

Abstract
Recent reports reveal increasing complexity of mechanisms underlying the bone sparing effects of sex steroids. This review focuses on mechanisms by which sex steroids attenuate endocortical and trabecular adult bone turnover, perhaps their most important property as bone mass regulators. Clearly, estrogen withdrawal increases osteoclast number and bone resorption; however, important open questions are the extent to which osteoblasts and their precursors are involved, and the relative contributions of the RANK/RANKL/OPG system, Fas ligand and Runx2. In addition to reviewing these aspects of estrogen action, we also discuss proskeletal effects of androgens on the adult male skeleton, including aromatization to estrogens and male-specific mechanisms. Detailed understanding of skeletal site- and gender-dependent mechanisms by which sex steroids protect the adult skeleton will provide the foundation for improved risk assessment, prevention and management of osteoporosis.
After decades of work on the bone sparing effects of sex steroids, discoveries are still continuously made, which provide new insight into the underlying mechanisms and offer opportunities for the development of therapeutic approaches to hypogonadism-induced osteoporosis. Here, we review and put in context recent findings regarding the bone-sparing effects of estrogens and androgens. We limit the review to adult trabecular and endocortical bone turnover, and do not discuss mechanisms by which sex steroids regulate longitudinal bone growth (reviewed in Vanderschueren et al., 2004) or periosteal expansion (reviewed in Vanderschueren et al., 2006). We also limit the discussion to cells of the osteoblast and osteoclast lineages, although other cell types, in particular immune cells, are also involved in the proskeletal properties of sex steroids (reviewed in Lorenzo et al., 2008).
Estrogens Attenuate Bone Turnover
Maintenance of bone tissue by estrogens has been attributed to both their anti-resorptive and anabolic effects (Riggs et al., 2002; Syed and Khosla, 2005). However, loss of bone mass in the first few years after menopause is associated not only with increased bone resorption but also with increased – not decreased – bone formation (Riggs et al., 2002; Syed and Khosla, 2005). Although estrogens have been shown to promote osteoblast commitment and survival (Khosla, 2008), multiple studies with women and animal models of postmenopausal osteoporosis documented increases in both bone resorption and bone formation based on serum and urinary markers as well as direct histological assessment of bone turnover (Gorai et al., 1997; Weitzmann et al., 2002; Eghbali-Fatourechi et al., 2003; Usui et al., 2004). Thus, the present review focuses on the attenuation of bone turnover by estrogens and the increased bone turnover that follows menopause.
A critical question remains: which are the primary and which are the secondary effects of estrogens in keeping bone turnover in check? Do estrogens primarily restrain proliferation and differentiation of cells in the monocyte/osteoclast lineage, with osteoblastic cells following suit via coupling mechanisms? Or, do estrogens primarily restrain proliferation and differentiation of cells in the osteoblast lineage, with attenuation of monocytes/osteoclasts as a secondary effect? A related but distinct issue is the identification of the cell type(s) in which estrogens activate estrogen receptor α (ERα)—the main receptor involved in their proskeletal properties (Lindberg et al., 2002; Sims et al., 2003)—and the downstream molecular events that ultimately restrain bone turnover and preserve bone mass.
Estrogens Attenuate Bone Turnover Via Regulation of Cell Proliferation in the Osteoblast Lineage
Although estrogen withdrawal clearly results in increased osteoclast number and accelerated bone resorption, early studies by Manolagas and colleagues (Jilka et al., 1998; Di Gregorio et al., 2001) suggested that the anti-resorptive action of estrogens was rooted in cells of the osteoblast lineage. Fibroblast and osteoblast colony formation assays using bone marrow stromal cells from ovariectomized (OVX) mice and mice receiving estradiol after OVX, suggested that the anti-resorptive effect of estrogens was mediated by attenuating cell proliferation early in the osteoblast lineage (Jilka et al., 1998; Di Gregorio et al., 2001). Furthermore, the effect of estrogen withdrawal on the marrow stromal cells persisted under conditions that suppressed osteoclastogenesis (Jilka et al., 1998). Thus, although bone multicellular units (BMU) initiate the microanatomical cycle of bone turnover with osteoclastic bone resorption, the regulation of bone turnover was assigned to osteoblast progenitors, with estrogens keeping the regulatory process in check. According to this concept, loss of estrogen unleashes the otherwise restrained proliferation and differentiation of mesenchymal progenitors, and the stimulated mesenchymal cells go on to fuel excessive bone turnover, a major contributor to bone loss under these conditions.
Estrogens Attenuate Osteoclast Differentiation and Survival Via Effects on Mesenchymal Cells
Within the osteoblast lineage, estrogens not only attenuate proliferation of early osteoblast progenitors (Jilka et al., 1998), but also influence cell-cell communications by which mesenchymal cells regulate osteoclastogenesis and bone resorption. Below we describe estrogen-responsive molecular mechanisms that regulate osteoclast differentiation and survival via osteoblasts and their precursors.
FASL
An important means by which mesenchymal cells regulate bone resorption is the production of Fas Ligand (FASL), which in turn binds to pro-apoptotic Fas receptors on osteoclasts. Treatment of osteoblasts with estradiol resulted in increased FASL expression and accelerated apoptosis of co-cultured osteoclasts (Krum et al., 2008). The enhancement of FASL expression was mediated by binding of ERα to an enhancer located 86-kb downstream of the FASL transcription start site (Krum et al., 2008). Earlier studies suggested that estrogens stimulated osteoclast apoptosis in a cell autonomous manner (Kameda et al., 1997) and that this occurred via stimulation of FASL expression in the osteoclast itself (Nakamura et al., 2007; Imai et al., 2009). Whereas a role for FASL is undisputed, the work described above (Krum et al., 2008) argues that mesenchymal cells are required, based on the inability of pure osteoclast cultures to respond to estrogens. Either way, an important mechanism underlying the proskeletal effect of estrogens has been established: they stimulate FASL expression (likely in mesenchymal cells) leading to promotion of osteoclast apoptosis, thereby limiting the number and thus the activity of osteoclasts. Indeed, estradiol did not induce osteoclast apoptosis in vitro in the presence of FASL neutralizing antibody, or when the osteoclasts were derived from mice with defective FASL signaling (Krum et al., 2008). Furthermore, such mice did not suffer bone loss after ovariectomy (Nakamura et al., 2007).
RANKL and OPG
Whereas Fas/FASL is the pathway most recently implicated in the bone-sparing effect of estrogens, previous studies provided ample evidence for the involvement of osteoblastic RANKL and its osteoclastic receptor RANK, activation of which is critical for osteoclast differentiation and function. Most notably, estradiol attenuates RANK/RANKL signaling by strongly stimulating OPG biosynthesis in osteoblasts (Hofbauer et al., 1999a). In addition, postmenopausal women have increased RANKL expression in pre-osteoblasts (as well as T- and B-cells) as compared either to premenopausal women or postmenopausal women on estrogen therapy (Eghbali-Fatourechi et al., 2003). Inhibition of osteoclastogenesis by estrogens was earlier attributed to reduced expression of proinflammatory cytokines including IL-1, IL-6, TNFα, M-CSF and PGE2, some of which function upstream of RANK/RANKL (Pacifici, 1996; Manolagas, 2000).
Runx2
Runx2 is the master regulator of osteoblast differentiation and bone formation (Ducy et al., 1997; Komori et al., 1997; Otto et al., 1997). Besides this classical role, Runx2 also promotes osteoblast-mediated osteoclastogenesis and bone resorption through stimulation of genes such as M-CSF, RANKL and MMP-13, (Liu et al., 2001; Geoffroy et al., 2002; Enomoto et al., 2003; Maruyama et al., 2007). It appears, therefore, that Runx2 activity has to be well regulated in order to support normal bone metabolism; sufficient Runx2 activity is necessary to promote bone formation, but this activity should not exceed certain limits (Fig. 1). Indeed, the activity of Runx2 is tightly regulated by a panel of co-repressors (Westendorf, 2006). A recent addition to this panel is ERα (Khalid et al., 2008). Thus, estrogens plausibly attenuate bone turnover, at least in part, by keeping Runx2 activity in check. At menopause, unleashing of Runx2 would then contribute to excessive bone resorption and loss of bone mass (Fig. 1).
Fig. 1.
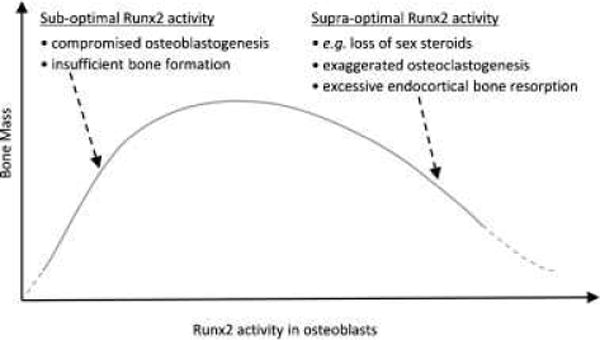
Proposed model for a biphasic relationship between bone mass and Runx2 activity.
Given its classical role in osteoblast differentiation and bone formation (Komori et al., 1997; Otto et al., 1997), the initial finding of increased bone resorption in mice whose osteoblasts overexpress Runx2 was unexpected (Liu et al., 2001). Initially, the increased bone resorption was attributed to abnormal composition of the bone matrix produced by the Runx2-overexpressing osteoblasts (Liu et al., 2001). However, a similar study published soon thereafter, with similar in vivo results, was accompanied by co-culture assays, which demonstrated increased ex vivo osteoclastogenic activity by Runx2-overexpressing osteoblasts (Geoffroy et al., 2002). This was the first clear demonstration of stimulated osteoclastogenesis by osteoblastic Runx2. Runx2-mediated osteoclastogenesis in a coculture assay, was demonstrated again more recently by viral transduction of osteoblasts with Runx2 (Maruyama et al., 2007). Moreover, these authors also over-expressed a Runx2 dominant negative (DN) form in osteoblasts, which resulted in a mirror effect with respect to wild type Runx2, namely reduced osteoclastogenesis in co-culture assays and reduced osteoclast number and increased bone mass in vivo (Maruyama et al., 2007). The investigators went on to show that overexpression of the Runx2-DN form rendered mice resistant to OVX-induced high turnover bone loss (Maruyama et al., 2007). Physiologically, estrogens may fulfill a similar function by activating ERα, which then directly interacts with and inhibits Runx2 (Khalid et al., 2008).
In summary, we discussed two concepts by which cells of the osteoblast lineage mediate proskeletal effects of estrogens: (1) estrogens restrain proliferation of early osteoblast progenitors, hence estrogen withdrawal results in accelerated proliferation of these mesenchymal cells, which in turn fuels excessive bone turnover; and (2) estrogens restrain signals emanating from cells of the osteoblast lineage that promote osteoclastogenesis and increase the osteoclast lifespan; therefore, after menopause the same number of osteoblast progenitors will more efficiently promote osteoclast formation and survival. Both these mechanisms (which are not mutually exclusive) ascribe the antiresorptive property of estrogens to ER signaling within osteoblasts and their progenitors.
Proskeletal Properties of Estrogens Require ERα Signaling in Osteoclasts
Female mice with osteoclast-specific ablation of ERα have a high bone turnover and low trabecular bone mass (Nakamura et al., 2007). This seminal report introduced a paradigm shift by ascribing the proskeletal effect of estrogens, at least in part, to cells in the osteoclast lineage. The osteoclast-specific ERα knockout mice do not suffer further trabecular bone loss at least until 2 weeks after OVX (but they do later), and they do not benefit from estrogen replacement (Nakamura et al., 2007; Millan et al., 2009). Interestingly, cortical bone in female and both trabecular and cortical bone in male mice lacking osteoclastic ERα were not affected by estrogen deprivation (see below).
In osteoclasts, activated ERα attenuates cell proliferation and thus bone resorption through several possible mechanisms. As described above, it is still unclear to what extent transcriptional activation of FASL is responsible for a cell autonomous pro-apoptotic activity of estradiol in osteoclasts (Nakamura et al., 2007; Krum et al., 2008; Imai et al., 2010). Other, yet unknown target genes with ERE-containing promoters and enhancers may be involved. Alternatively, ERα may attenuate expression of genes in osteoclasts in a non-classical fashion, for example by inhibiting NFκB (Ray et al., 1994), or by antagonizing JNK activity, thus limiting RANKL-mediated activation of AP-1 target genes (Srivastava et al., 2001). Be that as it may, the unexpected outcomes of osteoclast-specific ERα gene ablation (Nakamura et al., 2007) certainly set a novel concept in the field: ERα signaling in osteoclasts is involved in the proskeletal property of estrogens in females.
The landmark discovery of a requirement for ERα signaling in osteoclasts raised the question of whether the new paradigm should replace the old one. In other words, how complete is the evidence assigning the proskeletal role of estrogens to ERα signaling in cells of the osteoblast lineage? To date, only meeting abstracts have reported adverse effects of ERα ablation specifically in osteoblasts and their precursors (Lagerquist et al., 2008; Kondoh et al., 2009). Completion of these and other studies may lead to a unitary picture, whereby proskeletal properties of estrogens are assigned to ERα signaling in cells of both the osteoblast and the osteoclast lineages.
Androgens Protect the Skeleton Via Aromatization to Estrogens
The impact of androgens on bone turnover has been attributed to both aromatization-dependent effects mediated by ERα and aromatization-independent effects mediated by the Androgen Receptor (AR). The view ascribing bone-sparing effects of sex hormones to ER activation in females and AR activation in males was shattered with the landmark report of osteoporosis in a man with a mutation in the ERα gene (Smith et al., 1994) and the similar phenotype later described in men with mutations in the CYP19 gene (Bouillon et al., 2004; Gennari et al., 2004; Rochira et al., 2007; Lanfranco et al., 2008), whose product, aromatase, is the enzyme responsible for converting testosterone to estradiol. Men deficient in aromatase activity respond well to estrogen treatment, with suppression of bone resorption and increased bone mass (Carani et al., 1997; Bilezikian et al., 1998), whereas treatment with androgens has little to no effects (Carani et al., 1997). Similar to the clinical cases, male mice deficient in aromatase (ArKO) display a high bone turnover and low bone mass phenotype, which can be corrected by administration of estradiol (Oz et al., 2000; Miyaura et al., 2001). Both the “experiments of nature” and those with the ArKO mice highlight the aromatase-dependent proskeletal action of androgens.
Two groups investigated the role of aromatization in male bone turnover by experimentally treating healthy men with combinations of a GnRH analog (to induce hypogonadism) along with sex steroids and an aromatase inhibitor, thereby selectively restoring AR or ER signaling (Falahati-Nini et al., 2000; Leder et al., 2003). Based on serum and urinary markers of bone resorption and formation, both studies demonstrated that bone metabolism in the hypogonadal men was corrected to the highest degree when both androgen and estrogen signaling were restored. These results suggest that androgens preserve bone mass in adult men by activation of both AR and ERα. However, there were differences between the two studies, for example, the subjects’ age, which resulted in closer-to-normal bone metabolism with restoration of estrogen signaling in the study employing older men (Falahati-Nini et al., 2000) versus closer-to-normal bone metabolism with restoration of androgen signaling in the study employing younger men (Leder et al., 2003). Therefore, androgens may restrain bone turnover in older men primarily through aromatization and activation of ERα, while activation of AR may be more important early in life (Khosla and Riggs, 2003). The importance of ER signaling is further supported by recent clinical studies, suggesting a minimal threshold level of estradiol for the maintenance of bone mass and the prevention of fractures in men (Mellstrom et al., 2008; LeBlanc et al., 2009; Vandenput and Ohlsson, 2009; Khosla, 2010).
Aromatase-Independent Proskeletal Effects of Androgens: Evidence From Animal Models
As discussed above, the increased bone turnover and decreased bone mass in aromatase deficient male mice demonstrate the importance of estrogen signaling in protecting the male skeleton (Oz et al., 2000; Miyaura et al., 2001). Interestingly, however, bone loss between 9 and 32 weeks of age is less severe in the male as compared to the female knockout mice (where aromatase is also necessary for the conversion of testosterone to estradiol) (Miyaura et al., 2001). As is the case with the human studies, this observation again suggests that androgen signaling in males provides an additional mechanism of skeletal protection, especially in young adults. Earlier studies demonstrated that testosterone could protect against orchiectomy-induced bone loss even when ERα activation was not possible due to pharmacological blockade (Vandenput et al., 2002) or ERα gene ablation (Vandenput et al., 2001). Moreover, orchiectomy in ArKO mice resulted in further bone resorption and further bone loss beyond the adverse effects of aromatase deficiency alone (Matsumoto et al., 2006). These studies show that at least in mice, androgens significantly attenuate bone turnover and preserve bone mass independently of aromatization.
Conditional ablation of the AR in bone was recently described, providing direct evidence for AR-mediated proskeletal effects of androgens. Interpretation of earlier mouse studies, in which AR was globally deficient, was limited because the mice had low levels of circulating testosterone (Yeh et al., 2002; Kawano et al., 2003; Vandenput et al., 2004), and thus the observed increase in bone turnover and decreased bone mass were equally attributable to loss of ER signaling and loss of AR signaling. In contrast, sex organs and thus circulating testosterone were normal when the AR was ablated specifically in osteoblasts, allowing the investigators to attribute the observed phenotype to loss of AR signaling in bone. When AR was conditionally ablated by crossing AR-floxed with α1(I) Collagen-Cre mice (α1(I)-Cre;ARf/f), the bone loss could be attributed to decreased AR signaling in osteoblasts (Notini et al., 2007). Based on the reduction in trabecular number, the authors concluded that the bone loss was due to increased resorption (Notini et al., 2007). Interestingly, this phenotype was age dependent at the distal femur, again pointing to an age-dependent contribution of AR signaling to the control of bone metabolism (Notini et al., 2007). Despite minor differences with respect to the α1(I)-Cre;ARf/f mice (Notini et al., 2007), crossing the AR-floxed mice with Osteocalcin-Cre mice also resulted in bone loss, most likely secondary to increased bone turnover (Chiang et al., 2009). This skeletal phenotype was mirrored in male mice with osteoblast-specific AR overexpression, which exhibited reduced bone turnover and increased bone mass (Wiren et al., 2004).
Molecular Mechanisms by Which AR Restrains Bone Turnover
Much like ERα in adult women and men, the bone-sparing effects of the AR in adult men are thought to be exerted primarily by keeping bone turnover in check. This is likely mediated by AR signaling in cells of the osteoblast lineage, as suggested by the conditional ablation of AR in α1(I) collagen-and osteocalcin-expressing cells (Notini et al., 2007; Chiang et al., 2009). The question of whether AR signaling in osteoclasts also plays a role in the bone-sparing effect of androgens awaits results from mice with osteoclast-specific AR knockout. With regard to AR signaling in osteoblasts, and similar to the case of ERα, a key question remains. Does AR in males simply keep at bay the proliferation of osteoblastic cells, which otherwise fuel excessive bone remodeling? Or, does it also restrain osteoclastogenic signals emanating from osteoblasts and their precursors? In favor of the latter scenario, osteoclastogenesis was increased in co-cultures containing AR-deficient compared to wild type osteoblasts (Kawano et al., 2003).
In contrast to estrogens, androgens do not stimulate FASL expression, and male mice are not affected by osteoclast-specific ERα ablation (Nakamura et al., 2007), suggesting that both ERα and AR employ other molecular mechanisms to restrain bone turnover and maintain bone mass in males. Among such mechanisms, AR signaling in osteoblasts inhibits RANKL expression and IL6 production (Bellido et al., 1995; Hofbauer et al., 1999b; Kawano et al., 2003). The effect of AR activation on OPG expression remains controversial as both stimulation and repression have been reported (Hofbauer et al., 2002; Chen et al., 2004; Wiren et al., 2004). How AR inhibits the expression of these and/or other osteoblast-derived osteoclastogenic factors is not well understood. Among potential underlying mechanisms is one similar to that proposed above for ERα, namely inhibition of Runx2, a regulator of not only bone formation but also osteoblast-driven osteoclastogenesis (Liu et al., 2001; Geoffroy et al., 2002; Enomoto et al., 2003; Maruyama et al., 2007). Indeed, similar to ERα (Khalid et al., 2008), AR also directly interacts with and inhibits Runx2 (Baniwal et al., 2009). Both these inhibitory activities are tightly regulated, presumably allowing Runx2 to stimulate expression of bone anabolic genes during specific phases of osteoblast differentiation, while keeping Runx2 in check when it promotes expression of osteoclastogenic genes during other stages of differentiation. Notably, the interaction of Runx2 with AR is stronger than that with ERα as is the resulting inhibition of Runx2 (Khalid et al., 2008; Baniwal et al., 2009). Possibly related to this, Runx2’s PST domain is sufficient for interaction with ERα (Khalid et al., 2008), whereas interaction with AR occurs through simultaneous contacts with the PST and the DNA-binding domain of Runx2 (Baniwal et al., 2009; Fig. 2). Furthermore, unlike ERα, the interaction of AR with Runx2 in solution occurs in the absence of ligand. In the living cell, however, ligand is required in order to translocate the AR into the nucleus, where it then binds and inhibits Runx2 (Baniwal et al., 2009). Thus, in addition to activation of ERα after aromatization to estradiol, androgens have the capacity to restrain Runx2 activity and thus bone turnover by activation of the AR.
Fig. 2.
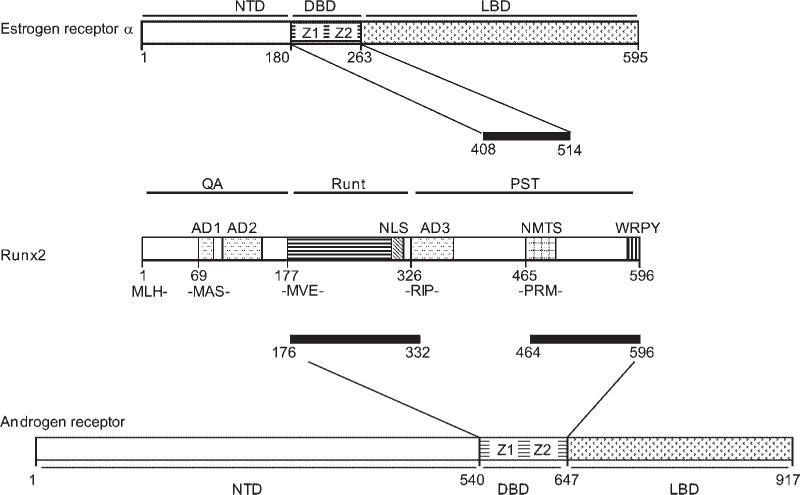
Comparison between ERα/Runx2 and AR/Runx2 interaction. Both ERα and AR interact with Runx2 via their respective DNA-binding domains (DBD). However, the interactions require different domains of Runx2 as shown by the thick horizontal lines (Khalid et al., 2008; Baniwal et al., 2009). Z1 and Z2 represent the zinc fingers at the DBDs of ERα and AR.
Conclusion
With the recent discovery of mechanisms underlying the bone-sparing effects of sex steroids, the picture appears increasingly complex. A recent breakthrough is the discovery of the central role of osteoclastic ERα in the anti-resorptive effect of estrogens in females, but interestingly not in males (Nakamura et al., 2007). Nevertheless, the old, simple concept of ‘estrogens for women, androgens for men’ is long shattered. Instead, aromatase-driven estradiol is now thought to be the predominant player in male bone mass regulation, with aromatization-independent AR action taking a back, yet still comfortable seat. It is important to remember the complexity and limitations of animal models from which we extract much of our knowledge. For example, the contribution of aromatase-independent androgen-mediated regulation of bone turnover is significantly more important in mice than in men (Carani et al., 1997; Oz et al., 2000; Miyaura et al., 2001; Khosla, 2007). In addition to species-dependence, the molecular mechanisms responsible for the bone-sparing effects of estrogens and androgens vary as a function of gender, age, skeletal site and the bone compartment. For instance, the dramatic role of osteoclastic ERα signaling in regulating trabecular bone turnover (Nakamura et al., 2007) is restricted to females. Sexual differences in the skeletal response to estrogens are further emphasized by preliminary results from mice with osteoblastic ERα knockout (Lagerquist et al., 2008; Kondoh et al., 2009). While estrogens affect trabecular bone primarily through osteoclastic ERα in females, their effect is mainly mediated by osteoblastic ERα in males (Nakamura et al., 2007; Kondoh et al., 2009). Bone compartment specificity is also demonstrated by the unaffected endocortical bone in osteoclastic ERα–KO mice (Nakamura et al., 2007). It appears therefore that estrogens inhibit endocortical bone resorption through osteoblastic ER signaling (Lagerquist et al., 2008). Interestingly, the endocortical bone surface is the very site that is specifically affected by over expression of Runx2 in osteoblasts (Liu et al., 2001; Geoffroy et al., 2002). This cellular and anatomical link between osteoblastic ERα signaling and Runx2-mediated bone resorption strongly supports the idea that unleashing of Runx2 activity upon estrogen loss (Khalid et al., 2008) is preferentially involved in postmenopausal endocortical bone resorption.
The complex bone phenotypes of newly developed mouse models may reflect a variety of molecular mechanisms selectively operative in different bone compartments, genders, ages and species. Understanding these complexities will offer opportunities for the development of novel approaches to treat osteoporosis. Early investigations of mouse models with global genetic ablation of receptors and enzymes involved in sex steroid action highlighted the limitations of such models in bone research, primarily due to the indirect effects of such genetic lesions through extraskeletal tissues (Syed and Khosla, 2005; Ohlsson and Vandenput, 2009). While evaluating the results of the more sophisticated tissue-specific knockout mouse models, we must keep in mind that these models too are complex and not without their own limitations. For example, the receptors are knocked out during early developmental stages, which may confound the interpretation of the results in the context of adult bone metabolism and their implementation to the understanding and the development of therapies for adult bone metabolic disease. Thus, along with expansion of our in vivo work, we should continue expanding our knowledge of sex steroid action using cell culture models, as well as biochemical and molecular approaches to bone biology. The results of such studies would then be tested in animal models and human subjects while paying close attention to lessons learned from mouse genetics.
Acknowledgments
We thank Dr. Sundeep Khosla (Mayo Clinic, Rochester, Minnesota) for making critical suggestions and comments. We thank the National Institutes of Health for grants DK071122 and AR047052 to B.F., and CA109147 to G.A.C. C.O. was supported by the Swedish Research Council, the Swedish Foundation for Strategic Research, the ALF/LUA research grant in Gothenburg, the Lundberg Foundation, the Torsten and Ragnar Söderberg’s Foundation, European Commission (HEALTH-F2-2008-201865-GEFOS) and the Novo Nordisk Foundation. S.K.B. is partly supported by the Innovative Chapter Research Award from Arthritis Foundation, USA. Y.G. is partly supported by a Meyer Young Investigator Fellowship from the Arthritis Foundation Southern California Chapter. BF holds the J. Harold and Edna L. LaBriola Chair in Genetic Orthopaedic Research at the University of Southern California.
Contract grant sponsor: National Institutes of Health;
Contract grant numbers: DK071122, AR047052, CA109147.
Contract grant sponsor: Swedish Research Council.
Contract grant sponsor: Swedish Foundation for Strategic Research.
Contract grant sponsor: ALF/LUA in Gothenburg.
Contract grant sponsor: Lundberg Foundation.
Contract grant sponsor: Torsten and Ragnar Söderberg’s Foundation.
Contract grant sponsor: Novo Nordisk Foundation.
Contract grant sponsor: European Commission;
Contract grant number: HEALTH-F2-2008-201865-GEFOS.
Contract grant sponsor: Arthritis Foundation.
Literature Cited
- Baniwal SK, Khalid O, Sir D, Buchanan G, Coetzee GA, Frenkel B. Repression of Runx2 by androgen receptor (AR) in osteoblasts and prostate cancer cells: AR binds Runx2 and abrogates its recruitment to DNA. Mol Endocrinol. 2009;23:1203–1214. doi: 10.1210/me.2008-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellido T, Jilka RL, Boyce BF, Girasole G, Broxmeyer H, Dalrymple SA, Murray R, Manolagas SC. Regulation of interleukin-6, osteoclastogenesis, and bone mass by androgens. The role of the androgen receptor. J Clin Invest. 1995;95:2886–2895. doi: 10.1172/JCI117995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilezikian JP, Morishima A, Bell J, Grumbach MM. Increased bone mass as a result of estrogen therapy in a man with aromatase deficiency. N Engl J Med. 1998;339:599–603. doi: 10.1056/NEJM199808273390905. [DOI] [PubMed] [Google Scholar]
- Bouillon R, Bex M, Vanderschueren D, Boonen S. Estrogens are essential for male pubertal periosteal bone expansion. J Clin Endocrinol Metab. 2004;89:6025–6029. doi: 10.1210/jc.2004-0602. [DOI] [PubMed] [Google Scholar]
- Carani C, Qin K, Simoni M, Faustini-Fustini M, Serpente S, Boyd J, Korach KS, Simpson ER. Effect of testosterone and estradiol in a man with aromatase deficiency. N Engl J Med. 1997;337:91–95. doi: 10.1056/NEJM199707103370204. [DOI] [PubMed] [Google Scholar]
- Chen Q, Kaji H, Kanatani M, Sugimoto T, Chihara K. Testosterone increases osteoprotegerin mRNA expression in mouse osteoblast cells. Horm Metab Res. 2004;36:674–678. doi: 10.1055/s-2004-826013. [DOI] [PubMed] [Google Scholar]
- Chiang C, Chiu M, Moore AJ, Anderson PH, Ghasem-Zadeh A, McManus JF, Ma C, Seeman E, Clemens TL, Morris HA, Zajac JD, Davey RA. Mineralization and bone resorption are regulated by the androgen receptor in male mice. J Bone Miner Res. 2009;24:621–631. doi: 10.1359/jbmr.081217. [DOI] [PubMed] [Google Scholar]
- Di Gregorio GB, Yamamoto M, Ali AA, Abe E, Roberson P, Manolagas SC, Jilka RL. Attenuation of the self-renewal of transit-amplifying osteoblast progenitors in the murine bone marrow by 17 beta-estradiol. J Clin Invest. 2001;107:803–812. doi: 10.1172/JCI11653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J Clin Invest. 2003;111:1221–1230. doi: 10.1172/JCI17215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto H, Shiojiri S, Hoshi K, Furuichi T, Fukuyama R, Yoshida CA, Kanatani N, Nakamura R, Mizuno A, Zanma A, Yano K, Yasuda H, Higashio K, Takada K, Komori T. Induction of osteoclast differentiation by Runx2 through receptor activator of nuclear factor-kappa B ligand (RANKL) and osteoprotegerin regulation and partial rescue of osteoclastogenesis in Runx2−/− mice by RANKL transgene. J Biol Chem. 2003;278:23971–23977. doi: 10.1074/jbc.M302457200. [DOI] [PubMed] [Google Scholar]
- Falahati-Nini A, Riggs BL, Atkinson EJ, O’Fallon WM, Eastell R, Khosla S. Relative contributions of testosterone and estrogen in regulating bone resorption and formation in normal elderly men. J Clin Invest. 2000;106:1553–1560. doi: 10.1172/JCI10942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennari L, Nuti R, Bilezikian JP. Aromatase activity and bone homeostasis in men. J Clin Endocrinol Metab. 2004;89:5898–5907. doi: 10.1210/jc.2004-1717. [DOI] [PubMed] [Google Scholar]
- Geoffroy V, Kneissel M, Fournier B, Boyde A, Matthias P. High bone resorption in adult aging transgenic mice overexpressing cbfa1/runx2 in cells of the osteoblastic lineage. Mol Cell Biol. 2002;22:6222–6233. doi: 10.1128/MCB.22.17.6222-6233.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorai I, Taguchi Y, Chaki O, Nakayama M, Minaguchi H. Specific changes of urinary excretion of cross-linked N-telopeptides of type I collagen in pre- and postmenopausal women: Correlation with other markers of bone turnover. Calcif Tissue Int. 1997;60:317–322. doi: 10.1007/s002239900235. [DOI] [PubMed] [Google Scholar]
- Hofbauer LC, Gori F, Riggs BL, Lacey DL, Dunstan CR, Spelsberg TC, Khosla S. Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: Potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology. 1999a;140:4382–4389. doi: 10.1210/endo.140.10.7034. [DOI] [PubMed] [Google Scholar]
- Hofbauer LC, Ten RM, Khosla S. The anti-androgen hydroxyflutamide and androgens inhibit interleukin-6 production by an androgen-responsive human osteoblastic cell line. J Bone Miner Res. 1999b;14:1330–1337. doi: 10.1359/jbmr.1999.14.8.1330. [DOI] [PubMed] [Google Scholar]
- Hofbauer LC, Hicok KC, Chen D, Khosla S. Regulation of osteoprotegerin production by androgens and anti-androgens in human osteoblastic lineage cells. Eur J Endocrinol. 2002;147:269–273. doi: 10.1530/eje.0.1470269. [DOI] [PubMed] [Google Scholar]
- Imai Y, Kondoh S, Kouzmenko A, Kato S. Minireview: Osteoprotective action of estrogens is mediated by osteoclastic estrogen receptor-{alpha} Mol Endocrinol. 2010 doi: 10.1210/me.2009-0238. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Youn MY, Kondoh S, Nakamura T, Kouzmenko A, Matsumoto T, Takada I, Takaoka K, Kato S. Estrogens maintain bone mass by regulating expression of genes controlling function and life span in mature osteoclasts. Ann NY Acad Sci. 2009;1173:E31–39. doi: 10.1111/j.1749-6632.2009.04954.x. [DOI] [PubMed] [Google Scholar]
- Jilka RL, Takahashi K, Munshi M, Williams DC, Roberson PK, Manolagas SC. Loss of estrogen upregulates osteoblastogenesis in the murine bone marrow. Evidence for autonomy from factors released during bone resorption. J Clin Invest. 1998;101:1942–1950. doi: 10.1172/JCI1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameda T, Mano H, Yuasa T, Mori Y, Miyazawa K, Shiokawa M, Nakamaru Y, Hiroi E, Hiura K, Kameda A, Yang NN, Hakeda Y, Kumegawa M. Estrogen inhibits bone resorption by directly inducing apoptosis of the bone-resorbing osteoclasts. J Exp Med. 1997;186:489–495. doi: 10.1084/jem.186.4.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano H, Sato T, Yamada T, Matsumoto T, Sekine K, Watanabe T, Nakamura T, Fukuda T, Yoshimura K, Yoshizawa T, Aihara K, Yamamoto Y, Nakamichi Y, Metzger D, Chambon P, Nakamura K, Kawaguchi H, Kato S. Suppressive function of androgen receptor in bone resorption. Proc Natl Acad Sci USA. 2003;100:9416–9421. doi: 10.1073/pnas.1533500100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalid O, Baniwal SK, Purcell DJ, Leclerc N, Gabet Y, Stallcup MR, Coetzee GA, Frenkel B. Modulation of Runx2 activity by estrogen receptor-alpha: Implications for osteoporosis and breast cancer. Endocrinology. 2008;149:5984–5995. doi: 10.1210/en.2008-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosla S. Estrogen and the death of osteoclasts: A fascinating story. Int Bone Mineral Soc. 2007;10:267–272. [Google Scholar]
- Khosla S. Estrogen and bone: Insights from estrogen-resistant, aromatase-deficient, and normal men. Bone. 2008;43:414–417. doi: 10.1016/j.bone.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosla S. Update in male osteoporosis. J Clin Endocrinol Metab. 2010;95:3–10. doi: 10.1210/jc.2009-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosla S, Riggs BL. Androgens, estrogens, and bone turnover in men. J Clin Endocrinol Metab. 2003;88:2352. doi: 10.1210/jc.2003-030054. author reply 2352–2353. [DOI] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- Kondoh S, Imai Y, Takada I, Tsuji N, Matsumoto T, Kato S. Osteoprotective estrogen action in males mediates osteoblastic estrogen receptor α. J Bone Miner Res. 2009;24 Available at http://www.asbmr.org/Meetings/AnnualMeeting/AbstractDetail.aspx?aid=ee2fb624-620c637-624c625e-8901-d8692a8921f8957b8903. [Google Scholar]
- Krum SA, Miranda-Carboni GA, Hauschka PV, Carroll JS, Lane TF, Freedman LP, Brown M. Estrogen protects bone by inducing Fas ligand in osteoblasts to regulate osteoclast survival. EMBO J. 2008;27:535–545. doi: 10.1038/sj.emboj.7601984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerquist MK, Hakansson C, Windahl SH, Borjesson AE, Jochems C, Antal MC, Krust A, Chambon P, Angel P, Carlsten H, Ohlsson C. Estrogen Receptor-alpha Expression in Mesenchymal Cells Is Crucial for the Bone Protective Effects of Estrogen. J Bone Miner Res. 2008;23:S36–S36. [Google Scholar]
- Lanfranco F, Zirilli L, Baldi M, Pignatti E, Corneli G, Ghigo E, Aimaretti G, Carani C, Rochira V. A novel mutation in the human aromatase gene: Insights on the relationship among serum estradiol, longitudinal growth and bone mineral density in an adult man under estrogen replacement treatment. Bone. 2008;43:628–635. doi: 10.1016/j.bone.2008.05.011. [DOI] [PubMed] [Google Scholar]
- LeBlanc ES, Nielson CM, Marshall LM, Lapidus JA, Barrett-Connor E, Ensrud KE, Hoffman AR, Laughlin G, Ohlsson C, Orwoll ES. The effects of serum testosterone, estradiol, and sex hormone binding globulin levels on fracture risk in older men. J Clin Endocrinol Metab. 2009;94:3337–3346. doi: 10.1210/jc.2009-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leder BZ, LeBlanc KM, Schoenfeld DA, Eastell R, Finkelstein JS. Differential effects of androgens and estrogens on bone turnover in normal men. J Clin Endocrinol Metab. 2003;88:204–210. doi: 10.1210/jc.2002-021036. [DOI] [PubMed] [Google Scholar]
- Lindberg MK, Weihua Z, Andersson N, Moverare S, Gao H, Vidal O, Erlandsson M, Windahl S, Andersson G, Lubahn DB, Carlsten H, Dahlman-Wright K, Gustafsson JA, Ohlsson C. Estrogen receptor specificity for the effects of estrogen in ovariectomized mice. J Endocrinol. 2002;174:167–178. doi: 10.1677/joe.0.1740167. [DOI] [PubMed] [Google Scholar]
- Liu W, Toyosawa S, Furuichi T, Kanatani N, Yoshida C, Liu Y, Himeno M, Narai S, Yamaguchi A, Komori T. Overexpression of Cbfa1 in osteoblasts inhibits osteoblast maturation and causes osteopenia with multiple fractures. J Cell Biol. 2001;155:157–166. doi: 10.1083/jcb.200105052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo J, Horowitz M, Choi Y. Osteoimmunology: Interactions of the bone and immune system. Endocr Rev. 2008;29:403–440. doi: 10.1210/er.2007-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolagas SC. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21:115–137. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
- Maruyama Z, Yoshida CA, Furuichi T, Amizuka N, Ito M, Fukuyama R, Miyazaki T, Kitaura H, Nakamura K, Fujita T, Kanatani N, Moriishi T, Yamana K, Liu W, Kawaguchi H, Komori T. Runx2 determines bone maturity and turnover rate in postnatal bone development and is involved in bone loss in estrogen deficiency. Dev Dyn. 2007;236:1876–1890. doi: 10.1002/dvdy.21187. [DOI] [PubMed] [Google Scholar]
- Matsumoto C, Inada M, Toda K, Miyaura C. Estrogen and androgen play distinct roles in bone turnover in male mice before and after reaching sexual maturity. Bone. 2006;38:220–226. doi: 10.1016/j.bone.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Mellstrom D, Vandenput L, Mallmin H, Holmberg AH, Lorentzon M, Oden A, Johansson H, Orwoll ES, Labrie F, Karlsson MK, Ljunggren O, Ohlsson C. Older men with low serum estradiol and high serum SHBG have an increased risk of fractures. J Bone Miner Res. 2008;23:1552–1560. doi: 10.1359/jbmr.080518. [DOI] [PubMed] [Google Scholar]
- Millan M, Almeida M, Ambrogini E, Han L, Warron A, Vyas K, Shelton R, Xiaohua Q, Weinstein R, Jilka R, O’Brien C, Manolagas SC. Deletion of the ERα from osteoclast precursors and their progeny abrogates the protective effects of estrogens in cancellous but not cortical bone. Denver, CO: American Society for Bone and Mineral Research; 2009. [Google Scholar]
- Miyaura C, Toda K, Inada M, Ohshiba T, Matsumoto C, Okada T, Ito M, Shizuta Y, Ito A. Sex- and age-related response to aromatase deficiency in bone. Biochem Biophys Res Commun. 2001;280:1062–1068. doi: 10.1006/bbrc.2001.4246. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, Harada Y, Azuma Y, Krust A, Yamamoto Y, Nishina H, Takeda S, Takayanagi H, Metzger D, Kanno J, Takaoka K, Martin TJ, Chambon P, Kato S. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. 2007;130:811–823. doi: 10.1016/j.cell.2007.07.025. [DOI] [PubMed] [Google Scholar]
- Notini AJ, McManus JF, Moore A, Bouxsein M, Jimenez M, Chiu WS, Glatt V, Kream BE, Handelsman DJ, Morris HA, Zajac JD, Davey RA. Osteoblast deletion of exon 3 of the androgen receptor gene results in trabecular bone loss in adult male mice. J Bone Miner Res. 2007;22:347–356. doi: 10.1359/jbmr.061117. [DOI] [PubMed] [Google Scholar]
- Ohlsson C, Vandenput L. The role of estrogens for male bone health. Eur J Endocrinol. 2009;160:883–889. doi: 10.1530/EJE-09-0118. [DOI] [PubMed] [Google Scholar]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Oz OK, Zerwekh JE, Fisher C, Graves K, Nanu L, Millsaps R, Simpson ER. Bone has a sexually dimorphic response to aromatase deficiency. J Bone Miner Res. 2000;15:507–514. doi: 10.1359/jbmr.2000.15.3.507. [DOI] [PubMed] [Google Scholar]
- Pacifici R. Estrogen, cytokines, and pathogenesis of postmenopausal osteoporosis. J Bone Miner Res. 1996;11:1043–1051. doi: 10.1002/jbmr.5650110802. [DOI] [PubMed] [Google Scholar]
- Ray A, Prefontaine KE, Ray P. Down-modulation of interleukin-6 gene expression by 17 beta-estradiol in the absence of high affinity DNA binding by the estrogen receptor. J Biol Chem. 1994;269:12940–12946. [PubMed] [Google Scholar]
- Riggs BL, Khosla S, Melton LJ., 3rd Sex steroids and the construction and conservation of the adult skeleton. Endocr Rev. 2002;23:279–302. doi: 10.1210/edrv.23.3.0465. [DOI] [PubMed] [Google Scholar]
- Rochira V, Zirilli L, Madeo B, Aranda C, Caffagni G, Fabre B, Montangero VE, Roldan EJ, Maffei L, Carani C. Skeletal effects of long-term estrogen and testosterone replacement treatment in a man with congenital aromatase deficiency: Evidences of a priming effect of estrogen for sex steroids action on bone. Bone. 2007;40:1662–1668. doi: 10.1016/j.bone.2007.01.022. [DOI] [PubMed] [Google Scholar]
- Sims NA, Clement-Lacroix P, Minet D, Fraslon-Vanhulle C, Gaillard-Kelly M, Resche-Rigon M, Baron R. A functional androgen receptor is not sufficient to allow estradiol to protect bone after gonadectomy in estradiol receptor-deficient mice. J Clin Invest. 2003;111:1319–1327. doi: 10.1172/JCI17246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, Williams TC, Lubahn DB, Korach KS. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. 1994;331:1056–1061. doi: 10.1056/NEJM199410203311604. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Toraldo G, Weitzmann MN, Cenci S, Ross FP, Pacifici R. Estrogen decreases osteoclast formation by down-regulating receptor activator of NF-kappa B ligand (RANKL)-induced JNK activation. J Biol Chem. 2001;276:8836–8840. doi: 10.1074/jbc.M010764200. [DOI] [PubMed] [Google Scholar]
- Syed F, Khosla S. Mechanisms of sex steroid effects on bone. Biochem Biophys Res Commun. 2005;328:688–696. doi: 10.1016/j.bbrc.2004.11.097. [DOI] [PubMed] [Google Scholar]
- Usui M, Yoshida Y, Tsuji K, Oikawa K, Miyazono K, Ishikawa I, Yamamoto T, Nifuji A, Noda M. Tob deficiency superenhances osteoblastic activity after ovariectomy to block estrogen deficiency-induced osteoporosis. Proc Natl Acad Sci USA. 2004;101:6653–6658. doi: 10.1073/pnas.0303093101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenput L, Ohlsson C. Estrogens as regulators of bone health in men. Nat Rev Endocrinol. 2009;5:437–443. doi: 10.1038/nrendo.2009.112. [DOI] [PubMed] [Google Scholar]
- Vandenput L, Ederveen AG, Erben RG, Stahr K, Swinnen JV, Van Herck E, Verstuyf A, Boonen S, Bouillon R, Vanderschueren D. Testosterone prevents orchidectomy-induced bone loss in estrogen receptor-alpha knockout mice. Biochem Biophys Res Commun. 2001;285:70–76. doi: 10.1006/bbrc.2001.5101. [DOI] [PubMed] [Google Scholar]
- Vandenput L, Swinnen JV, Van Herck E, Verstuyf A, Boonen S, Bouillon R, Vanderschueren D. The estrogen receptor ligand ICI 182,780 does not impair the bone-sparing effects of testosterone in the young orchidectomized rat model. Calcif Tissue Int. 2002;70:170–175. doi: 10.1007/s00223-001-2065-z. [DOI] [PubMed] [Google Scholar]
- Vandenput L, Swinnen JV, Boonen S, Van Herck E, Erben RG, Bouillon R, Vanderschueren D. Role of the androgen receptor in skeletal homeostasis: The androgen-resistant testicular feminized male mouse model. J Bone Miner Res. 2004;19:1462–1470. doi: 10.1359/JBMR.040505. [DOI] [PubMed] [Google Scholar]
- Vanderschueren D, Vandenput L, Boonen S, Lindberg MK, Bouillon R, Ohlsson C. Androgens and bone. Endocr Rev. 2004;25:389–425. doi: 10.1210/er.2003-0003. [DOI] [PubMed] [Google Scholar]
- Vanderschueren D, Venken K, Ophoff J, Bouillon R, Boonen S. Clinical review: Sex steroids and the periosteum–reconsidering the roles of androgens and estrogens in periosteal expansion. J Clin Endocrinol Metab. 2006;91:378–382. doi: 10.1210/jc.2005-1766. [DOI] [PubMed] [Google Scholar]
- Weitzmann MN, Roggia C, Toraldo G, Weitzmann L, Pacifici R. Increased production of IL-7 uncouples bone formation from bone resorption during estrogen deficiency. J Clin Invest. 2002;110:1643–1650. doi: 10.1172/JCI15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westendorf JJ. Transcriptional co-repressors of Runx2. J Cell Biochem. 2006;98:54–64. doi: 10.1002/jcb.20805. [DOI] [PubMed] [Google Scholar]
- Wiren KM, Zhang XW, Toombs AR, Kasparcova V, Gentile MA, Harada S, Jepsen KJ. Targeted overexpression of androgen receptor in osteoblasts: Unexpected complex bone phenotype in growing animals. Endocrinology. 2004;145:3507–3522. doi: 10.1210/en.2003-1016. [DOI] [PubMed] [Google Scholar]
- Yeh S, Tsai MY, Xu Q, Mu XM, Lardy H, Huang KE, Lin H, Yeh SD, Altuwaijri S, Zhou X, Xing L, Boyce BF, Hung MC, Zhang S, Gan L, Chang C. Generation and characterization of androgen receptor knockout (ARKO) mice: An in vivo model for the study of androgen functions in selective tissues. Proc Natl Acad Sci USA. 2002;99:13498–13503. doi: 10.1073/pnas.212474399. [DOI] [PMC free article] [PubMed] [Google Scholar]