Google Scholar]
- 80). Parker H, Albrett AM, Kettle AJ, Winterbourn CC. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J Leukoc Biol 2012; 91: 369-376 [DOI] [PubMed] [Google Scholar]
- 81). Steinberg BE, Grinstein S. Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci STKE 2007; 379: 11. [DOI] [PubMed] [Google Scholar]
- 82). Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 2007; 176: 231-241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83). Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA 2010; 107: 15880-15885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84). Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, Brinkmann V, Lorenz M, Bidzhekov K, Khandagale AB, Konrad I, Kennerknecht E, Reges K, Holdenrieder S, Braun S, Reinhardt C, Spannagl M, Preissner KT, Engelmann B. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med 2010; 16: 887-896 [DOI] [PubMed] [Google Scholar]
- 85). Nahrendorf M, Swirski FK. Immunology. Neutrophil-macrophage communication in inflammation and atherosclerosis. Science. 2015; 349: 237-238 [DOI] [PubMed] [Google Scholar]
- 86). Joshi MB, Lad A, Bharath Prasad AS, Balakrishnan A, Ramachandra L, Satyamoorthy K. High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS letters 2013; 587: 2241-2246 [DOI] [PubMed] [Google Scholar]
- 87). Menegazzo L, Ciciliot S, Poncina N, Mazzucato M, Persano M, Bonora B, Albiero M, Vigili de Kreutzenberg S, Avogaro A, Fadini GP. NETosis is induced by high glucose and associated with type 2 diabetes. Acta diabetologica 2015; 52: 497-503 [DOI] [PubMed] [Google Scholar]