

Abstract
Atypical E2F transcription factors (E2F7 and E2F8) function as key regulators of cell cycle progression and their inactivation leads to spontaneous cancer formation in mice. However, the mechanism of the tumor suppressor functions of E2F7/8 remain obscure. In this study we discovered that atypical E2Fs control tumor angiogenesis, one of the hallmarks of cancer. We genetically inactivated atypical E2Fs in epithelial and mesenchymal neoplasm and analyzed blood vessel formation in three different animal models of cancer. Tumor formation was either induced by application of 7,12-Dimethylbenz(a)anthracene/12-O-Tetradecanoylphorbol-13-acetate or by Myc/Ras overexpression. To our surprise, atypical E2Fs suppressed tumor angiogenesis in all three cancer models, which is in a sharp contrast to previous findings showing that atypical E2Fs promote angiogenesis during fetal development in mice and zebrafish. Real-time imaging in zebrafish displayed that fluorescent-labeled blood vessels showed enhanced intratumoral branching in xenografted E2f7/8-deficient neoplasms compared with E2f7/8-proficient neoplasms. DLL4 expression, a key negative inhibitor of vascular branching, was decreased in E2f7/8-deficient neoplastic cells, indicating that E2F7/8 might inhibit intratumoral vessel branching via induction of DLL4.
Introduction
During development, the formation of new blood vessels (vasculogenesis) and blood vessels derived thereof (angiogenesis) is a tightly regulated process, resulting in a quiescent stable vasculature composed of arterioles, venules and capillaries.1 Angiogenesis can be transiently induced from this quiescent vasculature, for example, during wound healing.2 During tumor development, however, angiogenesis is almost continuously turned on, enabling tumor progression.3 The unstable, leaky and highly unorganized tumor vasculature not only gains access to oxygen and nutrients, but also provides a way for cancer cells to disseminate to distal organs.2, 3 In this light, angiogenic therapies that normalize tumor vasculature have been put forward, as they relieve tumor hypoxia and inhibit metastasis,4, 5 and could also improve the efficacy of radiotherapy, chemotherapy and immunotherapy.5
The E2F family of transcription factors consists of eight genes, which encode proteins that are generally classified either as activator (E2F1–E2F3) or repressor (E2F4–8).6, 7 E2F factors are key regulators of the cell cycle,6, 7 and therefore suspected regulators of human cancer. E2F amplification, overexpression or deletion is observed in a wide array of human cancers,8 although the exact contribution of these events to cancer is largely unknown. Studies in mice however point to dual roles for E2Fs in tumor development, suggesting that the role of E2Fs in cancer may be cell type specific, and may depend on the oncogenic background.8
The atypical E2Fs, E2F7 and E2F8 (E2F7/8), are unique members of the E2F family of transcription factors as they contain two instead of one DNA binding domain, lack a Retinoblastoma and Dimerization Partner domain, and instead form hetero- or homo-dimers.6, 9 Interestingly, although E2Fs are key regulators of the cell cycle, embryonically lethal E2f7/8 double-knockout (DKO) mice do not display significant proliferative defects, but instead display vascular defects and widespread apoptosis, demonstrating critical functions for E2F7/8 beyond proliferation.9 Intriguingly, recent findings suggest a role for E2F7/8 in human cancer. For example, deregulation of E2F7/8 is observed in various cancers.8 And although E2F7/8 are classified as transcriptional repressors, suggesting tumor suppressive functions, E2F8 was found to be overexpressed in human lung and hepatocellular carcinoma, and to promote tumor growth of human lung and liver cancer cells in xenograft studies.10, 11 However, recent studies from our lab did reveal tumor suppressive roles for E2F7/8 in mouse models of liver and skin cancer. We demonstrated that conditional deletion of E2f7/8 in hepatocytes resulted in spontaneous formation of hepatocellular carcinomas and loss of E2f7/8 in keratinocytes accelerated carcinogen-induced squamous cell carcinoma formation.12, 13 However, the biological mechanisms through which E2F7/8 control tumor development are largely unknown. As tumor angiogenesis represents one of the hallmarks of cancer and atypical E2Fs regulate developmental angiogenesis,9, 14 here we investigated a potential role for E2F7/8 in tumor angiogenesis, utilizing mouse and zebrafish models of cancer. We discovered that cancer cell-specific loss of atypical E2Fs enhances tumor angiogenesis and branching of tumor blood vessels.
Results and discussion
E2f7/8-deficient skin tumors display enhanced angiogenesis
Recently, we demonstrated that keratinocyte-specific deletion of E2f7/8 enhances tumor growth and aggressiveness in a two-stage mouse skin carcinogenesis model.13 In this model, skin tumors were induced in wildtype (WT) and keratinocyte-specific E2f7/8 DKO mice by painting the back skin with application of 7,12-Dimethylbenz(a)anthracene and 12-O-Tetradecanoylphorbol-13-acetate. To investigate if E2F7/8 regulate tumor angiogenesis in these skin tumors, we performed immunohistochemical staining for Factor VIII, an established marker for blood vessels, in control and keratinocyte-specific E2f7/8 DKO papillomas. This analysis revealed enhanced tumor angiogenesis in E2f7/8 DKO versus WT papillomas (Figures 1a and b). A similar trend was observed in squamous cell carcinomas (not shown). Because transcriptional control of E2F1 is a key mechanism through which E2F7/8 regulate developmental processes, as shown for apoptosis9 and liver polyploidization,15 and because E2F1 itself regulates tumor angiogenesis16, 17, 18 and its expression was increased in E2f7/8 DKO papillomas (Figure 1c), we next explored if E2F7/8 regulate tumor vascularization in an E2F1-dependent manner. For this purpose tumor angiogenesis was examined in E2f1/7/8 triple knockout (TKO) papillomas. Notably, Factor VIII staining of TKO papillomas revealed that additional deletion of E2f1 did not significantly change the extent of tumor angiogenesis compared with DKO papillomas (Figures 1a and b). Therefore we conclude that E2F7/8 regulate angiogenesis during papilloma formation independent of E2F1, although it cannot be excluded that its loss is compensated for by other activator E2Fs.
Figure 1.

E2F7/8 repress tumor angiogenesis in a two-stage skin carcinogenesis model. (a) Immunohistochemical staining for Factor VIII in wildtype (WT), E2f7/8 double knockout (DKO) or E2f7/8/1 triple knockout (TKO) papillomas. Papillomas were obtained from a previously performed two-stage (application of 7,12-Dimethylbenz(a)anthracene/12-O-Tetradecanoylphorbol-13-acetate) skin carcinogenesis study.13 The brown staining shows factor VIII positive cells. Arrows indicate micro blood vessels (MBV) in each genetic group. (b) Quantification of MBV as depicted in (a). MBV were counted in 25, 21 and 13 fields in wildtype, DKO and TKO papillomas, respectively. Wildtype and DKO papillomas were harvested from six mice, TKO from three mice. Quantified data present the average±s.e.m. (c) qPCR analysis of Nrp1a, Vegfa, E2f1, E2f7 and E2F8 mRNA levels analyzed in wildtype (n=6) and DKO (n=9) papillomas. Quantified data present the average±s.d. compared with the indicated controls. **P<0.01; ***P<0.001; ns, not significant.
Recently, we showed that E2F7/8, in cooperation with HIF1, control vascular development through direct transcriptional stimulation of VEGFA,14 and motor neuron development through direct transcriptional repression of NRP1.19 As VEGFA and NRP1 both regulate embryonic and tumor angiogenesis,20, 21 E2F7/8 may regulate tumor angiogenesis through transcriptional control of these genes. Therefore we explored if Vegfa and Nrp1 mRNA levels were deregulated in the E2f7/8 DKO papillomas. However, despite the fact the we could confirm the stimulatory role of E2f7/8 in Vegfa expression in two primary keratinocyte cell lines in vitro (Supplementary Figures S1a, b), Vegfa and Nrp1 mRNA levels were not significantly changed in E2f7/8 DKO papillomas (Figure 1c), while the E2F7/8 repressed target E2f19 was significantly increased, confirming functional E2f7/8 deletion (Figure 1c). These data therefore show that E2F7/8 inhibit papilloma vascularization, and likely do so independent of VEGFA and NRP1.
Loss of E2F7/8 in Myc/Ras-induced sarcomas promotes tumor angiogenesis
To determine whether E2F7/8 can regulate tumor angiogenesis in a different oncogenic setting, we transformed WT and E2f7/8 DKO mouse embryonic fibroblasts (MEFs) with the Myc and Ras oncogenes, hereafter referred to as control MEFs and E2F7/8 DKO MEFs, respectively (Supplementary Figure S2). MEFs were grafted subcutaneously into athymic nude mice (Figure 2a). Microscopic analysis showed that the morphology did not differ between control and E2f7/8 DKO sarcomas (Figure 2b). To analyze the amount of angiogenesis within the grafts, we assessed the influx of endothelial cells (ECs) from the host into solid tumor mass. To this end, ECs were labeled with fluorescent Isolectin B4, an EC-specific marker, and were quantified by confocal microscopy. Similar to the skin cancer model, we found a marked increase in the number of ECs in E2f7/8 DKO compared with control tumors (Figures 2c and d). We also found reduced Vegfa mRNA levels (and increased E2f1 mRNA levels) in E2f7/8 DKO tumors (Figure 2e). From these data we conclude that E2f7/8 also inhibit tumor angiogenesis in a xenograft model for sarcomas driven by Myc and Ras oncogenes, and again likely do so independent of Vegfa which levels are reduced. To test if the enhanced tumor angiogenesis in E2f7/8 DKO tumors could result from enhanced EC migration (influx), an in vitro scratch assay was performed.22 In this experiment we assessed the potential of either E2f7/8 DKO and control MEFs-conditioned medium to close a scratch made in a confluent layer of human umbilical cord vein endothelial cells (HUVECs). HUVECs cultured in E2f7/8 DKO-conditioned medium showed a decreased closure of the scratch compared with controls (supplementary Fig. S3a, b). Similar results were obtained when HUVECs were replaced with WT MEFs or mouse embryonic ECs (not shown). These data suggest that loss of E2F7/8 does not stimulate tumor angiogenesis by enhanced secretion of a factor that stimulates endothelial migration.
Figure 2.

Absence of E2F7/8 in mouse xenograft tumors leads to increased tumor vascularization.(a) Schematic representation of xenograft positions and experimental setup. MEFs were injected with a total of one million cells subcutaneously into athymic nude mice. The diameter of the tumors was monitored every 2 days. After 8 days, when the first tumors reached a diameter of 1 cm, all mice were euthanized and tumors harvested. (b) Hematoxylin and eosin (H&E) staining of control and E2f7/8 DKO xenograft tumors. Dashed black line indicates tumor border. (c) Staining and quantification (d) of intratumoral endothelial cell (Isolectin B4) and nuclei (TO-PRO-3) of control and E2f7/8 DKO xenograft tumors (n=8 tumors per genotype). For each tumor, five fields (× 400 magnification) were quantified. White scale bars in (c) indicate 10 μM. (e) Messenger RNA expression of Vegfa, E2f1, Cdc6 and E2f7 in harvested E2f7/8 DKO and control xenografted MEF tumors (n=8 tumors per genotype). Quantitative PCR analysis was used to analyze mRNA levels. Messenger RNA levels in E2f7/8 DKO tumors are depicted compared with control tumors. (f) Quantification (similar as in (d)) of tumor volume, in vivo BrdU incorporation, and phospho-histone 3 (PH3)-positive and TUNEL-positive cells as determined in the outer border zone of control and E2f7/8 DKO xenograft tumors. All quantified data present the average±s.e.m. compared with the indicated controls. *P<0,05; ***P<0,005; ns, not significant; a.u., arbitrary units.
Although we previously demonstrated that genetic ablation of E2f7/8 enhances tumor growth in a carcinogen-induced skin cancer model,13 in this study E2f7/8 DKO MEF tumors were instead significantly smaller compared with controls (Figure 2f). And although E2F7/8 are critical regulators in preventing apoptosis in vivo,9 which could explain the reduced tumor size, quantification of terminal deoxynucleotidyl transferase dUTP nick end labeling-positive cells revealed a comparable incidence of apoptosis between control and E2f7/8 DKO grafts (Figure 2f). Therefore we next tested if tumor proliferation was affected. Strikingly, in vivo BrdU incorporation studies demonstrated a significant reduction in DNA replication in E2f7/8 DKO tumors, while staining of tumor sections with the mitosis marker phospho-histone 3 did not show differences between control and E2f7/8 DKO tumors (Figure 2f). This indicates that E2f7/8 DKO tumors proliferate slower, which likely contributes to their reduced size. Notably, mice deficient for activator E2Fs also have a delayed cell cycle progression.23 Importantly, although the growth rate of E2f7/8 DKO tumors was delayed (Figure 2f), the growth of E2f7/8 DKO MEFs in vitro was not delayed (Supplementary Figure S2a-b), arguing for a role of the tumor microenvironment. In this regard it is interesting to mention that the subcutaneous space, in which the MEFs are engrafted, is relatively poorly vascularized, and that transplanted tumors depend on HIF1 for their growth in this relatively hypoxic environment.24 And because E2F7/8 are critical mediators of HIF functions in vivo,14, 19 E2F7/8 may be required to support the growth of subcutaneously engrafted, hypoxic tumors. In response to genotoxic stress, however, E2F7/8 may limit the growth of tumors as we observed that E2f7/8 DKO skin tumors are larger in carcinogen-induced skin cancer.13 However, future experiments are required to address if E2F7/8 indeed differentially affect cellular proliferation in response to hypoxic or genotoxic stress, and whether this occurs in a HIF1-dependent manner. Another factor that could explain why E2f7/8 DKO papillomas are larger,13 and transplanted E2f7/8 DKO MEFs tumors smaller than their controls, may be the fact that E2f7/8 DKO papillomas benefit for 3 months from the increased tumor vascularization, whereas this is only eight days in the E2f7/8 DKO MEF tumors (Figure 2).
Inactivation of E2F7/8 stimulates hyperbranching of intratumoral blood vessels and decreases DLL4 expression
To investigate the tumor angiogenesis phenotype of E2f7/8-deficient tumors in more detail, we grafted Ras/Myc transformed control and E2f7/8 DKO MEFs into Tg(fli1:eGFP) zebrafish, in which endothelial cells (ECs) are labeled green. We used this xenograft zebrafish model to monitor in real-time tumor angiogenesis, and the dissemination and metastasis of neoplastic cells (Figure 3a).25 In line with the skin cancer and mouse xenograft tumor models, E2f7/8 DKO MEFs grafts triggered an increased influx of ECs, quantified as the amount of GFP signal within the boundaries of the tumor 48 h post injection (hpi) (Figure 3b). Tumor size measurements revealed that E2f7/8 DKO MEFs grafts were comparable to controls (Figure 3b). This is consistent with the absence of either genotoxic and hypoxic stress in this model. Furthermore, time-lapse imaging showed a continuous movement of tumor cells within the graft and, in addition, cells entering EC (GFP-positive) tube-like structures (Figure 3c, and supplementary movie). Metastatic cells were detected throughout the fish 48 hpi, but within the existing circulatory system and with increased density in the caudal vein (CV, Figure 3d). However, quantification of the number of cells present in the CV 48 hpi showed no difference in metastatic rate between E2f7/8 DKO compared with control MEFs (Figure 3d). Of note, we sporadically detected cells outside the vasculature (Figure 3e). These cells could give rise to metastases, although we cannot exclude the possibility that these DiI- or DiD-positive cells are macrophages that have scavenged fluorescent cell debris or phagocytized MEFs. Therefore we conclude that the absence of E2F7/8 in this xenograft zebrafish model does not impact the dissemination of neoplastic cells, but does enhance tumor angiogenesis, consistent with the findings from the mouse tumor studies (Figures 1 and 2).
Figure 3.

Absence of E2f7/8 in zebrafish xenograft tumors leads to increased tumor vascularization. (a) Schematic representation of xenograft positions and experimental setup. Labeled MEFs were injected 48 h post fertilization (hpf) in the perivitelline space on the yolk sac, ventrally of the sub intestinal vein (SIV) and posterior of the common cardinal vein (CCV or duct of Cuvier). Grafts tend to evoke an angiogenic response from the SIV, but frequently also from the CCV (3a’). In addition, neoplastic cells from the injection site are able to enter the circulation via the vessels formed by tumor angiogenesis and often attach or simply get stuck in the caudal vein (CV) region (3a’’). Reprinted with permission from Magliozzi, R. et al.31 (b) Representative images and quantification of tumor angiogenesis and size of control (Red; DiI; n=10) and E2f7/8 DKO tumors (Red; DiI; n=10) injected in Tg(fli1a:gfp) zebrafish. (c) Time-lapse series that shows metastasizing cell (dashed line) from the tumor into the vasculature. Insets display metastasizing cell only. (d) Representative image and quantification of MEFs metastasis in the trunk region. (e) Example of extravasation of (E2F7/8 DKO) MEFs into the surrounding tissue. Abbreviations: CV, caudal vein; CCV, common cardinal vein; hpi, hours post injection. All quantified data present the average±s.d. compared with the indicated controls in at least three independent experiments. ***P<0.05; ns, not significant.
Enhanced tumor angiogenesis can result not only from advanced capillary sprouting, but also from excessive vessel branching.3 For this reason we quantified the number of intratumoral blood vessel branching points in the zebrafish xenograft model. Notably, blood vessels in E2f7/8 DKO tumors show an almost four times higher number of branching points compared with control tumors (Figure 4a). As the Delta-like 4 (Dll4)-Notch pathway is a key regulator of blood vessel branching during developmental and tumor angiogenesis,26, 27 we tested whether components of this pathway were deregulated. We found that deletion of E2f7/8 both in vitro and in engrafted tumors, resulted in decreased expression of the Notch ligand Dll4, and of the Dll4/Notch1 target Hey1, however the latter did not reach statistical significance (Figures 4b and c). The observed increase in tumor angiogenesis upon E2f7/8 deletion is in line with the decreased expression of Dll4, as Dll4 is known to have a repressive function in blood vessel branching.1 Similar to the mouse tumor xenografts, Vegfa mRNA levels in the grafts were reduced (data not shown).
Figure 4.
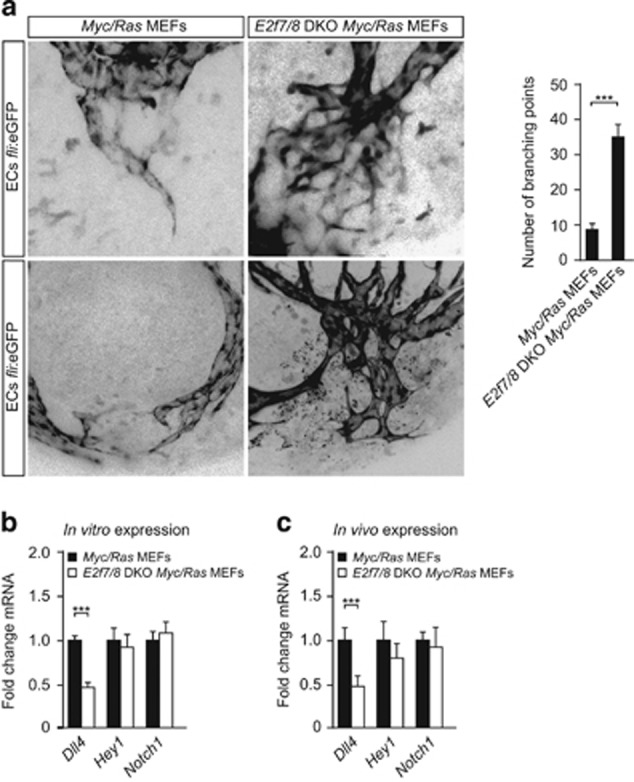
Loss E2f7/8 in xenografted MEF tumors in zebrafish leads to increased intratumoral blood vessel branching. Experimental setup similar as described in Figure 3a. (a) Representative image and quantification (n=10) of hyperbranching in control and E2f7/8 DKO tumors injected in Tg(fli1a:gfp) (black color; endothelial cells). (b) Indicated mRNA levels measured in control or E2f7/8 DKO MEFs cultured under standard conditions using qPCR. (c) Indicated mRNA levels measured in zebrafish xenografts of control (eight tumors) or E2f7/8 DKO MEFs (five tumors). All quantified data present the average±s.d. compared with the indicated controls. ***P<0.05; ns, not significant.
These data suggest that E2F7/8 regulate vessel branching through direct or indirect control of DLL4 expression. This hypothesis is supported by the observed decrease in Dll4 mRNA levels in cultured E2f7/8 DKO MEFs before grafting into animals (Figure 4b), and by the observation that multiple E2Fs (E2F1/4/6) bind the DLL4 promoter in various cell types (Supplementary Figure S4). Direct transcriptional control of the DLL4 locus by E2F7/8 seems unlikely as DLL4 was not identified in two recent chromatin immunoprecipitation-seq analysis for E2F7 (supplementary Fig. S4).19, 28 Although we recently identified E2F6 as a potential E2F7/8 target,19, 28 E2F6 mRNA and protein levels were not deregulated in E2f7/8 DKO MEFs (not shown). Therefore it is unlikely that E2F7/8 control DLL4 expression indirectly through E2F6.
Alternatively, the induction of E2F1 upon loss of E2F7/8 may reduce DLL4 expression through inhibition of HIF1. Namely, HIF1 has been reported to stimulate DLL4 expression,29 and to bind the DLL4 promoter (Supplementary Figure S4), whereas our previous data suggests competitive promoter binding between E2F1 and HIF1α,19 and reduced HIF1α protein levels upon overexpression of E2F1 (data not shown).
In conclusion, using three different tumor models, we have identified E2F7/8 as inhibitors of tumor angiogenesis (Figures 1b, 2d, 3b). And although we recently reported that E2F7/8 regulate developmental angiogenesis through transcriptional stimulation of VEGFA in cooperation with HIF1,14 this study strongly suggests that E2F7/8 regulate tumor angiogenesis independent of VEGFA, as we observed reduced Vegfa mRNA levels in E2f7/8 DKO MEFs (Supplementary Figure S2c), keratinocytes (Supplementary Figure S1b), and in engrafted tumors (Figure 2e), whereas tumor angiogenesis was enhanced in all three tumor models. Instead we found that E2F7/8 affect tumor angiogenesis at least in part through control of vessel branching, possibly through indirect control of DLL4 expression. However, we expect that this does not present a general mechanism of how E2F7/8 mediate tumor angiogenesis, as Dll4 expression was not deregulated in E2f7/8-deficient mouse papillomas (Supplementary Figure S1c). E2F7/8 may therefore mediate tumor angiogenesis through multiple mechanisms. Consistent with this notion is our previous observation that a variety of angiogenic factors are deregulated in E2f7/8-deficient mouse embryos and placentas.30 In line with the increasing understanding of E2Fs as genetic and cellular context-dependent factors, E2F7/8 may thus affect tumor angiogenesis in a context-dependent manner, as previously suggested for other players of the E2F pathway.30 Therefore it will be valuable to decipher these context-dependent mechanisms via which E2F7/8 mediate tumor angiogenesis and development, in order to fully understand the versatile role of E2F7/8 in cancer.
Acknowledgments
This study was financially supported by grants from the Dutch Cancer Society (UU2009–4353) to WJB, and (UU2013-5777) to BW and AdB. From the Association of International Cancer Research (09–0718) to WJB, and from the Netherlands Organization for Scientific Research (NWO-ALW 11-28) to AdB.
Footnotes
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
The authors declare no conflict of interest.
Supplementary Material
References
- Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 2007; 8: 464–478. [DOI] [PubMed] [Google Scholar]
- Chung AS, Lee J, Ferrara N. Targeting the tumour vasculature: insights from physiological angiogenesis. Nat Rev Cancer 2010; 10: 505–514. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674. [DOI] [PubMed] [Google Scholar]
- Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 2011; 91: 1071–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 2014; 26: 605–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregori J, Johnson DG. Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Curr Mol Med 2006; 6: 739–748. [DOI] [PubMed] [Google Scholar]
- Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene 2005; 24: 2810–2826. [DOI] [PubMed] [Google Scholar]
- Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer 2009; 9: 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ran C, Li E, Gordon F, Comstock G, Siddiqui H et al. Synergistic function of E2F7 and E2F8 is essential for cell survival and embryonic development. Dev Cell 2008; 14: 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Wang Q, Zong WY, Zheng DL, Wen YX, Wang KS et al. E2F8 contributes to human hepatocellular carcinoma via regulating cell proliferation. Cancer Res 2010; 70: 782–791. [DOI] [PubMed] [Google Scholar]
- Park SA, Platt J, Lee JW, Lopez-Giraldez F, Herbst RS, Koo JS. E2F8 as a novel therapeutic target for lung cancer. J Natl Cancer Inst 2015; 107: 10. 1093/jnci/djv151. Print 2015 Sep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent LN, Rakijas JB, Pandit SK, Westendorp B, Chen HZ, Huntington JT et al. E2f8 mediates tumor suppression in postnatal liver development. J Clin Invest 2016; 126: 2955–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurlings I, Martinez-Lopez LM, Westendorp B, Zijp M, Kuiper R, Tooten P et al. Synergistic functions of E2F7 and E2F8 are critical to suppress stress-induced skin cancer. Oncogene 2017. 829–839. [DOI] [PMC free article] [PubMed]
- Weijts BG, Bakker WJ, Cornelissen PW, Liang KH, Schaftenaar FH, Westendorp B et al. E2F7 and E2F8 promote angiogenesis through transcriptional activation of VEGFA in cooperation with HIF1. EMBO J 2012; 31: 3871–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit SK, Westendorp B, Nantasanti S, van Liere E, Tooten PC, Cornelissen PW et al. E2F8 is essential for polyploidization in mammalian cells. Nat Cell Biol 2012; 14: 1181–1191. [DOI] [PubMed] [Google Scholar]
- Merdzhanova G, Gout S, Keramidas M, Edmond V, Coll JL, Brambilla C et al. The transcription factor E2F1 and the SR protein SC35 control the ratio of pro-angiogenic versus antiangiogenic isoforms of vascular endothelial growth factor-A to inhibit neovascularization in vivo. Oncogene 2010; 29: 5392–5403. [DOI] [PubMed] [Google Scholar]
- Qin G, Kishore R, Dolan CM, Silver M, Wecker A, Luedemann CN et al. Cell cycle regulator E2F1 modulates angiogenesis via p53-dependent transcriptional control of VEGF. Proc Natl Acad Sci USA 2006; 103: 11015–11020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontemaggi G, Dell'Orso S, Trisciuoglio D, Shay T, Melucci E, Fazi F et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat Struct Mol Biol 2009; 16: 1086–1093. [DOI] [PubMed] [Google Scholar]
- de Bruin A, A Cornelissen PW, Kirchmaier BC, Mokry M, Iich E, Nirmala E et al. Genome-wide analysis reveals NRP1 as a direct HIF1alpha-E2F7 target in the regulation of motorneuron guidance in vivo. Nucleic Acids Res 2016; 44: 3549–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondi C, Ruhrberg C. Neuropilin signalling in vessels, neurons and tumours. Semin Cell Dev Biol 2013; 24: 172–178. [DOI] [PubMed] [Google Scholar]
- Djordjevic S, Driscoll PC. Targeting VEGF signalling via the neuropilin co-receptor. Drug Discov Today 2013; 18: 447–455. [DOI] [PubMed] [Google Scholar]
- Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc 2007; 2: 329–333. [DOI] [PubMed] [Google Scholar]
- Chen D, Pacal M, Wenzel P, Knoepfler PS, Leone G, Bremner R. Division and apoptosis of E2f-deficient retinal progenitors. Nature 2009; 462: 925–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouw B, Song H, Tihan T, Bosze J, Ferrara N, Gerber HP et al. The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell 2003; 4: 133–146. [DOI] [PubMed] [Google Scholar]
- He S, Lamers GE, Beenakker JW, Cui C, Ghotra VP, Danen EH et al. Neutrophil-mediated experimental metastasis is enhanced by VEGFR inhibition in a zebrafish xenograft model. J Pathol 2012; 227: 431–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 2006; 444: 1032–1037. [DOI] [PubMed] [Google Scholar]
- Hogan BM, Herpers R, Witte M, Helotera H, Alitalo K, Duckers HJ et al. Vegfc/Flt4 signalling is suppressed by Dll4 in developing zebrafish intersegmental arteries. Development 2009; 136: 4001–4009. [DOI] [PubMed] [Google Scholar]
- Westendorp B, Mokry M, Groot Koerkamp MJ, Holstege FC, Cuppen E, de Bruin A. E2F7 represses a network of oscillating cell cycle genes to control S-phase progression. Nucleic Acids Res 2012; 40: 3511–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez H, Fischer A, Winkler A, Hu CJ, Hatzopoulos AK, Breier G et al. Hypoxia-mediated activation of Dll4-Notch-Hey2 signaling in endothelial progenitor cells and adoption of arterial cell fate. Exp Cell Res 2007; 313: 1–9. [DOI] [PubMed] [Google Scholar]
- Bakker WJ, Weijts BG, Westendorp B, de Bruin A. HIF proteins connect the RB-E2F factors to angiogenesis. Transcription 2013; 4: 62–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magliozzi R, Low TY, Weijts BG, Cheng T, Spanjaard E, Mohammed S et al. Control of epithelial cell migration and invasion by the IKKbeta- and CK1alpha-mediated degradation of RAPGEF2. Dev Cell 2013; 27: 574–585. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.