

Abstract
Castration-resistant prostate cancer (CRPC) progresses rapidly and is incurable. Constitutively active androgen receptor splice variants (AR-Vs) represent a well-established mechanism of therapeutic resistance and disease progression. These variants lack the AR ligand-binding domain and, as such, are not inhibited by androgen deprivation therapy (ADT), which is the standard systemic approach for advanced PC. Signaling by AR-Vs, including the clinically relevant AR-V7, is augmented by Vav3, an established AR coactivator in CRPC. Using mutational and biochemical studies, we demonstrated that the Vav3 Diffuse B-cell lymphoma homology (DH) domain interacted with the N-terminal region of AR-V7 (and full length AR). Expression of the Vav3 DH domain disrupted Vav3 interaction with and enhancement of AR-V7 activity. The Vav3 DH domain also disrupted AR-V7 interaction with other AR coactivators: Src1 and Vav2, which are overexpressed in PC. This Vav3 domain was used in proof-of-concept studies to evaluate the effects of disrupting the interaction between AR-V7 and its coactivators on CRPC cells. This disruption decreased CRPC cell proliferation and anchorage-independent growth, caused increased apoptosis, decreased migration, and resulted in the acquisition of morphological changes associated with a less aggressive phenotype. While disrupting the interaction between FL-AR and its coactivators decreased N-C terminal interaction, disrupting the interaction of AR-V7 with its coactivators decreased AR-V7 nuclear levels.
Implications
This study demonstrates the potential therapeutic utility of inhibiting constitutively active AR-V signaling by disrupting coactivator binding. Such an approach is significant, as AR-Vs are emerging as important drivers of CRPC that are particularly recalcitrant to current therapies.
Keywords: Vav3, Vav2, AR-V7, castration resistant prostate cancer
INTRODUCTION
Prostate cancer (PC) is highly prevalent and a leading cause of cancer deaths in men in the United States (1). Advanced PC is treated systemically by androgen deprivation therapy (ADT), which has been the standard of care for nearly 80 years since the recognition that PC is dependent on androgen receptor (AR) signaling for survival and growth (2-4). Despite symptomatic benefits, the disease frequently recurs as castration-resistant prostate cancer (CRPC) (reviewed in 5; 6, 7). Since in the majority of cases, CRPC continues to rely on AR signaling, newer pharmacologic agents with improved capacity to block AR (enzalutamide) and androgen synthesis (abiraterone acetate) are employed (8,9). Despite these newer generation drugs, CRPC remains incurable.
One major mechanism underlying CRPC progression is the expression of constitutively active AR variants (AR-Vs) that lack the AR ligand binding domain and are therefore not readily targeted by current approaches (10-13; reviewed in 14). AR-Vs retain the potent transactivating N-terminal domain (NTD), which is unique in the nuclear receptor family because of the presence of dominant activation motifs (15-17). Multiple AR-Vs have been discovered, but AR-V7 (also known as AR3 or AR1/2/3/CE3) is the most well-studied since it is readily detectable in clinical specimens (18,19, reviewed in 20). AR-Vs confer castration resistance in vitro and in vivo, and their presence in PC tumors and circulating tumor cells denotes poor prognosis (10, 18, 21, 22, 23). A recent study by Antonarakis et al. 2017 (24), which was performed in 202 patients, underlines the clinical significance of AR-V7 in human PC samples by demonstrating a correlation between AR-V7 levels and therapeutic resistance to ADT.
AR-Vs bind as homodimers or as heterodimers with full-length (FL) AR to androgen response elements (AREs) in chromatin (25, 26). The extent to which AR-Vs regulate unique genes (compared to full length AR) to drive PC progression is under active investigation (27-30). Since AR-V activity is critical for CRPC cell survival and resistance to even the newest generation of AR-targeted therapies, these variants are attractive targets for CRPC treatment (31). However, since AR-Vs lack the AR LBD, designing specific, high-affinity drugs is a major challenge (31). An alternative approach is to impede the activity of AR-Vs by inhibiting their interaction with coactivators, many of which are overexpressed in CRPC (32-34).
We have previously demonstrated that AR and AR-V7 signaling is greatly enhanced by the coactivator Vav3 (35-37), a Rho GTPase guanine nucleotide exchange factor (GEF) (38). Much like levels of AR-Vs, levels of Vav3 mRNA increase during progression to castration resistance in PC cell models, xenografts, and the Nkx3.1; Pten mouse PC model (32, 35, 39, 40, 41). Importantly, Vav3 protein levels are elevated in metastatic CRPC human specimens and are prognostic for post-treatment disease recurrence (42). We have also shown that Vav3 confers castration resistance in vitro and in vivo (36, 37). Here, we identified the domains of Vav3 and AR-V7 that interact, generated a reagent to disrupt this interaction, and observed the biological impact resulting from this disruption. Further, we found that a closely related protein to Vav3, Vav2, is also overexpressed in human PC and enhanced AR and AR-Vs activity. We found that Vav protein interaction with the AR N-terminal Tau 5 domain is paradigmatic for other N-terminal interacting coactivators and was critical for AR/AR-V activity as well as CRPC cell survival, proliferation, and migration. This study provides proof-of-concept that disrupting the interaction between AR-Vs and their coactivators is a promising therapeutic strategy for CRPC.
MATERIALS AND METHODS
Cell culture and chemical reagents
The human PC cell lines LNCaP (ATCC catalog no. CRL 1740; batch F-11701), CWR-22Rv1 (CRL-2505, batch 4484055), and PC-3 (ATCC catalog no. CRL 1435; batch F-11154) were obtained from American Type Culture Collection (Manassas, VA). CWR-R1, LNAI, ALVA31, and C4-2B cells were generous gifts from Dr. Elizabeth M. Wilson (University of North Carolina, Chapel Hill, NC), Dr. Priyamvada Rai (University of Miami), Drs. Stephen Loop and Richard Ostensen (Department of Veteran Affairs Medical Center, Tacoma, WA), and Dr. Conor Lynch (Moffitt Cancer Center, Tampa, FL), respectively. LNCaP, 22Rv1, CWR-R1, PC3, and ALVA31 DH-FLAG or empty vector linked to FLAG (EV-FLAG) cells were pools derived following transduction with the corresponding construct and selection using 500 mg/mL of G418 (Sigma, St. Louis, MO). 22Rv1 and LNAI shVav2 cells were obtained from cells transduced with a PLKO.1 shVav2 plasmid and selected in 2.5 μg/mL puromycin (Sigma, St. Louis, MO). Cell culture media (RPMI-1640 and DMEM) were obtained from Cellgro by Mediatech, Inc. (Manassas, VA). Fetal bovine serum (FBS) was obtained from Atlanta Biologicals, Inc. (Lawrenceville, GA). LNCaP, ALVA31, 22Rv1, CWR-R1, and PC3 cell lines were cultured in RPMI supplemented with 100 IU/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine (Life Technologies, Inc.), and 10% FBS or 2% charcoal-stripped serum (CSS). C4-2B cells were cultured in DMEM supplemented with 100 IU/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine (Life Technologies, Inc.) and 10% FBS or 2% charcoal-stripped serum (CSS). R1881 (methyltrienolone) was purchased from PerkinElmer Life and Analytical Sciences (Boston, MA) and used at 1 nM. All cell lines were authenticated on February 2016 using STR (Genetica), and tested for mycoplasma contamination every 6 months using the Mycoplasma PCR Detection kit (Sigma, St. Louis, MO; MP0035-1KT). All cell lines used were negative for mycoplasma, bacteria and fungi contamination.
Plasmids
The following DNA constructs were generously provided: pcDNA3.1 ARv567es (Dr. Stephen Plymate, University of Washington), the constructs for the mammalian two-hybrid assay: Gal4DBD-ARLBD, VP16AD-ARTAD, and Gal4-TataLuc (Dr. Karen Knudsen, Thomas Jefferson University), the ARE luciferase (ARE-luc) (Dr. Zafar Nawaz, University of Miami), the MMTV and GRE luciferase plasmids (Dr. Mona Nemer, University of Ottawa, Canada), and the P5HB AR-V7 wild type and deletion mutants (Dr. Scott Dehm, University of Minnesota, Minneapolis, MN). The nucleotide sequence used to target Vav2 mRNA (CCGGCAAGTGAAACTGGAGGAATTTCTNGAGNAANTCCTCCAGTTTCACTTGTTTTTG) was cloned into a PLK0.1 vector.
Vav3ΔDH was created via site-directed mutagenesis with the following primers: F- Tcagcccaaatgtccagaaaatgagaatttgaaccaaccagttttgctttttggacgacctcagggaga, R- tctccctgaggtcgtccaaaaagcaaaactggttggttcaaattctcattttctggacatttgggctga. DH-FLAG was isolated from Vav3 with the following primers: F- AAGCGGCCGCATGGATTACAAGGATGACGACGATAA, R- AAGAATTCTATAGATAGCTGAAACTGTTTAATTTCACGAAGG.
Reporter gene assays and transfections
A dual plasmid Mouse Mammary Tumor Virus (MMTV)-luciferase system was used, in which one plasmid encodes wild type MMTV promoter while the control plasmid lacks androgen/glucocorticoid response elements (ΔGRE). Non-AR-driven transcriptional activity and transfection efficiency can be accounted for by utilizing the ΔGRE plasmid as a baseline control. All transfections were carried out using Lipofectamine (Invitrogen Life Technologies) according to the manufacturer’s instructions. For luciferase assays, cells were plated at a density of 3.0×105 cells in 35-mm dishes 16–20 h before transfection. Immediately before transfection, media were replaced with unsupplemented DMEM. For PC3 cells, each well was transfected with 1.6 μg of MMTV or ΔGRE reporter plasmids, and a combination of 250 ng pCMV-AR, pcDNA3.1ARv567, or p5Hb-AR-V7; and 500 ng of pIRES-egfp-Vav2, pIRES-egfp-Vav3 wild type, pIRES-egfp-Vav3 mutants, pIRES-SRC-1, or empty vector. DH interference was conducted in PC3, LNCaP, and C4-2B cells with 100 ng of pCMV-3tag1b-DH-FLAG or empty vector (pCMV-3tag1b-FLAG). For determining FL-AR N-C interaction (mammalian two-hybrid assay), PC3 cells were transfected with 500 ng of Gal4DBD-ARLBD, VP16ADARTAD, and Gal4-Tata-Luc. DH interference was conducted in PC3, LNCaP, and C4-2B with 100ng of pCMV-3tag1b-DH-FLAG or empty vector (pCMV-3tag1b-FLAG). After a 6-h incubation with DNA/lipid complexes, cells were re-fed with RPMI supplemented with 2% (CSS) and treated with vehicle or 1 nM R1881. Cells were harvested 48 h after transfection, lysed, and assayed for luciferase activity using the Promega luciferase assay kit (Promega Corp., Madison, WI). Luciferase assays in CWR-R1, LNCaP, C4-2B, and 22Rv1 were performed with the addition of a five-minute DNA incubation with PLUS reagent (Invitrogen Life Technologies). The experiments performed using the dual reporter plasmid MMTV or ΔGRE were analyzed by normalizing the reads of luciferase activitiy of each well to the protein amounts, and the activities from cells expressing the MMTV plasmids to those from cells expressing the ΔGRE control plasmid.]
Luciferase activities were normalized to the protein amounts, and the activities from cells expressing the MMTV plasmids were normalized to those from cells expressing the ΔGRE control plasmid.
Cellular Fractionation
Cells were plated at 2×106 cells per 100 mm dish and grown in 5% CSS for 72 h. Cellular fractionation was performed with the Nuclear and Cytoplasmic Extraction Reagents (NE-PER #78833) according to the manufacturer’s protocol (Thermo Scientific,Waltham, MA). Ten μg of each protein sample were subjected to western blot analysis as described below.
Immunoblotting
Cellular proteins were extracted and separated in 10-12% SDS-PAGE gels, and western blot analyses were performed as previously described (36). The antibodies used were: anti-AR (N-20; 1:1000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti-AR-V7 (1:500; Precision Antibody), anti-Histone (1:1000; Santa Cruz), anti-SOD (1:1000; Santa Cruz), anti-Cleaved PARP (1:1000; Cell Signaling), anti-Vav3 (1:1000, Cell Signaling), anti-Vav2 (Santa Cruz), anti-actin (1:500; Santa Cruz), or anti-FLAG (1:1000, Sigma).
Densitometry was performed using ImageJ software (43).
Cell proliferation assay (trypan blue)
Cells were plated at an initial density of 20,000 per well in 24-well dishes. After 5 days, cells were trypsinized and viable cells were counted by trypan blue exclusion using a hemocytometer.
RNA isolation and reverse transcriptase quantitative RT-qPCR
Total RNA was collected using Trizol according to the manufacturer’s protocol (Life Technologies), and isolated using Direct-zol RNA MiniPrep Plus (Zymo Research, Catalog number R2072). Total RNA was reverse transcribed using a cDNA Reverse Transcription kit (Applied Biosystems, Catalog number 4368814) as per the manufacturer’s protocol. TaqMan probes from Applied Biosystems for FKBP5, UBE2C and GAPDH were used.
Cell proliferation assay and apoptosis assay (Incucyte)
For growth assays, cells were plated in 96-well plates at 5,000 or 7,500 cells/well and transfected with 2% (v/v) of non-perturbing nuclear-restricted green fluorescent label (IncuCyte NucLight Green BacMam 3.0, Essen Bioscience). For apotosis assays, cells were plated in 96-well plates at 10,000 cells/well and transfected with 1% (v/v) apoptosis marker reagent, which is cleaved by activated caspase 3/7, releasing a green fluorescent label (IncuCyte Caspase-3/7 Apoptosis Assay Reagent). After 2 h, cells were incubated in an Incucyte Zoom (Essen Bioscience), acquiring phase and green fluorescent images were obtained at 10X magnification every 2 h. Incucyte Zoom software was used to analyze and graph the results.
Soft Agar Assays
Soft agar assays were performed as previously described (3). ALVA31, CWR-R1, and 22Rv1 plates were incubated for 2, 3, or 4 weeks respectively. Colonies were stained with 0.005% crystal violet and counted using the Bio-Rad Geldoc system.
Immunoprecipitation
HEK293 cells were plated at 3.5×106 cells/100 mm dish. Cells were transfected with 5 μg of PQCXIP AR-V7 or AR and pIRES-egfp-Vav3-myc or pIRES-Vav3-DPC-myc utilizing a calcium-phosphate transfection kit according to the manufacturer’s instructions (ClonTech). For DH interference, 22Rv1 and CWR-R1 cells were plated at 3×106 cells/100 mm dish and transfected with 10 μg DH-FLAG or EV using Lipofectamine reagent and Plus reagent (Invitrogen Life Technologies). After 48 hours, cells were lysed and immunoprecipitation was performed as previously described (36), using nonspecific mouse IgG (2 μg, Santa Cruz), monoclonal mouse anti-myc (2 μg, Invitrogen) or anti-FLAG (2 μg, Sigma), nonspecific rabbit IgG (2 μg, Santa Cruz), rabbit polyclonal anti AR-N20 (2 μg, Santa Cruz), rabbit polyclonal anti-Vav2 (2 μg, Santa Cruz), or rabbit polyclonal anti-Vav3 (2 μg, Millipore).
Immunofluorescence, imaging, and analysis
C4-2B cells were plated on glass cover slips in 24-well plates at 20,000 cells/well, and transfected with DH-FLAH or FLAG empty vector. After 48 hours, cells were fixed in 4% paraformaldehyde for 1 h, permeabilized in 0.2% TrixonX-100 for 10 minutes, and then incubated with Alexa Fluor 594-Phalloidin conjugate (Molecular Probes, Life Technologies). The coverslips were mounted using SlowFade Gold antifade reagent containing DAPI (Molecular Probes, Life Technologies) and imaged using a fluorescent microscope. Images were analyzed and cell body and protrusion lengths were measured using ImageJ software (NIH, Bethesda, MD, USA).
Analysis of human samples datasets
The GSE56701 dataset was analyzed using Galaxy (44, 45) and the pipeline was modeled using Tophat aligner and Cufflinks to analyze mapped transcripts. GSE29650, GSE3325, and GSE6099 datasets were analyzed using the GEO2R online tool from ncbi.nih.gov. The TCGA dataset (provisional dataset for prostate adenocarcinoma) was analyzed using cbioportal.org to build the Kaplan-Meier curves.
Migration assays
22Rv1 and C4-2B cells were serum-starved overnight and seeded at 20,000 cells/well in the top chamber of Boyden Chambers (8 μm pore size, BD Biosciences, San Jose, CA, USA), and placed in 24-well plates. Media supplemented with 10% FBS was placed in the lower chambers as a chemoattractant. After 18 h, cotton wool was used to remove non-migratory cells from the top chambers. Cells on the lower surface of the membrane were fixed in ice-cold methanol for 20 minutes, then stained with 0.01% crystal violet, and counted using a light microscope.
Statistical analysis
Data were graphed and analyzed using Prism 7 (GraphPad) and Statistica 8.0 (Statsoft). Data were tested for normality (Shapiro-Wilk test) and homogeneity of variances (Levene’s test). When both assumptions were met, data were tested for significance (p<0.05) using a two-tailed Student’s T-test (two groups) or Analysis of Variances (ANOVA) (three or more groups). Otherwise, Welch’s correction or non-parametric statistical analyses were used: Mann-Whitney’s test (two groups) and Kruskal-Wallis (three or more groups).
RESULTS
We previously showed that Vav3 increases the transcriptional activity of AR splice variants, including AR-V7, and that an ectopically expressed FLAG-Vav3 fusion protein interacts with AR-V7 (36). To determine if endogenously expressed Vav3 interacted with AR-V7, we performed co-immunoprecipitations in the human CRPC cell line 22Rv1, which expresses both AR-V7 and Vav3. We observed that endogenous Vav3 and AR-V7 were present in the same immunocomplexes, and also found that Vav3 was co-immunoprecipitated with full length (FL) AR (Fig. 1A).
Figure 1. The Vav3 central region [Dbl homology, pleckstrin homology, cysteine rich domain (DPC)] interacts endogenously with full length AR and AR-V7 to enhance FL-AR and AR-V7 transcriptional activities.
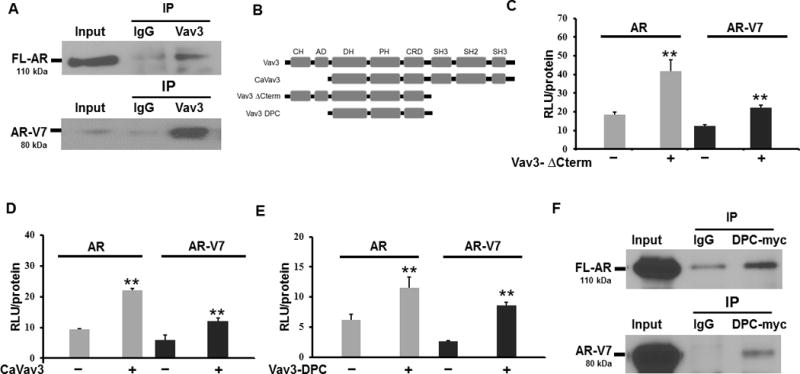
A) Co-immunoprecipitations were in 22Rv1 cell lysates with antibodies against Vav3 (or rabbit IgG as a control). Immuno-complexes were immunoblotted with antibodies against AR or AR-V7. B) A schematic of the subdomains of Vav3 and Vav3 mutants is depicted with: CH = calponin homology domain, AD = acidic domain, DH = Diffuse B-Cell lymphoma homology (GEF) domain, PH = Pleckstrin homology domain, CRD = cysteine rich domain, SH2 = Src homology 2 domain, SH3 = Src homology 3 domain. C) The AR-negative PC cell line PC3 was transfected with the reporter plasmid ARE-Luc, and full length (FL)-AR or AR-V7, as well as Vav3ΔCterm (panel C), CaVav3 (panel D) or Vav3-DPC (panel E) [or equivalent amounts of the corresponding empty vector (EV)]. Cells transfected with FL-AR were treated with 1nM of R1881. Luciferase activity was determined. Data represent one of three experiments performed in triplicate plotting the mean RLU/protein ± SE. Significance was determined using a two-tailed Student’s T-test. F) HEK293 cells were transfected with Vav3-DPCmyc and FL-AR or Vav3-DPCmyc and AR-V7, and co-immunoprecipitation was performed using antibodies to myc or mouse IgG as control. Immunocomplexes were subjected to western blotting and probed with an antibody to the AR N-terminus or myc. A representative experiment of three independent experiments is shown. (** p value < 0.01)
We next sought to identify the minimal necessary and sufficient regions of Vav3 required to enhance AR-V7 activity. We performed reporter gene assays using PC3 cells, a human AR-null PC cell line, transfected with an ARE-Luciferase reporter plasmid plus cDNAs encoding either FL-AR or AR-V7 and Vav3 truncation mutants. The N-terminus, C-terminus, or both of Vav3 were deleted to produce CaVav3, Vav3ΔCterm, and Vav3DPC, respectively (Fig. 1B). As expected based on our previous work, these three Vav3 mutants all retained the capacity for androgen-inducible co-activation of FL-AR (35, 37, 41, 46), but additionally, we found that these three Vav3 truncation mutants enhanced the ligand-independent activity of AR-V7 (Fig. 1C-E). Thus, the Vav3 DPC truncation mutant consisting of the DH, PH and CRD regions retained the capacity to enhance AR-V7. Furthermore, Vav3-DPC interacted with both FL-AR and AR-V7 in co-immunoprecipitation experiments (Fig. 1F).
To refine further the Vav3 functional domains needed to enhance AR-V7 transcriptional activity, we generated two additional Vav3 mutants: Vav3ΔCRD, that lacks the Cysteine-rich Domain (CRD); and Vav3ΔDH, which lacks the DH domain, where the GEF catalytic activity resides (Fig. 2A). We found that Vav3ΔCRD retained the capacity to enhance the activities of both FL-AR and AR-V7 to the same extent as wild type Vav3 (Fig. 2B). However, Vav3ΔDH was ineffective at enhancing either FL-AR or AR-V7 transcriptional activity compared to wild type Vav3 (Fig. 2C), while there were no significant differences between Vav3 and Vav3ΔDH expression levels (Supplemental Figure 1A). These data indicate that while Vav3-mediated co-activation of FL-AR and enhancement of AR-V7 is GEF-independent (35,36), an intact GEF domain (DH domain) was needed.
Figure 2. Vav3 interacts with the TAU5 region of AR-V7 to enhance full length FL-AR and AR-V7 transcriptional activities in a Vav3 DH domain-dependent manner.
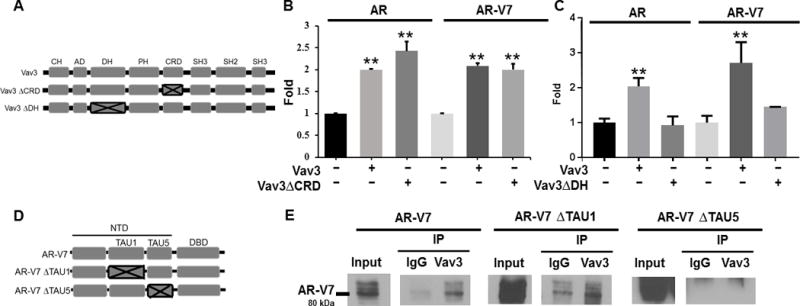
A) A schematic of the subdomains of Vav3 and Vav3 mutants is depicted with: CH = calponin homology domain, AD = acidic domain, DH = Diffuse B-Cell lymphoma homology (GEF) domain, PH = Pleckstrin homology domain, CRD = cysteine rich domain, SH2 = Src homology 2 domain, SH3 = Src homology 3 domain. PC3 cells (AR-negative) were transfected with FL-AR or AR-V7, the reporter plasmid ARE-Luc, and a Vav3 mutant lacking the CRD (Vav3ΔCRD) (panel B), Vav3 mutant lacking the DH domain (Vav3ΔDH) (panel C), or equivalent amounts of the corresponding empty vector. Cells transfected with FL-AR were treated with R1881 (1nM). Luciferase activity was determined 48 h after transfection for all panels. Data in panels B and C are compilations of 3-4 experiments performed in triplicate plotting the mean fold ± SEM. Significance was determined using a two-tailed Student’s T-test. D) A schematic of AR-V7 and deletion mutants is depicted with: NTD = N-terminal domain, AF-1 = Activation Function-1, TAU1 = Transactivation unit 1, TAU5 = Transactivation unit 5, DBD = DNA-binding domain. E) The human PC AR-null cell line ALVA-31 was transfected with AR-V7 or its deletion mutants: AR-V7 ΔTAU1 or AR-V7 ΔTAU5; and co-immunoprecipitations were performed using anti-Vav3 antibody or rabbit IgG antibody as a control. Immunocomplexes were immunoblotted probing for AR-V7. A representative experiment of two independent experiments is shown. (** p value < 0.01).
Conversely, we mapped the region in AR that interacted with Vav3. Since AR-Vs lack the ligand-binding domain (LBD) and the AR N-terminal domain (NTD) is known to possess strong activation functions, we focused on the AR NTD. Within the NTD, the activation function-1 (AF-1) region is essential for AR transactivation and for interaction with several co-regulators (reviewed in 47; 48). We examined the possibility that Vav3 would interact with the AF-1 region, which contains Transactivation Unit 1 (TAU1) and Transactivation Unit 5 (TAU5). We expressed AR-V7 or its deletion mutants lacking either TAU1 or TAU5 (Fig. 2D), in the AR-null but Vav3-expressing human PC cell line ALVA-31, and performed co-immunoprecipitations with endogenous Vav3. We found that AR-V7 lacking TAU5 was not co-immunoprecipitated with Vav3 whereas AR-V7 lacking TAU1 retained interaction with Vav3. These data indicate that Vav3 interaction with AR-V7 requires TAU5.
Because Vav3 required its DH domain to interact with FL-AR and AR-V7, we examined if the DH domain of Vav3 was sufficient for this interaction. By performing co-immunoprecipitations, we determined that FL-AR and Vav3 DH domain linked to FLAG (DH-FLAG), and AR-V7 and DH-FLAG, were present in the same protein complexes (Fig. 3A and 3B). Once we established that both FL-AR and AR-V7 interacted with the Vav3 DH domain, we postulated that overexpressing this domain would interfere with FL-AR:Vav3 and AR-V7:Vav3 interactions and that disrupting these interactions would reduce Vav3-mediated enhancement of FL-AR and AR-V7 transcriptional activities. We found that expressing the Vav3 DH domain disrupted FL-AR:Vav3 and AR-V7:Vav3 interactions, as shown by co-immunoprecipitations (Fig. 3C). As expected, disruption of these interactions greatly reduced Vav3-mediated augmentation of FL-AR and blocked AR-V7 transcriptional activities (Fig. 3D). Expression of Vav3 DH domain did not affect endogenous Vav3 levels (Fig. 3E). These data show that expression of the Vav3 DH domain was sufficient to disrupt FL-AR:Vav3 and AR-V7:Vav3 physical interactions and enhancement of AR (FL and variant) transcriptional activities.
Figure 3. The Vav3 DH domain is sufficient for full length FL-AR and AR-V7 interaction, disrupts FL-AR:Vav3 and AR-V7:Vav3 interaction, and blocks Vav3 enhancement of FL-AR and AR-V7 transcriptional activities.
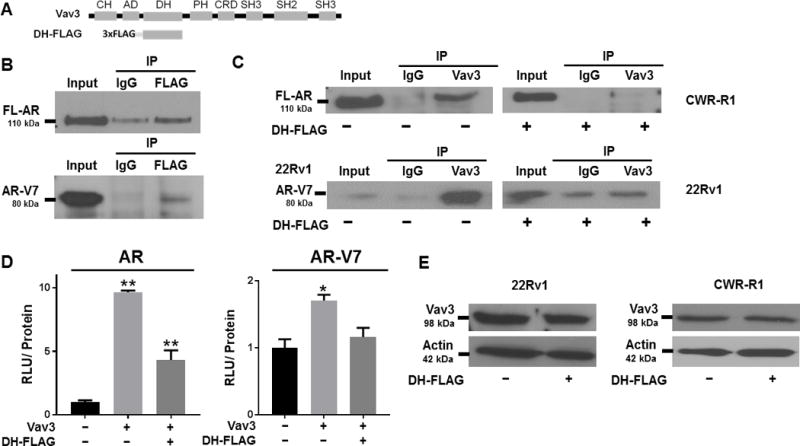
A) A schematic of Vav3 and the Vav3 DH domain linked to FLAG is depicted with: CH = calponin homology domain, AD = acidic domain, DH = Diffuse B-Cell lymphoma homology (GEF) domain, PH = Pleckstrin homology domain, CRD = cysteine rich domain, SH2 = Src homology 2 domain, SH3 = Src homology 3 domain. B) PC3 cells (AR-negative) were transfected with FL-AR and DH-FLAG, and HEK293 cells were transfected with AR-V7 and DH-FLAG, and co-immunoprecipitations were performed using antibodies to mouse IgG control or FLAG antibody. Immunocomplexes were immunoblotted with an anti-AR N-terminal antibody. A representative experiment of two independent experiments is shown. C) CWR-R1 and 22Rv1 cells were transfected with DH-FLAG or the FLAG vector, and co-immunoprecipitations were performed using antibodies to rabbit IgG control or Vav3. Immunocomplexes were immunoblotted with antibodies against FL-AR in CWR-R1 and AR-V7 in 22Rv1. A representative experiment of 2 independent experiments is shown. (** p value < 0.01, * p value < 0.05). D) PC3 cells were transfected with FL-AR or AR-V7, the dual plasmid luciferase reporter system: MMTV or ΔGRE, and a combination of Vav3 or empty vector, and DH-FLAG or FLAG empty vector, and luciferase activity was determined. Data represent 3 independent experiments performed in triplicate, showing the mean ± SE. Non-parametric Kruskal Wallis test were performed, for FL-AR: p value = 0.01; for AR-V7: p value = 0.05. E) 22Rv1 and CWR-R1 cells stably expressing DH-FLAG or its EV control were harvested and immunoblotting was performed using an anti-VAV3 antibody and anti-actin as the loading control.
To extend our work on the role of Vav family proteins on AR signaling in PC, we sought to determine the role of Vav2 (Fig. 4A), which is closely related to Vav3 in terms of primary sequence and structure (49). To assess the possible relevance of Vav2 with respect to AR-V7 signaling in PC, we evaluated whether these genes were coexpressed in PC patient samples by querying two existing independent human datasets. The first dataset, from Hornberg et al., 2011 (GSE29650) (50), contained microarray data of 10 bone metastases samples from different PC patients with relatively high levels of AR-V7. Computational analysis revealed that Vav2, as well as Vav3 mRNAs, were present in all the AR-V7-high samples at significant, detectable levels (Fig. 4B). The coexpression of Vav2, Vav3, and AR-V7 was confirmed in a second dataset of PC patient samples from Antonarakis et al., 2014 (GSE56701) (51) in AR-V7-expressing circulating tumor cells (data not shown). The Vav2 gene is amplified in 10% of patient samples [Prostate Adenocarcinoma (Broad/Cornell, Nat Genet 2012, and TCGA dataset prostate adenocarcinoma)] and is overexpressed in 34% of PC patients samples in [Prostate Adenocarcinoma (MSKCC, Cancer Cell 2010) ]. Moreover, analysis of the Varambally et al. 2005 dataset (GSE3325) (52) revealed that Vav2 mRNA expression levels were elevated in primary PC patient samples compared to levels in normal prostate samples, and Vav2 mRNA levels were further elevated in metastatic PC samples (Fig. 4C). Similar results were obtained from the Tomlins et al., 2007 dataset (GSE6099) (53) (data not shown). Interestingly, patients who presented higher levels of Vav2 at the time of prostate biopsy also exhibited decreased disease-free survival (DFS) (p value = 0.001) (Fig. 4D), and reduced overall survival (data not shown, p value = 0.0113) (TCGA dataset prostate adenocarcinoma, from Cbioportal.org).
Figure 4. Expression of Vav2, a member of the Vav family, is elevated in human CRPC samples, and Vav2 interacts endogenously with and promotes the transcriptional activities of full length FL-AR and AR-V7.
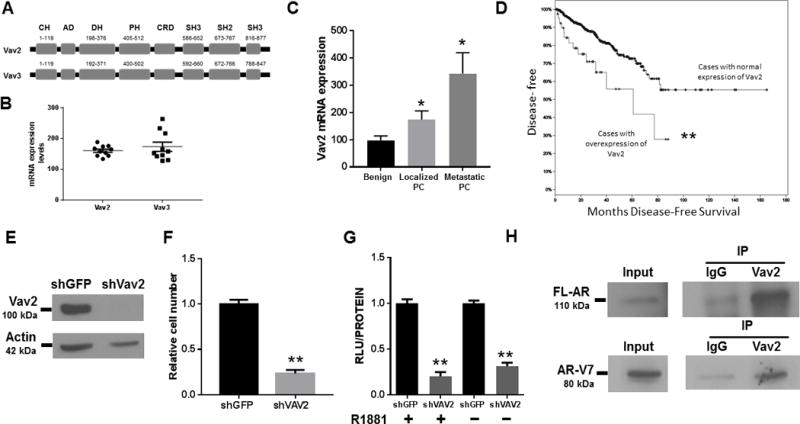
A) A schematic of Vav2 and Vav3 structure with the amino acid position of each domain. CH = calponin homology domain, AD = acidic domain, DH = Diffuse B-Cell lymphoma homology (GEF) domain, PH = Pleckstrin homology domain, CRD = cysteine rich domain, SH2 = Src homology 2 domain, SH3 = Src homology 3 domain. B) Vav3 and Vav2 mRNA levels are co-expressed in human CRPC bone metastases which contain relatively high AR-V7 levels (dataset of Hornberg et al., 2011 (50)). C) Vav2 mRNA levels are elevated in PC and metastatic PC compared to benign tissue (dataset of Varambally et al., 2005). Kruskal Wallis test was performed, p = 0.02. D) Vav2 overexpression is prognostic for decreased disease-free survival. The Kaplan-Meier curve was built using the TCGA Prostate Adenocarcinoma dataset (n= 499). The upper curve denotes cases with no abnormal expression of Vav2, while the lower curve represents the cases in which Vav2 mRNA levels are upregulated (z-score threshold ± 2.0). P-value = 0.001. E) Immunoblotting was performed on 22Rv1 cell lysate using an anti-VAV2 antibody and anti-actin as the loading control. Data shown represent 1 of 2 independent experiments. F) Stable Vav2 depletion in 22Rv1 cells (vs control shGFP) decreased cell number. Data shown represent 2 independent experiments performed in quintuplicate. Independent Student’s T-test, p value < 0.001. G) 22Rv1 cells were transfected with the dual plasmid luciferase reporter system: MMTV or ΔGRE described in Materials and Methods. Luciferase activity was determined 48 h after transfection. Data shown represent 3 independent experiments performed in triplicate, showing the mean ± SE, and normalized to their shGFP controls. Unpaired T-test, p value = 0.001 in the presence of androgen; and, p value < 0.001 in the absence of androgen. H) 22Rv1 cells were harvested and co-immunoprecipitations were performed using antibodies to rabbit IgG control or Vav2. Immunocomplexes were immunoblotted with antibodies against FL-AR or AR-V7 (** p value < 0.01, * p value < 0.05).
Because of the potential significance of Vav2 in PC, we examined the effect of depleting Vav2 on 22Rv1 cell number and FL-AR/AR-V7 activity. Stable depletion of Vav2 using shRNA (Fig. 4E) reduced cell number (Fig. 4F) and decreased AR ligand-dependent and ligand-independent transcriptional activity (Fig. 4G). A similar result for ligand-dependent AR activity was observed in the CRPC cell line LNAI, a derivative of androgen-dependent LNCaP cells (data not shown). Co-immunoprecipitations in 22Rv1 cells revealed that endogenous Vav2 was co-immunoprecipitated with endogenous FL-AR and AR-V7 (Fig. 4H). Similar results were obtained in an additional CRPC cell line, CWR-R1 (data not shown).
As was observed for Vav3, we found that expressing the Vav3 DH domain disrupted Vav2-mediated enhancement of FL-AR and AR-V7 transcriptional activities (Fig. 5A). We examined the effects of the Vav3 DH domain against a distinct and well-characterized AR coactivator, SRC-1 which is known to modulate AR and AR-V7 activities (54-56). Expression of the Vav3 DH domain blocked AR transcriptional enhancement by SRC-1 (Fig. 5B). Expression of only the Vav3 DH domain caused decreased FL-AR transcriptional activity (Fig. 5C) in LNCaP, a human cell line that contains high levels of the coactivator SRC-1 (54).
Figure 5. Vav3 DH domain disrupts the enhancement of FL-AR and AR-V7 by multiple coactivators, decreases FL-AR N-C interaction and AR-V7 nuclear levels.
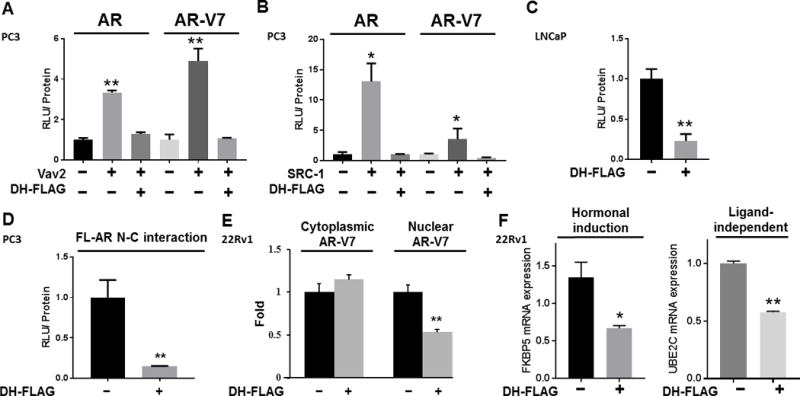
A) AR-null PC3 cells were transfected with a combination of FL-AR or AR-V7, DH-FLAG or FLAG empty vector, Vav2 or empty vector, and the dual luciferase reporter system: MMTV or ΔGRE. Luciferase activity was determined. Experiments were performed in triplicate, showing the mean ± SE (Kruskal-Wallis, p value < 0.01). B) PC3 cells were transfected with a combination of FL-AR or AR-V7, DH-FLAG or empty vector, SRC-1 or empty vector, and the dual luciferase reporter system: MMTV or ΔGRE. Luciferase activity was determined 48 h after transfection. Experiments were performed in triplicate, showing the mean ± SE. (Kruskal-Wallis, p value < 0.05) C) LNCaP cells were transfected with MMTV or ΔGRE and luciferase activity was measured as described previously. Experiments were performed in triplicate, showing the mean ± SE (Kruskal-Wallis, p value = 0.01). D) PC3 cells were transfected with vectors encoding AR fusion proteins to asses AR N/C interaction in a mammalian two-hybrid assay. The plasmids contained AR LBD linked to Gal4 DNA-binding domain (Gal4DBD-ARLBD) and AR N-terminal domain fused to the transcriptional activation domain of VP16 (VP16AD-ARTAD). Cells were also transfected with the reporter plasmid Gal4-Tata-Luc and DH-FLAG or its empty vector linked to FLAG. Cells were treated with 1nM of R1881 and luciferase activity was measured. Data represents one experiment of two performed in triplicate, showing the mean ± SE (Independent Student’s T-test, p value = 0.01). E) 22Rv1 cells stably expressing DH-FLAG or its empty vector linked to FLAG were grown in 5% charcoal stripped-serum for 72 hours. Cells were fractionated and immunoblotted for AR-V7, histone and SOD. Protein levels were normalized to SOD (cytoplasmic fraction) or histone (nuclear fraction). Protein levels were quantified from four independent experiments and plotted as the mean ± SE (Student’s T-test, p value<0.01). F)The expression of FKBP5 and UBE2C was examined in 22Rv1 cells transfected with DH-FLAG or EV by RT-qPCR analysis and normalized to GAPDH mRNA. Hormonal induction was the ratio of FKBP5 mRNA levels from cells stimulated with R1881 compared to vehicle control treated cells (Unpaired T-test, p value = 0.03). For UBE2C quantification, cells described above were cultured in 2% CSS (Unpaired T-test, p value < 0.01). A representative experiment of two independent experiments performed in triplicate is shown. (** p value < 0.01, * p value < 0.05).
We pursued the mechanism underlying the decrease in FL-AR and AR-V7 transcriptional activities in cells expressing the Vav3 DH domain to determine if effects were specific to FL-AR and AR-V7. The androgen-inducible transcriptional activity of FL-AR depends on the interaction of the AR LBD (C-terminus) with its N-terminus, a phenomenon known as the N/C interaction (57). This intramolecular interaction is influenced by co-regulators. For example, our lab showed that Vav3 potently increases FL-AR N/C interaction (37,46). We performed mammalian two-hybrid assays with two AR fusion proteins: the AR LBD linked to the Gal4 DNA-binding domain (Gal4DBD-ARLBD) and the AR N-terminus fused to the transcriptional activation domain of VP16 (VP16AD-ARTAD). When AR N/C interaction occurs, both fusion proteins interact, causing transcription of the reporter plasmid Gal4-Tata-Luc. We found that expression of the Vav3 DH domain reduced FL-AR N/C interaction (Fig. 5D).
Since the splice variant AR-V7 lacks the C-terminal LBD and is constitutively active, AR-V7 activity relies on AR-V7 presence in the nucleus, where it can enhance the transcription of downstream targets. We previously found that Vav3 increases nuclear levels of AR-V7 (36). 22Rv1 cells stably expressing DH-FLAG or empty vector linked to FLAG as a control were established. Subcellular fractionation of DH-FLAG-overexpressing cells exhibited 50% less nuclear AR-V7 levels compared to cells expressing FLAG only as a control (Fig. 5E). These data indicate that the interaction of AR-V7 with its coactivators plays an essential role in determining nuclear levels of AR-V7.
Moreover, expression of DH-FLAG decreased androgen-inducible AR target gene (FKBP5) expression as well as ligand-independent expression of the AR-V7 target gene UBE2C (Fig. 5F).
Coactivators enhance FL-AR and AR-V7 transcriptional activity, while promoting an oncogenic transcriptional program that is thought to drive PC progression and metastasis (56, 58– 61). Therefore, we used the Vav3 DH domain for proof-of-concept experiments to study the cellular effects of disrupting the interaction of FL-AR and AR-V7 with endogenous coactivators that interact with both the AR and AR-Vs N-terminal domain. We generated LNCaP, 22Rv1, CWR-R1, PC3 (AR-null), and ALVA 31 (AR-null) cell lines stably expressing Vav3 DH-FLAG or its empty vector linked to FLAG. DH-FLAG was expressed at similar levels for CRPC and AR-null cell lines (Supplemental Fig. 1 A). Cell proliferation was measured by two techniques: in real-time using a live-cell imaging microscope (Incucyte Zoom, Essen Bioscience) and by tyrpan blue exclusion using a hemocytometer. While LNCaP, 22Rv1, and CWR-R1 expressing Vav3 DH-FLAG exhibited decreased proliferation (Fig. 6A and Supplemental Fig. 1C), PC3 (Fig. 6A and Supplemental Fig. 1D) and ALVA31 (Supplemental Fig. 1C) cell viability was not affected, suggesting that the effects of the Vav3 DH domain were specific for AR. Expression of DH-FLAG did not decrease FL-AR (Supplemental Fig. 1E), AR-V7 (Supplemental Fig. 1E and Supplemental Fig. 1F), Vav3 (Supplemental Fig. 1F), or Vav2 levels (Supplemental Fig. 1G). We found that the observed difference in cell viability was due to an increase in apoptosis in cells expressing DH-FLAG, as measured using an apoptotic marker activated by Caspase 3/7 in CWR-R1 (Fig. 6B). As expected, no difference in apoptosis was observed in PC3 cells expressing DH-FLAG or its empty vector FLAG control (Fig. 6B). We also examined PARP cleavage and quantified the number of dead cells in 22Rv1, CWR-R1, and ALVA 31 cell lines. In accordance with our previous experiments, expression of DH-FLAG increased cell death (Supplemental Fig. 2A and 2B) in 22Rv1 and CWR-R1 expressing DH-FLAG compared to controls. In contrast, there was no difference in cell viability between AR-null ALVA 31 cells expressing DH-FLAG or FLAG control. This suggests that interfering with the coactivation of FL-AR and AR-V7 decreases cell growth by promoting apoptosis.
Figure 6. Expression of Vav3 DH domain inhibits anchorage dependent and anchorage independent growth, and induces apoptosis in AR-expressing cells.
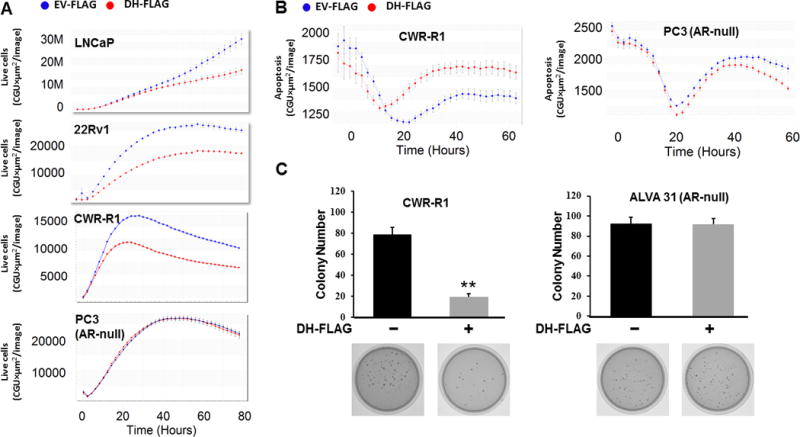
A) The androgen-dependent cell line LNCaP, the CRPC cell lines 22Rv1 and CWR-R1, and the AR-null PC3 cell line stably expressing DH-FLAG or empty vector linked to FLAG (EV-FLAG) were transfected with a non-perturbing nuclear restricted green fluorescent label and incubated in an Incucyte Zoom (Essen Bioscience), acquiring phase and green fluorescent images. Graphs show the mean ± SE for the 12 replicates for each condition. B) CWR-R1 and PC3 cells stably expressing DH-FLAG or empty vector linked to FLAG (EV-FLAG) were transfected with an apoptosis marker reagent, which is cleaved by activated caspase 3/7, releasing a green fluorescent label. Cells were then incubated in an Incucyte Zoom, acquiring phase and green fluorescent images. Graphs show the mean ± SE for the 6 replicates for each condition. C) CWR-R1 and the AR-null PC cell line ALVA31 stably expressing DH-FLAG or empty vector were assessed for soft agar growth. Average colony number per plate ± SEM was plotted. Data represent two independent experiments performed in triplicate. (Student’s T-test, p value<0.01).
Soft agar assays in 22Rv1 (data not shown) and CWR-R1 (Fig. 6C) cells showed that cells expressing DH-FLAG exhibited decreased anchorage-independent growth than the control cells, while the AR-null cell line ALVA 31 showed no difference in anchorage-independent growth upon expression of the DH domain (Fig. 6C). Hence, disrupting the interaction between FL-AR and AR-V7 with their coactivators decreases anchorage-dependent and -independent growth, and increases apoptosis.
To determine the role of the interaction between AR and coactivators in cell aggressiveness, we examined cell migration and morphology. Using Boyden chamber assays, we found that disrupting the interaction between FL-AR and AR-V7 with their coactivators reduced cell motility in 22Rv1 (Fig. 7A) and C4-2B (data not shown) cells, and changed cell morphology (Fig. 7B). As visualized using fluorescent phalloidin staining for F-actin, data were quantified by measuring the length of total protrusions for each cell, normalized to cell body length. We found that disrupting the interaction between AR and its coactivators rendered cells more rounded and with shorter protrusions (Fig. 7C), consistent with less motile cells and a less aggressive phenotype.
Figure 7. Disruption of coactivator interactions with FL-AR or AR-V7 decreases cell migration and introduces morphological changes.
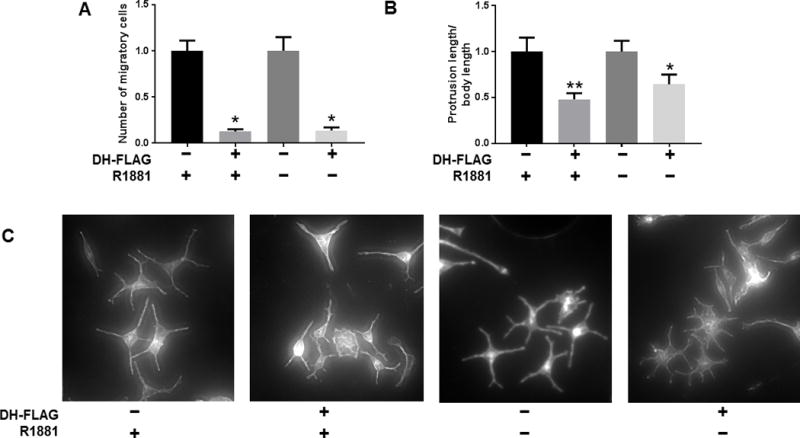
A) 22Rv1 cells stably expressing DH-FLAG or empty vector linked to FLAG were seeded in Boyden Chambers for migration assays. Cells expressing DH-FLAG were normalized to their corresponding EV controls. Data show one experiment out of two performed in triplicate, showing the mean ± SE. For conditions with R1881: Independent T-test, p value = 0.01; for conditions without R1881: Independent T-test, p value = 0.02. B) The CRPC cell line C4-2B transiently expressing DH-FLAG or empty vector linked to were kept in vehicle or 1nM of R1881, and stained for Phalloidin immunofluorescence and DAPI. The total length of protrusions for each cell was measured and divided by cell body length. The average ± SE is shown. Cells expressing DH-FLAG were normalized to their EV controls. For conditions with R1881: n= 13, Mann Whitney test, p value = 0.008; for conditions without R1881: n = 15, Mann Whitney test, p value = 0.02. C) Representative images of C4-2B at 10X stained with Phalloidin. (** p value < 0.01, * p value < 0.05).
DISCUSSION
As expression and function of AR-V7 (and other constitutively active AR variants) is a key mechanism underlying resistance to current PC treatments (10, 18, 21-24), it is imperative to develop novel AR inhibitors that target the N-terminal domain, which is present in both FL-AR and AR splice variants (reviewed in 51). Since AR-Vs may function either as homodimers or heterodimers with FL-AR (13, 22) and promote ligand-independent AR activity (13, 25, 26), therapeutic modalities must address both possible modes of AR-V action. Targeting the AR DNA-binding domain (DBD), common to FL and AR-Vs, has been pursued. Given that the DBD is the most conserved domain across the entire steroid/nuclear receptor family, generating selective inhibitors is challenging. The AF-1 region, located in the N-terminal domain, is also shared by FL AR and AR-Vs and is critical for AR transcriptional activity (48). Efforts to target this region are also underway. However, the NTD has a disordered structure making it difficult to design selective inhibitors. Our results support the importance of channeling research efforts towards targeting the AR AF-1 region, since this would disrupt AR interaction with coactivators, in turn preventing FL-AR N-C interaction and decreasing AR-V7 nuclear levels, leading to decreased cell proliferation and acquisition of an aggressive phenotype.
Here, we show for the first time that Vav3 interacted via its DH domain with endogenous FL-AR and AR-V7 to enhance AR transcriptional activity. The Vav3 binding site on AR was mapped to the TAU5 region in the AR AF-1 domain, although additional interaction sites are possible. We also identified a novel AR coactivator: Vav2, another member of the Vav family, which is upregulated in PC human samples, and is prognostic for poor outcome. Like Vav3, Vav2 enhanced and interacted with endogenous FL-AR and AR-V7. We used the expression of the Vav3 DH domain, which consists of 178 amino acids and adopts a distinct structure, in a proof-of-principle approach to evaluate the effects of disrupting FL-AR and AR-V7 interaction with ectopic or endogenously expressed coactivators in multiple PC cell lines. Disruption of these critical interactions decreased AR-expressing PC anchorage-dependent and anchorage-independent proliferation, increased apoptosis and decreased migration accompanied by morphological changes. Most importantly, these effects were specific to PC cells expressing AR, since expression of the Vav3 DH domain had no effect in two distinct AR-null PC cell lines. These results are in agreement with those reported by Nakka et al., 2013 (56); who demonstrated that disrupting the FL-AR and the p160 coactivator interface is a sound therapeutic approach in CRPC. Our results extend these findings by demonstrating the importance of targeting AR N-terminal-interacting coactivators to reduce AR and AR-V7 transcriptional activity, PC cell growth, survival, and migration. Mechanistically, we found that FL-AR interaction with coactivators depended on the well-characterized interaction between the AR amino and carboxyl termini (N-C interaction (critical for its activation and transcriptional activity). On the other hand, disruption of AR-V7 interaction with coactivators led to decreased AR-V7 nuclear levels. Since disrupting AR-V7 interaction with coactivators decreased AR-V7 nuclear levels but did not increase AR-V7 cytoplasmic levels, coactivators interacting with the TAU5 region of AR-V7 may increase AR-V7 stability in the nucleus. This increased stability may be achieved by protecting AR-V7 ubiquitination sites from detection, delaying AR-V7 degredation in the nucleus, and thus prolonging its transcriptional activity. In addition, AR-V7 interaction with coactivators such as Vav2 and Vav3 may also disrupt and compete with inhibitory factors, such as FOXO1, for the AR-V7 TAU5 binding site (62).
Peptidomimetic conjugates (MCPs) are peptoids that display bioactive ligands and are resistant to proteases (63, 64). Such conjugates have been used to antagonize AR. MCPs, such as MCP6, prevent intermolecular interactions between AR and its coactivators by changing the AR conformation (64). However, MCP6 shows no effect in the human CRPC cell line 22Rv1. The reason MCP6 fails to inhibit AR-V-containing 22Rv1 may be that MCP6 competes for androgen binding to the AR LBD (64), and may not interfere with binding of coactivators to the NTD. Similarly, the peptidomimetics used by Ravindranathan et al., 2013 (65) disrupt the interaction between FL-AR LBD and its coactivators. However, these peptidomimetics are unable to inhibit AR-V7 activity. Our results support the need for novel inhibitors against the AR NTD with the capacity to target both FL-AR as well as AR-Vs and that are effective even in the setting of overexpression of AR coactivators, as we have modeled here.
The current study indicates that targeting the interaction between FL-AR or AR-V7 and their coactivators is sufficient to inhibit PC and CRPC proliferation, as well as survival and migration, and thereby could serve as a therapeutic modality in advanced disease.
Supplementary Material
Acknowledgments
Drs. Scott Dehm, Elizabeth M. Wilson, Maria Mudryi, Zafar Nawaz, Mona Nemer, Karen Knudsen, and Stephen Plymate generously provided reagents and/or cells. We are grateful to Drs. Cale Fahrenholtz, Fayi Wu (University of Miami) and Leah Lyons (Nova Southeastern University) for helpful advice. We thank Dr. Vladlen Slepak for use of the fluorescence microscope. Research performed in this manuscript was supported by NIH grant CA132200 (to KLB), NIH pre-doctoral fellowship F30AG038275 (SOP), and developmental funds from the Sylvester Comprehensive Cancer Center (KLB).
Grants: R01CA132200 (KLB), Sylvester Comprehensive Cancer Center Developmental funds (KLB), F30AG038275 (SOP).
Footnotes
DISCLOSURE STATEMENT: The authors have nothing to disclose.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer research. 1941;1:293–297. doi: 10.3322/canjclin.22.4.232. [DOI] [PubMed] [Google Scholar]
- 3.Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends in Endocrinology & Metabolism. 2010;21:315–324. doi: 10.1016/j.tem.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taplin ME, Balk SP. Androgen receptor: a key molecule in the progression of prostate cancer to hormone independence. Journal of cellular biochemistry. 2004;91:483–190. doi: 10.1002/jcb.10653. [DOI] [PubMed] [Google Scholar]
- 5.Lonergan PE, Tindall DJ. Androgen receptor signaling in prostate cancer development and progression. Journal of carcinogenesis. 2011;10:20. doi: 10.4103/1477-3163.83937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nacusi LP, Tindall DJ. Androgen receptor abnormalities in castration-recurrent prostate cancer. Expert review of endocrinology & metabolism. 2009;4:417–422. doi: 10.1586/eem.09.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohler JL, Gregory CW, Ford OH, Kim D, Weaver CM, Petrusz P, et al. The androgen axis in recurrent prostate cancer. Clinical cancer research. 2004;10:440–448. doi: 10.1158/1078-0432.ccr-1146-03. [DOI] [PubMed] [Google Scholar]
- 8.De Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. New England Journal of Medicine. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. New England Journal of Medicine. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 10.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. New England Journal of Medicine. 2014;371:1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer research. 2013;73:483–489. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clinical cancer research. 2011;17:5913–5925. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proceedings of the national academy of sciences. 2010;107:16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ware KE, Garcia-Blanco MA, Armstrong AJ, Dehm SM. Biologic and clinical significance of androgen receptor variants in castration resistant prostate cancer. Endocrine-related cancer. 2014;21:T87–T103. doi: 10.1530/ERC-13-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dehm SM, Regan KM, Schmidt LJ, Tindall DJ. Selective Role of an NH2-Terminal WxxLF Motif for Aberrant Androgen Receptor Activation in Androgen Depletion–Independent Prostate Cancer Cells. Cancer research. 2007;67:10067–10077. doi: 10.1158/0008-5472.CAN-07-1267. [DOI] [PubMed] [Google Scholar]
- 16.He B, Gampe RT, Kole AJ, Hnat AT, Stanley TB, An G, et al. Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Molecular cell. 2004;16:425–438. doi: 10.1016/j.molcel.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 17.Kemppainen JA, Lane MV, Sar M, Wilson EM. Androgen receptor phosphorylation, turnover, nuclear transport, and transcriptional activation. Specificity for steroids and antihormones. Journal of Biological Chemistry. 1992;267:968–974. [PubMed] [Google Scholar]
- 18.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion–resistant growth. Cancer research. 2009;69:2305–2313. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jernberg E, Thysell E, Ylitalo EB, Rudolfsson S, Crnalic S, Widmark A, et al. Characterization of prostate cancer bone metastases according to expression levels of steroidogenic enzymes and androgen receptor splice variants. PloS one. 2013;8:e7740. doi: 10.1371/journal.pone.0077407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo J. Development of AR-V7 as a putative treatment selection marker for metastatic castration-resistant prostate cancer. Asian journal of andrology. 2016;18:580. doi: 10.4103/1008-682X.178490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Del Re M, Biasco E, Crucitta S, Derosa L, Rofi E, Orlandini C, et al. The detection of androgen receptor splice variant 7 in plasma-derived exosomal RNA strongly predicts resistance to hormonal therapy in metastatic prostate cancer patients. European Urology. 2017;71:680–687. doi: 10.1016/j.eururo.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 22.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. The Journal of clinical investigation. 2010;120:2715–2730. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, True LD, et al. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PloS one. 2011;6:e27970. doi: 10.1371/journal.pone.0027970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antonarakis E, Lu C, Luber B, Wang H, Chen Y, Zhu Y, et al. Clinical Significance of Androgen Receptor Splice Variant-7 mRNA Detection in Circulating Tumor Cells of Men With Metastatic Castration-Resistant Prostate Cancer Treated With First- and Second-Line Abiraterone and Enzalutamide. Urologic Oncology. 2017;35:1–9. doi: 10.1200/JCO.2016.70.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao B, Qi Y, Zhang G, Xu D, Zhan Y, Alvarez X, et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget. 2014;5:1646–1656. doi: 10.18632/oncotarget.1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu D, Zhan Y, Qi Y, Cao B, Bai S, Xu W, et al. Androgen receptor splice variants dimerize to transactivate target genes. Cancer research. 2015;75:3663–3671. doi: 10.1158/0008-5472.CAN-15-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. Journal of Biological Chemistry. 2012;287:19736–19749. doi: 10.1074/jbc.M112.352930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer research. 2012;72:3457–3462. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu J, Lonergan PE, Nacusi LP, Wang L, Schmidt LJ, Sun Z, et al. The cistrome and gene signature of androgen receptor splice variants in castration resistant prostate cancer cells. The Journal of urology. 2015;193:690–698. doi: 10.1016/j.juro.2014.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shafi AA, Putluri V, Arnold JM, Tsouko E, Maity S, Roberts JM, et al. Differential regulation of metabolic pathways by androgen receptor (AR) and its constitutively active splice variant, AR-V7, in prostate cancer cells. Oncotarget. 2015;6:31997. doi: 10.18632/oncotarget.5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan SC, Selth LA, Li Y, Nyquist MD, Miao L, Bradner JE, et al. Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic acids research. 2015:gkv262. doi: 10.1093/nar/gkv262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong Z, Liu Y, Lu S, Wang A, Lee K, Wang LH, et al. Vav3 oncogene is overexpressed and regulates cell growth and androgen receptor activity in human prostate cancer. Molecular Endocrinology. 2006;20:2315–2325. doi: 10.1210/me.2006-0048. [DOI] [PubMed] [Google Scholar]
- 33.Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, Wilson EM. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer research. 2001;61:4315–4319. [PubMed] [Google Scholar]
- 34.Yuan X, Balk SP. Mechanisms mediating androgen receptor reactivation after castration. In: Elsevier, editor. Urologic Oncology: Seminars and Original Investigations. Vol. 27. 2009. pp. 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lyons LS, Burnstein KL. Vav3, a Rho GTPase guanine nucleotide exchange factor, increases during progression to androgen independence in prostate cancer cells and potentiates androgen receptor transcriptional activity. Molecular endocrinology. 2006;20:1061–1072. doi: 10.1210/me.2005-0346. [DOI] [PubMed] [Google Scholar]
- 36.Peacock SO, Fahrenholtz CD, Burnstein KL. Vav3 enhances androgen receptor splice variant activity and is critical for castration-resistant prostate cancer growth and survival. Molecular Endocrinology. 2012;26:1967–1979. doi: 10.1210/me.2012-1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao S, Lyons LS, Fahrenholtz CD, Wu F, Farooq A, Balkan W, Burnstein KL. A novel nuclear role for the Vav3 nucleotide exchange factor in androgen receptor coactivation in prostate cancer. Oncogene. 2012;31:716–727. doi: 10.1038/onc.2011.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Movilla N, Bustelo XR. Biological and regulatory properties of Vav-3, a new member of the Vav family of oncoproteins. Molecular and cellular biology. 1999;19:7870–7885. doi: 10.1128/mcb.19.11.7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Banach-Petrosky W, Jessen WJ, Ouyang X, Gao H, Rao J, Quinn J, et al. Prolonged exposure to reduced levels of androgen accelerates prostate cancer progression in Nkx3. 1; Pten mutant mice. Cancer research. 2007;67:9089–9096. doi: 10.1158/0008-5472.CAN-07-2887. [DOI] [PubMed] [Google Scholar]
- 40.Holzbeierlein J, Lal P, LaTulippe E, Smith A, Satagopan J, Zhang L, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. The American journal of pathology. 2004;164:217–227. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lyons LS, Rao S, Balkan W, Faysal J, Maiorino CA, Burnstein KL. Ligand-independent activation of androgen receptors by Rho GTPase signaling in prostate cancer. Molecular endocrinology. 2008;22:597–608. doi: 10.1210/me.2007-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin KT, Gong J, Li CF, Jang TH, Chen WL, Chen HJ, Wang LH. Vav3-rac1 signaling regulates prostate cancer metastasis with elevated Vav3 expression correlating with prostate cancer progression and posttreatment recurrence. Cancer research. 2012;72:3000–3009. doi: 10.1158/0008-5472.CAN-11-2502. [DOI] [PubMed] [Google Scholar]
- 43.Abràmoff MD, Magalhães PJ, Ram SJ. Image processing with ImageJ. Biophotonics international. 2004;11:36–42. [Google Scholar]
- 44.Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome biology. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blankenberg D, Kuster GV, Coraor N, Ananda G, Lazarus R, Mangan M, et al. Galaxy: a web-based genome analysis tool for experimentalists. Current protocols in molecular biology. 2010:19–10. doi: 10.1002/0471142727.mb1910s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu F, Peacock SO, Rao S, Lemmon SK, Burnstein KL. Novel interaction between the co-chaperone Cdc37 and Rho GTPase exchange factor Vav3 promotes androgen receptor activity and prostate cancer growth. Journal of Biological Chemistry. 2013;288:5463–5474. doi: 10.1074/jbc.M112.390963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocrine reviews. 2007;28:778–808. doi: 10.1210/er.2007-0019. [DOI] [PubMed] [Google Scholar]
- 48.McEwan IJ. Molecular mechanisms of androgen receptor-mediated gene regulation: structure-function analysis of the AF-1 domain. Endocrine-related cancer. 2004;11:281–293. doi: 10.1677/erc.0.0110281. [DOI] [PubMed] [Google Scholar]
- 49.Bustelo XR. Regulatory and signaling properties of the Vav family. Molecular and cellular biology. 2000;20:1461–1477. doi: 10.1128/mcb.20.5.1461-1477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hörnberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, et al. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PloS one. 2011;6:e19059. doi: 10.1371/journal.pone.0019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Antonarakis ES, Chandhasin C, Osbourne E, Luo J, Sadar MD, Perabo F. Targeting the N-terminal domain of the androgen receptor: a new approach for the treatment of advanced prostate cancer. The oncologist. 2016;21:1427–1435. doi: 10.1634/theoncologist.2016-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer cell. 2005;8:393–406. doi: 10.1016/j.ccr.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 53.Tomlins SA, Mehra R, Rhodes DR, Cao X, Wang L, Dhanasekaran SM, et al. Integrative molecular concept modeling of prostate cancer progression. Nature genetics. 2007;39:41–51. doi: 10.1038/ng1935. [DOI] [PubMed] [Google Scholar]
- 54.Agoulnik IU, Vaid A, Bingman WE, Erdeme H, Frolov A, Smith CL, et al. Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer research. 2005;65:7959–7967. doi: 10.1158/0008-5472.CAN-04-3541. [DOI] [PubMed] [Google Scholar]
- 55.Bevan CL, Hoare S, Claessens F, Heery DM, Parker MG. The AF1 and AF2 domains of the androgen receptor interact with distinct regions of SRC1. Molecular and cellular biology. 1999;19:8383–8392. doi: 10.1128/mcb.19.12.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakka M, Agoulnik IU, Weigel NL. Targeted disruption of the p160 coactivator interface of androgen receptor (AR) selectively inhibits AR activity in both androgen-dependent and castration-resistant AR-expressing prostate cancer cells. The international journal of biochemistry & cell biology. 2013;45:763–772. doi: 10.1016/j.biocel.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Askew EB, Minges JT, Hnat AT, Wilson EM. Structural features discriminate androgen receptor N/C terminal and coactivator interactions. Molecular and cellular endocrinology. 2012;348:403–410. doi: 10.1016/j.mce.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Attard G, Richards J, de Bono JS. New strategies in metastatic prostate cancer: targeting the androgen receptor signaling pathway. Clinical Cancer Research. 2011;17:1649–1657. doi: 10.1158/1078-0432.CCR-10-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Halkidou K, Gnanapragasam VJ, Mehta PB, Logan IR, Brady ME, Cook S, et al. Expression of Tip60, an androgen receptor coactivator, and its role in prostate cancer development. Oncogene. 2003;2:2466–2477. doi: 10.1038/sj.onc.1206342. [DOI] [PubMed] [Google Scholar]
- 60.Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an overview. Endocrine reviews. 2002;23:175–200. doi: 10.1210/edrv.23.2.0460. [DOI] [PubMed] [Google Scholar]
- 61.Ueda T, Mawji NR, Bruchovsky N, Sadar MD. Ligand-independent activation of the androgen receptor by interleukin-6 and the role of steroid receptor coactivator-1 in prostate cancer cells. Journal of Biological Chemistry. 2002;277:38087–38094. doi: 10.1074/jbc.M203313200. [DOI] [PubMed] [Google Scholar]
- 62.Bohrer LR, Liu P, Zhong J, Pan Y, Angstman J, Brand LJ, et al. FOXO1 binds to the TAU5 motif and inhibits constitutively active androgen receptor splice variants. The Prostate. 2013;73:1017–1027. doi: 10.1002/pros.22649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Levine PM, Garabedian MJ, Kirshenbaum K. Targeting the Androgen Receptor with Steroid Conjugates: Miniperspective. Journal of medicinal chemistry. 2014;57:8224–8237. doi: 10.1021/jm500101h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Y, Dehigaspitiya DC, Levine PM, Profit AA, Haugbro M, Imberg-Kazdan K, et al. Multivalent Peptoid Conjugates Which Overcome Enzalutamide Resistance in Prostate Cancer Cells. Cancer Research. 2016;76:5124–5132. doi: 10.1158/0008-5472.CAN-16-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ravindranathan P, Lee TK, Yang L, Centenera MM, Butler L, Tilley WD, et al. Peptidomimetic targeting of critical androgen receptor–coregulator interactions in prostate cancer. Nature communications. 2013;4:1923. doi: 10.1038/ncomms2912. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.