

Abstract
Deregulation of microRNAs (miRs) contributes to progression and metastasis of prostate and other cancers. miR-23b and -27b, encoded in the same miR cluster (miR-23b/-27b), are downregulated in human metastatic prostate cancer compared with primary tumors and benign tissue. Expression of miR-23b/-27b decreases prostate cancer cell migration, invasion and results in anoikis resistance. Conversely, antagomiR-mediated miR-23b and -27b silencing produces the opposite result in a more indolent prostate cancer cell line. However, neither miR-23b/-27b expression or inhibition impacts prostate cancer cell proliferation suggesting that miR-23b/-27b selectively suppresses metastasis. To examine the effects of miR-23b/-27b on prostate cancer metastasis in vivo, orthotopic prostate xenografts were established using aggressive prostate cancer cells transduced with miR-23b/-27b or non-targeting control miRNA. Although primary tumor formation was similar between miR-23b/-27b-transduced cells and controls, miR-23b/-27b expression in prostate cancer cells decreased seminal vesicle invasion and distant metastases. Gene-expression profiling identified the endocytic adaptor, Huntingtin-interacting protein 1-related (HIP1R) as being downregulated by miR-23b/-27b. Increased HIP1R expression in prostate cancer cells inversely phenocopied the effects of miR-23b/-27b overexpression on migration, invasion and anchorage-independent growth. HIP1R rescued miR-23b/-27b-mediated repression of migration in prostate cancer cells. HIP1R mRNA levels were decreased in seminal vesicle tissue from mice bearing miR-23b/-27b-transduced prostate cancer cell xenografts compared with scrambled controls, suggesting HIP1R is a key functional target of miR-23b/-27b. In addition, depletion of HIP1R led to a more rounded, less mesenchymal-like cell morphology, consistent with decreased metastatic properties. Together, these data demonstrate that the miR-23b/-27b cluster functions as a metastasis-suppressor by decreasing HIP1R levels in pre-clinical models of prostate cancer.
Introduction
Metastasis is responsible for the majority of deaths attributed to prostate cancer (PC).1 Therefore, there is a critical need to understand the metastatic process and identify new therapies targeted to disseminated disease. MicroRNAs (miRs) regulate an expanding list of complex biological processes, including cell proliferation, migration and invasion, and are implicated in all stages of cancer.2–6 MiRs may themselves act as tumor suppressors, oncogenes or even specifically as metastasis suppressors or activators.7–9 Therefore miRs possess both diagnostic and therapeutic potential,10,11 and investigation of their role in PC metastasis is an active area of research.12
MiR-23b and -27b are components of the miR-23b-27b-24-1 cluster located on human chromosome 9. Roughly 40% of human miRs are organized in evolutionarily conserved clusters, which are coordinately transcribed as discrete polycistronic pri-miRs.13 Two components of the miR-23b-27b-24-1 cluster, hereafter termed miR-23b/-27b are specifically downregulated in aggressive human PC clinical samples compared with primary tumors and benign tissue.6,14–17 We previously identified miR-23b/-27b as a selective suppressor of key metastatic processes in vitro including cell invasion, migration and anchorage-independent survival; however, miR-23b/-27b does not affect cell proliferation.18
Here we demonstrate that miR-23b/-27b expression in aggressive PC cells implanted orthotopically significantly limited their in vivo capacity for local invasion and the formation of distant metastases. We provide novel evidence that implicates Huntingtin-interacting protein 1-related (HIP1R), a clathrin-binding protein and endocytic adaptor, as a functional target of miR-23b/-27b. To that end, the silencing of HIP1R expression resulted in the inhibition of PC metastatic processes.
Results
miR-23b/-27b decreases local invasion and distant metastasis of human PC cells in vivo
Previous experiments have shown that the ectopic expression of miR-23b/-27b decreases metastatic characteristics in vitro, whereas silencing miR-23b and -27b with antagomiRs has the opposite effect.18 Therefore, we examined the impact of miR-23b/-27b expression on metastasis of PC cells in vivo. For these studies, we chose an orthotopic PC xenograft model, which recapitulates the entire metastatic cascade including loss of cell–cell contact, invasion, intravasation, extravasation and tumor growth at secondary sites.19,20 PC3-ML Luciferase cells (PC3-ML Luc),21 a metastatic human PC cell line, were transduced with either lentivirus-expressing miR-23b/-27b or a scrambled miR control. We confirmed that miR-23b/-27b inhibited the migration of the luciferase-expressing PC3-ML cells in vitro as had been previously observed for the parental PC3-ML line (Figure 1a).18 PC3-ML Luc miR-23b/-27b and scrambled control cells were injected into the ventral prostates of nude mice. Tumor formation was monitored by IVIS imaging. Bioluminescence imaging at the experimental end point (day 40) revealed primary tumor formation in 15/19 mice receiving control cells and 14/19 mice receiving miR-23b/-27b expressing cells. On the basis of IVIS imaging, the mice bearing xenografts of PC cells transduced with miR-23b/-27b showed decreased metastases compared with controls (Figure 1b). However, metastases could not be quantified effectively given the strong signal coming from the primary tumor. Therefore, the prostates were removed from the mice and luminescence measured ex vivo. Post mortem ex vivo IVIS imaging of prostates showed no significant difference in bioluminescence, consistent with a lack of effect of miR-23b/-27b on primary tumor growth between the two groups (PC3-ML Luc cells transduced with miR-23b/-27b versus control; Figure 1c). In addition, prostate weights were comparable between mice xenografted with miR--23b/-27b or scrambled expressing cells (data not shown). Although no statistically significant effect of miR expression was seen on the growth of the primary tumor, mice bearing miR-23b/-27b transduced PC3-ML Luc cell xenografts displayed significantly less local invasion to the seminal vesicles and decreased distal metastases (Figures 1d and e). At the end of the experiment, the number of metastases per mouse was quantified (as measured by distinct photon regions of interest) and found to be significantly decreased in xenografts from miR-23b/-27b-transduced compared with control cells (Figure 1f). These metastases appeared largely confined to inguinal, lumbar, caudal and splenic lymph nodes as observed during autopsy (data not shown). Histological analysis was performed on prostate and seminal vesicle tissue by hematoxylin and eosin staining confirming that tumor invasion into seminal vesicles was largely restricted to the control group (Figure 1g). Overall, these data demonstrate that miR-23b/-27b decreased local invasion and distal metastasis of PC cells in vivo.
Figure 1.

MiR-23b/-27b decreases local invasion and distal metastasis in an in vivo preclinical model of prostate cancer. (a) PC3-ML Luc cells transduced with miR-23b/-27b or scrambled control were tested in Matrigel invasion assays as described in Materials and methods section. (b) PC3-ML Luc cells expressing miR-23b/-27b or scrambled control were injected into the prostate ventral lobes of nude mice. IVIS imaging was performed at day 40; shown are the representative images of mice from both groups (scrambled n = 15, miR-23b/-27b n = 14). Post mortem ex vivo imaging was performed on isolated prostate (c) and seminal vesicles (d). (e) Distal metastasis was defined as all bioluminescence signal remaining post mortem after removal of prostates and seminal vesicles. (f) Individual metastases were identified as distinct photon regions of interest (ROIs; ± s.e.m.) using Living Image software on IVIS images of mice after removal of prostates and seminal vesicles. (g) Representative images of hematoxylin and eosin immunostaining of formalin-fixed tissues. *P<0.05, **P<0.01, ***P<0.001, NS, not significant.
miR-23b/-27b decreases expression of HIP1R in PC cells
To identify possible targets of miR-23b/-27b, microarray analyses were performed using PC3-ML cells transduced with miR-23b/-27b versus scrambled control, as well as LNCaP, a more indolent PC cell line with relatively high miR-23b/-27b levels, transfected with miR-23b and -27b antagomiRs versus control antagomiRs. PC3-ML miR-23b/-27b-expressing cells exhibited ∼ 25-fold greater levels of miR-23b compared with cells receiving the scrambled control, as determined by quantitative real-time PCR (Supplementary Figure S1a). LNCaP transfected with antagomiRs to miR-23b and -27b had an almost 60% decrease in miR-23b/-27b (Supplementary Figure S1b). Gene-expression changes were analyzed and priority was given to genes that were significantly decreased in PC3-ML miR-23b/-27b transduced cells compared with controls and increased in LNCaP cells transfected with miR-23b and -27b antagomiRs versus controls. We also considered whether candidate target genes contained predicted 3′-untranslated region (UTR)-binding sites for both miR-23b and miR-27b, the existence of multiple putative miR-binding sites, and whether the candidate gene was known to be expressed in human PC.
Using quantitative real-time PCR, we validated several candidates in two independent aggressive PC cell lines, ALVA31 and PC3-ML, ectopically expressing miR-23b/-27b, as well as in the less aggressive LNCaP transfected with miR-23b and miR-27b antagomiRs. The candidate miR-23b/-27b target, HIP1R was inversely correlated with miR-23b/-27b expression in PC3-ML, ALVA31 and LNCaP cells (Figure 2a). Similarly, HIP1R protein levels were inversely correlated with miR-23b/-27b in PC cells (Figure 2b).
Figure 2.

HIP1R is downregulated by miR-23b/-27b in prostate cancer cells. PC3-ML and ALVA31 cells were transduced with miR-23b/-27b or scrambled control, and LNCaP cells were transfected with antagomiRs to both miR-23b and -27b or control antagomiRs. (a) RNA was isolated and reverse transcriptase quantitative PCR (RT-qPCR) was performed. Values are of HIP1R mRNA levels normalized to β-actin mRNA control. (b) Protein lysates were collected, and western blot analysis was performed for HIP1R and actin. Blot shown is representative of three independent experiments. (c) PC3-ML and ALVA31 cells, transduced as described above, and LNCaP cells were additionally transfected with luciferase reporter constructs containing either the wild-type 3′-UTR of HIP1R or an empty vector (EV) control. Forty-eight hours following transfection, luciferase assays were performed on cell lysates. Mean ratios of relative luciferase units (± s.e.m.) of three independent experiments performed in triplicate are shown. Relative luciferase units were obtained by normalization to Renilla luciferase. ***P<0.001.
To determine if HIP1R was a direct target of miR-23b/-27b, a luciferase reporter vector containing the wild-type 3′-UTR of HIP1R was utilized. Overexpression of miR-23b/-27b (as compared with scrambled control) in PC3-ML or ALVA31 cells resulted in similar HIP1R 3′-UTR-related luciferase activities as the control luciferase vector lacking the 3′-UTR (Figure 2c). In addition, the wild-type HIP1R 3′-UTR luciferase and vector constructs were introduced into LNCaP cells. Consistent with experiments in which miR-23b/-27b was overexpressed, endogenous miR-23b/-27b had no effect on HIP1R 3′-UTR-regulated luciferase activity compared with the vector alone (Figure 2c). Thus, although miR-23b/-27b regulated HIP1R mRNA and protein levels, the miR cluster did not appear to directly bind and suppress the 3′-UTR of HIP1R in PC cells. These results suggest that miR-23b/-27b indirectly regulated HIP1R expression by potentially targeting an as of yet unidentified gene(s) that acts upstream of HIP1R and regulates HIP1R expression. Interestingly, the level of decrease in HIP1R protein expression greatly exceeded the level of silencing of HIP1R mRNA in the miR-23b/-27b-transduced cells. This suggests that additional post-transcriptional regulatory mechanisms may be used that decrease HIP1R protein levels.
In addition, analysis of publicly available data sets from Oncomine.org revealed that HIP1R mRNA levels are increased in metastatic compared with organ-confined PC (Figure 3) consistent with our observations that miR-23b/-27b selectively impacts metastasis.18
Figure 3.

HIP1R mRNA is significantly upregulated in human metastatic prostate cancer compared with primary site disease in multiple data sets. Prostate cancer mRNA analyses from Oncomine were surveyed for HIP1R in primary prostate cancer as compared with sites of metastasis. HIP1R levels, graphed as log2 median-centered ratio, are listed for three separate significant studies: Grasso Prostate Statistics,43 primary site (59 samples), metastasis (35 samples). Chandran Prostate Statistics,44 primary site (10 samples), metastasis (21 samples). Yu Prostate Statistics,45 primary site (64 samples), metastasis (24 samples). **P<0.01, ***P<0.001.
Ectopic expression of HIP1R increases invasion and migration of poorly aggressive human PC cells
We previously demonstrated that inhibition of miR-23b/-27b using specific antagomiRs increases migration and invasion of the poorly migratory and less invasive PC cell line, LNCaP.18 Using Boyden chamber assays, we found that ectopic expression of HIP1R increased LNCaP cell motility compared with cells transfected with empty vector control (Figure 4a). In addition, matrigel invasion assays revealed that HIP1R increased the invasiveness of LNCaP cells (Figure 4b), whereas expression of HIP1R had no effect on proliferation (Figure 4c), or cell viability (Supplementary Figure S3). Therefore, overexpression of the miR-23b/-27b target HIP1R in LNCaP cells reproduced the phenotype observed when miR-23b/-27b levels were reduced in this cell line consistent with a potential role of HIP1R down-regulation by miR-23b/-27b in metastasis suppression.
Figure 4.

HIP1R increases invasion and migration, but not proliferation of the relatively indolent prostate cancer cell line LNCaP. (a) Boyden Chamber (b) Matrigel Invasion and (c) proliferation assays were performed as described on LNCaP cells transfected with pIRES-HIP1R or pIRES-empty vector (EV). Representative images are shown for Boyden chamber assays. For all, the mean cell number (± s.d.) of a representative experiment (n = 3) performed in triplicate is shown. *P<0.05, **P<0.01.
HIP1R depletion reduces the metastatic phenotype of aggressive human PC cells
To evaluate further the importance of HIP1R on PC invasion and migration, we depleted HIP1R in PC3-ML and ALVA31 cells using a short-hairpin RNA (shRNA) retroviral construct (shHIP1R) and examined the potential of these cells for invasion and migration. PC3-ML and ALVA31, transduced with an shRNA construct against GFP (shGFP) were used as controls. HIP1R protein levels were greatly decreased in cells stably expressing shHIP1R as compared with shGFP controls as measured by western blot analysis (Figure 5a). We observed decreased migration and invasion of both PC3-ML and ALVA31 cells transduced with shHIP1R compared with the shGFP-transduced controls (Figures 5b and c) thereby phenocopying the effects of miR-23b/-27b over-expression in ALVA31 and PC3-ML cells.18 Consistent with the lack of miR-23b/-27b effects on cell proliferation observed previously,18 HIP1R knockdown did not alter the proliferation, or cell viability of these PC cell lines (Figure 5d and Supplementary Figure S3, respectively). Knockdown of HIP1R resulted in decreased ALVA31 and PC3-ML growth in soft agar (Figure 5e), as was observed in these cells expressing miR-23b/-27b.18 Taken together, this evidence suggests that suppression of PC invasion and migration by miR-23b/-27b may be mediated through the downregulation of HIP1R.
Figure 5.
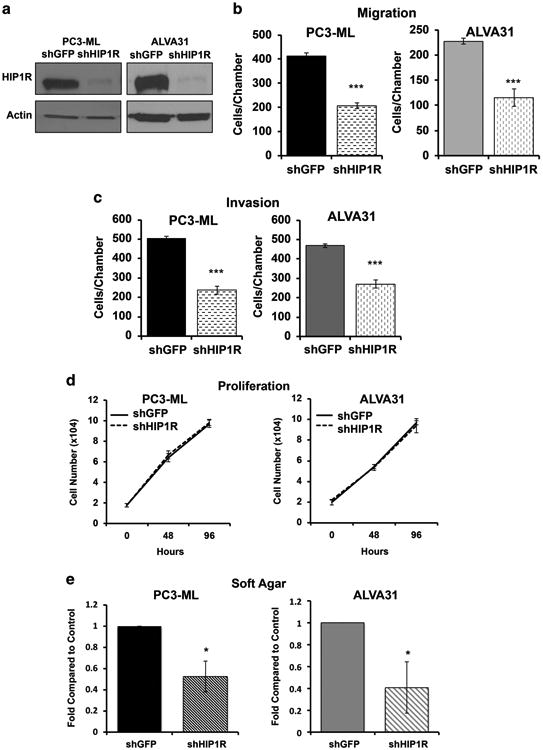
Depletion of HIP1R decreases invasion, migration, and anchorage-independent growth of aggressive prostate cancer cells. PC3-ML and ALVA-31 cells were stably transduced with either a short-hairpin against HIP1R (shHIP1R) or GFP as a control (shGFP). (a) Cellular lysates were collected and HIP1R and actin were detected by western blotting. (b) Boyden Chamber, (c) Matrigel Invasion and (d) proliferation assays were also performed for these cells. The mean cell number (± s.d.) of a representative experiment (n = 3) performed in triplicate is shown for b–d. (e) Anchorage-independent growth was analyzed for cells depleted of HIP1R using a soft agar assay. Results of three experiments were averaged, and graphed relative to shGFP control (±s.e.m.). *P<0.05, ***P<0.001.
HIP1R rescues miR-23b/-27b-mediated suppression of aggressive PC cell migration
To determine whether miR-23b/-27b exerts metastasis suppressing effects through downregulation of HIP1R, we introduced a HIP1R cDNA construct into PC3-ML or ALVA31 cells expressing miR-23b/-27b or scrambled control. This HIP1R construct is not susceptible to miR-23b/-27b-mediated depletion (Supplementary Figure S2). We found that HIP1R expression rescued miR-23b/-27b-mediated suppression of PC cell migration of both PC3-ML (Figure 6a) and ALVA31 cell lines (Figure 6b). As restoration of HIP1R expression attenuated the metastasis suppressor effects of miR-23b/-27b in PC cells, modulating HIP1R expression levels appears to be a key mechanism of action of these miRs.
Figure 6.

HIP1R rescues miR-23b/-27b-mediated suppression of migration in aggressive prostate cancer cells. (a) PC3-ML and (b) ALVA31 cells transduced with either miR-23b/-27b or scrambled control were transfected with pIRES-HIP1R or a pIRES-empty vector (EV) control. Forty-eight hours following transfection, cells were serum-starved overnight then seeded in Boyden Chambers for migration assays. The mean cell number (± s.d.) of a representative experiment (n = 3) performed in triplicate is shown. *P<0.05, **P<0.01, NS, not significant.
HIP1R modifies cell morphology in aggressive PC lines
On the basis of HIP1R's well-recognized role as an endocytic adaptor protein that binds clathrin light chain and coordinates actin reorganization with vesicle dynamics,22 we posited that depletion of HIP1R would affect cellular morphology, specifically involving F-actin. Phalloidin staining (binds to F-actin) was performed to determine possible effects of HIP1R depletion on cell morphology. In PC3-ML and ALVA31 shHIP1R cells compared with the shGFP controls, we observed altered cell shape (Figures 7a and b). We quantified these changes by calculating the ratio of the long axis to the short axis of cells stably expressing shHIP1R or shGFP controls and found that HIP1R knockdown lead to rounder (less elongated) cells.
Figure 7.

HIP1R knockdown in aggressive prostate cancer cells results in decreased cell elongation. (a) PC3-ML and (b) ALVA31 cells transduced with shHIP1R or shGFP were grown for 5 days in supplemented media, then stained for Phalloidin immunofluorescence. Cell shape was measured by determining long axis versus short axis and graphed (± s.e.m.), n≥30 cells per condition. Mann–Whitney's test was used to analyze cell axes ratios. **P<0.01, ***P<0.001.
Decreased HIP1R mRNA is linked to miR-23b/-27b-mediated anti-invasive actions in vivo
To solidify HIP1R as a relevant target for the anti-invasive properties of mir-23b/-27b in vivo, we evaluated expression of HIP1R mRNA in the seminal vesicles of mice bearing orthotopic xenografts. As expected, miR-23b and miR-27b levels were higher in the samples derived from mice xenografted with miR-23b/-27b-transduced PC cells (as compared with scrambled controls; Figure 8a). Further, HIP1R mRNA was significantly decreased in the seminal vesicle tissues expressing miR-23b and -27b (Figure 8b). These data provide further strong support for miR-23b/-27b-mediated suppression of HIP1R leading to the anti-invasive effects of this miR cluster in vivo.
Figure 8.

Decreased HIP1R mRNA is linked to miR-23b/-27b-mediated anti-invasive actions in vivo. Seminal vesicle tissue from mice bearing PC3-ML Luc orthotopic prostate xenografts expressing miR-23b/-27b or a scrambled control were homogenized in Trizol and assayed for mRNA. (a) RT-qPCR analysis was performed using Taqman probes to miR-23b and -27b. Samples were normalized to SNU6, and controlled to vehicle tissue. (b) HIP1R human-specific Taqman probes were used for RT-qPCR analysis, and normalized to GAPDH mRNA. Mann–Whitney non-parametric test was used to determine significance. *P<0.05, **P<0.01, ***P<0.001.
Discussion
We explored the potential for the miR-23b/-27b cluster to serve as a metastasis suppressor in vivo to complement our previous studies demonstrating that miR-23b/-27b decreases migration, invasion and anoikis resistance in multiple PC cell lines. We also sought to understand the mechanism responsible for the anti-metastatic action of this miR cluster in PC, as similar metastasis suppressing miRs have been described for aggressive breast cancer.23,24 Expression of miR-23b/-27b in the metastatic human PC cell line PC3-ML was sufficient to decrease seminal vesicle invasion and metastasis in an orthotopic xenograft model of PC. We chose this preclinical model as PC3-ML cells reproduce the full metastatic cascade following implantation into the prostates (orthotopic xenografts) of immunocompromised mice. Orthotopic xenografting of these cells results in reliable local invasion to seminal vesicles and lymph node metastasis, as is common in aggressive cases of human PC.25 Thus, the orthotopic xenograft model faithfully reproduces these aspects of the disease in humans. A limitation of this model is that PC3-ML cells do not typically metastasize to bone as will PC cells following intracardiac or intratibia injection. Yet, such current preclinical models of PC bone metastasis circumvent early steps in the metastatic process. That miR-23b/-27b decreased PC seminal vesicle invasion and distal metastases, coupled with our previous in vitro data,18 provides strong support for the contention that the miR-23b/-27b cluster functions as a metastasis suppressor.
To understand the mechanisms of miR-23b/-27b-mediated metastasis suppression in PC, we sought to identify miR-repressed target genes that exert pro-metastatic activities. Through microarray analyses of two PC cell lines, expressing miR-23b/-27b or transfected with miR-23b and -27b antagomiRs, followed by qPCR validation, we identified HIP1R as a novel target of miR-23b/-27b in PC cells. Data mining from multiple microarray studies of human PC samples indicates that HIP1R is over-expressed in metastases compared with PC (Figure 3). Although HIP1R is a predicted target of miR-23b/-27b, luciferase-reporter assays suggested miR-23b/-27b may not directly regulate HIP1R via its 3′-UTR. Although 3′-UTR binding is the canonical mechanism of miRNA target binding, it is possible that the essential regulatory sequences of HIP1R lie outside this region.26
This less common localization of miRNA-targeting sequences has been shown to be the case for other mRNAs regulated by miRNAs, such as the regulation of Nanog by miR-296.27 However, we were able to express a recombinant version of HIP1R that lacks the 3′-UTR in the presence of miR-23b/-27b suggesting that there are no miR-23b/-27b-binding sites in the coding sequence of the gene. Therefore, HIP1R mRNA may not be a direct target of miR-23b/-27b. Instead, it is possible that miR-23b/-27b targets an as of yet unidentified gene, which lies upstream of HIP1R. For example, if miR-23b/-27b directed the silencing of a transcription factor that was required for the transcription of HIP1R, the silencing of this transcription factor would result in a concomitant decrease in HIP1R expression at both the mRNA and protein levels. Nevertheless, our data support HIP1R downregulation as a key component of miR-23b/-27b anti-metastatic actions.
HIP1R is best recognized as an endocytic adaptor with roles in clathrin-mediated endocytosis and vesicle trafficking.28 We found that depletion of HIP1R in aggressive PC cell lines recapitulated the anti-metastatic effects of miR-23b/-27b, including decreased migration, invasion, and anchorage-independent growth. Ectopic HIP1R overexpression led to increased invasion and migration of relatively indolent PC cells. Rescue experiments demonstrated that restoring expression of HIP1R attenuated the anti-migratory effect of miR-23b/-27b in aggressive PC cells. In addition, HIP1R mRNA expression was decreased in the seminal vesicles from mice bearing orthotopic xenografts of PC cells expressing miR-23b/-27b compared with the scrambled control. Together these data indicate that the anti-metastatic actions of miR-23b/-27b in PC cells is due, at least in large part, to repression of HIP1R levels.
HIP1R shares 56% identity at the amino acid level to Huntingtin-interacting protein 1 (HIP1) and is also closely related to the yeast endocytic protein Sla2p.29,30 All three proteins participate in endocytosis by coordinating interactions between clathrin, membrane phospholipids and actin.31 On the basis of biochemical, genetic and knock-out mouse models, HIP1 and HIP1R have partially overlapping functions in endocytosis and other cellular processes.29,32,33 HIP1, which is also overexpressed in PC, is associated with poor outcome,34,35 yet hip1 was not identified as a miR-23b/-27b-regulated gene in our microarray analyses.
Endocytosis is exceedingly complex, involving recognition of receptors and cell-adhesion molecules at the plasma membrane, assembly of clathrin-coated pits, vesicle scission, cargo internalization and intracellular trafficking of endocytic vesicles (in which signaling may proceed) followed by cargo recycling or degradation.36 The mechanisms by which HIP1R promoted invasion, migration and anoikis resistance are currently unknown.
However, the demonstrated role of HIP1R as an adaptor of clathrin-mediated endocytosis provides several novel possibilities. HIP1R may increase migratory and invasive properties of PC cells by promoting internalization of E-cadherin and/or integrins at sites of cell contact or focal adhesions, regulating recycling of membrane proteins, or modulating the internalization, trafficking, stability and/or signaling of growth factor/cytokine receptors. As deregulation of endocytosis is increasingly recognized as an important driver of cancer progression,36 future studies will address whether HIP1R involvement in this key pathway leads to metastasis.
Materials And Methods
Cell culture
The human PC cell line ALVA31 was provided by Drs Stephen Loop and Richard Ostensen at the Department of Veteran Affairs Medical Center, Tacoma, WA, USA.18,37 LNCaP were obtained from and authenticated by American Type Culture Collection, Manassas, VA, USA. PC3-ML were provided by Dr Alessandro Fatatis at Drexel University College of Medicine,38,39 and were authenticated by Genetica DNA Labs, Cincinnati, OH, USA, as previously described.21 All cells were passaged and maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, penicillin/streptomycin (Gibco/Life Technologies, Grand Island, NY, USA), and L-glutamine (Gibco/Life Technologies). PC3-ML cells stably expressing luciferase21 were cultured in RPMI as described above, in 300 μg/ml G418 (Corning, Corning, NY, USA). shGFP and shHIP1R knockdown cells were derived from ALVA31 or PC3-ML cells transduced with the corresponding pLKO.1 shRNA construct and selected over 48 h using 2.5 μg/ml puromycin. Cell cultures were maintained at 37 °C in a humidified atmosphere of 5% CO2.
RNA isolation and reverse transcriptase quantitative RT-qPCR
Total RNA was collected using Trizol according to the manufacturer's protocol (Life Technologies). In all, 500 ng of total RNA were used for reverse transcription using cDNA Archive Kit as per the manufacturer's protocol (Applied Biosystems, Foster City, CA, USA). A total of, 100 ng of cDNA were used for quantitative PCR of HIP1R and 1 ng for 18S rRNA Using the following primers HIP1R (forward 5′-AGATGCTGTGCGGAGGATTGAG-3′; reverse 5′-TGCAGGCTAGTGGATGTCGTCA-3′) and 18S (forward 5′-ACCCG TTGAACCCCATTCGTCA-3′; reverse 5′-GCCTCACTAAACCATCCAATCGG-3′). Quantitative real-time PCR was performed using iQ SYBR Green Supermix (Bio-RAD, Hercules, CA, USA) and run on the ABI Prism 7700 (Applied Biosystems by Life Technologies). The comparative threshold cycle method was used to determine relative mRNA expression levels. Human specific HIP1R Taqman probes, HS00952362_g1, were used for seminal vesicle tissue (Applied Biosystems by Life Technologies).
For all miR-level determinations, total RNA was isolated using Trizol as described above. The TaqMan stemloop RT-qPCR method using the TaqmanH miRNA reverse transcription kit and the TaqmanH miRNA assays (Applied Biosystems by Life Technologies) was used to assess the expression of miR-23b and 27b. Small nuclear RNA U6 served as the internal control for quantification of miRs.
Immunoblotting
Cellular proteins were extracted and separated on 8% SDS-PAGE gels, and western blot analyses were performed using standard procedures as previously described.40 Western blotting of β-actin on the same membrane was used as a loading control. The antibodies used were anti-HIP1R (EMD Millipore, Billerica, MS, USA; AB9882) and anti-β-actin (Santa Cruz Biotechnology, Inc., Dallas, TX, USA; 1616).
Plasmids and lentiviral production
The human miR-23b/-27b precursor (pMIRNA-23b/-27bGFP) and scrambled control (pMIRNA-Scrambled-GFP) cloned into lentiviral vectors (Systems Biosciences, Mountain View, CA, USA) were transfected into LentiX 293T cells (Clontech, Mountain View, CA, USA) and viral particles isolated as previously described.18 GFP expression in transduced ALVA31 and PC3-ML cells was determined by flow cytometry, performed on an Accuri C6 cytometer using CFlow software according to the manufacturer's instructions. pLKO.1 and pLKO.1 shGFP viral vectors were kindly provided by Dr Priyamvada Rai (University of Miami). The nucleotide sequences used to target HIP1R mRNA were obtained from the RNAi Consortium (TRC) human lentiviral shRNA libraries, available from Sigma-Aldrich Company (Sigma-Aldrich, St Louis, MO, USA). To generate oligonucleotides for cloning into pLKO.1, the sense and antisense sequences used to target HIP1R were inserted into the following oligonucleotide sequences: Forward 5′-CCGGGCCCGTCAGATTTGAACGAATCTCGAGATTCGTTCAAATCT GACGGGCTTTTTG-3′ and reverse 5′-AATTCAAAAAGCCCGTCAGATTTGAAC GAATCTCGAGATTCGTTCAAATCTGACGGGC-3′. The oligonucleotides were annealed by heating to 95 °C followed by slow cooling to room temperature. The annealed oligos were ligated into AgeI- and EcoRI-digested pLKO.1 vector. For selection of stable cell line derivatives, cells were transduced with the appropriate constructs 24 h after seeding. Forty-eight hours following transduction, cells were selected in 2.5 μg/ml puromycin for a subsequent 48–72 h. Plasmids pEZXMT01 miRNA 3′-UTR target-expression clones for HIP1R (HmiT021900-MT01) and miRNA Target clone control vector for pEZX-MT01(CmiT000001-MT01) were purchased from GeneCopoeia (Rockville, MD, USA). HIP1R-6myc was a gift from Dr David Drubin (University of California, Berkeley, CA, USA).41
Cell transfections and luciferase assays
Chemically modified antisense nucleotides (antagomiRs) against miR-23b and -27b or control antagomiRs were purchased from Applied Biosystems and transfected at 50 nM each using Lipofectamine 2000 (Life Technologies) according to the manufacturer's protocol. For luciferase-reporter assays, cells were transiently transfected with pEZX-MTO1-HIP1R′3UTR or pEZX-MTO1-EV control-reporter plasmid. Firefly luciferase activities were measured using the Dual Luciferase Assay (Promega, Madison, WI, USA) 48 h after transfection and the results were normalized to Renilla luciferase levels.
Microarray
Gene-expression profiling was performed using Illumina HumanHT-12 V4 microarrays with PC3-ML cells expressing miR-23b/-27b or scrambled control and LNCaP cells transfected with antagomiR-23b plus antagomiR-27b or control antagomiRs. Three independent replicates for each treatment were used. The preparation of the probes and the microarrays were done according to the manufacturer's protocol. Results were analyzed using Partek Genomics Suite 6.6 software (Partek, St Louis, MO, USA). Only genes with a P-value of ≤0.05 and a fold change of 1.5-times or more were considered for further analysis.
Cell proliferation and viability assays
ALVA31, PC3-ML (with stable knockdown of shGFP or shHIP1R) and LNCaP cells (expressing HIP1R or empty vector control) were seeded at an initial density of 10 000 cells/well in a 6-well plate. At the indicated time points, the cells were trypsinized and viable cells (those that exclude trypan blue) were counted using a hemocytometer. Viability assays were performed using CellTiter-Glo Luminescent Cell Viability Assay (Promega) according to the manufacturer's specifications. Cells were seeded at a density of 1000 cells/well in a 96-well plate, and luminometer readings were taken at 1, 3 and 5 days.
Migration and invasion assays
Boyden Chamber Migration and Matrigel Invasion Assays (BD Biosciences, San Jose, CA, USA) were performed using ALVA31 and PC3-ML cells 72 h after transduction with miR-23b/-27b or scrambled control and LNCaP cells 72 h after transfection with antagomiR-23b/-27b or control antagomiR, as previously described.18 Briefly, cells were serum starved for 16 h then 50 000 cells were seeded into the top chambers of the transwell apparatus with Matrigel-coated membranes (24-well insert, 8-mm pore size). Medium supplemented with 10% FBS was used as chemoattractant in the lower chambers. After incubation at 37 °C for 48 h, the top chambers were wiped with cotton wool to remove non-migratory or non-invasive cells. Cells on the lower surface of the membrane were then fixed with cold methanol, stained with 0.01% crystal violet and all cells were counted under a microscope using a light microscope.
Soft agar assays
The capacity of ALVA31 or PC3-ML cells transduced with shHIP1R or shGFP control to grow in soft agar was evaluated as previously described.18,42
Orthotopic xenografts
Studies involving animals were conducted under a protocol approved by the University of Miami Animal Care and Use Committee. PC3-ML Luc cells were transduced with either scrambled or miR-23b/-27b expressing lentivirus as described above. Only cell populations with >95% transduction efficiencies (determined using flow cytometry for GFP as described previously18) were used for implantation. PC3-ML Luc (5× 105) cells expressing miR-23b/-27b or scrambled control were injected into the ventral prostates of 6-week-old athymic nude mice, randomly assigned to experimental groups (Harlan Laboratories, Indianapolis, IN, USA). Injections were performed using a repeating dispenser (Hamilton, Reno, NV, USA) with a tumor take of 70 and 75%, respectively. Surgical sites were sealed with sutures and 9 mm wound clips. Power analysis assumed a 30% decrease in treated cells, with an α=0.05, β= 0.2, and accounting for 20% attrition. Investigators were not blinded in these studies.
Ex vivo imaging
Bioluminescence imaging on the IVIS Spectrum in vivo Imaging System (Perkin Elmer, Waltham, MA, USA) began at 7 days post injection when wound clips were removed, and continued once weekly until time of killing at day 40. D-Luciferin (Invitrogen, Carlsbad, CA, USA) was injected intraperitoneally (150 mg/kg) in sterile PBS 5–10 min before imaging. Following the day 40 imaging time point, mice were killed and the prostate and seminal vesicles were excised. Mice (lacking prostates and seminal vesicles) were reimaged to determine distal metastasis signal, read as photon flux (photons/s/cm2). Prostate and seminal vesicles were placed into individual wells of a 24-well plate in D-Luciferin to extend the signal life and imaged. Bioluminescence imaging emission, read as Photon Flux (photons/s), was quantified using Living Image Software (Perkin Elmer). Tissues were subsequently rinsed with PBS and fixed in 10% buffered formalin for 24 h. After fixation, samples were dehydrated, embedded in paraffin, sectioned at 4 μm. The sections were stained with hematoxylin and eosin and were examined using a Nikon Microphot-FXA microscope and images were captured using a Nikon Coolpix 4300 digital camera at × 200 magnification (Nikon, Tokyo, Japan).
Immunofluorescence
For phalloidin staining to assess cell morphology, cells were plated on glass cover slips in 24-well plates and incubated for 5 days at 37 °C in 5% CO2. Plated cells were fixed in 4% paraformaldehyde for 30 min, permeabilized for 10 min of in 0.2% TrixonX-100, and incubated with Alexa Fluor 594 Phalloidin conjugate (Molecular Probes, Life Technologies) in PBS for 30 min. Coverslips were mounted using SlowFade Gold antifade reagent containing DAPI (Molecular Probes, Life Technologies).
Imaging and analysis
Confocal microscopy was done using a Plan Apo N 60×/1.42 Oil objective on the Olympus FluoView FV1000 confocal microscope (Olympus, Tokyo, Japan). Confocal image stacks were acquired at 800 × 800 pixel resolution and a step size of 0.5–2.0 μm. 2D image reconstruction, as well as long-versus short-axis calculations were performed using ImageJ (NIH, Bethesda, MD, USA). Image merging was done using Adobe Photoshop CS6 (Adobe, San Jose, CA, USA).
Statistical analysis
All normally distributed data were tested for significance using two-tailed Student's t-test. Data with non-normal distribution were log10 transformed and t-tests were performed. Non-parametric Mann–Whitney's tests were done for cell-axis statistical analysis, and mRNA expression levels from seminal vesicle tissue. Oncomine (Compendia Bioscience, Ann Arbor, MI, USA) was used for analysis and visualization.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Funding provided by DOD pre-doctoral fellowship W81XWH-11-1-0314 (RAI/MAR), NIH R01CA132200 and Women's Cancer Association of the University of Miami (KLB). We thank UM investigators Drs Cale Fahrenholtz, Stephanie Peacock and Ning Zhao, as well as Annie Greene and Govindi Samaranayake for helpful technical assistance and guidance. Dr Michael Henry at the University of Iowa and Dr David Drubin at University of California at Berkeley generously provided the PQCXIN-luciferase and HIP1R-6myc constructs respectively.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Lagos-Quintana M, Rauhurt R, Lendeckel W, Tuschi T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 3.Baines AT, Xu D, Der CJ. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem. 2011;3:1787–1808. doi: 10.4155/fmc.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalmay T, Edwards DR. MicroRNAs and the hallmarks of cancer. Oncogene. 2006;25:6170–6175. doi: 10.1038/sj.onc.1209911. [DOI] [PubMed] [Google Scholar]
- 5.Ambs S, Prueitt RL, Yi M, Hudson RS, Howe TM, Petrocca F, et al. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res. 2008;68:6162–6170. doi: 10.1158/0008-5472.CAN-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porkka KP, Pfeiffer MJ, Waltering KK, Vessella RL, Tammela TL, Visakorpi T. MicroRNA expression profiling in prostate cancer. Cancer Res. 2007;67:6130–6135. doi: 10.1158/0008-5472.CAN-07-0533. [DOI] [PubMed] [Google Scholar]
- 7.Coppola V, Maria RD, Bonci D. MicroRNAs and prostate cancer. Endocr Relat Cancer. 2009;17:F1–F17. doi: 10.1677/ERC-09-0172. [DOI] [PubMed] [Google Scholar]
- 8.Hurst DR, Edmonds MD, Welch DR. Metastamir: the field of metastasis-regulatory microRNA is spreading. Cancer Res. 2009;69:7495–7498. doi: 10.1158/0008-5472.CAN-09-2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dykxhoorn DM. MicroRNAs and metastasis: little RNAs go a long way. Cancer Res. 2010;70:6401–6406. doi: 10.1158/0008-5472.CAN-10-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gandellini P, Folini M, Zaffaroni N. Towards the definition of prostate cancer-related microRNAs: where are we now? Trends Mol Med. 2009;15:381–390. doi: 10.1016/j.molmed.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Shi XB, Tepper CG, White RW. microRNAs and prostate cancer. J Cell Mol Med. 2008;12:1456–1465. doi: 10.1111/j.1582-4934.2008.00420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang YL, Wu S, Jiang B, Yin FF, Zheng SS, Hou SC. Role of microRNAs in prostate cancer pathogenesis. Clin Genitourin Cancer. 2015;13:261–270. doi: 10.1016/j.clgc.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 13.Altuvia Y, Landgraf P, Lithwick G, Elefant N, Pfeffer S, Aravin A, et al. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res. 2005;33:2697–2706. doi: 10.1093/nar/gki567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuse M, Kojima S, Enokida H, Chiypmaru T, Yoshino H, Nohata N, et al. Tumor suppressive microRNAs (miR-222 and miR-31) regulate molecular pathways based on microRNA expression signature in prostate cancer. J Hum Genet. 2012;57:691–699. doi: 10.1038/jhg.2012.95. [DOI] [PubMed] [Google Scholar]
- 15.Martens-Uzunova ES, Jalava SE, Dits NF, van Leenders GJ, Møller S, Trapman J, et al. Diagnostic and prognostic signatures from the small non-coding RNA transcriptome in prostate cancer. Oncogene. 2012;31:978–991. doi: 10.1038/onc.2011.304. [DOI] [PubMed] [Google Scholar]
- 16.Sun T, Yang M, Chen S, Balk S, Pomerantz M, Hsieh CL, et al. The altered expression of MiR-221/-222 and MiR-23b/-27b is associated with the development of human castration resistant prostate cancer. Prostate. 2012;72:10. doi: 10.1002/pros.22456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun T, Wang Q, Balk S, Brown M, Lee GSM, kantoff P. The role of microRNA-221 and -222 in androgen-independent prostate cancer cell lines. Cancer Res. 2009;69:3356–3363. doi: 10.1158/0008-5472.CAN-08-4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishteiwy RA, Ward TM, Dykxhoorn DM, Burnstein KL. The microRNA-23b/-27b cluster suppresses the metastatic phenotype of castration-resistant prostate cancer cells. PLoS One. 2012;7:e52106. doi: 10.1371/journal.pone.0052106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stephenson RA, Dinney CP, Gohji K, Ordonez NG, Killion JJ, Fidler IJ. Metastatic model for human prostate cancer using orthotopic implantation in nude mice. J Natl Cancer Inst. 1992;84:951–957. doi: 10.1093/jnci/84.12.951. [DOI] [PubMed] [Google Scholar]
- 20.Liu W, Vivian CJ, Brinker AE, Hampton KR, Lianidou E, Welch DR. Microenvironmental influences on metastasis suppressor expression and function during a metastatic cell's journey. Cancer Microenviron. 2014;7:117–131. doi: 10.1007/s12307-014-0148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yates TJ, Lopez LE, Lokeshwar SD, Ortiz N, Kallifatidis G, Jordan A, et al. Dietary supplement 4-methylumbelliferone: an effective chemopreventive and therapeutic agent for prostate cancer. J Natl Cancer Inst. 2015;107:7. doi: 10.1093/jnci/djv085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brett TJ, Legendre-Guillemin V, McPherson PS, Fremont DH. Structural definition of the F-actin-binding THATCH domain from HIP1R. Nat Struct Mol Biol. 2006;13:121–130. doi: 10.1038/nsmb1043. [DOI] [PubMed] [Google Scholar]
- 23.O'Day E, Lal A. MicroRNAs and their target gene networks in breast cancer. Breast Cancer Res. 2010;12:201. doi: 10.1186/bcr2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Howe EN, Cochrane DR, Richer JK. Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 2011;13:R45. doi: 10.1186/bcr2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.D'Amico AV, Whittington R, Malkowicz SB, Schultz D, Schnall M, Tomaszewski JE, et al. A multivariate analysis of clinical and pathological factors that predict for prostate specific antigen failure after radical prostatectomy for prostate cancer. J Urol. 1995;154:131–138. [PubMed] [Google Scholar]
- 26.Thompson DW, Bracken CP, Goodall GJ. Experimental strategies for microRNA target identification. Nucleic Acids Res. 2011;39:6845–6853. doi: 10.1093/nar/gkr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature. 2008;455:1124–1128. doi: 10.1038/nature07299. [DOI] [PubMed] [Google Scholar]
- 28.Engqvist-Goldstein AE, Zhang CX, Carreno S, Barroso C, Heuser JE, Drubin DG. RNAi-mediated Hip1R silencing results in stable association between the endocytic machinery and the actin assembly machinery. Mol Biol Cell. 2004;15:1666–1679. doi: 10.1091/mbc.E03-09-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gottfried I, Ehrlich M, Ashery U. The Sla2p/HIP1/HIP1R family: similar structure, similar function in endocytosis? Biochem Soc Trans. 2010;38:187–191. doi: 10.1042/BST0380187. [DOI] [PubMed] [Google Scholar]
- 30.Seki N, Muramatsu M, Sugano S, Suzuki Y, Nakagawara A, Ohhira M, et al. Cloning, expression analysis, and chromosomal localization of HIP1R, an isolog of huntingtin interacting protein (HIP1) J Hum Genet. 1998;43:268–271. doi: 10.1007/s100380050087. [DOI] [PubMed] [Google Scholar]
- 31.Boettner DR, Chi RJ, Lemmon SK. Lessons from yeast for clathrin-mediated endocytosis. Nat Cell Biol. 2012;14:2–10. doi: 10.1038/ncb2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hyun TS, Rao DS, Saint-Dic D, Michael LE, Kumar PD, Bradley SV, et al. HIP1 and HIP1r stabilize receptor tyrosine kinases and bind 3-phosphoinositides via epsin N-terminal homology domains. J Biol Chem. 2004;279:14294–14306. doi: 10.1074/jbc.M312645200. [DOI] [PubMed] [Google Scholar]
- 33.Hyun TS, Li L, Oravecz-Wilson KI, Bradley SV, Provot MM, Munaco AJ, et al. Hip1-related mutant mice grow and develop normally but have accelerated spinal abnormalities and dwarfism in the absence of HIP1. Mol Cell Biol. 2004;24:4329–4340. doi: 10.1128/MCB.24.10.4329-4340.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rao DS, Hyun TS, Kumar PD, Mizukami IF, Rubin MA, Lucas PC, et al. Huntingtin-interacting protein 1 is overexpressed in prostate and colon cancer and is critical for cellular survival. J Clin Invest. 2002;110:351–360. doi: 10.1172/JCI15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Yu W, Cai Y, Ren C, Ittmann MM. Altered fibroblast growth factor receptor 4 stability promotes prostate cancer progression. Neoplasia. 2008;10:847–856. doi: 10.1593/neo.08450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mosesson Y, Mills GB, Yarden Y. Derailed endocytosis: an emerging feature of cancer. Nat Rev Cancer. 2008;8:835–850. doi: 10.1038/nrc2521. [DOI] [PubMed] [Google Scholar]
- 37.Loop SM, Rozanski TA, Ostenson RC. Human primary prostate tumor cell line, ALVA-21: a new model for studying the hormonal regulation of prostate tumor cell growth. Prostate. 1993;22:93–108. doi: 10.1002/pros.2990220202. [DOI] [PubMed] [Google Scholar]
- 38.Wang M, Stearns M. Isolation and characterization of PC-3 human prostatic tumor sublines which preferentially metastasize to select organs in S.C.I.D mice. Differentiation. 1991;48:115–125. doi: 10.1111/j.1432-0436.1991.tb00250.x. [DOI] [PubMed] [Google Scholar]
- 39.D'Ambrosio J, Fatatis A. Osteoblasts modulate Ca2+ signaling in bone-metastatic prostate and breast cancer cells. Clin Exp Metastasis. 2009;26:955–964. doi: 10.1007/s10585-009-9286-3. [DOI] [PubMed] [Google Scholar]
- 40.Peacock SO, Fahrenholtz CD, Burnstein KL. Vav3 enhances androgen receptor splice variant activity and is critical for castration-resistant prostate cancer growth and survival. Mol Endocrinol. 2012;26:1967–1979. doi: 10.1210/me.2012-1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Engqvist-Goldstein AE, Kessels MM, Chopra VS, Hayden MR, Drubin DG. An Actin-binding protein of the Sla2/Huntingtin interacting protein 1 family is a novel component of clathrin-coated pits and vesicles. J Cell Biol. 1999;147:1503–1518. doi: 10.1083/jcb.147.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lyons LS, Rao S, Balkan W, Faysal J, Maiorino CA, Burnstein KL. Ligand-independent activation of androgen receptors by Rho GTPase signaling in prostate cancer. Mol Endocrinol. 2008;22:597–608. doi: 10.1210/me.2007-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grasso CS, Wu Y, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chandran UR, Ma C, Dhir R, Bisceglia M, Lyons-Weiler M, Liang W, et al. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer. 2007;7:64. doi: 10.1186/1471-2407-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu YP, Landsittel D, Jing L, Ren B, Liu L, McDonald C, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22:2790–2799. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.