

Abstract
The worldwide unrestrained emission of carbon dioxide (CO2) has caused serious environmental pollution and climate change issues. For the sustainable development of human civilization, it is very desirable to convert CO2 to renewable fuels through clean and economical chemical processes. Recently, electrocatalytic CO2 conversion is regarded as a prospective pathway for the recycling of carbon resource and the generation of sustainable fuels. In this review, recent research advances in electrocatalytic CO2 reduction are summarized from both experimental and theoretical aspects. The referred electrocatalysts are divided into different classes, including metal–organic complexes, metals, metal alloys, inorganic metal compounds and carbon‐based metal‐free nanomaterials. Moreover, the selective formation processes of different reductive products, such as formic acid/formate (HCOOH/HCOO−), monoxide carbon (CO), formaldehyde (HCHO), methane (CH4), ethylene (C2H4), methanol (CH3OH), ethanol (CH3CH2OH), etc. are introduced in detail, respectively. Owing to the limited energy efficiency, unmanageable selectivity, low stability, and indeterminate mechanisms of electrocatalytic CO2 reduction, there are still many tough challenges need to be addressed. In view of this, the current research trends to overcome these obstacles in CO2 electroreduction field are summarized. We expect that this review will provide new insights into the further technique development and practical applications of CO2 electroreduction.
Keywords: carbon cycle, catalytic mechanisms, electrochemical CO2 reduction, electrocatalysts, renewable fuels
1. Introduction
The urgent energy crisis and serious global warming problem represent two major challenges of the world. In the past decades, tremendous efforts have been made to relieve these issues. Most energy consumed by human society was derived from nonrenewable fossil fuels.1, 2 Carbon dioxide (CO2) is an extremely disturbing greenhouse gas released from the excessive use of fossil fuels. The CO2 emission problems have drawn intensive attention and increasing investments for more than 30 years. CO2 gas produced on the Earth should be equal to the amount consumed, so that the concentration of CO2 in atmosphere can remain unchanged to realize eco‐environmental stability and a favorable transition toward a sustainable society. Decreasing CO2 emissions and further regenerating CO2 into carbonaceous fuels and chemicals by mimicking the photosynthesis process of green plants would be an excellent method to relieve our demands on high‐polluting fossil energy and provide indispensable resources for industrial applications.3, 4, 5
Compared to the geological sequestration of CO2, to convert waste CO2 gas into hydrocarbons is recognized as a more worthwhile approach owing to its high‐efficiency utilization and recycling of carbon sources. To achieve this goal, the traditional catalytic processes of CO2 absorption, activation, and conversion still suffer from certain drawbacks, such as high energy demands for the transfer of CO2 molecules to active sites, low conversion rate to obtain high‐value carbonaceous chemicals, and so on. So far, various methods have been adopted to convert CO2 into other chemicals, such as: (1) biologic transformation with microalgae outdoor‐pool/photobioreactor or biocatalysis;6, 7, 8, 9 (2) chemical transformation through organic reactions or mineralization/carbonatation;10, 11, 12 (3) photocatalytic or electrocatalytic reduction;13, 14, 15, 16 and (4) other techniques like hydrogenation, dry reforming, and so on.17, 18, 19, 20 It is worthy mentioned that the realization of CO2 reduction by electrochemical catalysis has attracted great attention owing to the unique merits,21, 22, 23, 24, 25, 26 as follows: (1) the CO2 electroreduction system can be employed for practical application; (2) the electrocatalytic process under mild conditions is moderate and controllable; (3) the products of electrochemical reduction can be adjusted by reaction parameters, such as redox potential, reaction temperature, electrolyte, etc.; (4) through the optimization of electrocatalysts, the by‐products of CO2 reduction can be minimized to a low content; (5) electric power, as the drive force, can be attained with other renewable energy sources (such as solar power, wind power, and so on) without any additional CO2 generation.
CO2 molecules are very inert and stable, because the carbon atoms in CO2 are at the highest oxidation state. Therefore, it is necessary to develop efficient electrocatalysts for promoting the kinetically sluggish CO2 reduction process. The routes of electrochemical CO2 reduction can be realized through multiple electron transfer in aqueous solution with suitable electrocatalysts. The various possible products formed through different pathways in a schematic electrocatalytic cell are presented in Scheme 1 . CO2 can be converted into small carbonaceous molecules with high energy density, such as formic acid (HCOOH), carbon monoxide (CO), methanol (CH3OH), methane (CH4), and so on. Based on a thermodynamic study, a variety of half‐reactions and their corresponding electrode potentials versus standard hydrogen electrode (SHE) in aqueous solution (pH = 7, at 25 °C, 1 atm, and 1.0 m concentration of other solutes) are listed in Table 1 .15, 27 It is very likely that a mixture composed of gaseous products (CO, CH4, etc.) and liquid products (HCOOH, CH3OH, C2H5OH, etc.) would be formed in the electrochemical cell, rather than a single product. The employed electrocatalysts and the applied electrode potential are crucial to the efficiency and selectivity of CO2 reduction. This brings some serious pending technological challenges, such as high cost, inferior efficiency, low product selectivity, and fast degradation of electrocatalytic activity.28, 29, 30 Especially, due to the inadequate selectivity and stability of existing electrocatalysts, the state‐of‐the‐art techniques are still unable to adequately meet the requirements for large‐scale industrial application.
Scheme 1.
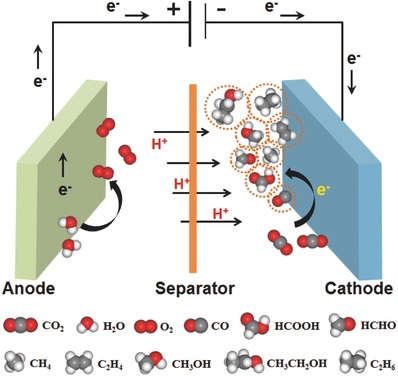
Illustration of the electrochemical CO2 reduction process and the possible products generated in an electrochemical reaction cell.
Table 1.
Electrochemical potentials of possible CO2 reduction reactions in aqueous solutions for the production of different hydrocarbon fuels
| Possible half‐reactions of electrochemical CO2 reduction | Electrode potentials (V vs SHE) at pH 7 |
|---|---|
| CO2 (g) + e− → *COO− | −1.90 |
| CO2 (g) + 2H+ + 2e− → HCOOH (l) | −0.61 |
| CO2 (g) + H2O (l) + 2e− → HCOO− (aq) + OH− | −0.43 |
| CO2 (g) + 2H+ + 2e− → CO (g) + H2O (l) | −0.53 |
| CO2 (g) + H2O (l) + 2e− → CO (g) + 2OH− | −0.52 |
| CO2 (g) + 4H+ + 2e− → HCHO (l) + H2O (l) | −0.48 |
| CO2 (g) + 3H2O (l) + 4e− → HCHO (l) + 4OH− | −0.89 |
| CO2 (g) + 6H+ (l) + 6e− → CH3OH (l) + H2O (l) | −0.38 |
| CO2 (g) + 5H2O (l) + 6e− → CH3OH (l) + 6OH− | −0.81 |
| CO2 (g) + 8H+ + 8e− → CH4 (g) + 2H2O (l) | −0.24 |
| CO2 (g) + 6H2O (l) + 8e− → CH4 (g) + 8OH− | −0.25 |
| 2CO2 (g) + 12H+ + 12e− → C2H4 (g) + 4H2O (l) | 0.06 |
| 2CO2 (g) + 8H2O (l) + 12e− → C2H4 (g) + 12OH− | −0.34 |
| 2CO2 (g) + 12H+ + 12e− → CH3CH2OH (l) + 3H2O (l) | 0.08 |
| 2CO2 (g) + 9H2O (l) + 12e− → CH3CH2OH (l) + 12OH− (l) | −0.33 |
In the past decades, numerous efforts have been made to ameliorate the electrocatalysts and reaction conditions to overcome the above obstacles.15, 16, 26, 31, 32, 33 In recent years, as the demand of clean energy is increasing worldwide, the research of CO2 electroreduction is progressing very rapidly. Therefore, a comprehensive review includes various aspects, such as electrocatalysts categories, product selectivity, stability, as well as challenges and perspectives, is needed for summarizing the recent advances and promoting the further development in this field.
2. Electrocatalysts for Electrocatalytic CO2 Reduction
The electrocatalysts applicable to CO2 reduction can be classified into different types, basically inorganic and organic species. Since the 1970s, some metal–organic complexes have been applied as a class of typical homogeneous electrocatalysts, because their special coordinative structures and active centers can tightly bind with CO2 molecules.34 The electrocatalysts based on metal–organic complexes have attracted significant attention for decades due to the remarkable selectivity, but also have some unpopular disadvantages, such as complicate synthesis processes, low reduction activity, and toxic effects.35, 36 Heterogeneous metal electrocatalysts have been developed later, accompanying with some advantageous characteristics, such as low toxicity, facile synthesis processes and superior electrocatalytic activity.37, 38 Inorganic metal compounds (metal oxides, chalcogenides, etc.) and carbon‐based materials have also been employed as emergent electrocatalysts. The following sections will introduce the development of these representative electrocatalysts of CO2 reduction in recent five years.
2.1. Metal–Organic Complexes
2.1.1. Metal–Macrocyclic Complexes
Macrocyclic ligands can be divided into different classes, such as phthalocyanine, porphyrin, cyclam, and so on. In 1970s, Meshitsuka et al. first reported the utilization of metal–macrocyclic complexes composed of transition metal atom (Co or Ni) and phthalocyanine ligands for CO2 electroreduction.34 Since then, numerous of researches related to metal–macrocyclic complexes have been come forth. Acted as an applicable and desirable “Trash to Treasure” approach,39 the greenhouse gas CO2 can be effectively transferred into carbon monoxide (CO) using different kinds of Fe‐porphyrin molecules, as illustrated in Figure 1 a. Typically, iron 5,10,15,20‐tetrakis(2′,6′‐dihydroxylphenyl)‐porphyrin (Fe TDHPP) could achieve a stable electrocatalytic performance over 4 h for CO generation with a Faradaic yield above 90%, attributing to the high local proton concentration of phenolic hydroxyl. A cobalt‐protoporphyrin electrocatalyst loaded on pyrolytic graphite electrode can convert CO2 mainly into CO in acidic conditions,40 showing high electrocatalytic activity comparable to other porphyrin‐based molecules in previous reports at a lower overpotential (0.5 V). The pH‐dependent activity and selectivity are shown in Figure 1b. Besides, a composite electrode prepared by the electrodeposition of [Cu(cyclam)](ClO4)2 complex (cyclam = 1,4,8,11‐tetraazacyclotetradecane) can reduce CO2 into HCOOH with a Faradaic efficiency of 90% in dimethyl formamide (DMF)/H2O mixture (97:3, v/v).41
Figure 1.
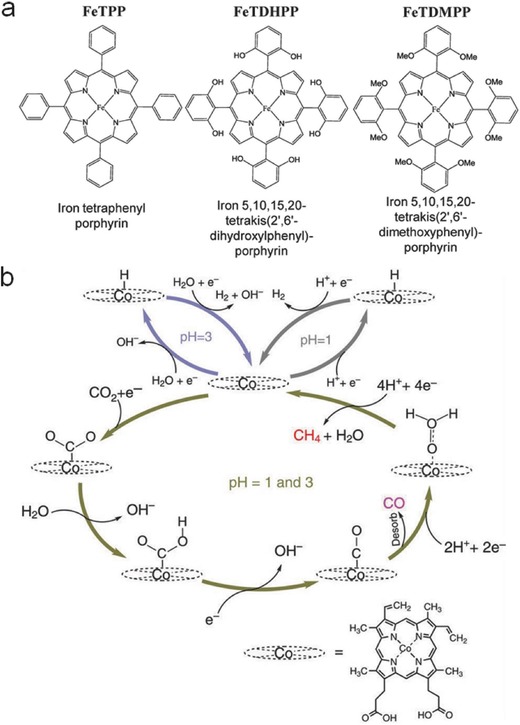
Metal–macrocyclic complexes as electrocatalysts for CO2 reduction. a) Investigated iron porphyrins. Reproduced with permission.39 Copyright 2016, American Association for the Advancement of Science. b) Schematic mechanism of the electrochemical CO2 reduction using Co protoporphyrin. Reproduced with permission.40 Copyright 2015, Macmillan Publishers Limited.
2.1.2. Metal–Bipyridine Complexes
Bipyridine (bpy) complexes with earth‐abundant metal atoms were also considered as promising molecular electrocatalysts for reducing CO2 to CO or hydrocarbons, such as HCOOH. There are many transition metals explored in this group, such as Ru, Cu, W, Mo, Mn, Re, Cr, and so on.42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52 For instance, a metal complex composed of Ru atom and 6,6′‐dimesityl‐2,2′‐bipyridine (mesbpy) ligands was applied to the generation of CO with high turnover frequency and Faradaic efficiency in the presence of Brønsted acids.51 The results benefited from the inhibition of Ru—Ru bond formation as well as the synergistic redox response between bipyridine ligands and Ru metal. Similarly, a manganese (Mn) based complex electrocatalyst composed of mesbpy ligands shows good performance at low overpotentials (0.3–0.45 V) with the assistance of Lewis acid (especially Mg2+ cations).52 The electrocatalytic mechanism of [Mn(mesbpy)(CO)3]− for converting CO2 into CO was detailed presented in Figure 2 a.
Figure 2.
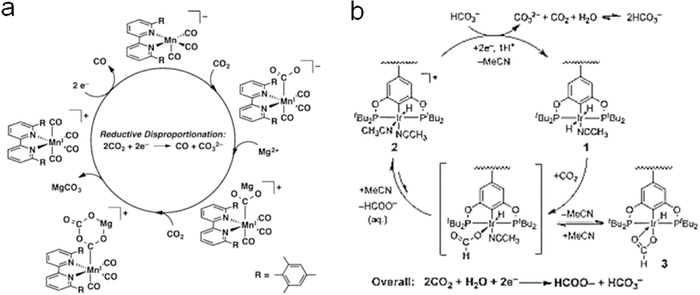
a) Redox mechanism of [Mn(mesbpy)(CO)3]− and Mg2+ at −1.5 V versus Fc+/0 for electroreduction of CO2 to CO. Reproduced with permission.52 Copyright 2016, American Chemical Society. b) Proposed mechanism for electroreduction of CO2 to HCOO− using iridium pincer dihydride electrocatalyst. Reproduced with permission.54
2.1.3. Other Metal–Organic Complexes
Some other organic–ligand based complexes have also been investigated as molecular electrocatalysts. Donovan et al. synthesized two new Zn(II) complexes with phosphine groups and evaluated their ability to reduce CO2 to CO.53 Kang et al. reported an iridium pincer dihydride electrocatalysts adopted to reduce CO2 to formates (HCOO−),54 exhibiting high efficiency, selectivity and turnover numbers (≈54200), of which mechanism is shown in Figure 2b. Besides, molecular electrocatalysts with other components, such as biscarbene pincer,55 N‐heterocyclic carbene,56 polyaniline,57 (R,R)‐Trost‐bis‐ProPhenol ([BPP]),58 4‐v‐tpy, 6‐v‐tpy,59 oxalate,60 and hydride,61 were also studied. Inspired by these analogous researches, the future extensive exploration of metal complex catalysts is to be expected.
2.2. Metals
Metal electrocatalysts for CO2 reduction can be divided into three groups based on the different reaction routes and main products (CO, HCOO−, hydrocarbons, alcohols, and so on), as illustrated in Figure 3 . Sn and Pb metals are classified as the same class, because they mainly generate HCOO− in aqueous solution since CO2•− intermediates can be easily desorbed from the surface of Sn and Pb.62 In comparison, Au, Ag, Pd, Zn, and Bi can tightly bind with *COOH intermediates, but can hardly bind with the generated *CO species, hence this class of metals tends to generate CO as the predominant product.63 Specially, Cu metal is individually divided into the third class, because Cu is in favor of binding *CO intermediates and converting it into alcohols or other hydrocarbons from *COH or *CHO intermediates through dimerization pathways.64 It is also worthy mentioned that some other metals like Pt, Ni have lower hydrogen evolution overpotentials and strong binding capability with *CO intermediates,65, 66 therefore the H2 evolution reaction (HER) will be the predominant process in the presence of water. Based on the chief principle of catalytic process concerning metallic catalysts, the concept of electronic structure should be introduced to pursue more anticipative performance.67 The key factor underlying the catalytic mechanism is that the interaction between adsorbate (CO2 molecules in this case) and metal surface are enormously determined by the d‐band levels of the catalyst itself. By adjusting the location of the d band centers, the bonding strength of adsorbed intermediates (*COOH, *CO, etc.) and Gibbs free energy (ΔG) consumed in rate‐determining steps would be optimized to enhance the catalytic performance. Hence, achieving satisfying activities of metallic catalysts relies on the adjustment of d band levels through lots of approaches,68, 69 such as particle size optimization, surface modification, and exposure of different crystal planes/active sites (such as terraces, edges or corners), etc. The recent progresses on different classes of metal catalysts for CO2 electroreduction are introduced in detail below.
Figure 3.
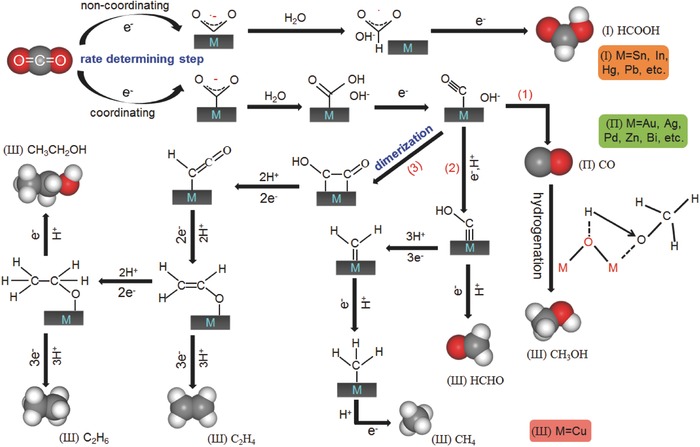
Schematic mechanism of different metal electrocatalysts for CO2 reduction reaction in aqueous solution.
2.2.1. Sn and Pb
Sn and Pb, as group IV metals, are categorized as the first class of metal catalysts. In most cases, Sn and Pb mainly produce HCOO− or formic acid owing to their weak bonding with CO2•− intermediates.62 In recent years, there are some researches in the electrochemical performance of Sn and Pb electrodes with different parameters, such as electrocatalyst sizes, surface modification and reaction conditions. In regard of the effect of particulate sizes, Castillo et al. showed that smaller Sn nanoparticles (NPs) were helpful to overcome the mass transfer limitation of CO2 onto the electrode surface and reached an enhanced Faradaic efficiency for HCOO− generation.70 Some surface modifications on Sn electrodes were also carried out to improve the activity and selectivity. It was reported that rationally designed SnOx‐derived Sn electrodes could electroreduce CO2 into HCOOH with superior Faradaic efficiencies and high production rates at relatively low overpotentials.71, 72, 73, 74, 75 The results indicated that the CO2•− intermediates were preferably stabilized on the surface of Sn with abundant oxygen species rather than on bare Sn electrodes. Wu et al. investigated the obvious difference of HCOOH generation rates using Sn electrode in Na2SO4 and KHCO3 electrolytes, respectively, emphasizing the nonnegligible effect of reaction conditions.76
Recently, Zhu et al. fabricated Pb electrodes for CO2 electroreduction,77 showing high partial current density and Faradaic efficiency of HCOOH production in an ionic liquid/acetonitrile/H2O ternary electrolyte. The above‐mentioned researches about earth‐abundant electrodes like Sn and Pb may provide a feasible pathway for the noteworthy yield of HCOOH through the optimization of metal catalysts and electrolytes.
2.2.2. Au, Ag, Pd, Zn, and Bi
The second class of metals, such as Au, Ag, Pd, Zn, and Bi has aroused intense attention for the specific selectivity of CO generation. The following works have verified that ligand‐protected Au clusters and NPs with various sizes, exposed planes or special morphologies can exhibit distinctive catalytic activity and selectivity.78, 79, 80, 81 Kauffman et al. reported Au25 clusters could effectively realize the reduction of CO2 into CO with ≈100% Faradaic efficiency, indicating a reversible interaction between CO2 and Au25.78 Monodisperse Au NPs with size‐dependent electrocatalytic activity were also synthesized to achieve superior Faradaic efficiency for selective CO production (Figure 4 a–d).79 The extraordinary selectivity of CO strongly depends on the binding energies of different reaction intermediates on active sites. The competitive processes of CO and H2 generation are displayed as following
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
Figure 4.
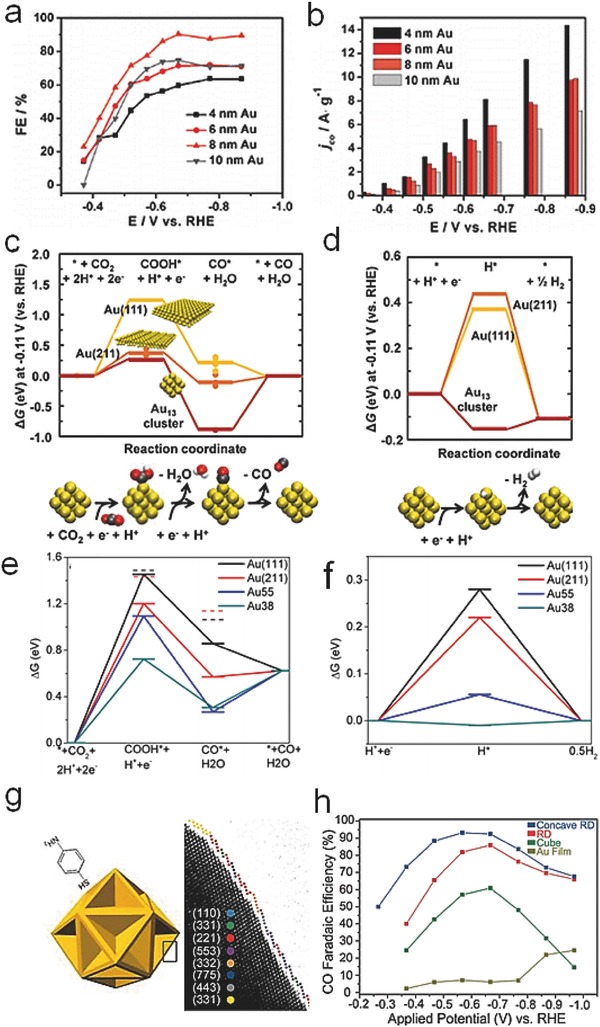
a) Potential‐dependent Faradaic efficiencies of different Au NPs (4, 6, 8, 10 nm) during electrocatalytic reduction of CO2 to CO. b) Current densities (mass activity) for electrocatalytic reduction of CO2 to CO on the Au NPs with different sizes at various applied potentials. Free energy diagrams for electrochemical reduction of c) CO2 to CO and d) protons to hydrogen on Au (111), Au (211), and a 13‐atom Au cluster at −0.11 V (vs RHE), respectively. Reproduced with permission.79 Copyright 2013, American Chemical Society. Free energy diagrams for electrochemical reduction of e) CO2 to CO and f) H+ to H2 on Au(111), Au(211), Au55 NPs, and Au38 NPs at 0 V versus RHE. Reproduced with permission.80 Copyright 2014, American Chemical Society. g) Morphological model of concave rhombic dodecahedron Au NPs with different exposed facets. h) Faradaic efficiencies of different Au NPs and Au film for CO production at applied potential (vs RHE). Reproduced with permission.81 Copyright 2015, American Chemical Society.
The facilitated stabilization (Equation (1)) and reduction (Equation (2)) of *COOH as well as the fast desorption of CO molecules (Equation (3)) together contributed to the high CO yield. In addition, the HER process (Equations (4) and (5)) as a major side reaction was effectively suppressed. Moreover, on the surface of relatively small Au NPs, the increased low‐coordinated sites and active edge sites could contribute to the stabilization of *COOH intermediates and the production of CO rather than the competitive HER process.80 According to the free energy (ΔG) diagrams of CO2 electroreduction to CO (Figure 4e,f), the overpotential mainly results from the step of COOH* formation. Due to the presence of an optimum ratio of edge sites on tiny‐sized Au NPs (Au38), the energy barrier between CO2 and *COOH was decreased, thus could realize a lower overpotential for higher CO yield. Besides, concave rhombic dodecahedron Au NPs (Figure 4g) were synthesized to explore the importance of high‐index planes, such as (332) and (775) facets, achieving good activity and high stability (Figure 4h).81
The effects of surface modifications on Au NPs have also been investigated. Feng and co‐workers deposited Au NPs with a relatively high density of grain boundaries on carbon nanotubes (Au/CNTs), which could improve the catalytic activity for CO generation by stabilizing unique active surfaces.82 The linear correlation between the reduction activity and the density of grain boundaries indicated the edges of grain boundaries could act as active sites for stronger adsorption of *COOH intermediates. These results provide new insights to control sizes, exposed facets and morphology of metal nanocrystals for improving the performance of CO2 reduction.
Ag metal as an appropriate candidate presents outstanding selectivity for CO generation. Ag disk electrodes with the assistance of imidazolium‐based ionic liquids exhibited selective CO production, owing to the immobilization of CO2•− intermediates by C4‐ and C5‐protons on imidazolium rings.83 Guo et al. prepared bovine serum albumin‐capped Ag nanoclusters demonstrating a high Faradaic efficiency up to 75% for selective CO evolution in dimethylformamide aqueous solution.84
Carrying out the CO2 reduction reaction with a bulk Ag electrode in ion liquid solutions or organic electrolytes is not conducive to large‐scale industrial applications. Luckily, Ag nanostructures with optimized size, structure and surface modification can also realize enhanced properties. Nanosized Ag electrodes possess abundant active sites and can achieve highly selective CO production at an overpotential lower than bulk electrode or flat surface.85, 86 Since more low‐coordinated atoms exposed on the surface of smaller sized Ag, it could promote the formation of Ag‐COOH bonds to stabilize *COOH intermediates (Figure 5 a). Lu et al. prepared nanoporous Ag NPs with highly curved surface (Figure 5b), which could achieve ≈92% of Faradaic yield for CO production.87 The strong adsorption of *COO− intermediates on nanoporous Ag can lead to a rapid first‐electron transfer step superior to that on polycrystalline Ag (Figure 5c). The density functional theory (DFT) calculations and experimental tests of CO2 reduction on the surface of nanosized Ag were also investigated.88 The abundant exposed active edge/corner sites of Ag NPs could decrease the activation energy barrier of electron transfer, thus beneficial for the yield and selectivity of CO production (Figure 5d).
Figure 5.
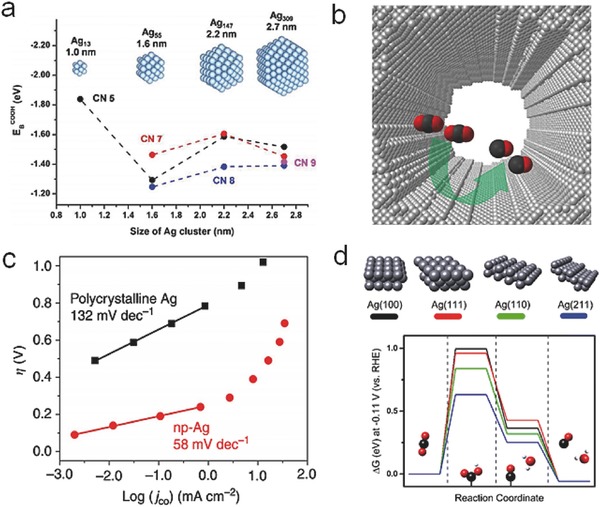
a) DFT calculation results on the binding energies of *COOH intermediates as a function of the size of Ag NPs. Reproduced with permission.86 Copyright 2015, American Chemical Society. b) Schematic diagram of nanoporous Ag (scale bar, 500 nm). c) The partial current density of CO production under different overpotentials on polycrystalline silver and nanoporous Ag, respectively. Reproduced with permission.87 Copyright 2014, Macmillan Publishers Limited. d) Free energy diagrams for the electroreduction of CO2 to CO on flat (Ag(100) and Ag(111)) and edge (Ag(221) and Ag(110)) sites. Reproduced with permission.88 Copyright 2015, American Chemical Society.
The catalytic activity of Ag nanocatalysts can be further improved by surface modifications. To form an active surface layer on Ag electrode, oxidation–reduction method,89 electrochemical deposition,90 and anodization treatment91 can be utilized. Through surface modifications, the derived Ag electrocatalysts can exhibit higher specific surface area and stronger adsorption of *COOH and *COO− intermediates, thus leading to higher activity and suppression of H2 evolution. The modified Ag nanocatalysts can realize a Faradaic yield as high as 90% for CO production at relatively low overpotentials.
Pd‐based electrocatalysts have also been studied for CO2 reduction. Owing to the poor catalytic activity of polycrystalline Pd foil,92 efforts have been made to construct nanostructural Pd for enhancing the activity and Faradaic yield. Novel Pd/C nanocatalyst93 exhibited high mass activities (50–80 mA mg−1) for HCOO− generation due to the formation of PdHx through a rapid electrohydrogenation step. Gao et al. explored the size‐dependent electrocatalytic activity of Pd/C NPs for generating CO,94 showing high Faradaic efficiency up to 91.2% at −0.89 V (vs RHE) using 3.7 nm Pd NPs, which was comparable to that of Au or Ag (Figure 6 a). The ratios of corner, edge, or terrace active sites can be modified by different sizes and morphologies of Pd NPs. According to the DFT calculation results (Gibbs free energy diagrams in Figure 6b), the steps of CO2 adsorption, *COOH formation, and *CO removal prefer to occur on smaller Pd NPs with a higher ratio of corner and edge sites. However, compared to Au and Ag metals, Pd NPs would be more easily deactivated by the poisoning of adsorbed CO after a period of reaction time at excessive overpotential.
Figure 6.
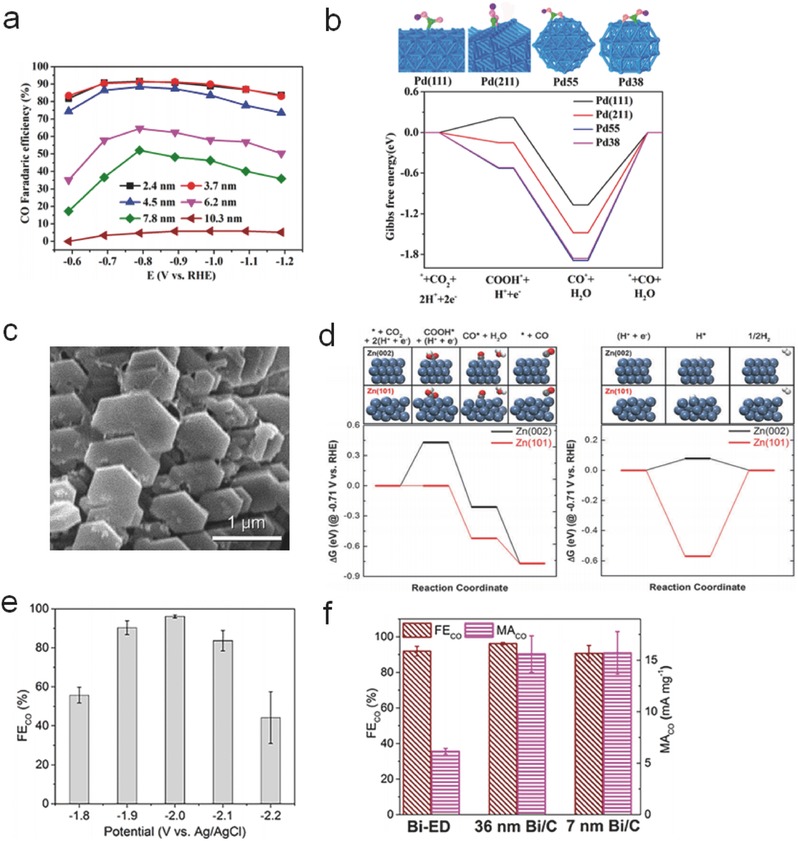
a) Applied potential dependence of Faradaic efficiencies for CO production over Pd NPs with different sizes. b) Adsorption of *COOH (top) and DFT results on the free energy for CO2 reduction to CO (bottom) on Pd(111), Pd(211), Pd55, and Pd38. Reproduced with permission.94 Copyright 2015, American Chemical Society. c) SEM image of hierarchical hexagonal Zn. d) Free‐energy diagrams of CO2 reduction (left) and HER (right) on Zn (002) and Zn (101). Reproduced with permission.99 (e) Faradaic efficiencies of CO production under different applied potentials on 36 nm freshly reduced Bi/C. f) Faradaic efficiencies and mass activities of CO production on electrodeposited Bi films (Bi‐ED), 36 or 7 nm freshly reduced Bi/C by hydrazine (36 nm Bi/C or 7 nm Bi/C). Reproduced with permission.100 Copyright 2016, American Chemical Society.
Zn, as a low‐cost and earth‐abundant metal, has been regarded as a promising electrocatalyst with high selectivity of CO production. However, the stability and catalytic activity are the predominant obstacles to be overcome. Previously, Hattori and co‐workers have explored bulk Zn electrocatalyst, which can successfully convert CO2 molecules into CO with a considerable current density.95, 96 Nevertheless, the efficiency of bulk Zn is hard to be improved due to the rapid oxidation on the surface. Nanosized Zn, such as nanostructured Zn dendrites synthesized by an electrodeposition approach,97 presented higher activities than bulk ones, owing to the minimization of surface oxide layer. With the assistance of NaCl electrolyte, nanoscale Zn could generate CO with a Faradaic efficiency of 93% since the adsorption of Cl− ions on Zn surface is conducive to the formation of *COO− intermediates.98 However, the stability was not satisfying due to the inevitable oxidization during electrolysis. Recently, hierarchical hexagonal Zn (Figure 6c) showed a Faradaic efficiency of 85.4% for selective CO production over 30 h.99 DFT calculation revealed that the exposed Zn (101) facet favored the stabilization of *COOH intermediates (Figure 6d). The researches indicated that superior catalytic performances could be achieved by the design of morphologic structure, especially exposed facets.
Due to earth‐abundant, cheap and pollution‐free features, Bi metal also has been applied for reducing CO2 to CO. The Bi NPs activated through hydrazine treatment performed the highest Faradaic yield of CO production (96.1%) in acetonitrile based electrolyte (Figure 6e,f).100 In another case, nanostructured Bi nanoflakes were directly grown on Cu film by a pulse electrodeposition method,101 showing a large number of edge/corner sites and unexpected Faradaic efficiency of 79.5% for HCOO− generation.
2.2.3. Cu
Cu as the third group metal can generate high value‐added carbonaceous compounds at low cost.102, 103, 104 However, poor selectivity and activity degradation are two remaining challenges for practical application. Great efforts are still needed to reduce the overpotential, optimize the selectivity, and stability.105, 106 Many experimental factors such as morphology, surface modification, crystal planes, and active sites can lead to different reaction pathways and various products.
Diverse morphologies (such as NPs, nanowires, nanocubes, etc.) of Cu nanocrystals have been investigated for the generation of HCOO−, CO, hydrocarbons, and alcohols. Cu nanopillars exhibited a Faradaic efficiency of 28% for the yield of HCOOH at −0.5 V (vs RHE).107 Porous hollow Cu fibers108 and Cu nanowires109 were employed to achieve distinct electrocatalytic selectivity of CO at low overpotentials. Cu NPs loaded on glassy carbon (n‐Cu/C) achieved a Faradaic efficiency up to ≈80% for CH4 generation, with four‐times higher energy efficiency than Cu foil (Figure 7 a–d).110 As displayed in Figure 7e, a detailed reaction mechanism of CO2 reduction using n‐Cu/C was described. The *CO2 − intermediates formed by a one electron‐transfer pre‐equilibrium step were strongly absorbed on the active surface of Cu. Then, *CO2 − reacted with another CO2 molecule to yield a *CO2–CO2 − intermediate by C−O coupling, realizing CH4 production through the formation and further reduction/hydrogenation of *CO intermediates. By utilizing different nanostructured Cu, like NPs,111 nanocubes,112 and nanowires,113 other hydrocarbons and alcohols (such as ethylene, ethane, n‐propanol, and ethanol) could generate through CO dimerization pathways (Figure 3).
Figure 7.
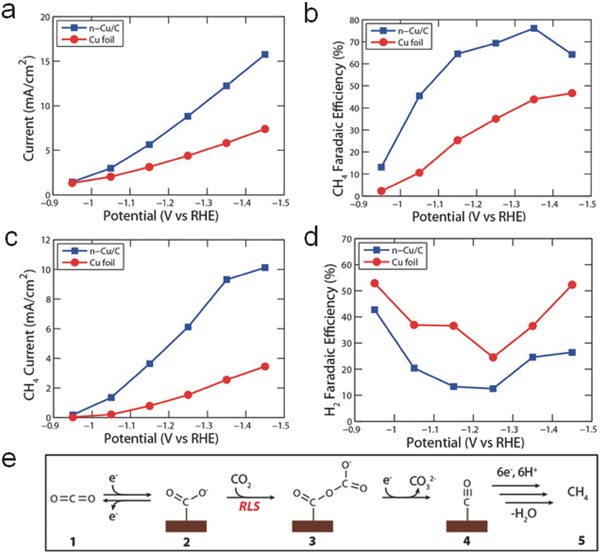
Comparison of current densities and Faradaic efficiencies of n‐Cu/C and copper foil. a) Total current density of n‐Cu/C and copper foil. b) Faradaic efficiencies for CH4 generation. c) Methanation current densities. d) Faradaic efficiencies for H2 generation, showing suppressed H2 evolution on n‐Cu/C catalyst. e) Proposed mechanism for the electrochemical reduction of CO2 to CH4, including the rate‐limiting step (RLS), consistent with the electrochemical data and known intermediates identified in the literature. Reproduced with permission.110 Copyright 2014, American Chemical Society.
Surface modification of Cu electrodes has also been investigated in recent years. To achieve high selectivity toward C2 hydrocarbon products (such as C2H4 and C2H5OH), oxygen plasma‐activated Cu114 and oxygen‐derived Cu mesoporous foam115 have shown high Faradaic efficiency. With an increased local pH value on the oxidized Cu surfaces, the CO dimerization would be promoted, which is beneficial to the yield of ethylene, as illustrated in Figure 8 a. Cu nanowires modified by amino acid were also tested (Figure 8b).116 The results confirmed that the introduced —NH3 + group on Cu surface can benefit the stabilization of *CHO intermediates and the subsequent hydrogenation reaction for producing C2 and C3 hydrocarbons.
Figure 8.
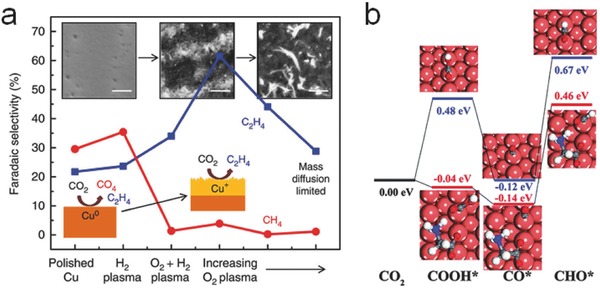
a) Hydrocarbon selectivity of plasma‐treated Cu foils. Reproduced with permission.114 Copyright 2016, the Author, published under CC‐BY 4.0 license. b) The DFT calculated free energy change of CO2 and CO protonation without glycine (blue lines) and with glycine (red lines). Reproduced with permission.116 Copyright 2016, The Royal Society of Chemistry.
Previous works have discovered some intimate relationships between product selectivity and exposed lattice planes or active sites of Cu electrodes. Cu nanocatalysts with different exposed crystal facets (Cu (111), Cu (211), and Cu (100)) could lead to multiple products under the same condition.112, 117 Densely packed (111) facets preferred to generated HCOOH, while highly stepped (211) facets was superior for CH4 generation. Close‐packed (100) facets performed the most favorable selectivity for C2 hydrocarbons instead of C1 products, by means of a sequential electron–proton transfer and the reduction of ethylene oxide (C2H3O) intermediates, as illustrated in Figure 9 a.118 Moreover, an optimal ratio of edge sites over (100) planes played a crucial role toward CO2 reduction and C2H4 production, as verified by the experimental results using Cu nanocubes of different sizes (Figure 9b).119 Therefore, it is meaningful to utilize Cu as a model electrocatalyst for the studies of CO2 reduction.
Figure 9.
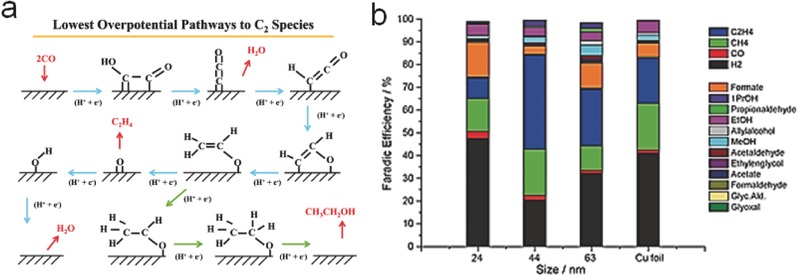
a) Schematic illustration of the species involved in the reaction pathways to generate C2H4 (blue) and C2H5OH (green). Reproduced with permission.118 b) Bar graph reporting the Faradaic efficiencies for each product produced by Cu foil and Cu nanocubes with different sizes at −1.1 V versus RHE. The glassy carbon signal has been subtracted. Reproduced with permission.119
2.3. Metal Alloys
Metal alloys can enhance the electrocatalytic reaction kinetics and selectivity of CO2 reduction by adjust the binding capability of active intermediates (such as *COOH and *CO). For example, a novel PdxPt(100− x )/C electrocatalyst was reported to convert CO2 into HCOOH at ≈0 V (vs RHE), which considerably approached the theoretical equilibrium potential of 0.02 V (vs RHE).120 However, the high cost and low stability of noble metals still need to be resolved.
The introduction of nonnoble‐metals into the alloy electrocatalysts can minimize the cost and improve the performances. Recently, some reports have investigated the activity of Cu alloys. Compared to Au or Cu NPs, nanosized Au3Cu alloys assembled into ordered monolayers121 showed higher Faradaic efficiency for CO production (Figure 10 a). Both the electronic effect and geometric effect of AumCun alloys should be taken into consideration for the selective CO production and the desorption ability of *COOH. The higher d‐band levels of Cu can enhance the binding capability of *COOH and *CO, which is conducive to the production of hydrocarbons. However, when referred to the geometric effect, Cu atoms next to the Au—C bonds can further stabilize *COOH and lead to the generation of CO (Figure 10b–d). Therefore, an appropriate content of Cu in Au–Cu alloys can promote CO production. Rasul et al. developed a Cu–In alloy electrocatalyst,122 which could selectively convert CO2 into CO with a Faradaic efficiency of 95% as well as negligible H2 or HCOOH evolution. It is because the intact Cu corner sites and the surface of In both promote a strong binding capability of *COO− superior to that of *H intermediates. Recently, a Cu–Sn bimetallic electrode achieved a high Faradaic efficiency over 90% for CO productivity by introducing an optimal amount of Sn.123 As the ratio of Sn atoms increased, the multifold sites on Cu were disturbed, thus inhibited the adsorption of *H on catalyst surface.
Figure 10.
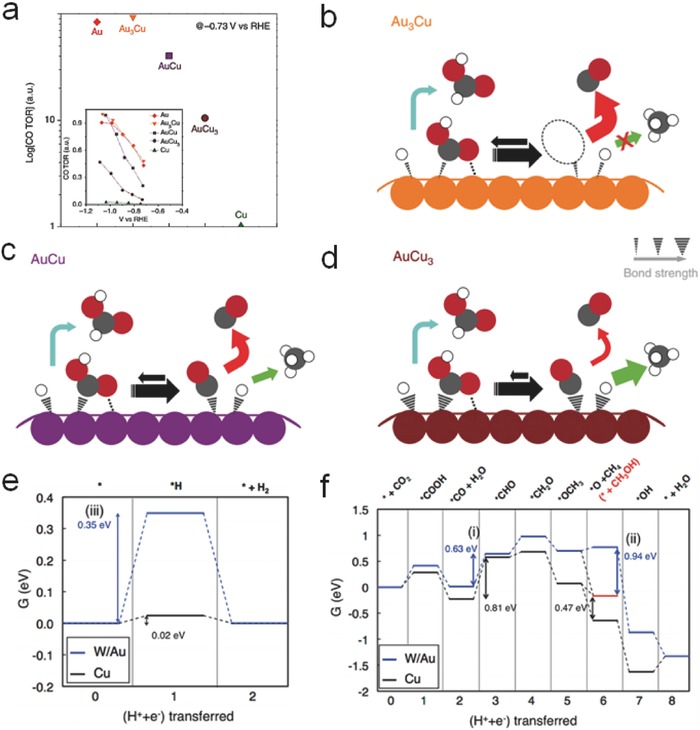
a) Relative turnover rates (TORs) for CO generation and (b–d) proposed mechanism for CO2 reduction on the Au–Cu bimetallic NPs. Reproduced with permission.121 Copyright 2014, Macmillan Publishers Limited. Free energy diagrams for e) H2 evolution and f) CO2 electroreduction to CH4 or CH3OH on W/Au and Cu electrodes. Reproduced with permission.125 Copyright 2014, American Chemical Society.
Some high‐value hydrocarbon compounds can be generated using other metal alloys. Torelli et al. prepared Ni–Ga films for the production of methane, ethylene, and ethane.124 The *COOH intermediates could be tightly bound on the surface of Ni and the introduction of Ga can weaken the Ni–CO interaction, therefore Ni and Ga synergistically increased the yields of C2 hydrocarbons and avoided the poisoning of CO on the catalyst surface. Analyzed by a computational calculation method (Figure 10e,f), W–Au alloy was regarded as a suitable candidate to decrease the overpotential for *COO− formation and suppress unfavorable *H adsorption for methanol production,125 possibly followed a pathway: CO2 → *COO− → COads → CHOads → CH3Oads → methanol. Sun et al. developed a Mo–Bi bimetallic electrocatalyst with high CH3OH selectivity, which achieved a maximum Faradaic efficiency of 71.2% in acetonitrile with the assistance of ion liquids.126 In brief, these works exhibited the possibility to realize low‐cost and highly active alloy electrocatalysts for the scalable CO2 electroreduction.
2.4. Inorganic Metal Compounds
2.4.1. Metal Oxides
Metal‐oxide‐based electrocatalysts have gradually got attention due to their decent energy efficiency and selectivity for CO2 electroreduction, although the instability is still a big problem.
According to DFT calculation results, HCOOH, methane, or methanol could be produced at different conditions with RuO2 electrocatalyst by adjusting the *CO coverage.127 Pb2O cathode exhibited a Faradaic efficiency of 60% for HCOOH generation in KHCO3 aqueous solution.128 However, owing to the high cost or toxicity, it is impracticable to used RuO2 or Pb2O for CO2 reduction. Instead, other earth‐abundant and low‐toxicity metals, such as Sn, Co, Ni, and Ti, have been considered as alternative electrocatalysts.
SnOx/CNT cathodes performed a 60% Faradaic efficiency with 25% energy efficiency for HCOOH formation.129 Xie and co‐workers synthesized ultrathin Co3O4 layers with 1.72 nm thickness as an effective electrocatalyst, showing an optimum 64.3% Faradaic efficiency for HCOO− production after 20 h reaction.130 Later, the same group prepared partially oxidized Co 4‐atomic‐layers with an average thickness of 0.84 nm (Figure 11 a–d), and achieved a ultrahigh HCOO− selectivity of 90.1% over 40 h.131 The Co based atomic layers possessed abundant active sites, and the increased charge density near Fermi level could improve electronic conductivity. The process of HCOO− production was occurred as below
| (6) |
| (7) |
| (8) |
| (9) |
Figure 11.
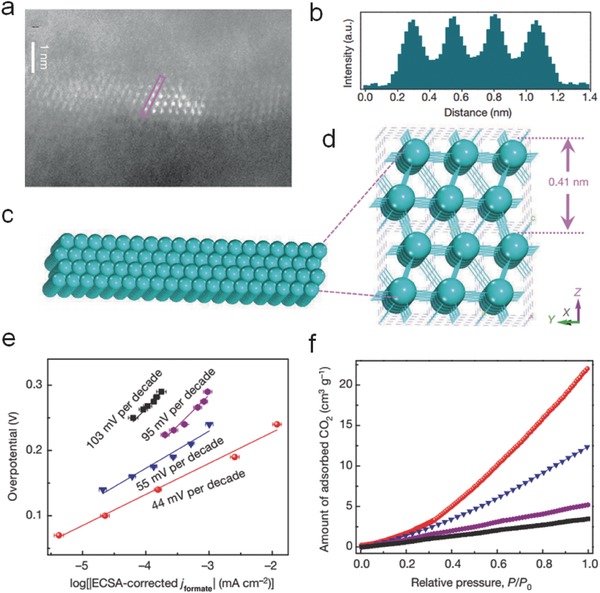
a) Lateral high‐angle annular dark‐field scanning transmission electron microscopy (HAADF‐STEM) image of partially oxidized Co 4‐atom‐thick layers and (b) the corresponding intensity profile along the pink rectangle in (a). c,d) Corresponding crystal structures. e) Electrochemical active surface area (ECSA) corrected Tafel plots for HCOO− production. f) CO2 adsorption isotherms of partially oxidized Co 4‐atom‐thick layers (red), Co 4‐atom‐thick layers (blue), partially oxidized bulk Co (violet) and bulk Co (black). Reproduced with permission.131 Copyright 2016, Macmillan Publishers Limited.
The Tafel slopes (≈59 mV dec−1, Figure 11e) and preferable CO2 adsorption capability (Figure 11f) of partially oxidized Co 4‐atomic‐layers indicated the good properties for CO2 activation and *COO− intermediate stabilization.
Some other metal oxides were also investigated. NiO showed a Faradaic efficiency up to 35.2% for syngas (CO and H2) products.132 Nanostructured TiO2 films were applied for CO2 reduction in acetonitrile electrolyte.133 The oxygen vacancies (or Ti3+ species) on TiO2 films were identified as active sites, which could strongly bind CO2 molecules and promote the generation of *COO− intermediates, resulting in the generation of methanol as primary product. These results indicated that the introduction of oxidation states in certain metallic catalysts can greatly improve the performance for CO2 electrochemical reduction.
2.4.2. Metal Chalcogenides
Interestingly, some chalcogenides of transition metals (such as Fe, Mo, W) were found to be available catalysts for electrocatalytic CO2 reduction. The reaction intermediates can be bound to different active sites on the surface of metal chalcogenides, therefore the limitation of linear‐scale relations between the binding energies of reaction intermediates and specific metals can be broken. It is also worth noting that the different edge sites of metal chalcogenides can perform different duties for the generation of varied products. In 2011, porous ternary chalcogels of Ni–Fe4S4 and Co–Fe4S4 with high surface area and high charge mobility were applied to improve the electrocatalytic activity for CO and CH4 production.134
Nørskov and co‐workers investigated the active edge sites of MoS2, MoSe2, and Ni‐doped MoS2 (Ni–MoS2) simulated by DFT method.135 The *COOH and *CHO intermediates prefer to attach to bridging S or Se atoms, while *CO intermediates trend to bind with the edge sites of metal atoms (Figure 12 a). All edges were involved in CO evolution, while the S edges of Ni–MoS2 and the Mo edges of MoSe2 could further turn CO to hydrocarbons or alcohols. Inspired by this, a cost‐effective MoSx electrocatalyst was introduced, which can produce syngas (CO and H2) at a low overpotential of ≈290 mV and achieve a maximum Faradaic efficiency of 85.1% for CO yield with the assistance of reduced graphene oxide (rGO) and polyethylenimine (PEI) (Figure 12b,c).136 In 2016, Nørskov and co‐workers found that the combination of dopant metal sites (*CO binding sites) and S binding sites (*COOH, *CHO, and *COH binding sites) on metal‐doped MoS2 can provide two different linear‐scaling relationships, which synergistically result in enhanced CO2 reduction performance than pristine Mo metal.137 The as‐prepared Co‐doped MoS2 could achieve better electrocatalytic ability for methanol production than pristine MoS2.138
Figure 12.
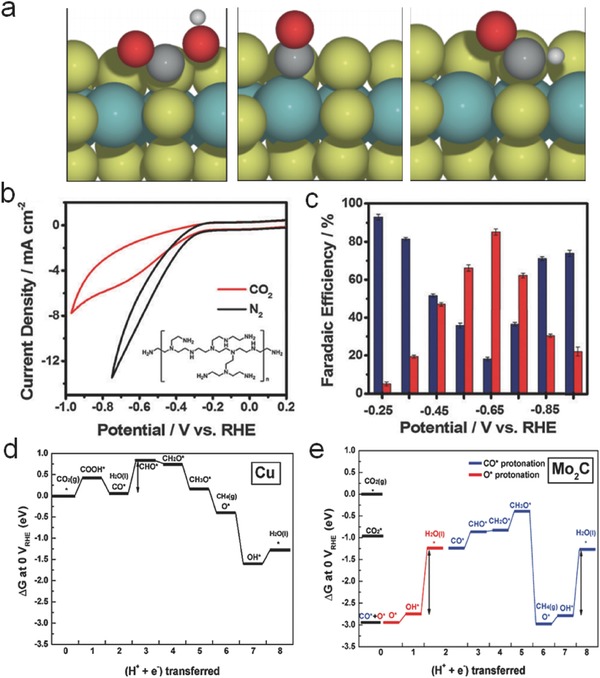
a) Binding configurations of *COOH, *CO, and *CHO on the Mo edge of MoS2. *COOH and *CHO preferably bind to the bridging S atoms, while *CO binds to the Mo atoms. Reproduced with permission.135 b) Cyclic voltammograms (CVs) of rGO–PEI–MoSx modified glassy carbon electrode in N2‐saturated and CO2‐saturated 0.5 m aqueous NaHCO3 solution, respectively. Inset: Structure of PEI. c) Faradaic efficiency for CO (red bars) and H2 (blue bars) production at different applied potentials. Reproduced with permission.136 Copyright 2016, The Royal Society of Chemistry. Free energy diagrams of CO2 conversion to CH4 over d) Cu (211) and e) Mo2C (100) surfaces at 0 V (vs RHE), respectively. The most endergonic step in the overall process is designated with an arrow. Reproduced with permission.141 Copyright 2016, American Chemical Society.
Compared to MoS2 and MoSe2, WSe2 has the lowest work function that can facilitate rapid electron transfer during CO2 reduction. Hence, WSe2 was considered as another promising candidate for CO production with high current density, which can perform a current density 60 times higher than that of Ag NPs under the same condition.139 Thanks to the low work function and high d‐electron density on the W‐terminated edge sites, the *COOH and *CO intermediates were more stable on WSe2 than Ag, resulting in the easier formation of CO on WSe2 at low overpotentials. We expect that further efforts devoted to metal chalcogenides will be very helpful to the research of carbon fixation in the future.
2.4.3. Metal Carbides
Transition metal carbides are another class of promising catalysts with low cost, favorable carbophobic and oxophilic properties. In 2015, to understand the relationships between the binding energies of reaction intermediates and active sites of metal carbides, Wannakao et al. studied the CO2 reduction mechanism of Fe‐, Co‐, and Pt‐doped W carbides through DFT method.140 The results showed that the d‐band center of transition metal was related to the adsorption energies, which relatively influenced the binding site preferences and geometries of the active intermediates. Therefore, the electron structure of W carbide catalysts could be tuned by metal doping to improve the carbophilicity and oxophilicity for the optimized activity and selectivity.
Kim et al. reported that Mo2C could convert CO2 into CH4 at an onset potential of −0.55 V (vs RHE), of which free energetics of CO hydrogenation was less than that of conventional Cu metal.141 The DFT‐calculated free energies for CO2 reduction to CH4 on Cu(211) surface are displayed in Figure 12d, which shows that the major energy‐consuming step for CH4 generation is the protonation of adsorbed *CO (−0.74 V vs RHE). In contrast, CO2 molecules were preferably adsorbed on Mo2C (100) surface (Figure 12), followed by the dissociation of C—O bonds at the initial reaction stage before protonation. Once the *O intermediates were generated by the C—O bond fission, the protonation was easily accessible because of a lower potential demand (about −0.20 V vs RHE). Hence, other than Cu, the rate‐limiting factors on Mo2C surface for selective CH4 production were determined to be the *OH removal and the nonelectrochemical C—O bond scission. The new insights on metal oxides/chalcogenides/carbides opened a new field for the design of low cost catalysts and new theoretical foundation for electrochemical CO2 reduction.
2.5. Carbon‐Based Metal‐Free Electrocatalysts
Metals like Pd, Au, Ag, and Cu have been popularly employed as electrocatalysts for CO2 reduction. However, there are some issues to be resolved, such as relatively costly price, high overpotential, and inferior selectivity. To address these problems, carbon‐based nanomaterials like carbon nanotubes, graphene, carbon fibers, and porous carbon have been considered as potential alternatives, which could bring about decent activity and low cost. However, when compared to the field of oxygen reduction reaction and water splitting, the relevant investigations of carbon‐based electrocatalysts for CO2 reduction are still quite few.
Nitrogen‐doped CNTs (NCNTs) could realize effective CO2 capture and high product selectivity for CO generation at a significantly decreased overpotential than pristine CNTs.142, 143 Compared to pristine CNTs, the introduction of pyridinic‐N into bamboo‐shaped NCNTs (Figure 13 a) led to higher electrical conductivity and achieved a Faradaic efficiency of 80% for CO generation.144 Among the different N defects (Figure 13b), pyridinic‐N sites exhibited the highest binding capability with CO2 molecules and the lowest absolute overpotential (0.20 V) for *COOH formation, which promoted the CO formation. Other than pyridinic‐N, the existence of quaternary‐N or pyrrolic‐N could stabilize the radical active intermediates and lower the reduction barriers.145 Moreover, with the help of PEI cocatalyst, NCNTs were employed for converting CO2 into HCOO− with an overpotential of ≈0.54 V (vs NHE), attributing to the strong stabilization of the *COO− intermediates.146
Figure 13.
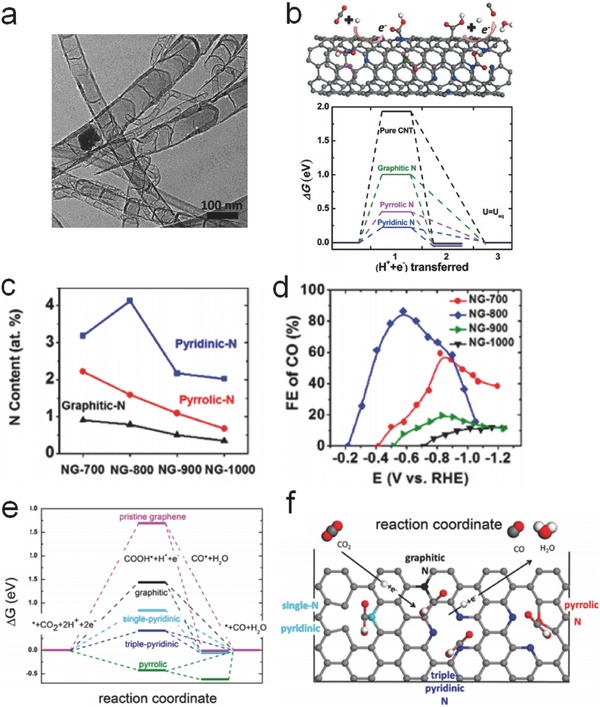
a) TEM image of bamboo‐shaped NCNTs. b) Schematic of CO formation on NCNTs and free‐energy diagram at equilibrium potential for CO2 reduction on pyridinic‐N, pyrrolic‐N, and graphitic‐N defects compared to original CNTs. Reproduced with permission.144 c) The corresponding N functionality content and d) Faradaic efficiency of CO production versus applied potential on N‐doped graphene with different doping temperatures (700–1000 °C). e) Free energy diagrams of electrocatalytic CO2 conversion on N‐doped graphene and f) schematic of nitrogen defects and CO2 reduction mechanism. Reproduced with permission.150 Copyright 2015, American Chemical Society.
N‐doped graphene has also been investigated for CO and HCOO− 147, 148 as well as CH4.149 The pyridinic‐N or pyrrolic‐N species resulted in stronger CO2 adsorption and lower energy barrier for the formation of *COOH intermediates. Similarly, the incorporation of pyridinic‐N defects into 3D graphene foam can also lower the free energy barrier to form adsorbed *COOH and facilitate the CO yield (Figure 13c,d).150 The corresponding free energy diagrams for selective CO generation on different sites of N‐doped graphene and pristine graphene through the lowest energy‐consuming pathway are explicitly shown in Figure 13e. The excess overpotential is resulted from the uphill barrier of the first electron‐transfer rate‐determine step for *COOH formation. The *COOH intermediates have good affinity with N defects, and the free energy barrier for *COOH adsorption decreases significantly on pyridinic‐ or pyrrolic‐N sites rather than graphitic‐N sites (Figure 13f). Subsequently, the second proton‐coupled electron transfer becomes thermodynamically easier for the formation of adsorbed *CO (or CO). Interestingly, it was found that boron‐doped graphene can efficiently generate the exclusive product of HCOO− at low overpotentials.151 GO/CNTs composite has also been reported for converting CO2 to CO, showing higher selectivity and activity than noble metals (Au and Ag).152 However, the need of adding ionic liquids makes it difficult to be used for large‐scale applications.
Some other carbon‐based materials, such as carbon fibers,153 metal‐doped nitrogenated carbon black,154 N‐doped diamond,155 nanoporous carbon,156 B‐doped diamond,157 and Cu NPs/B‐doped diamond158 were found can achieve CO, CH4, HCOO−, or HCHO formation with high selectivity and low overpotential due to their preferable adsorption of CO2 and suitable binding capability of active intermediates. The study of metal‐free carbon‐based electrocatalysts has opened a new door for developing cheaper alternatives instead of precious noble metals.
3. Product Selectivity in Electrocatalytic CO2 Reduction
The electrocatalysts play an important role to the product selectivity of CO2 reduction. Different electrocatalysts have shown diverse tendencies of generating specific carbonaceous compounds, such as HCOOH/HCOO−, CO, formaldehyde (HCHO), hydrocarbons, and alcohols, with almost unavoidable H2 evolution as side reaction. However, the product selectivity of CO2 reduction seems quite complicated and closely related to the reaction conditions and pathways. Not only the electrocatalyst, the intricate reaction steps can also be influenced by many other parameters, such as applied potential, electrolyte, pH value, temperature, and pressure. To ensure our readers can quickly find valuable recapitulative data from related literatures, Table 2 summarized a series of representative experimental results obtained from different electrocatalysts and reaction conditions (electrolytes and applied potentials), together with the associated information of measured selectivities and activities.
Table 2.
The representative examples of electrochemical CO2 reduction with different electrocatalysts, reaction conditions and selectivities
| Electrocatalyst | Electrolyte | Applied potential [V] | Major products [Faradaic efficiency, %] | Current density/mass activity | Ref. |
|---|---|---|---|---|---|
| 1. Selective production of HCOO−/HCOOH | |||||
| [Cu(cyclam)](ClO4)2 complex | DMF/H2O (97:3 v/v) | −2.0 (vs Fc/Fc+) | HCOOH (90%) | 1 mA cm−2 | 41 |
| Gas‐diffusion layer/CNT/Ir complex/polyethylene glycol | 0.5 m LiClO4/0.1 m NaHCO3/1% v/v MeCN | −1.40 (vs RHE) | HCOO− (83%) | 15.6 mA cm−2 | 54 |
| Pd‐polyaniline/CNTs | 0.1 m KHCO3 | −0.80 (vs SCE) | HCOO− (83%) | – | 57 |
| SnOx/Sn | 0.1 m KHCO3 | −1.36 (vs RHE) | HCOO− (71.6%) | 17.1 mA cm−2 | 71 |
| Nanostructured Sn | 0.1 m NaHCO3 | −1.80 (vs SCE) | HCOO− (93.6%) | 10.2 mA cm−2 | 72 |
| Sn | Ion liquids/H2O/MeCN | −2.30 (vs Ag/AgCl) | HCOOH (92.0%) | 32.1 mA cm−2 | 77 |
| Pb | HCOOH (91.6%) | 37.6 mA cm−2 | |||
| Pd NPs | 0.5 m NaHCO3 | −0.35 (vs RHE) | HCOO− (88%) | 3.45 mA cm−2 | 93 |
| Bi nanoflakes | 0.1 m KHCO3 | −0.40 (vs RHE) | HCOO− (79.5%) | – | 101 |
| Bi/BiOCl | 0.5 m KHCO3 | −1.50 (vs SCE) | HCOO− (≈92%) | 3.7 mA mg−1 | 161 |
| Cu pillars | 0.1 m KHCO3 | −0.50 (vs RHE) | HCOOH (28.7%) | ≈1.3 mA cm−2 | 107 |
| Cu nanofoam | 0.5 m KHCO3 | −1.50 (vs Ag/AgCl) | HCOOH (37%) | – | 162 |
| Ag–Sn alloy | 0.5 m NaHCO3 | −0.80 (vs RHE) | HCOOH (≈80%) | ≈16 mA cm−2 | 165 |
| PdxPt(100− x )/C | 0.1 m KH2PO4/0.1 m K2HPO4 | −0.40 (vs RHE) | HCOOH (88%) | ≈5 mA cm−2 | 120 |
| SnO2 porous nanowires | 0.1 m KHCO3 | −0.80 (vs RHE) | HCOO− (80%) | (−1.0 V) 10 mA cm−2 | 163 |
| Mesoporous SnO2 nanosheets/carbon paper | 0.5 m NaHCO3 | −1.60 (vs Ag/AgCl) | HCOO− (≈87%) | 50 mA cm−2 | 164 |
| Pb2O | 0.5 m KHCO3/NaHCO3 | −2.0 (vs Co3O4) | HCOOH (60%/50%) | – | 128 |
| Co3O4 atomic layers | 0.1 m KHCO3 | −0.88 (vs SCE) | HCOO− (64.3%) | 0.68 mA cm−2 | 130 |
| Partially oxidized Co atomic layers | 0.1 m Na2SO4 | −0.85 (vs RHE) | HCOO− (90.1%) | 10.59 mA cm−2 | 131 |
| PEI‐NCNTs/glassy carbon | 0.1 m KHCO3 | −1.80 (vs SCE) | HCOO− (85%) | 7.2 mA cm−2 | 146 |
| N‐doped graphene/carbon paper | 0.5 m KHCO3 | −0.84 (vs RHE) | HCOO− (73%) | 7.5 mA cm−2 | 148 |
| Boron‐doped graphene | 0.1 m KHCO3 | −1.40 (vs SCE) | HCOO− (66%) | 2 mA cm−2 | 151 |
| N‐doped nanodiamond/Si | 0.5 m NaHCO3 | −1.0 (vs RHE) |
HCOO− (13.6%) CH3COO− (77.6%) |
0.75 mA cm−2 | 155 |
| 2. Selective production of CO | |||||
| Fe TDHPP | DMF/2 m H2O | −1.16 (vs RHE) | CO (94%) | 0.31 mA cm−2 | 39 |
| Co protoporphyrin–pyrolytic graphite | Perchlorate solution (pH = 3) | −0.60 (vs RHE) | CO (60%) | 0.08 mA cm−2 | 40 |
| COF‐366‐Co | 0.5 m KHCO3 | −0.67 (vs RHE) | CO (90%) | 80 mA mg−1 (Co) | 201 |
| [Ru(‐6,6′‐dimesityl‐2,2′‐bipyridine)(CO)2Cl]0 | 0.1 m TBAPF6/MeCN | ≈−2.2 V (vs Fc/Fc+) | CO (95%) | – | 51 |
| Au‐1,3‐bis(2,4,6‐trimethylphenyl)imidazol‐2‐ylidene complex | 0.1 m KHCO3 | −0.57 (vs RHE) | CO (83%) | ≈2 mA cm−2 | 56 |
| Au NPs | 0.5 m KHCO3 | −0.67 (vs RHE) | CO (90%) | – | 79 |
| Au rhombic dodecahedrons | 0.5 m KHCO3 | −0.57 (vs RHE) | CO (93%) | – | 81 |
| Au/CNTs | 0.5 m NaHCO3 | −0.50 (vs RHE) | CO (≈94%) | ≈15 A g−1 (Au) | 82 |
| Au nanowires | 0.5 m KHCO3 | −0.35 (vs RHE) | CO (94%) | 1.84 A g−1 (Au) | 198 |
| Oxide‐derived Au | 0.5 m NaHCO3 | −0.35 (vs RHE) | CO (>96%) | 2–4 mA cm−2 | 199 |
| 6 µm thick highly porous Ag | 0.5 m KHCO3 | −0.50 (vs RHE) | CO (82%) | 10.5 mA cm−2 | 85 |
| Ag NPs | 0.5 m KHCO3 | −0.75 (vs RHE) | CO (79.2%) | 1 mA cm−2 | 86 |
| Nanoporous Ag | 0.5 m KHCO3 | −0.60 (vs RHE) | CO (≈92%) | ≈18 mA cm−2 | 87 |
| Ag nanocorals | 0.1 m KHCO3 | −0.60 (vs RHE) | CO (95%) | 6.62 mA cm−2 | 89 |
| Oxide‐derived Ag | 0.1 m KHCO3 | −0.80 (vs RHE) | CO (89%) | 1.15 mA cm−2 | 200 |
| Pd NPs | 0.1 m KHCO3 | −0.89 (vs RHE) | CO (91.2%) | 23.9 A g−1 (Pd) | 94 |
| Pd icosahedra/C | 0.1 m KHCO3 | −0.80 (vs RHE) | CO (91.1%) | – | 166 |
| Zn dendrites | 0.5 m NaHCO3 | −1.10 (vs RHE) | CO (79%) | – | 97 |
| Zn foil | 0.5 m NaCl | −1.60 (vs SCE) | CO (93%) | – | 98 |
| Hexagonal Zn | 0.5 m KHCO3 | −0.95 (vs RHE) | CO (85.4%) | 9.5 mA cm−2 | 99 |
| Surface activated Bi NPs | MeCN/[bmim][OTf] | −2.0 (vs Ag/AgCl) | CO (96.1%) | 15.6 mA mg−1 (Bi) | 100 |
| Cu fibers | 0.3 m KHCO3 | −0.40 (vs RHE) | CO (75%) | ≈9 mA cm−2 | 108 |
| Cu nanowires | 0.1 m KHCO3 | −0.40 (vs RHE) | CO (61.8%) | 1 mA cm−2 | 109 |
| Au3Cu alloy | 0.1 m KHCO3 | −0.73 (vs RHE) | CO (64.7%) | 3 mA cm−2 | 121 |
| Ordered AuCu NPs | 0.1 m KHCO3 | −0.77 (vs RHE) | CO (80%) | – | 167 |
| Cu–In alloy | 0.1 m KHCO3 | −0.60 (vs RHE) | CO (85%) | ≈0.75 mA cm−2 | 122 |
| Cu–Sn alloy | 0.1 m KHCO3 | −0.60 (vs RHE) | CO (>90%) | 1 mA cm−2 | 123 |
| Oxide‐derived Cu | CO (63%) | 2.1 mA cm−2 | |||
| TiO2 film | MeCN/0.1 m TEAP | −1.8 (vs Ag/AgCl) | CO (90%) | – | 133 |
| rGO–PEI–MoSx | 0.5 m NaHCO3 | −0.65 (vs RHE) | CO (85.1%) | 55 mA cm−2 | 136 |
| WSe2 nanoflakes | 50 vol%/50 vol% EMIMBF4/H2O | −0.164 (vs RHE) | CO (24%) | 18.95 mA cm−2 | 139 |
| MoSeS alloy monolayers | 4 mol%/96 mol% EMIMBF4/H2O | −1.15 (vs RHE) | CO (45.2%) | 43 mA cm−2 | 168 |
| NCNTs | 0.1 m KHCO3 | −1.05 (vs RHE) | CO (80%) | – | 144 |
| N‐doped graphene foam | 0.1 m KHCO3 | −0.58 (vs RHE) | CO (≈85%) | ≈1.8 mA cm−2 | 150 |
| 3. Selective production of HCHO | |||||
| Boron‐doped diamond | MeOH electrolyte | −1.70 (vs Ag/AgCl) | HCHO (74%) | 97.5 µA cm−2 | 157 |
| Cu NPs/boron‐doped diamond | (10 × 10−6 m) H2O/bmim‐PF6 | −1.3 (vs RHE) | HCOOH and HCHO (>80%) | 5.1 mA cm−2 | 158 |
| 4. Selective production of methane and ethylene | |||||
| Cu–porphyrin complex | 0.5 m KHCO3 | −0.976 (vs RHE) | CH4 and C2H4 (44%) | 13.2 mA cm−2 (CH4) | 176 |
| 8.4 mA cm−2 (C2H4) | |||||
| Cu NPs supported on glassy carbon | 0.1 m NaHCO3 | −1.25 (vs RHE) | CH4 (80%) | ≈9 mA cm−2 | 110 |
| Cu nanowires | 0.1 m KClO4 | −1.10 (vs RHE) | C2H6 (20.3%) | 4–5 mA cm−2 | 113 |
| 0.1 m KHCO3 | C2H6 (17.4%) | ||||
| 0.1 m K2HPO4 | C2H6 (10%) | ||||
| Plasma‐treated Cu foil | 0.1 m KHCO3 | −0.90 (vs RHE) | C2H4 (60%) | – | 114 |
| Cu foam | 0.5 m NaHCO3 | −0.80 (vs RHE) | C2H4, C2H6 (55%) | – | 115 |
| Glycine/Cu nanowires | 0.1 m KHCO3 | −1.90 (vs Ag/AgCl) | C2H4, C2H6, C3H6 (34.1%) | ≈11 mA cm−2 | 116 |
| Cu nanocubes [44 nm] | 0.1 m KHCO3 | −1.1 (vs RHE) | C2H4 (41%) | ≈5.5 mA cm−2 | 119 |
| Cu NPs | 0.1 m KHCO3 | −1.1 (vs RHE) | CH4 (57%) | 23 mA cm−2 | 174 |
| C2H4 (<20%) | |||||
| CO (<5%) | |||||
| HCOOH (<5%) | |||||
| Pd–Au alloy | 0.1 m KH2PO4/0.1 m K2HPO4 | −0.60 (vs RHE) | CO (30.9%) | – | 203 |
| −1.40 (vs RHE) | CH4 (2%) | ||||
| −1.40 (vs RHE) | C2 hydrocarbons (0.7%) | ||||
| −1.40 (vs RHE) | C3 hydrocarbons (0.3%) | ||||
| −1.30 (vs RHE) | 1‐Butene (0.16%) | ||||
| Cu2Pd alloy | 0.1 m TBAPF6/CH3CN/1 m H2O | −1.8 (vs Ag/AgNO3) | CH4 (51%) | ≈6 mA cm−2 | 177 |
| NixGay alloy | 0.1 m NaHCO3 | −0.48 (vs RHE) | CH4 (>2%) | (−1.18 V) 140 µA cm−2 | 124 |
| C2H4 (1.3%) | (−1.18 V) 100 µA cm−2 | ||||
| Cu2O/Cu | 0.1 m KHCO3 | −0.98 (vs RHE) | C2H4 (42.6%) | 13.3 mA cm−2 | 179 |
| C2H5OH (11.8%) | 3.7 mA cm−2 | ||||
| C3H7OH (5.4%) | 1.7 mA cm−2 | ||||
| Mo2C | 0.1 m KHCO3 | –1.10 (vs RHE) | CH4 (29%) | >30 mA cm−2 | 141 |
| H2 (≈39%) | |||||
| N‐doped carbon | [bmim]BF4/H2O | −1.4 (vs RHE) | CH4 (93.5%) | 1.42 mA cm−2 | 149 |
| Pyridinic‐N rich graphene/Cu | 0.5 m KHCO3 | −0.90 (vs RHE) | C2H4 (19%) | 7.7 A g−1 | 202 |
| 5. Selective production of alcohols | |||||
| Enzymes | Phosphate buffer solution | −1.20 (vs Ag/AgCl) | CH3OH (≈10%) | – | 180 |
| [4‐(3‐Phenoxy‐2,2‐bis(phenoxymethyl)propoxy)pyridine]@Cu–Pd | 0.5 m KCl | −0.04 (vs RHE) | CH3OH (26%) | 21 mA cm−2 | 204 |
| −0.64 (vs RHE) | C2H5OH (12%) | – | |||
| Cu nanocrystals | 0.1 m KHCO3 | −0.95 (vs RHE) | C3H7OH | 1.74 mA cm−2 | 111 |
| Mo–Bi alloy | 0.5 m [bmim]BF4/MeCN | −0.70 (vs RHE) | CH3OH (71.2%) | 12.1 mA cm−2 | 126 |
| Cu2O | 0.1 m KHCO3 | −0.99 (vs RHE) | C2H4 (34–39%) | 30–35 mA cm−2 | 178 |
| C2H5OH (9–16%) | |||||
| Cu2O | 0.5 m KHCO3 | −2.0 (vs Co3O4) | C2H5OH (96.2%) | 4.5 mA cm−2 | 185 |
| Cu2O/multiwalled CNT | 0.5 m NaHCO3 | −0.80 (vs RHE) | CH3OH (38.0%) | 7.5 mA cm−2 | 188 |
| Oxidized Cu | 0.5 m KHCO3 | −1.10 (vs SCE) | CH3OH (38.0%) | – | 189 |
3.1. Selective Production of Formic Acid/Formates
As early as 1870, HCOOH/HCOO− generations have been realized from electrocatalytic reduction of CO2 molecules in aqueous solution.159 From electrokinetic perspective, the rate‐determining step was determined to be the hydrogenation step, which realized the bonding of electrogenerated surface hydrogen onto CO2 molecules. The obtained *COOH intermediate was then reduced by one electron to generate the HCOO− product. As aforementioned, some metal–organic complexes have been reported to mainly produce HCOOH/HCOO− products with the assistance of ion liquids.41, 44 Afterward, numerous efforts have been made for the large‐scale conversion of CO2 to HCOOH/HCOO− with economic practicability.160 As the first class of metals, Sn, In, Hg, Pb, and Bi based catalysts can facilitate HCOOH/HCOO− generation,101, 161 due to the easy desorption of *COO− intermediates on the surface. On the other hand, Cu foam with higher surface roughness can present an ultrahigh HCOOH production rate among Cu‐based electrodes, and also can suppress the formation of *CO intermediates and inhibit the evolution of dimeric products (CH4 and C2H4).162 Some metal oxides have been found to be outstanding candidates for generating HCOOH/HCOO−, for example, partially oxidized Co 4‐atomic‐layer131 could realize a HCOO− selectivity of 90% owing to its good stabilization of *COO− intermediates. Sn, SnOx, as well as Sn–Ag alloys also can facilitate selective *COO− protonation for HCOO− formation.71, 72, 73, 74, 75, 163, 164, 165 Besides, carbon nanomaterials like NCNTs, graphene, and others presented enhanced current density and high Faradaic efficiency for acid production, mainly attributed to the preferable CO2 adsorption and stabilization of reduced *COO− intermediates.147, 151, 155
3.2. Selective Production of Carbon Monoxide
Two common issues have seriously influenced the reductive activity for CO formation: (1) the conversion of CO2 to *COOH is hindered by the weak *COO− binding; (2) the release of CO gas from the electrocatalyst surface is suppressed by strong binding of *CO. Aiming at these problems, the effects of morphology, active sites and exposed facets of electrocatalysts (such as Au, Ag, Bi, Zn, Pd, and metal oxides) have been experimentally and computationally investigated.63, 166 Many researchers reported the synthesis of Cu–M (M = Au, In, Sn) and other metallic alloys for converting CO2 to CO with a low overpotential.121, 122, 123, 167 With the assistance of ionic liquid, ternary transition metal dichalcogenides (MoSeS)168 as well as some metal complexes like Fe/Co porphyrins,39, 40, 169 Mn bipyridines,52 and Zn phosphines,53 have been found to exhibit preferable catalytic activity for generating CO. Carbon materials were also utilized for selective and stable CO2 reduction into CO. NCNTs as a durable electrocatalyst also showed ultralow overpotential (−0.18 V) and selectivity (80%) for CO production.144
3.3. Selective Production of Formaldehyde
A binuclear cobalt complex, [Co2BPP], also undertook a four‐electron reduction pathway for HCHO generation below −1.0 V (vs NHE).58 In 1995, [M(4‐v‐tpy)2]2+ or [M(6‐v‐tpy)2]2+ (M = Cr, Ni, Co, Fe, Ru, or Os) complexes were employed to reduce CO2 into formaldehyde (HCHO) as the dominate product.59 Boron‐doped diamond, an ideal catalyst with high overpotential for hydrogen evolution, exhibited superior Faradaic efficiency of 74% for HCHO generation in methanol and aqueous solutions.157 Moreover, Boron‐doped diamond decorated with Cu NPs showed a high current density of ≈5.1 mA cm−2 at −1.3 V (vs NHE), and the highest Faradaic efficiency for HCHO evolution was expected to be 80% under optimized reaction conditions.158
3.4. Selective Production of Methane and Ethylene
For the yield of high‐energy‐density hydrocarbon products, such as CH4 and C2H4, reaction processes with six or more electron transfer and multiple intermediate steps at higher overpotentials are normally required. Nonprecious Cu metal as a promising electrocatalyst possesses high activities and Faradaic efficiencies for methanation/vinylation, thus can produce higher value‐added hydrocarbon products, mainly methane (CH4), ethylene (C2H4) in considerable amounts.170, 171, 172, 173, 174, 175, 176 Cu‐based alloys177 and Cu oxides178, 179 also have been qualified for the formation of hydrocarbons. Some other adopted electrocatalysts such as organic metal complexes, metal alloys, metal carbides, and carbon materials for selective generation of hydrocarbons are listed in Table 2.
3.5. Selective Production of Alcohols
The production of alcohols in CO2 electrocatalytic reduction suffers from low yield and poor selectivity. Many catalysts, such as enzymes, metals, metal alloys, metal oxides/chalcogenides, and CNTs have been used for generating alcohols, such as CH3OH and C2H5OH.
Interestingly, bio‐electrocatalytic reduction of CO2 promoted by immobilized enzymes realized a Faradaic efficiency of ≈10% for CH3OH generation.180 Pyridinium (PyrH+) cations on Pt interface were employed to generate CH3OH at low overpotentials.181 Among Pt3Co alloy nanostructures with different morphologies, Pt3Co octapods displayed the highest TOF number of 758 h−1 for CH3OH production.182 Mo–Bi bimetallic chalcogenide with the help of ion liquids reached a high Faradaic efficiency of 71.2% for selective CH3OH production.126 Notably, N‐doped graphene/CNTs were theoretically predicted to be a good electrocatalyst for effective CH3OH production at the applied potential from −1.29 to −0.49 V.183
There are very few existing optional electrocatalysts for the selective production of C2H5OH. Remarkably, Cu and CuxO based catalysts have shown special selectivity for reduction of CO2 to C2/C3 compounds (including C2H5OH, n‐propanol), owing to the favorable d‐band levels.109, 111, 113, 118, 119, 178, 184, 185, 186, 187, 188, 189 Ullah et al. demonstrated that Ir/Ru oxide could efficiently convert of CO2 into different valuable organic molecules (ethanol as the major product; methanol, acetone and acetaldehyde as the minor products in the liquid phase).190 It should be noted that photo‐assisted electroreduction of CO2 can also selectively generate alcohols through the aid of diversified photocathodes.191, 192, 193, 194, 195, 196
4. Challenges and Perspectives
The above sections intensively summarized the recent considerable progress in CO2 electroreduction. However, to scale up the technology for practical and commercial applications, some inevitable challenges still need to be resolved. First, the kinetically sluggish multielectron transfer process during CO2 reduction reaction require excessive overpotential, which usually leads to relatively low energy efficiency and high power consumption.
Secondary, the product selectivity of existing electrocatalytic CO2 reduction systems is not satisfying so far. Although the aforementioned catalysts like metal complexes, Sn, Pb, Au, and Ag can make contributes to the generation of specific products, such as CO or HCOOH/HCOO−, it is still difficult to selectively produce desirable chemicals with higher commercial value, such as C2 or longer chain chemicals.
Third, the activity degradation is a serious problem usually originated from the instability of catalysts, especially nonnoble metal catalysts. Generally, the cathodic degradation and the inactivation of reaction sites are responsible for activity decay. As the reaction lasts for a long time, the inert intermediates or poisonous by‐products preferably deposit on catalyst surface and go against further catalysis process. Moreover, hydrogen evolution is inevitable at high applied potential because of the polarization of electrode, and thus gas bubbles form drastically on the surface of cathode. This phenomenon may decrease the effective area of electrocatalyst and accelerate the cathodic degradation.197
Last but not least, solid fundamental theory and optimized standard experimental systems are still absent. It is hard to precisely predict the performance of specific electrocatalysts by unsubstantial theoretical study. Besides, the reaction systems and conditions in the literatures are varied, which is detrimental to the reciprocal evaluation and comparison of different experimental cases.
In view of this, more targeted efforts should be made to improve the fundamental research of CO2 electroreduction. In the aspect of catalysts, it is known that many metals and compounds have been investigated, and new catalysts are burgeoning. Even so, the exploration of high‐performance catalysts is far from sufficient. For example, a whole lot of alloys and intermetallic compounds with distinctive component combinations are still waiting to be inspected. When it referred to carbon materials, the intrinsic catalytic activity, decent conductivity and high‐adjustable surface state make these materials considered as promising catalysts and support materials. What is more, it is very meaningful to design novel composite catalysts composed of different materials to play a cooperative and synergistic effect, which can definitely lead to the further enhancement of overall performance.
Moreover, to expose more desirable active sites, the morphology control, crystalline/defect engineering and surface modification of nanosized catalysts should be studied more thoroughly. Ideally, the structure of catalyst should also fulfill the requirements of high specific surface area, high carrier mobility and good long‐term durability simultaneously.
Expect for the catalyst itself, more attention should be paid on the establishment of more reliable theoretical calculation and essential electrochemical methods that can be used to understand the chemical absorption/desorption steps, the breakage/reconstruction of C=O bonds, the rate determining factor and the competition of reaction pathways. The detailed operating conditions, such as the reactor design, adopted electrolytes, separators, temperature and pressure, should be carefully compared. The experimental results combined with solid instrumental characterizations (aberration‐corrected transmission electron microscopy, electron paramagnetic resonance, scanning tunneling microscopy, synchrotron radiation X‐ray absorption spectroscopy, and so on) can come in handy to explore favorable electrocatalyst structures/components and active centers.
In brief, electrocatalytic reduction of CO2 into carbonic fuels and chemicals is aimed at alleviating energy and environmental problems. To overcome the critical challenges in this field, consecutive achievements like enhanced reduction activity, higher valid product efficiencies and super stability will be attained through proper research advances. Given further fundamental progress in CO2 reduction, the facile and clean recycling of carbon resources for renewable fuels and high‐value chemicals is expected to be realized in the future.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by National Key Research and Development Program of China (2017YFA0208200, 2016YFB0700600), National Key Basic Research Program (2015CB659300), Projects of NSFC (21403105, 21573108), China Postdoctoral Science Foundation (2015M580413), Natural Science Foundation of Jiangsu Province (BK20160647), Fundamental Research Funds for the Central Universities (020514380107), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Biographies
Wenjun Zhang received her B.S. degree in Chemistry from Anhui University, P.R. China (2015). She is now pursing his Ph.D. degree under the supervision of Prof. Zhong Jin at School of Chemistry and Chemical Engineering, Nanjing University. Her main interest is the design and fabrication of nanocatalysts for electrochemical and photoelectrochemical reduction of carbon dioxide.

Zhong Jin is a professor at School of Chemistry and Chemical Engineering, Nanjing University. He received his B.S. (2003) and Ph.D. (2008) in chemistry from Peking University. He worked as a postdoctoral scholar at Rice University (2008–2010) and Massachusetts Institute of Technology (2010–2014). Now he leads a research group at Nanjing University, making efforts on the study of novel functional nanomaterials and devices for efficient energy conversion and storage.

Zhang W. J., Hu Y., Ma L. B., Zhu G. Y., Wang Y. R., Xue X. L., Chen R. P., Yang S. Y., Jin Z., Adv. Sci. 2018, 5, 1700275 https://doi.org/10.1002/advs.398
References
- 1. Shakun J. D., Clark P. U., He F., Marcott S. A., Mix A. C., Liu Z. Y., Otto‐Bliesner B., Schmittner A., Bard E., Nature 2012, 484, 49. [DOI] [PubMed] [Google Scholar]
- 2. Arora V. K., Scinocca J. F., Boer G. J., Christian J. R., Denman K. L., Flato G. M., Kharin V. V., Lee W. G., Merryfield W. J., Geophys. Res. Lett. 2011, 38, 387. [Google Scholar]
- 3. Centi G., Quadrelli E. A., Perathoner S., Energy Environ. Sci. 2013, 6, 1711. [Google Scholar]
- 4. Kondratenko E. V., Mul G., Baltrusaitis J., Larrazábal G. O., Pérez‐Ramírez J., Energy Environ. Sci. 2013, 6, 3112. [Google Scholar]
- 5. Wang W., Wang S. P., Ma X. B., Gong J. L., Chem. Soc. Rev. 2011, 40, 3703. [DOI] [PubMed] [Google Scholar]
- 6. Chen C. Y., Yeh K. L., Aisyah R., Lee D. J., Chang J. S., Bioresour. Technol. 2011, 102, 71. [DOI] [PubMed] [Google Scholar]
- 7. Lam M. K., Lee K. T., Biotechnol. Adv. 2012, 30, 673. [DOI] [PubMed] [Google Scholar]
- 8. Savile C. K., Lalonde J. J., Curr. Opin. Biotechnol. 2011, 22, 818. [DOI] [PubMed] [Google Scholar]
- 9. Alissandratos A., Easton C. J., Beilstein J. Org. Chem. 2015, 11, 2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fiorani G., Guo W. S., Kleij A. W., Green Chem. 2015, 17, 1375. [Google Scholar]
- 11. Quadrelli E. A., Centi G., Duplan J. L., Perathoner S., ChemSusChem 2011, 4, 1194. [DOI] [PubMed] [Google Scholar]
- 12. López‐Periago A. M., Fraile J., López‐Aranguren P., Vega L. F., Domingo C., Chem. Eng. J. 2013, 226, 357. [Google Scholar]
- 13. Tu W. G., Zhou Y., Zou Z. G., Adv. Mater. 2014, 26, 4607. [DOI] [PubMed] [Google Scholar]
- 14. Habisreutinger S. N., Schmidt‐Mende L., Stolarczyk J. K., Angew. Chem., Int. Ed. 2013, 52, 7372. [DOI] [PubMed] [Google Scholar]
- 15. Benson E. E., Kubiak C. P., Sathrum A. J., Smieja J. M., Chem. Soc. Rev. 2009, 38, 89. [DOI] [PubMed] [Google Scholar]
- 16. Costentin C., Robert M., Savéant J. M., Chem. Soc. Rev. 2013, 42, 2423. [DOI] [PubMed] [Google Scholar]
- 17. Saeidi S., Amin N. A. S., Rahimpour M. R., J. CO2 Util. 2014, 5, 66. [Google Scholar]
- 18. Jadhav S. G., Vaisya P. D., Bhanage B. M., Joshi J. B., Chem. Eng. Res. Des. 2014, 92, 2557. [Google Scholar]
- 19. Pakhare D., Spivey J., Chem. Soc. Rev. 2014, 43, 7813. [DOI] [PubMed] [Google Scholar]
- 20. Shi L., Yang G. H., Tao K., Yoneyama Y., Tan Y. S., Tsubaki N., Acc. Chem. Res. 2013, 46, 1838. [DOI] [PubMed] [Google Scholar]
- 21. Jones J. P., Prakash G. K. S., Olah G. A., Isr. J. Chem. 2014, 54, 1451. [Google Scholar]
- 22. Inglis J. L., Maclean B. J., Pryce M. T., Vos J. G., Coord. Chem. Rev. 2012, 256, 2571. [Google Scholar]
- 23. Mao X. W., Hatton T. A., Ind. Eng. Chem. Res. 2015, 54, 4033. [Google Scholar]
- 24. Kauffman D. R., Thakkar J., Siva R., Matranga C., Ohodnicki P. R., Zeng C. J., Jin R. C., ACS Appl. Mater. Interfaces 2015, 7, 15626. [DOI] [PubMed] [Google Scholar]
- 25. Qiao J. L., Liu Y. Y., Hong F., Zhang J. J., Chem. Rev. 2014, 43, 631. [DOI] [PubMed] [Google Scholar]
- 26. Najafabadi A. T., Int. J. Energy Res. 2013, 37, 485. [Google Scholar]
- 27. Innocent B., Pasquier D., Ropital F., Hahn F., Léger J.‐M., Kokoh K. B., Appl. Catal. B: Environ. 2010, 94, 219. [Google Scholar]
- 28. Spinner N. S., Vega J. A., Mustain W. E., Catal. Sci. Technol. 2012, 2, 19. [Google Scholar]
- 29. Kwon Y., Lee J., Electrocatalysis 2010, 1, 108. [Google Scholar]
- 30. Hori Y., Mod. Aspects Electrochem. 2008, 42, 89. [Google Scholar]
- 31. Kortlever R., Shen J., Schouten K. J. P., Calle‐Vallejo F., Koper M. T. M., J. Phys. Chem. Lett. 2015, 6, 4073. [DOI] [PubMed] [Google Scholar]
- 32. Wang W. H., Himeda Y., Muckerman J. T., Manbeck G. F., Chem. Rev. 2015, 115, 12936. [DOI] [PubMed] [Google Scholar]
- 33. Lim R. J., Xie M. S., Sk M. A., Lee J. M., Fisher A., Wang X., Lim K. H., Catal. Today 2014, 233, 169. [Google Scholar]
- 34. Meshitsuka S., Ichikawa M., Tamaru K., J. Chem. Soc. Chem. Commun. 1974, 5, 158. [Google Scholar]
- 35. Schneider J., Jia H. F., Muckerman J. T., Fujita E., Chem. Soc. Rev. 2012, 41, 2036. [DOI] [PubMed] [Google Scholar]
- 36. Kang P., Chen Z. F., Brookhart M., Meyer T. J., Top Catal. 2015, 58, 30. [Google Scholar]
- 37. Faber M. S., Jin S., Energy Environ. Sci. 2014, 7, 3519. [Google Scholar]
- 38. Zeng M., Li Y. G., J. Mater. Chem. A 2015, 3, 14942. [Google Scholar]
- 39. Costentin C., Drouet S., Robert M., Savéant J. M., Science 2012, 338, 90. [DOI] [PubMed] [Google Scholar]
- 40. Shen J., Kortlever R., Kas R., Birdja Y. Y., Morales O. D., Kwon Y. K., Ledezma‐Yanez I., Schouten K. J. P., Mul G., Koper M. T. M., Nat. Commun. 2015, 6, 809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huan T. N., Andreiadis E. S., Heodkamp J., Simon P., Derat E., Cobo S., Royal G., Bergmann A., Strasser P., Dau H., Artero V., Fontecave M., J. Mater. Chem. A 2015, 3, 3901. [Google Scholar]
- 42. Haghighi F. H., Hadadzadeh H., Farrokhpour H., Serri N., Abdi K., Rudbari H. A., Dalton Trans. 2014, 43, 11317. [DOI] [PubMed] [Google Scholar]
- 43. Sampson M. D., Nguyen A. D., Grice K. A., Moore C. E., Rheingold A. L., Kubiak C. P., J. Am. Chem. Soc. 2014, 136, 5460. [DOI] [PubMed] [Google Scholar]
- 44. Ponnurangam S., Yun C. M., Chernyshova I. V., ChemElectroChem 2016, 3, 74. [Google Scholar]
- 45. Franco F., Cometto C., Sordello F., Minero C., Nencini L., Fiedler J., Gobetto R., Nervi C., ChemElectroChem 2015, 2, 1372. [Google Scholar]
- 46. Tory J., Setterfield‐Price B., Dryfe R. A. W., Hartl F., ChemElectroChem 2015, 2, 213. [Google Scholar]
- 47. Liyanage N. P., Dulaney H. A., Huckaba A. J., Jurss J. W., Delcamp J. H., Inorg. Chem. 2016, 55, 6085. [DOI] [PubMed] [Google Scholar]
- 48. Riplinger C., Sampson M. D., Ritzmann A. M., Kubiak C. P., Carter E. A., J. Am. Chem. Soc. 2014, 136, 16285. [DOI] [PubMed] [Google Scholar]
- 49. Smieja J. M., Kubiak C. P., Inorg. Chem. 2010, 49, 9283. [DOI] [PubMed] [Google Scholar]
- 50. Grice K. A., Saucedo C., Inorg. Chem. 2016, 55, 6240. [DOI] [PubMed] [Google Scholar]
- 51. Machan C. W., Sampson M. D., Kubiak C. P., J. Am. Chem. Soc. 2015, 137, 8564. [DOI] [PubMed] [Google Scholar]
- 52. Sampson M. D., Kubiak C. P., J. Am. Chem. Soc. 2016, 138, 1386. [DOI] [PubMed] [Google Scholar]
- 53. Donovan E. S., Barry B. M., Larsen C. A., Wirtz M. N., Geiger W. E., Kemp R. A., Chem. Commun. 2016, 52, 1685. [DOI] [PubMed] [Google Scholar]
- 54. Kang P., Zhang S., Meyer T. J., Brookhart M., Angew. Chem., Int. Ed. 2014, 53, 8709. [DOI] [PubMed] [Google Scholar]
- 55. Sheng M. L., Jiang N., Gustafson S., You B., Ess D. H., Sun Y. J., Dalton Trans. 2015, 44, 16247. [DOI] [PubMed] [Google Scholar]
- 56. Cao Z., Kim D., Hong D. C., Yu Y., Xu J., Lin S., Wen X. D., Nichols E. M., Jeong K., Reimer J. A., Yang P. D., Chang C. J., J. Am. Chem. Soc. 2016, 138, 8120. [DOI] [PubMed] [Google Scholar]
- 57. Zhao C. C., Yin Z. S., Wang J. L., ChemElectroChem 2015, 2, 1974. [Google Scholar]
- 58. Netto C. G. C. M., Toma H. E., Eur. J. Inorg. Chem. 2013, 2013, 5826. [Google Scholar]
- 59. Sende J. A. R., Arana C. R., Hernández L., Potts K. T., Keshevarz‐K M., Abruña H. D., Inorg. Chem. 1995, 24, 3339. [Google Scholar]
- 60. Angamuthu R., Byers P., Lutz M., Spek A. L., Bouwman E., Science 2010, 327, 313. [DOI] [PubMed] [Google Scholar]
- 61. Tang Q., Lee Y., Li D. Y., Choi W., Liu C. W., Lee D., Jiang D. E., J. Am. Chem. Soc. 2017, 139, 9728. [DOI] [PubMed] [Google Scholar]
- 62. Li H., Oloman C., J. Appl. Electrochem. 2006, 36, 1105. [Google Scholar]
- 63. Zhu D. D., Liu J. L., Qiao S. Z., Adv. Mater. 2016, 28, 3423. [DOI] [PubMed] [Google Scholar]
- 64. Montoya J. H., Shi C., Chan K., Nørskov J. K., J. Phys. Chem. Lett. 2015, 6, 2032. [DOI] [PubMed] [Google Scholar]
- 65. Durst J., Siebel A., Simon C., Hasché F., Herranz J., Gasteiger H. A., Energy Environ. Sci. 2014, 7, 2255. [Google Scholar]
- 66. Gong M., Wang D. Y., Chen C. C., Hwang B. J., Dai H. J., Nano Res. 2016, 9, 28. [Google Scholar]
- 67. Greeley J., Nørskov J. K., Mavrikakis M., Annu. Rev. Phys. Chem. 2002, 53, 319. [DOI] [PubMed] [Google Scholar]
- 68. Andrews E. M., Flake J., Fang Y., ECS Trans. 2015, 66, 67. [Google Scholar]
- 69. Back S., Yeom M. S., Jung Y. S., ACS Catal. 2015, 5, 5089. [Google Scholar]
- 70. Castillo A. D., Alvarez‐Guerra M., Solla‐Gullón J., Sáez A., Montiel V., Irabien A., Appl. Energy 2015, 157, 165. [Google Scholar]
- 71. Chen Y. H., Kanan M. W., J. Am. Chem. Soc. 2012, 134, 1986. [DOI] [PubMed] [Google Scholar]
- 72. Zhang S., Kang P., Meyer T. J., J. Am. Chem. Soc. 2014, 136, 1734. [DOI] [PubMed] [Google Scholar]
- 73. Zhang R., Lv W. X., Lei L. X., Appl. Surf. Sci. 2015, 356, 24. [Google Scholar]
- 74. Cui C. N., Han J. Y., Zhu X. L., Liu X., Wang H., Mei D. H., Ge Q. F., J. Catal. 2016, 343, 257. [Google Scholar]
- 75. Won D. H., Choi C. H., Chung J., Chung M. W., Kim E. H., Woo S. I., ChemSusChem 2015, 8, 3092. [DOI] [PubMed] [Google Scholar]
- 76. Wu J. J., Risalvato F. G., Ke F. S., Pellechia P. J., Zhou X. D., J. Electrochem. Soc. 2012, 159, F353. [Google Scholar]
- 77. Zhu Q. G., Ma J., Kang X. C., Sun X. F., Liu H. Z., Hu J. Y., Liu Z. M., Han B. X., Angew. Chem., Int. Ed. 2016, 55, 9012. [DOI] [PubMed] [Google Scholar]
- 78. Kauffman D. R., Alfonso D., Matranga C., Qian H. F., Jin R. C., J. Am. Chem. Soc. 2012, 134, 10237. [DOI] [PubMed] [Google Scholar]
- 79. Zhu W. L., Michalsky R., Metin Ö., Lv H. F., Guo S. J., Wright C. J., Sun X. L., Peterson A. A., Sun S. H., J. Am. Chem. Soc. 2013, 135, 16833. [DOI] [PubMed] [Google Scholar]
- 80. Mistry H., Reske R., Zeng Z. H., Zhao Z. J., Greeley J., Strasser P., Cuenya B. R., J. Am. Chem. Soc. 2014, 136, 16473. [DOI] [PubMed] [Google Scholar]
- 81. Lee H. E., Yang K. D., Yoon S. M., Ahn H. Y., Lee Y. Y., Chang H., Jeong D. H., Lee Y. S., Kim M. Y., Nam K. T., ACS Nano 2015, 9, 8384. [DOI] [PubMed] [Google Scholar]
- 82. Feng X. F., Jiang K. L., Fan S. S., Kanan M. W., J. Am. Chem. Soc. 2015, 137, 4606. [DOI] [PubMed] [Google Scholar]
- 83. Lau G. P. S., Schreier M., Vasilyev D., Scopelliti R., Grätzel M., Dyson P. J., J. Am. Chem. Soc. 2016, 138, 7820. [DOI] [PubMed] [Google Scholar]
- 84. Guo S. X., Macfarlane D. R., Zhang J., ChemSusChem 2016, 9, 80. [DOI] [PubMed] [Google Scholar]
- 85. Zhang L., Wang Z. Y., Mehio N., Jin X. B., Dai S., ChemSusChem 2016, 9, 428. [DOI] [PubMed] [Google Scholar]
- 86. Kim C., Jeon H. S., Eom T. Y., Jee M. S., Kim H. J., Friend C. M., Min B. K., Hwang Y. J., J. Am. Chem. Soc. 2015, 137, 13844. [DOI] [PubMed] [Google Scholar]
- 87. Lu Q., Rosen J., Zhou Y., Hutchings G. S., Kimmel Y. C., Chen J. G. G., Jiao F., Nat. Commun. 2014, 5, 149. [DOI] [PubMed] [Google Scholar]
- 88. Rosen J., Hutchings G. S., Lu Q., Rivera S., Zhou Y., Vlachos D. G., Jiao F., ACS Catal. 2015, 5, 4293. [Google Scholar]
- 89. Hsieh Y. C., Senanayake S. D., Zhang Y., Xu W. Q., Polyansky D. E., ACS Catal. 2015, 5, 5349. [Google Scholar]
- 90. Sun K., Wu L., Qin W., Zhou J. G., Hu Y. F., Jiang Z. H., Shen B. Z., Wang Z. J., J. Mater. Chem. A 2016, 4, 12616. [Google Scholar]
- 91. Zhou L. Q., Ling C., Jones M., Jia H. F., Chem. Commun. 2015, 51, 17704. [DOI] [PubMed] [Google Scholar]
- 92. Hori Y., Wakebe H., Tsukamoto T., Koga O., Electrochim. Acta 1994, 39, 1833. [Google Scholar]
- 93. Min X. Q., Kanan M. W., J. Am. Chem. Soc. 2015, 137, 4701. [DOI] [PubMed] [Google Scholar]
- 94. Gao D. F., Zhou H., Wang J., Miao S., Yang F., Wang G. X., Wang J. G., Bao X. H., J. Am. Chem. Soc. 2015, 137, 4288. [DOI] [PubMed] [Google Scholar]
- 95. Ikeda S., Hattori A., Maeda M., Ito K., Noda H., Electrochemistry 2000, 68, 257. [Google Scholar]
- 96. Ikeda S., Hattori A., Ito K., Noda H., Electrochemistry 1999, 67, 27. [Google Scholar]
- 97. Rosen J., Hutchings G. S., Lu Q., Forest R. V., Moore A., Jiao F., ACS Catal. 2015, 5, 4586. [Google Scholar]
- 98. Quan F. J., Zhong D., Song H. C., Jia F. L., Zhang L. Z., J. Mater. Chem. A 2015, 3, 16409. [Google Scholar]
- 99. Won D. H., Shin H., Koh J., Chung J., Lee H. A., Kim H. J., Woo S. I., Angew. Chem., Int. Ed. 2016, 55, 9297. [DOI] [PubMed] [Google Scholar]
- 100. Zhang Z. Y., Chi M. F., Veith G. M., Zhang P. F., Lutterman D. A., Rosenthal J., Overbury S. H., Dai S., Zhu H. Y., ACS Catal. 2016, 6, 6255. [Google Scholar]
- 101. Kim S., Dong W. J., Gim S., Sohn W., Park J. Y., Yoo C. J., Jang H. W., Lee J. L., Nano Energy 2017, 39, 44. [Google Scholar]
- 102. Lee J. Y., Tak Y. S., Electrochim. Acta 2001, 46, 3015. [Google Scholar]
- 103. Liu S. P., Zhao M., Gao W., Jiang Q., ChemSusChem 2016, 9, 1. [Google Scholar]
- 104. Tang W., Peterson A. A., Varela A. S., Jovanov Z. P., Bech L., Durand W. J., Dahl S., Nørskov J. K., Chorkendorff I., Phys. Chem. Chem. Phys. 2012, 14, 76. [DOI] [PubMed] [Google Scholar]
- 105. Li Y. W., Sun Q., Adv. Energy Mater. 2016, 6, 1600463. [Google Scholar]
- 106. Mistry H., Behafarid F., Reske R., Varela A. S., Strasser P., Cuenya B. R., ACS Catal. 2016, 6, 1075. [Google Scholar]
- 107. Chung J. H., Won D. H., Koh J. K., Kim E. H., Woo S. I., Phys. Chem. Chem. Phys. 2016, 18, 6252. [DOI] [PubMed] [Google Scholar]
- 108. Kas R., Hummadi K. K., Kortlever R., Wit P. D., Milbrat A., Luiten‐Olieman M. W. J., Benes N. E., Koper M. T. M., Mul G., Nat. Commun. 2016, 7, 10748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Raciti D., Livi K. J., Wang C., Nano Lett. 2015, 15, 6829. [DOI] [PubMed] [Google Scholar]
- 110. Manthiram K., Beberwyck B. J., Alivisatos A. P., J. Am. Chem. Soc. 2014, 136, 13319. [DOI] [PubMed] [Google Scholar]
- 111. Ren D., Wong N. T., Handoko A. D., Huang Y., Yeo B. S., J. Phys. Chem. Lett. 2016, 7, 20. [DOI] [PubMed] [Google Scholar]
- 112. Roberts F. S., Kuhl K. P., Nilsson A., Angew. Chem., Int. Ed. 2015, 54, 5179. [DOI] [PubMed] [Google Scholar]
- 113. Ma M., Djanashvili K., Smith W. A., Angew. Chem., Int. Ed. 2016, 55, 6680. [DOI] [PubMed] [Google Scholar]
- 114. Mistry H., Varela A. S., Bonifacio C. S., Zegkinoglou L., Sinev I., Choi Y. W., Kisslinger K., Stach E. A., Yang J. C., Strasser P., Cuenya B. R., Nat. Commun. 2016, 7, 12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Dutta A., Rahaman M., Luedi N. C., Mohos M., Broekmann P., ACS Catal. 2016, 6, 3804. [Google Scholar]
- 116. Xie M. S., Xia B. Y., Li Y. W., Yan Y., Yang Y. H., Sun Q., Chan S. H., Fisher A., Wang X., Energy Environ. Sci. 2016, 9, 1687. [Google Scholar]
- 117. Durand W. J., Peterson A. A., Studt F., Abild‐Pedersen F., Nørskov J. K., Surf. Sci. 2011, 605, 1354. [Google Scholar]
- 118. Calle‐Vallejo F., Koper M. T. M., Angew. Chem., Int. Ed. 2013, 52, 7282. [DOI] [PubMed] [Google Scholar]
- 119. Loiudice A., Lobaccaro P., Kamali E. A., Thao T., Huang B. H., Ager J. W., Buonsanti R., Angew. Chem., Int. Ed. 2016, 55, 5789. [DOI] [PubMed] [Google Scholar]
- 120. Kortlever R., Peters I., Koper S., Koper M. T. M., ACS Catal. 2015, 5, 3916. [Google Scholar]
- 121. Kim D., Resasco J., Yu Y., Asiri A. M., Yang P. D., Nat. Commun. 2014, 5, 4948. [DOI] [PubMed] [Google Scholar]
- 122. Rasul S., Anjum D. H., Jedidi A., Minenkov Y., Cavallo L., Takanabe K., Angew. Chem., Int. Ed. 2015, 54, 2146. [DOI] [PubMed] [Google Scholar]
- 123. Sarfraz S., Garcia‐Esparza A. T., Jedidi A., Cavallo L., Takanabe K., ACS Catal. 2016, 6, 2842. [Google Scholar]
- 124. Torelli D. A., Francis S. A., Crompton J. C., Javier A., Thompson J. R., Brunschwig B. S., Soriaga M. P., Lewis N. S., ACS Catal. 2016, 6, 2100. [Google Scholar]
- 125. Back S., Kim H., Jung Y., ACS Catal. 2015, 5, 965. [Google Scholar]
- 126. Sun X. F., Zhu Q. G., Kang X. C., Liu H. Z., Qian Q. L., Zhang Z. F., Han B. X., Angew. Chem., Int. Ed. 2016, 55, 6771. [DOI] [PubMed] [Google Scholar]
- 127. Karamad M., Hansen H. A., Rossmeisl J., Nørskov J. K., ACS Catal. 2015, 5, 4075. [Google Scholar]
- 128. Yadav V. S. K., Purkait M. K., RSC Adv. 2015, 5, 40414. [Google Scholar]
- 129. Zhao C. C., Wang J. L., Goodenough J. B., Electrochem. Commun. 2016, 65, 9. [Google Scholar]
- 130. Gao S., Jiao X. C., Sun Z. T., Zhang W. H., Sun Y. F., Wang C. M., Hu Q. T., Zu X. L., Yang F., Yang S. Y., Liang L., Wu J., Xie Y., Angew. Chem., Int. Ed. 2016, 55, 698. [DOI] [PubMed] [Google Scholar]
- 131. Gao S., Lin Y., Jiao X. C., Sun Y. F., Luo Q. Q., Zhang W. H., Li D. Q., Yang J. L., Xie Y., Nature 2016, 529, 68. [DOI] [PubMed] [Google Scholar]
- 132. Bashir S. M., Hossain S. S., Rahman S. U., Ahmed S., Hossain M. M., Electrocatalysis 2015, 6, 544. [Google Scholar]
- 133. Ramesha G. K., Brennecke J. F., Kamat P. V., ACS Catal. 2014, 4, 3249. [Google Scholar]
- 134. Yuhas B. D., Prasittichai C., Hupp J. T., Kanatzidis M. G., J. Am. Chem. Soc. 2011, 133, 15854. [DOI] [PubMed] [Google Scholar]
- 135. Chan K., Tsai C., Hansen H. A., Nørskov J. K., ChemCatChem 2014, 6, 189. [Google Scholar]
- 136. Li F. W., Zhao S. F., Chen L., Khan A., Macfarlane D. R., Zhang J., Energy Environ. Sci. 2016, 9, 216. [Google Scholar]
- 137. Hong X., Chan K., Tsai C., Nørskov J. K., ACS Catal. 2016, 6, 4428. [Google Scholar]
- 138. Peng H., Lu J., Wu C. X., Yang Z. X., Chen H., Song W. J., Li P. Q., Yin H. Z., Appl. Surf. Sci. 2015, 353, 1003. [Google Scholar]
- 139. Asadi M., Kim K., Liu C., Addepalli A. V., Abbasi P., Yasaei P., Phillips P., Behranginia A., Cerrato J. M., Haasch R., Zapol P., Kumar B., Klie R. F., Abiade J., Curtiss L. A., Salehi‐Khojin A., Science 2016, 353, 467. [DOI] [PubMed] [Google Scholar]
- 140. Wannakao S., Artrith N., Limtrakul J., Kolpak A. M., ChemSusChem 2015, 8, 2745. [DOI] [PubMed] [Google Scholar]
- 141. Kim S. K., Zhang Y. J., Bergstrom H., Michalsky R., Peterson A., ACS Catal. 2016, 6, 2003. [Google Scholar]
- 142. Wu J. J., Yadav R. M., Liu M. J., Sharma P. P., Tiwary C. S., Ma L. L., Zou X. L., Zhou X. D., Yakobson B. I., Lou J., Ajayan P. M., ACS Nano 2015, 9, 5364. [DOI] [PubMed] [Google Scholar]
- 143. Jiao Y., Zheng Y., Smith S. C., Du A. J., Zhu Z. H., ChemSusChem 2014, 7, 435. [DOI] [PubMed] [Google Scholar]
- 144. Sharma P. P., Wu J. J., Yadav R. M., Liu M. J., Wright C. J., Tiwary C. S., Yakobson B. I., Lou J., Ajayan P. M., Zhou X. D., Angew. Chem., Int. Ed. 2015, 54, 13701. [DOI] [PubMed] [Google Scholar]
- 145. Xu J. Y., Kan Y. H., Huang R., Zhang B. S., Wang B. L., Wu K. H., Lin Y. M., Sun X. Y., Li Q. F., Centi G., Su D. S., ChemSusChem 2016, 9, 1085. [DOI] [PubMed] [Google Scholar]
- 146. Zhang S., Kang P., Ubnoske S., Brennaman M. K., Song N., House R. L., Glass J. T., Meyer T. J., J. Am. Chem. Soc. 2014, 136, 7845. [DOI] [PubMed] [Google Scholar]
- 147. Liu Y. J., Zhao J. X., Cai Q. H., Phys. Chem. Chem. Phys. 2016, 18, 5491. [DOI] [PubMed] [Google Scholar]
- 148. Wang H. X., Chen Y. B., Hou X. L., Ma C. Y., Tan T. W., Green Chem. 2016, 18, 3250. [Google Scholar]
- 149. Sun X. F., Kang X. C., Zhu Q. G., Ma J., Yang G. Y., Liu Z. M., Han B. X., Chem. Sci. 2016, 7, 2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Wu J. J., Liu M. J., Sharma P. P., Yadav R. M., Ma L. L., Yang Y. C., Zou X. L., Zhou X. D., Vajtai R., Yakobson B. I., Lou J., Nano Lett. 2016, 16, 466. [DOI] [PubMed] [Google Scholar]
- 151. Sreekanth N., Nazrulla M. A., Vineesh T. V., Sailaja K., Phani K. L., Chem. Commun. 2015, 51, 16061. [DOI] [PubMed] [Google Scholar]
- 152. Zhu Q. G., Ma J., Kang X. C., Sun X. F., Hu J. Y., Yang G. Y., Han B. X., Sci. China Chem. 2016, 59, 551. [Google Scholar]
- 153. Kumar B., Asadi M., Pisasale D., Sinha‐Ray S., Rosen B. A., Haasch R., Abiade J., Yarin A. L., Salehi‐Khojin A., Nat. Commun. 2013, 4, 94. [Google Scholar]
- 154. Varela A. S., Sahraie N. R., Steinberg J., Ju W., Oh H. S., Strasser P., Angew. Chem., Int. Ed. 2015, 54, 10758. [DOI] [PubMed] [Google Scholar]
- 155. Liu Y. M., Chen S., Quan X., Yu H. T., J. Am. Chem. Soc. 2015, 137, 11631. [DOI] [PubMed] [Google Scholar]
- 156. Li W. L., Seredych M., Rodríguez‐Castellón E., Bandosz T. J., ChemSusChem 2016, 9, 606. [DOI] [PubMed] [Google Scholar]
- 157. Nakata K., Ozaki T., Terashima C., Fujishima A., Einaga Y., Angew. Chem., Int. Ed. 2014, 53, 871. [DOI] [PubMed] [Google Scholar]
- 158. Yang N. J., Gao F., Nebel C. E., Anal. Chem. 2013, 85, 5764. [DOI] [PubMed] [Google Scholar]
- 159. Royer M. E., C. R. Hebd. Seances Acad. Sci. 1870, 70, 731. [Google Scholar]
- 160. Agarwal A. S., Zhai Y. M., Hill D., Sridhar N., ChemSusChem 2011, 4, 1301. [DOI] [PubMed] [Google Scholar]
- 161. Zhang H., Ma Y., Quan F. J., Huang J. J., Jia F. L., Zhang L. Z., Electrochem. Commun. 2014, 46, 63. [Google Scholar]
- 162. Sen S., Liu D., Palmore G. T. R., ACS Catal. 2014, 4, 3091. [Google Scholar]
- 163. Kumar B., Atla V., Brian J. P., Kumari S., Nguyen T. Q., Sunkara M., Spurgeon J. M., Angew. Chem., Int. Ed. 2017, 56, 3645. [DOI] [PubMed] [Google Scholar]
- 164. Li F. W., Chen L., Knowles G. P., MacFarlane D. R., Zhang J., Angew. Chem., Int. Ed. 2017, 129, 520. [Google Scholar]
- 165. Luc W., Collins C., Wang S. W., Xin H. L., He K., Kang Y. J., Jiao F., J. Am. Chem. Soc. 2017, 139, 1885. [DOI] [PubMed] [Google Scholar]
- 166. Huang H. W., Jia H. H., Liu Z., Gao P. F., Zhao J. T., Luo Z. L., Yang J. L., Zeng J., Angew. Chem., Int. Ed. 2017, 129, 3648. [Google Scholar]
- 167. Kim D., Xie C. L., Becknell N., Yu Y., Karamad M., Chan K., Crumlin E. J., Nørskov J. K., Yang P. D., J. Am. Chem. Soc. 2017. [DOI] [PubMed] [Google Scholar]
- 168. Xu J. Q., Li X. D., Sun Y. F., Ju Z. Y., Yao T., Wang C. M., Ju H. X., Zhu J. F., Wei S. Q., Xie Y., Angew. Chem., Int. Ed. 2017. [Google Scholar]
- 169. Maurin A., Robert M., J. Am. Chem. Soc. 2016, 138, 2492. [DOI] [PubMed] [Google Scholar]
- 170. Lum Y. W., Kwon Y., Lobaccaro P., Chen L., Chark E. L., Bell A. T., Ager J. W., ACS Catal. 2016, 6, 202. [Google Scholar]
- 171. Varela A. S., Ju W., Reier T., Strasser P., ACS Catal. 2016, 6, 2136. [Google Scholar]
- 172. Chen Z. Z., Zhang X., Lu G., Chem. Sci. 2015, 6, 6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Koo Y. M., Malik R., Alvarez N., White l., Shanov V. N., Schulz M., Collins B., Sankar J., Yun Y. H., RSC Adv. 2014, 4, 16362. [Google Scholar]
- 174. Reske R., Mistry H., Behafarid F., Cuenya B. R., Strasser P., J. Am. Chem. Soc. 2014, 136, 6978. [DOI] [PubMed] [Google Scholar]
- 175. Baturina O. A., Lu Q., Padilla M. A., Xin L., Li W. Z., Serov A., Artyushkova K., Atanassov P., Xu F., Epshteyn A., Brintlinger T., Schuette M., Collins G. E., ACS Catal. 2014, 4, 3682. [Google Scholar]
- 176. Weng Z., Jiang J. B., Wu Y. S., Wu Z. S., Guo X. T., Materna K. L., Liu W., Batista V. S., Brudvig G. W., Wang H. L., J. Am. Chem. Soc. 2016, 138, 8076. [DOI] [PubMed] [Google Scholar]
- 177. Zhang S., Kang P., Bakir M., Lapides A. M., Dares C. J., Meyer T. J., Proc. Natl. Acad. Sci. USA 2015, 112, 4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Ren D., Deng Y. L., Handoko A. D., Chen C. S., Malkhandi S., Yeo B. S., ACS Catal. 2015, 5, 2814. [Google Scholar]
- 179. Handoko A. D., Ong C. W., Huang Y., Lee Z. G., Lin L. L., Panetti G. B., Yeo B. S., J. Phys. Chem. C 2016, 120, 20058. [Google Scholar]
- 180. Schlager S., Dumitru L. M., Haberbauer M., Fuchabauer A., Neugebauer H., Hiemetsberger D., Wagner A., Portenkirchner E., Sariciftci N. S., ChemSusChem 2016, 9, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Ertem M. A., Konezny S. J., Araujo C. M., Batista V. s., J. Phys. Chem. Lett. 2013, 4, 745. [DOI] [PubMed] [Google Scholar]
- 182. Khan M. U., Wang L. B., Liu Z., Gao Z. H., Wang S. P., Li H. L., Zhang W. B., Wang M. L., Wang Z. F., Ma C., Zeng J., Angew. Chem., Int. Ed. 2016, 55, 9548. [DOI] [PubMed] [Google Scholar]
- 183. Chai G. L., Guo Z. X., Chem. Sci. 2016, 7, 1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Song Y., Peng R., Hensley D. K., Bonnesen P. V., Liang L. B., Wu Z. L., Meyer H. M. III, Chi M. F., Ma C., Sumpter B. G., Rondinone A. J., Chem. Select. 2016, 1, 6055. [Google Scholar]
- 185. Yadav V. S. K., Purkait M. K., Energy Fuels 2015, 29, 6670. [Google Scholar]
- 186. Chi D. H., Yang H. P., Du Y. F., Lv T., Sui G. J., Wang H., Lu J. X., RSC Adv. 2014, 4, 37329. [Google Scholar]
- 187. Gutiérrez‐Guerra N., Moreno‐lópez L., Serrano‐Ruiz J. C., Valverde J. L., Lucas‐Consuegra A. D., Appl. Catal., B 2016, 188, 272. [Google Scholar]
- 188. Malik M. I., Malaibari Z. O., Atieh M., Abussaud B., Chem. Eng. Sci. 2016, 152, 468. [Google Scholar]
- 189. Le M., Ren M., Zhang Z., Sprunger P. T., Kurtz R. L., Flake J. C., J. Electrochem. Soc. 2011, 158, E45. [Google Scholar]
- 190. Ullah N., Ali I., Jansen M., Omanovic S., Can. J. Chem. Eng. 2015, 93, 55. [Google Scholar]
- 191. Li P. Q., Xu J. F., Jing H., Wu C. X., Peng H., Lu J., Yin H. Z., Appl. Catal., B 2014, 156, 134. [Google Scholar]
- 192. Li P. Q., Sui X. N., Xu J. F., Jing H., Wu C. X., Peng H., Lu J., Yin H. Z., Chem. Eng. J. 2014, 247, 25. [Google Scholar]
- 193. Hursán D., Kormányos A., Rajeshwar K., Janáky C., Chem. Commun. 2016, 52, 8858. [DOI] [PubMed] [Google Scholar]
- 194. Yang Z. X., Xu J. F., Wu C. X., Jing H., Li P. Q., Yin H. Z., Appl. Catal., B 2014, 156, 249. [Google Scholar]
- 195. Boston D. J., Pachón Y. M. F., Lezna R. O., Tacconi N. R. D., MacDonnell F. M., Inorg. Chem. 2014, 53, 6544. [DOI] [PubMed] [Google Scholar]
- 196. Ma K., Yehezkeli O., Park E., Cha J. N., ACS Catal. 2016, 6, 6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Chiacchiarelli L. M., Zhai Y., Frankel G. S., Agarwal A. S., Sridhar N., J. Appl. Electrochem. 2012, 42, 21. [Google Scholar]
- 198. Zhu W. L., Zhang Y. J., Zhang H. Y., Lv H. F., Li Q., Michalsky R., Peterson A. A., Sun S. H., J. Am. Chem. Soc. 2014, 136, 16132. [DOI] [PubMed] [Google Scholar]
- 199. Chen Y. H., Li C. W., Kanan M. W., J. Am. Chem. Soc. 2012, 134, 19969. [DOI] [PubMed] [Google Scholar]
- 200. Ma M., Trześniewski B. J., Xie J., Smith W. A., Angew. Chem., Int. Ed. 2016, 55, 9748. [DOI] [PubMed] [Google Scholar]
- 201. Lin S., Diercks C. S., Zhang Y. B., Koenienko N., Nichols E. M., Zhao Y. B., Paris A. R., Kim D. H., Yang P. D., Yaghi O. M., Chang C. J., Science 2015, 349, 6253. [DOI] [PubMed] [Google Scholar]
- 202. Li Q., Zhu W. L., Fu J. J., Zhang H. Y., Wu G., Sun S. H., Nano Energy 2016, 24, 1. [Google Scholar]
- 203. Kortlever R., Peters I., Balemans C., Kas R., Kwon Y., Mul G., Koper M. T. M., Chem. Commun. 2016, 52, 10229. [DOI] [PubMed] [Google Scholar]
- 204. Yang H. P., Qin S., Yue Y. N., Liu L., Wang H., Lu J. X., Catal. Sci. Technol. 2016, 6, 6490. [Google Scholar]